
DNA Damage Response-Associated Cell Cycle Re-Entry and Neuronal Senescence in Brain Aging and Alzheimer’s Disease
Abstract
Chronological aging is by far the strongest risk factor for age-related dementia and Alzheimer’s disease. Senescent cells accumulated in the aging and Alzheimer’s disease brains are now recognized as the keys to describing such an association. Cellular senescence is a classic phenomenon characterized by stable cell arrest, which is thought to be applicable only to dividing cells. Emerging evidence indicates that fully differentiated post-mitotic neurons are also capable of becoming senescent, with roles in contributing to both brain aging and disease pathogenesis. The key question that arises is the identity of the upstream triggers and the molecular mechanisms that underly such changes. Here, we highlight the potential role of persistent DNA damage response as the major driver of senescent phenotypes and discuss the current evidence and molecular mechanisms that connect DNA repair infidelity, cell cycle re-entry and terminal fate decision in committing neuronal cell senescence.
INTRODUCTION
Over a hundred years ago, the first seminal report of the patient Auguste D. as described by Doctor Alois Alzheimer has once suggested that Alzheimer’s disease (AD) is a rare cause of presenile dementia. It was not until almost 60 years later, when the classic pathological features of neurofibrillary tangles and senile plaques that were previously described by Dr. Alzheimer were found as well in many aged patients who developed dementia late in life, that the concept of the disease and its prevalence had undergone dramatic shifts [1]. It is estimated that the global number of people with dementia will triple in five years’ time, from the 57.4 million cases reported in 2019 to almost 153 million cases predicted in 2025 [2]. AD is the most common form of dementia, characterized by early and prominent deficits in episodic memory, with varying degrees of executive, language, and visuospatial impairment in clinical presentations, along with the pathological depositions of senile plaques and neurofibrillary tangles in the brain [3]. Molecular-based analyses performed since the early 1980s have subsequently identified that the tangles described by Dr. Alzheimer are indeed paired-helical insoluble filaments of hyperphosphorylated tau proteins inside neurons [4, 5]; whereas the plaques are mainly composed of extracellular deposits of misfolded amyloid-β (Aβ) proteins [6, 7]. Since then, for the past two decades, the amyloid cascade [8, 9] and tau hypotheses [10–12] have become the mainstream explanations for the pathogenesis of this disease. However, unsuccessful attempts at testing anti-Aβ-targeting drugs for treating AD over a decade or so [13, 14] and the recently halted clinical trial aimed at antagonizing tau in a similar fashion [15] indicated that the disease is far more complicated than we could have imagined [16]. In the wake of these failures from the clinical trials, different voices are now urging to reassess the roles of both amyloid and tau in the aspects of disease causality and to re-consider other important factors that may contribute to the onset and progression of this multifactorial disease [16–18]. One recent emerging aspect is the recognition of pathological cell aging as a new etiological framework for these age-related neurodegenerative disorders.
Chronological aging is by far the strongest risk factor for age-related cognitive decline and dementia, and the prevalence of dementia increases with age [19]. However, significant variation in the rate of aging is found across individuals. Subjective age, on the other hand, is the psychological feeling of age relative to their chronological age, and this may capture such individual differences as well as modulate the risk of dementia [20, 21]. At the tissue level, physiological aging can be considered as a progressive loss of physiological integrity, leading to an impaired body homeostasis and therefore increased vulnerability to death [22]. Accumulation of senescent cells is a major contributor to tissue aging. Within the brain, these cells are well-documented and their presence is associated with a variety of age-related diseases such as Parkinson’s disease (PD) and AD [23]. Their emergences are known to be induced by various cellular stimuli, including heightened oxidative stress [24, 25], replicative exhaustion [26, 27], protein aggregation [28, 29], loss of proper proteostasis and autophagy functions [30, 31]. While many of these upstream stressors are seemingly unrelated; at some points downstream, they converge by inducing DNA damage, which can exacerbate the upstream stressors in return, forming a vicious cycle [24, 27, 28, 31].
The phenomenon of cellular senescence is classically described as a process leading to permanent cell cycle arrest. At the beginning, it intends to only describe the changes occurring in dividing cells, but the potential of neurons and other fully differentiated cell types in the brain to senesce has been neglected due to their post-mitotic status. Growing evidence now indicates that these cells are indeed capable of senescence. It was originally thought that cell senescence was simply a cell-autonomous arrest program without any influences on its surroundings [32]. Now it has become clear that these cells can acquire a so-called senescence-associated secretory phenotype (SASP) to induce sterile inflammation, thereby imposing unexpected threats to their neighboring cells [29, 33]. Previous characterization of senescent neurons consistently revealed markers of sustained DNA damage response (DDR) and evidence of cell cycle re-activation [30, 31, 34, 35], suggesting these events are potential upstream drivers of such changes and responses. This review will therefore center on the likelihood of sustained DDR as the driver of cellular senescence in mature post-mitotic neurons.
In the discussion, we will start with an overview of general evidence on how senescent cells are the building blocks of tissue aging, followed by a specific analysis of evidence supporting such a phenomenon among different brain cells in aging and AD brains. Before going into the key discussion, we will provide evidence that persistent DDR is the unifying trigger of cell senescence in general. Afterwards, we will discuss why neurons are particularly susceptible to persistent DDR, which is associated with cell cycle re-activation. Based on these arguments, we will explain how cellular senescence could be achieved in post-mitotic neurons.
ACCUMULATION OF SENESCENT CELLS IS A MAJOR CONTRIBUTOR TO TISSUE AGING AND PATHOGENESIS
Senescent cells differ from normal cells in many aspects. By definition, these are cells that have committed to a permanent state of cell cycle arrest with altered gene expression profiles that facilitate their acquisition of various morphological and functional changes. Many of these cells, even from different tissue origins, exhibit common features, such as a flattened and enlarged morphology, the formation of senescence-associated heterochromatin foci, lipofuscin granules, and the ability to release an array of soluble factors by the acquisition of SASP [36].
Since cells are considered as the fundamental building units of tissues and organs, it is expected that aged cells, known as senescent cells, will at least in part contribute exclusively to the normal aging process. However, early investigation into the roles of senescent cells in the context of tissue homeostasis and functioning also led to the discovery that they indeed have important roles in many physiological aspects of life [37]. For instance, senescent cells are formed as a part of the normal developmental mechanism in embryonic growth and patterning [38]. As part of the body defense against any malignant transformation of pre-neoplastic cells, cellular senescence is also a safeguarding mechanism that halts the proliferation of these cells [39]. During tissue repairing, on the other hand, cell senescence also plays a positive role as a morphogenetic force to promote wound healing and repair [40]. In addition to these “not-so-frequent” events, cellular senescence is also a part of the body’s daily regulation of insulin homeostasis as it promotes insulin secretion by the pancreatic beta cells and regulates the maturation of pancreatic tissue [41]. These findings together sparked years of debates on whether this cellular phenomenon truly represents aging, or even contributes to age-related disease pathogenesis. However, mounting evidence does indicate that heightened accumulation of senescent cells is indeed associated with a wide variety of age-related pathological conditions, such as cancer [42], cardiovascular disease [43], diabetes [44], osteoporosis [45], and dementia including AD [35, 46]. In order to explain this contradictory discrepancy, it is suggested that the duration of senescent cells’ persistence is a major determining factor of their nature in shaping tissue homeostasis [47–49]. Senescent cell populations, as a part of those physiological processes, only exist transiently in the respective tissues and are actively cleared out by the immune system upon fulfilling their purposes [48–53]. In contrast, those thought to be associated with tissue aging and disease pathogenesis are the ones that persist, leading to the escalation of their populations over time [54]. It is proposed that a malfunctioning and aged immune system which fails to promptly remove these cells may contribute to that [55, 56], but others also suggested that senescent cells emerged from persistent DDR may become resistant to apoptosis [57]. The latter appears to be caused by an upregulation of the anti-apoptotic Bcl-2 family of genes, which blocks the PUMA/NOXA regulated apoptotic pathways [58–61]. Moreover, sustained activation of cell cycle inhibitors like p21 is also known to inhibit p53-mediated apoptosis [62]. Another property of these cells that contributes to tissue aging is the acquisition of SASP, which seems to be a downstream feature of sustained DDR [63, 64]. SASP is characterized by the hypersecretion of various signaling factors, such as an array of pro-inflammatory cytokines like IL-6, IL-8, and TNF-α chemokines and extracellular matrix proteases [65]. This contributes to the low-grade sterile inflammation and modulations of the tissue architecture that are frequently found in age-related conditions even in the absence of exogenous pathogens.
EVIDENCE OF SENESCENT CELL ACCUMULATION IN THE AGING AND AD BRAIN
Similar to other organ systems, multiple functional domains of the brain deteriorate progressively during aging, which manifests as declines in learning and memory, attention, speed of decision making, sensory perception, and motor coordination [66, 67]. The aging brain indeed reveals similar hallmarks of aging that are also evident in other tissues [68]. These include mitochondrial dysfunction, accumulation of oxidatively damaged macromolecules, dysregulated energy metabolism, impaired autophagy, proteasome functions and stress response signaling, compromised DNA repair, and chronic low-grade inflammation [68]. Many of these are indeed common to the molecular signatures of senescent cells. Almost universally, senescent cells are characterized by metabolic changes, mitochondrial and lysosomal impairment and accumulation, persistent DDR, and increased secretion of trophic factors and cytokines [69]. As chronological aging is the major risk factor for neurodegenerative diseases [70], it is therefore reasonable to argue that the accumulation of senescent cells plays major roles in contributing to age-related pathological changes in the organ. The adult brain is made up of multiple brain cell types, mainly neurons, oligodendrocytes, astrocytes, microglia, and endothelial cells. Cellular senescence among these cells is found in both aged brains [31, 71, 72] and in relevant brain regions of age-related diseases like AD [73, 74], PD [75, 76], Huntington’s disease [76, 77], and amyotrophic lateral sclerosis [78, 79], exerting negative effects on the brain homeostasis and functioning [80].
Focusing on AD, senescent astrocytes are one of the well-characterized cellular species. A number of in vitro and in vivo evidence revealed that human and mouse astrocytes could become senescent upon the exposure to multiple types of stress, including Aβ1–42. In brain tissues harvested from aged individuals and patients with AD, a significant increase in the number of senescent astrocytes is found, and they could be identified by the classic senescence-associated β-galactosidase (SA-β-gal) staining and p16INK4A nuclear signals. These cells also exhibit the classic markers of SASP, including the upregulation of p38MAPK and the release of a number of cytokines [81, 82]. Evidence of senescent microglia is also recently reported in human AD brains. While sustained proliferation of these cells is a hallmark of AD, it is suggested that this change ultimately promotes replicative senescence. Senescent microglia are characterized by positive SA-β-gal staining and a transcriptomic signature resembling that of disease-associated microglia, with a loss in their ability to prevent the progression of amyloid-related pathologies [26]. Apart from glia, cellular senescence is also recently reported in the endothelial cells of intact microvessels located in the dorsolateral prefrontal cortex of AD brains, which are associated with vascular dysfunction and tau pathologies [83].
It was once thought that the phenomenon of cellular senescence is irrelevant to post-mitotic cells in the brain, as the fundamental criterion that involves the transition from a cell cycling to a permanently cell cycle arrested state does not readily apply [84]. However, pioneering work led by the von Zglinicki laboratory first used the term “senescence” to describe the related changes in neurons and pointed out their accumulation in the aging brain [84]. In aged mice of 22–24 months of age, around 20–40% of all terminally differentiated neurons located in the cortex, the hippocampus and the gut are senescent. Among the Purkinje cell population of the cerebellum, the percentage is even higher, reaching around 40–80%. These cells are not only characterized by the classic SA-β-gal staining, but also by markers indicating a secretory response involving the activation of proinflammatory p38MAPK and heightened expression of IL-6 [34]. In human AD brains, distinct heterochromatic structures designated as senescence-associated heterochromatic foci are reported in the granule cells of the hippocampus [85]. A recent single-nucleus transcriptome profiling study also revealed that among the 2% senescent cells identified from around 140,000 nuclei derived from 76 postmortem human brains with various levels of AD pathology, more than 97% of them are excitatory neurons with overlapping neurofibrillary tangle tau pathology [86]. Although the findings of earlier studies did not explicitly use the term “senescent neurons” in their descriptions, they indeed matched the modern hallmarks and definition of cellular senescence. For instance, abnormal expressions of checkpoint regulators like the INK4 family of proteins and p21 have been reported in tangle-bearing neurons and neuritic components of plaques [87]. Others reported that in pyramidal neurons located inside the hippocampus, so as neurons being positive for neurofibrillary tangles and granulovacuolar degeneration, are aberrantly expressing cell cycle regulator p16 [88]. In another study which aimed at examining the regional, cellular, and subcellular localization of active and phosphorylated p38MAPK (pp38) in the brain also reported pp38 immunoreactivity in hippocampal neurons bearing early neurofibrillary pathologies [89]. Together, these independent findings from different laboratories confirmed the possibility of cellular senescence responses in post-mitotic neurons. Many of these cells tend to be associated with tau pathologies, despite the molecular mechanisms underlying this transformation remain ill-defined.
PERSISTENT DNA DAMAGE RESPONSE IS THE UNIFYING TRIGGER OF CELL SENESCENCE
Length of the telomere is not the determining factor
In order to better understand the molecular changes underlying cellular senescence, it is essential to understand its history. Cellular senescence was first described in the 1960s by Leonard Hayflick and Paul Moorhead as the irreversible proliferative arrest of fibroblasts following prolonged culturing in dishes [90]. This phenomenon is later known as replicative senescence and has become a major focus in the field of biogerontology [90, 91]. Since then, replicative senescence has also been reported in vivo across different mammalian species, ranging from the proliferating cells of embryonic tissues [38, 92] to many of those in adults, including endothelial cells [93], lymphocytes [94], and vascular smooth muscle cells [95]. The term “Hayflick limit” is therefore coined, referring to the finite replicative capacity of any diploid dividing cells [96], which also varies considerably among different cell types [97]. At the molecular level, it is generally believed that replicative senescence resulted from successive telomere shortening events that occur during each round of cell division. This model was built upon the findings from in vitro experiments with fibroblasts initially [98], as well as both in vitro and in vivo studies in other proliferative cell types [99–102]. Despite this, the direct relationship between telomere shortening and organismal aging remains inconclusive. Some studies indicated that only weak linkages between telomere lengths and the age of sample donors are present [103–105]; whereas many others failed to find any meaningful correlations at all [106–110]. While it seems logical to assume that organisms with a longer lifespan should correspondingly possess longer telomeres, a comparative study involving sixty mammalian species however revealed that many short-lived species tended to have longer telomeres instead [111]. These findings are indeed important, as they suggest that if senescent cells contribute to tissue and organismal aging, they should be formed independently of changes in telomere length— the side effect of cell cycling. In other words, a pre-existing cycling status is not necessary to induce cellular senescence. Therefore, this implied that terminally differentiated cells, even if by default they do not actively engage in the cell cycle, could still become senescent by other means. Their existence has already been reported in many different organs, including the adult brain [84].
Difficult-to-repair DNA damage is the underlying trigger
We now understand that the integrity of telomere sequences rather than their length is more likely the major driver of cell senescence, but how is it related to senescence in non-dividing cells? Telomeres located at the chromosome ends are organized into lariat-like structures known as t-loops, which are stabilized by a specialized 6-subunit protein complex known as shelterin [112, 113]. During each round of cell division, telomere shortening results in a loss of shelterin proteins [114], resulting in the destabilization of t-loop conformation and the exposure of telomere ends— a form of DNA lesion [113, 115]. Consistent with that, deletion of shelterin components also leads to DDR at the telomeric regions [115], which activates the p53 transcription factor [116] that governs the fate of cells. Depending on the duration and severity of the stress, cells can either undergo repair and survival, cell death, or senescence [117]. DDR in mammalian cells is facilitated by multiple lesion sensing and repair pathways [118]. By virtue, they have evolved into an interactive network [119, 120] so that even when the primary repair pathways against certain types of lesions become unavailable, secondary mechanisms are always standing by to avoid leaving damage unattended at potentially important regions within the genome, which could lead to unpredictable consequences. However, sometimes the backup repair process may not function as effectively as they are supposed to and may even introduce additional errors to the old lesion [121, 122]. The skewed tendency towards a senescent fate as a dominant outcome of telomere lesions is indeed related to the relatively limited DNA repair capacity at telomeres as compared to the rest of the genome [123–125]. This outcome is caused by the actions of the telomeric repeat-binding factor (TRF) proteins in the shelterin complex, which impede non-homologous end joining repair (NHEJ) as a way to prevent the risk of telomeric end-to-end fusions [125, 126]. Because of this, DNA damage at telomeric regions is often managed by another pathway of choice known as homologous recombination (HR) [127, 128], which may sometimes introduce additional deletions and irreversible damage to the site of repair, leading to an unresolved DDR that stabilizes the senescence response [127, 129]. Conversely, blocking the onset of HR, but not NHEJ, prevented cellular senescence despite the fact that multiple dysfunctional telomeres could be left unattended [129].
The above findings related to telomere biology indeed have important implications. They highlight that any circumstances that lead to unresolved DDR or the persistence of repair intermediates signifying unsuccessful DNA repair could lead to cellular senescence, regardless of the cycling status of a cell [84, 130–132]. In fact, mounting evidence has already hinted at the presence of robust DDR in post-mitotic senescent cells [84]. Around 40 years after the initial description by Hayflick and Moorhead, pioneering work led by Olivier Toussaint and others demonstrated that cellular senescence can also be induced by exposing cells to chronic but non-lethal dosages of stress [133], which they coined the phenomenon as stress-induced senescence [133]. Various forms of stressors that are capable of inducing so are identified, including oxidants, UV irradiation, chemotherapeutic agents and reactive oxygen species (ROS). While these agents seem to act by different mechanisms, it is arguable that the majority of them could at some point introduce damage randomly to the genome, both directly and indirectly [133, 134]. Chronic exposure to these agents likely takes on the senescence effect by increasing the chance of unfaithful DNA repair. This confirms the idea that unresolved DDR is a key driver of cell senescence.
THE NEURONAL GENOME IS CONSTANTLY UNDER STRESS
Persistent DDR is probably the most consistently observed feature of senescent cells [84]. As alluded to above, this could be triggered by either telomeric or non-telomeric DNA damage, and the infidelity of their repair. As mentioned, this concept will help explain how cellular senescence could be achieved in post-mitotic cells. In the following sections, we will discuss why neurons are susceptible to DDR infidelity, priming the initiation of signals leading to cellular senescence response.
Sources of DNA damage and their roles in neuronal function
The adult mammalian brain is inherently incapable of regeneration [135]. Fully differentiated cells in the brain, particularly the mature neurons, are expected to survive through the entire life of the organism, suggesting that the maintenance of neuronal genomic stability is of paramount importance in sustaining brain health [136]. However, the genomes of these cells are constantly under threat imposed by their own physiological activities. The neuronal demand for energy for sustaining intra- and intercellular communications is enormous [137], which is supported mainly by the intense activities of oxidative phosphorylation reactions in the mitochondria [138]. This process also generates a huge amount of free radicals and ROS as byproducts that ultimately introduce oxidative DNA damage [139]. Despite so, an optimal level of ROS produced by the NADPH oxidase is also found to be essential for maintaining synaptic plasticity by modulating long-term potentiation and depression in multiple brain regions [140]. A recent study also suggested that within the somato-dendritic compartment, a related NADPH oxidase called dual oxidase (DUOX) is required for the neural activity-regulated generation of H2O2, which in turn modulates neuronal dendritic growth and adaptive plasticity [141]. The duality in the role of ROS is a double-edged sword which clearly puts neurons at a heightened risk of DNA damage.
Robust neuronal activities, on the other hand, are associated with enhanced transcription of long genes that are necessary for the synaptic modulation and ion transport [142], which may lead to the production of RNA:DNA hybrids (R-loops) at these gene loci [143]. Significant increases in R-loop signals over gene bodies and age-associated broadening of R-loop peak signals are found in neurons [143]. Their existence may be deemed detrimental, particularly at times when collisions with the replication forks or transcription complexes occur [144], and their quantities are associated with a progressive loss in the expressions of these long genes [143]. In addition, single- and double-stranded breaks are frequently formed by the actions of neuronal DNA topoisomerase which are necessary to resolve any topological stress incurred during active gene transcription [145]. For instance, during learning and memory consolidation, immediate early genes such as the activity-regulated cytoskeleton-associated protein (ARC), fos proto-oncogene (FOS), and neuronal PAS domain protein-4 (NPAS4) genes are rapidly and transiently expressed in response to neuronal activities [146–148]. To turn on these genes, rapid DNA demethylation pathways have been proposed [149, 150], and it likely involves the GADD45 family of DNA repair proteins for guiding the removal of 5-methylcytosine by either the base- (BER) or nucleotide excision repair (NER) pathway [151–154]. Furthermore, the generation of targeted DNA double-stranded breaks (DSBs) within the FOS and NPAS4 promoters is also found to be sufficient to induce their expressions in response to neuronal activities, so the topological constraints imposed by the pre-existing chromain structure can be resolved. Such kind of activity-dependent DSB formation is suggested to be mediated by type II topoisomerase β, as knockdown of which attenuates all these events following neuronal stimulation [155]. At the neural circuitry level, increasing neuronal activity at one brain region can lead to increased neuronal DSBs in other brain regions within the relevant networks as well [156]. For instance, visual stimuli to one of the eyes in mice results in a specific increase in levels of γH2AX foci— a DSB marker— in the stimulated contralateral visual cortex (V1) but not in the unstimulated ipsilateral V1, and that is associated with the induction of FOS expression in neurons to a similar extent [156]. More importantly, these foci signals are transient only due to the efficient repair, and more likely, the formation of DSBs in this sense is a natural process that facilitates the extensive remodeling and changes in gene expression involved in information processing, learning andmemory [157].
Additional sources of DNA damage during brain aging and disease pathogenesis
During the processes of aging, additional sources of DNA damage could emerge as well. Recent evidence revealed that age-associated epigenetic alterations in the neuronal genome can directly result in the de-repression of the long interspersed element-1 family of transposable elements, causing unexpected DNA rearrangements and lesions [158]. On the other hand, the proteolytic activity of lysosomes declines with age [159], and its failure may indirectly lead to DNA damage [160, 161]. In many late-onset neurological diseases, the misfolded proteins which are constantly produced throughout life start to accumulate, which predominantly occurs in the adult or aged brains [162]. In the context of AD, the formation and deposition of Aβ may in turn serve as an additional source of DSBs and oxidative DNA damage to neurons [163], which could be exacerbated during the disease progression. Another possible linkage between lysosomal activities and DNA damage is that lysosomes contain Dnase2a which facilitates the clearance of damaged extranuclear DNA via autophagy, thereby preventing the accumulation of cytosolic DNA [164]. In the brain, the loss in Dnase2a expression has been associated with higher loads of senescent neurons [165]. Since the onset of senescence is often accompanied by the progressive remodeling of chromatin, a phenomenon called “DNA segments with chromatin alterations reinforcing senescence” (DNA-SCARS) occurs [166], which is unexpectedly a lysosomal-dependent process [167]. Moreover, the formation of DNA-SCARS is associated with a constitutive DDR, as marked by persistent signals of γH2AX, 53BP1 and the gradual activation of p53 [64].
Together, this evidence highlights that the neuronal genome is constantly under stress, and this may gradually increase with age for all sorts of reasons. Indeed, a transcriptome-wide profiling study of prefrontal cortex tissues harvested from individuals ranging from 26 to 106 years old revealed that the accumulation of oxidative DNA damage is remarkedly increased from 40 years of age onwards [168]. More importantly, these markers are selectively enriched at the promoters of genes that are critical for shaping cognition, memory formation, and neuronal survival, which are also associated with diminished gene expressions [168]. The association between random DNA damage with specific effects on brain aging and dysfunction is further supported by a more recent genome-wide mapping study, which precisely pointed out that CpG dinucleotides and demethylated sites located near neuron-specific enhancers are vulnerable regions of DNA single-stranded breaks [169]. At the cellular level, human neurons are found to take on somatic mutations as they age from 4 months to 82 years. These mutations accumulate with age in individual neurons, but when brought together the accumulated divergence of genomes across the brain may affect functions eventually [170, 171].
Limited flexibility in DNA repair
If DNA damage is inevitable even in healthy and functional neurons, one might expect that these cells should be equipped with a powerful and versatile DNA repair network. However, the nature of these cells tells us that this is unlikely to be the case. In most proliferating mammalian cells, five major DNA repair pathways are available, including the BER, NER, mismatch repair (MMR), HR, and NHEJ [172]. Each of them has its own advantages in effectively handling different types of DNA lesions while they partially overlap and function as an intercalating network, backing up the repair of one another [172]. For non-dividing cells, they are by default excluded from the repair mechanisms involving the usage of the more accurate, S and G2-phase dependent HR on DSB repair [173], hinting that fully differentiated neurons in the brain are facing such limitations. Instead, these cells rely heavily on the alternative but less accurate NHEJ-centric mechanism as the major pathway for handling DSBs. One exception is that on occasions when the DSBs are formed at regions where active transcription activities are found, an RNA-templated HR-mediated repair mechanism could be utilized. In contrast to the classic mechanism which takes place during the late S-G2 phases, this is a replication-independent recombinational repair predominantly occurring in the G0–G1 phase of the cell cycle, which depends on the nascent RNA generated during active transcription. This pathway is evident in post-mitotic neurons, serving as a high-fidelity DNA repair option for actively transcribed genes that are essential for sustaining the normal functioning and survival of these cells [174].
Such repair inflexibilities, together with the genomic stress imposed by various sources, render neurons selectively vulnerable to an unexpected loss in the functions of DNA repair machineries [175]. In genetic diseases resulting from mutations in DNA repair genes, neurological complications are commonly found [176, 177]. For example, xeroderma pigmentosum group A (XPA) is caused by mutations of the XPA gene, the product of which plays a central role in NER [176]. Around 30% of XPA patients are found to suffer from mild-to-severe degrees of intellectual disability, deafness, and seizures [178]. In Cockayne syndrome resulting from mutations in either the ERCC8 or ERCC6 gene involved in repairing transcription-coupled DNA lesions, many patients suffer from accelerated aging, severe photosensitivity, and impaired development of the nervous system [179]. In ataxia telangiectasia (A-T) where the ATM gene that facilitates DSB repair is mutated, Purkinje neuron degeneration is found in all the patients, leading to cerebellar atrophy and ataxia [180]. These, together with the findings from mouse models, where neurological symptoms are observed when DNA repair factors like ligase IV and XRCC4 of the NHEJ mechanism are knocked out [181], well-demonstrate that DNA repair deficiencies have a causative role in initiating neurodegeneration.
SUSTAINED DNA DAMAGE RESPONSE RESETS NEURONS INTO A “PSEUDO-CYCLING” STATUS
Persistent repair intermediates but not unattended DNA lesions are likely the troublemakers
One logical speculation on the direct consequences of defects in DNA repair is simply the accumulation of unattended DNA lesions. However, evidence indicated that even under normal conditions, neurons and other post-mitotic cells readily adopt a selective repair approach, in which genes that are actively transcribed are repaired more robustly than other elements in the genome [182]. Such an unexpected deviation from the ideology of effective global DNA repair hinted that unattended lesions in the genome may not immediately impose danger as long as they are not located on essential genes. This notion is indeed supported by a recent single neuronal nuclei whole-genome sequencing analysis, which revealed that neurons from neurologically normal individuals have somatic single-nucleotide variant count numbers correlated with age [170, 183], serving as another evidence that unattended DNA lesions are normally tolerated among these cells. On the contrary, unsuccessfully repaired damage— which indicates the type of DNA lesions that has already been recognized by the cellular repair system despite an incomplete and unsuccessful repair— could lead to the persistent activation of unresolvable DDR, which in turn triggers the aberrant reactivation of the cell cycle machinery, and hence the degeneration of neurons [184]. In the same vein, some studies revealed that the attenuation of DDR via knocking down or inhibiting DR effectors after the initial injury results in unexpected neuroprotective effects [185–187].
The risk of emerging this kind of unsuccessful DNA repair, just like the case of repair at telomeric regions, can be heightened when choices of repair pathways become limited. Such a kind of intrinsic limitation is well experienced by the post-mitotic neurons even under physiological conditions, as mentioned in the previous section (Limited flexibilities of DNA repair). Apart from this, another major risk is obviously the compromised DNA repair capacity resulting from either reduced expressions or activities of repair proteins. DDR dysfunction in neurons is indeed well-documented in the pathogenesis of common neurodegenerative disorders [188]. Reduced levels and activities of DNA repair proteins are reported in AD brains. These include the ATM [189], BRCA1 [157], DNA-PKcs [190], and the MRE11 complex [191] of the DSB repair network; so as the OGG, NEIL1, and POLB needed for correcting base-related lesions [192, 193], and many others. Considering that DNA repair pathways are generally interconnected, defects in the primary repair choice could potentially be backed up by others. While on many occasions such kinds of arrangements could be observed, an error-free and faithful repair is not always a guarantee. One example is the activation of HR which serves as a local support of repair when the primary NHEJ repair is intentionally suppressed at the telomeric regions [127, 128]. Although HR is thought to be a better choice of repair in general as template-mediated repair is logically more accurate, the repetitiveness of the DNA sequence at the telomeric region has made the homologous form of repair more vulnerable to additional deletions and irreversible damage, which may ultimately evolve into an unresolvable DDR [127, 129]. In other locations of the genome, sometimes an error-prone repair called the microhomology-mediated end joining pathway is turned on as a surrogate repair mechanism for DSBs when the NHEJ mechanism becomes defective. This kind of repair is often associated with deletions flanking the break sites and this may even heighten the chance of undesirable chromosome translocations and rearrangements [194]. In post-mitotic neurons, DSB is the major type of lesion that is believed to be tackled mainly by the NHEJ pathway [173]. However, signs of activated HR, for instance, phosphorylated ATM [195], phosphorylated checkpoint kinase-2 [196], foci of BRCA1, and the RAD family of proteins [197], are readily detectable in healthy neurons as well. As mentioned before, these signals can be caused by their routine participation in lesion repair located adjacent to actively transcribed sites, which utilize nascent RNA-transcripts as the repair template [174]. Alternatively, they could be rooted in the classic mechanism involving the sister chromatid as a template, where HR acts as a secondary pathway of choice when NHEJ is not readily available. In the latter situation, it is plausible that unexpected lingering and unresolved errors are introduced when the DNA template has to be sought from the sister chromatid alignments. Nevertheless, reduced neuronal NHEJ activities and protein levels of DNA-PKcs and KU have already been reported in AD [190]. Equally detrimental, other studies also reported that neuronal activities or levels of proteins involved in HR are compromised as well [157, 189], hinting at a heightened chance of HR repair going awry. Together, these imply that the competence to successfully repair DSBs in AD neurons, by any means, is compromised. This echoes with the observation that the immunosignals of γH2AX— a post-translational histone modification that is widely used as a marker of DSBs— are persistently increased in neurons located in the hippocampus and the frontal cortex of mild cognitive impairment and AD patients [198]. As γH2AX is a phosphorylation product of cellular DNA lesion sensors like ATM and ATR, such signals indeed indicate repair intermediates rather than free, unattended ends of DSBs [199, 200]. The accumulation of unresolved repair intermediates, in other words, sustained DDR, should therefore be sensibly addressed for their roles in disease progression.
Sustained DDR brings back the “cell cycle” pre-requisite of cellular senescence
Being long-lived and terminally differentiated, mature neurons at some points are strictly post-mitotic, but it is also suggested that they are never free from cell cycle-related events. Mounting evidence indicates that even in normal situations, mature neurons undergo DDR that is closely linked to the cell cycle regulatory mechanism [187, 201, 202]. In all eukaryotes, DSBs can be repaired through NHEJ or HR. In post-mitotic neurons, NHEJ is the predominant pathway of choice which comprises both the canonical NHEJ (c-NHEJ) and alternative NHEJ (a-NHEJ). The c-NHEJ is primarily facilitated by a number of associated factors including the KU70/80 (KU), X-ray repair cross complementing 4 (XRCC4), DNA ligase 4 (LIG4), and DNA-dependent protein kinase catalytic subunit (DNA-PKcs). During physiological conditions, neurons may exit their resting state (G0 phase) and re-initiate events in the G1 phase, at which point the c-NHEJ pathway repair machinery can be recruited to directly ligate the ends without the use of extensive homology [201]. However, in situations like AD when the levels and activities of KU-associated machineries are diminished [203], this could result in the de-repression of the shared machineries required for the initiation of a-NHEJ and HR [204]. Both involve the recruitment of ATM, Artemis, and Retinoblastoma-binding protein 8 (CtIP) [205].
Previous studies suggested that the pathway choice between a-NHEJ and HR is likely dependent on the cell cycle status. If the recruitment of factors occurs in the G1 phase, activation of a-NHEJ predominates [205], which would likely allow the lesions to be resolved by minimal resection followed by direct annealing of microhomologous sequences [206, 207]. However, if the recruitment process happens in the S or G2 phases, activation of HR prevails [205, 208] but the outcome of end resection could be completely different. Unlike the minimal resection that occurs in a-NHEJ, HR involves additional endo- and exonucleases, such as DNA replication helicase/nuclease-2 (DNA2), exonuclease-1 (EXO1) and meiotic recombination-11 (MRE11) proteins that remove several kilobases from the 5’ terminus of the DSB [209], creating a long range single-stranded DNA (ssDNA) resection for subsequent strand invasion by the homologous sister strand [207]. The ssDNA created is then rapidly covered by the ssDNA-binding replication protein A (RPA), which briefly recruits and activates the ataxia telangiectasia and RAD3 related (ATR) [210, 211]. In proliferating cells, the subsequent replacement with RAD51 recombinase (RAD51) protein on the ssDNA followed by homology search would occur to complete the HR repair [212]. However, in post-mitotic neurons, such downstream events are unlikely to be fulfilled due to their cell cycle constraints.
In light of the existence of such crosstalks between the ATM and ATR-dependent signaling, some studies also suggested that the ATM-dependent mechanism is mainly activated by sublethal DSB lesions. This signaling mediates their proper repair in neurons by allowing a “brief” G1 phase re-engagement where a-NHEJ could be initiated, and after the repair, these cells would likely return to the G0 resting phase [201]. On the contrary, the ATR-dependent pathway is mainly hyperactivated in response to replicative stress (i.e., S-phase) [213, 214], which is logically a more devastating situation for neurons. The “replication-like” stress is reported in neurons which attempt to transit from G1 to S phase upon unrepairable lesions or when facing the exhaustion of repair machineries [215, 216]. Subsequent activation of ATR signaling would entail the transition through the G2-M cell cycle checkpoint via Chk1 phosphorylation [214, 217]. If this occurs in proliferating cells, this will lead to high-fidelity HR repair and full recovery will likely follow [218]. However, in mature neurons, the initiation of the classic HR repair machinery is hardly effective due to their fully differentiated status. It is plausible that the delayed processing of DSBs in these cells eventually results in high levels of resected single stranded DNA, which further enhance ATR-dependent signaling in a vicious cycle [132]. Evidence of S-phase re-engagement and activated ATR signaling has been reported in neurons of human AD brains [219]. Recent studies suggested that early S-phase re-engagement in neurons is indeed a protective mechanism in preventing immediate cell death induced by the exposure to Aβ [73] or the introduction of SV40 large T antigen [220]. Together, these findings clearly illustrate how persistent repair intermediates are strong inducers of cell cycle reactivation in neurons, priming them to proceed to permanent cell cycle arrest (i.e., cellular senescence) at times if needed.
THE CHOICE OF TERMINAL FATES BETWEEN IMMEDIATE CELL DEATH AND CELL SENESCENCE
Observation: Death row is not the only possible outcome
Indeed, the association between cell cycle re-entry and neurodegeneration is well-established, and the population of these cell cycle re-activated neurons is also correlated with the decline in brain cognitive function [221]. In patients with AD and even those with mild cognitive impairment, evidence of neuronal cell cycle re-entry has been reported, which seems to be associated with the levels of hyperphosphorylated tau and neurofibrillary tangles [222]. In non-diseased healthy brains, low levels of cell cycle re-activated neurons are found occasionally [221], but their number can be escalated by exposing them to disease-associated conditions, such as hyperglycemia [223], hyperinsulinemia [35], stroke [224, 225], and traumatic brain injuries [226]. Markers indicating activated G1 to S phases are frequently observed in these cells. Occasionally, evidence of partial DNA replication and G2 phase markers is also reported [222, 227]. Despite so, signs of mitotic phase entry and successful events of cell division have never been observed, instead most neurons are found to be arrested at the G1-S and G2-M cell cycle checkpoints [222, 228].
The fate of cells arrested at cycling checkpoints initiated by the DDR is mainly either survival from successful repair or cell death from failing to do so [229]. A number of cell death mechanisms related to DNA damage and DDR have been reported in neurons. These include apoptosis, autophagy-dependent cell death, and necroptosis [230]. Apoptosis has been widely studied as a response to severe DNA damage, in particular, this mechanism involves the rapid activation of p53 signaling axis in neurons [231]. The relationship between autophagy-dependent cell death and DDR is also recently addressed [232], as multiple DNA repair pathways, including the HR, BER, NER, and MMR are at some point regulated by autophagy [233, 234]. Regarding necroptosis, a type of regulated cell death mechanism which displays features of both apoptosis and necrosis, recent studies also suggested that DNA damage is a potential upstream trigger [235], and crosstalks with autophagy in neurons as well [236, 237]. However, it appears that the severity of DNA damage as well as the strength of cell cycle progression signaling could both interfere with the fate of these neurons. As illustrated above, low levels of DNA damage promote the entry into G1 phase, which will then be likely handled by NHEJ; whereas high levels of damage or the incompetence of NHEJ may lead to S phase re-entry through the onset and activation of HR. This may then result in the emergence of secondary damage formed during the strand invasion process and partial replication of DNA, thereby promoting cell death. Apart from these classic findings, emerging evidence also suggests that some of these cell cycle re-activated neurons could persist in the brain for months or even years [238]. Many of these cells appeared to be functionally active, despite the fact that they might have deviated from their original physiology [239]. Indeed, other studies also suggested that neurons are way more resilient than we could have imagined, as their longevity is not limited by the maximum lifespan of the organism from which they originated. Rather, if they are transplanted into another organism with a longer lifespan, they could outlive their original host [240]. This does not only suggest that neuronal survival and chronological aging are coincidental but separable processes, but also hints that neurons are inherently well-equipped to survive after being put in unexpected situations. Together, these findings indicate that in contrast to the simple and acute execution of cell death and removal, an unexpected “pro-survival” phase may sometimes emerge, so that these neurons can last chronically in tissues or even escape from the death row (Fig. 1).
Fig. 1
Three major cell fates are found in terminally differentiated neurons in response to DNA insults. Successful repair results in a quiescent outcome, and the neurons maintain their normal physiology as a part of a healthy brain. In contrast, sustained damage resulted from unsuccessful repair or unattended lesions may lead to neuronal death and senescence, contributing to brain aging and disease pathogenesis.
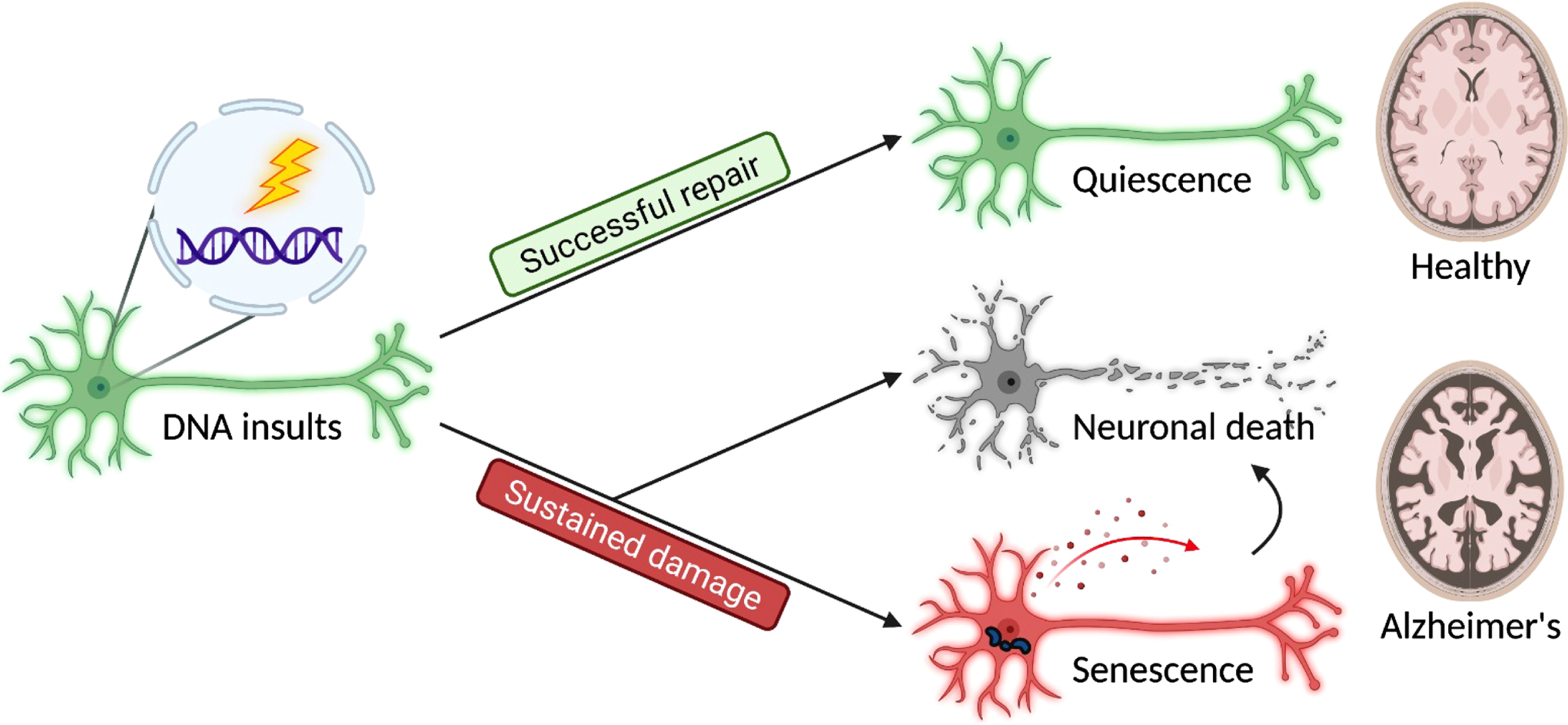
Permanent cell cycle arrest following cell cycle re-entry in neurons
Now it becomes clear that one possible mechanism underlying the anti-apoptotic properties acquired by these cells is the commitment to cellular senescence [239]. At the molecular level, despite how checkpoint activation and repair are coordinated to determine the cell fate of neurons after initiation of DDR still remains largely unknown, studies performed in other cellular systems suggested that the decision to irreversibly exit the cell cycle (i.e., cellular senescence) can be established quickly within hours after the DDR is triggered in the G2 phase. In contrast to that scenario, a substantially longer time for the repair is allowed when the damage is detected in other phases of the cell cycle [241]. Other evidence also echoed that the permanent cell cycle exit decision from the G2 phase is marked by the p53-p21-dependent entrapment of cyclin B1/Cdk into the nucleus, where the latter serves as a final trigger of a senescence response [241–243]. This outcome is indeed downstream of and dependent on the ATR-mediated DDR signaling [132, 244]. The linkage between ATR signaling and cell senescence is evident. In ATM-deficient cells, activation of ATR in the absence of DNA breaks is sufficient to promote cell cycle arrest, and if the signal persists, it triggers p53-dependent senescence [245]. In neurons, ATR is indispensable for preventing the S phase-dependent neuronal death in vivo, which allows the “cell cycle events” to be sustained in the affected neurons for weeks to years before their cell death is observed [246]. If our prediction is correct, persistent repair intermediates will likely trigger neuronal senescence through a sustained ATR signaling axis. While the direct evidence related to this hypothesis currently lacks, transcriptome profiling of neurons characterized with persistent DDR did reveal an induction of gene expression patterns mapped to the ATR-dependent DDR and APC/C cycle regulatory complex and p53 signaling [247] (Fig. 2).
A proposed model
Combining the current understanding of the mechanistic and consequential heterogeneity in DDR and our prediction, we have proposed a model describing the fate of neurons upon DSBs. The neuronal genome is constantly subjected to various kinds of stress. Upon sensing the DSB lesions, DDR is elicited. It is likely that NHEJ is elicited only when the NHEJ core proteins, including KUs, XRCC4, LIG4, and DNA-PKcs, are present. This leads to a temporary re-engagement into the G1 phase for the c-NHEJ, and to a lesser extent, the a-NHEJ (Figs. 2 and 3A). As both variations of NHEJ are error-prone, by chance, somatic mutations could be generated and left behind. Since neurons are inherently well-equipped to survive after DNA insults, when the mutations are tolerable, DDR will be resolved, followed by neurons returning to the quiescent and homeostatic G0 phase (Figs. 1 and 2). However, if mutations are intolerable, particularly those on essential genes for cell functioning and survival, then non-homeostatic fates prevail(Fig. 2).
Fig. 2
A proposed decision roadmap leading to the different cell fates of neurons in the face of DSB challenge. For the details, please refer to the “A proposed model” section.
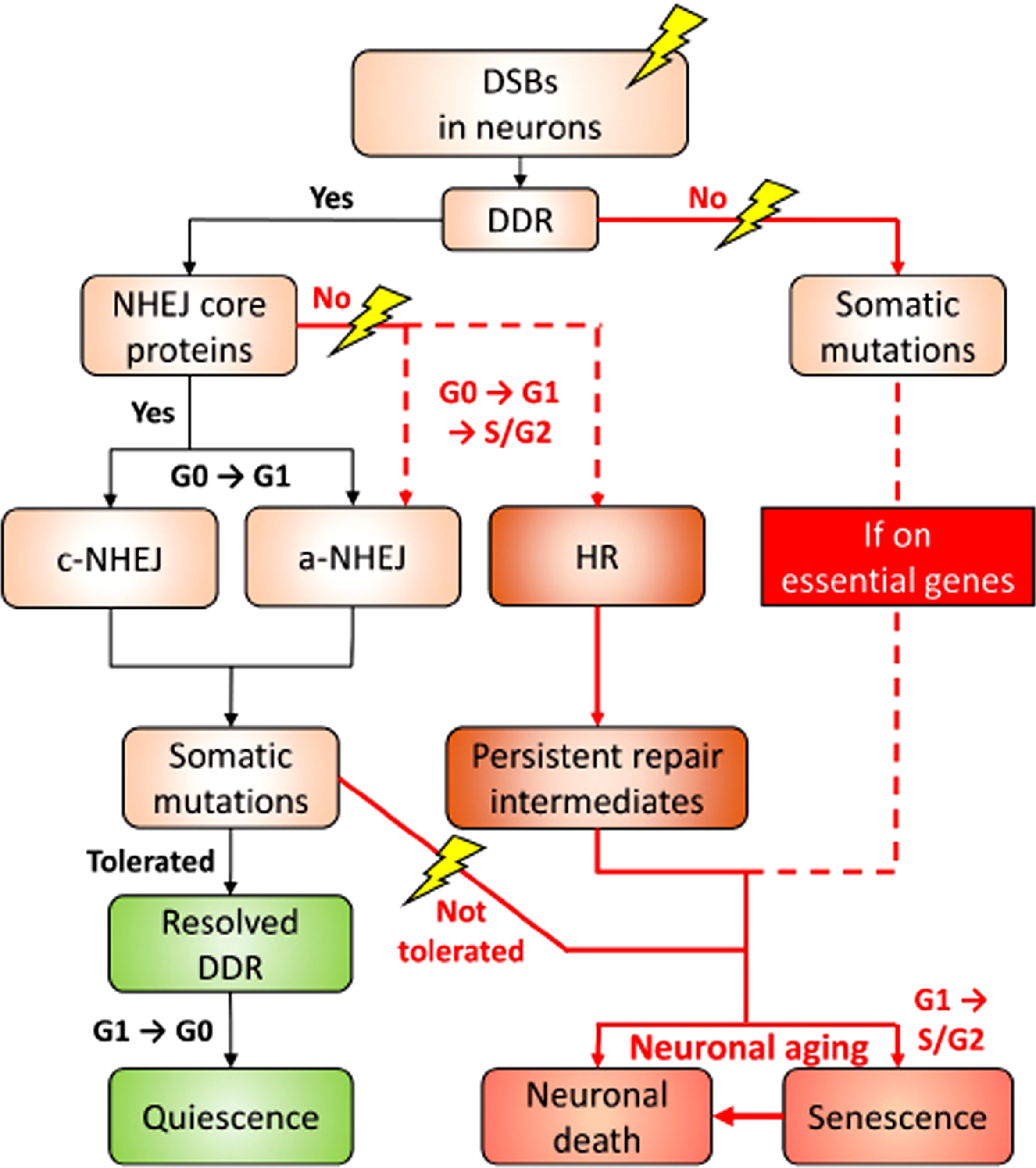
Fig. 3
Mechanistic insights into how neuronal senescence is achieved from DNA damage responses. A) The competition and preferences of DSB repair pathways under healthy and diseased conditions. In healthy brains, c-NHEJ is the predominant pathway of DSB repair. Other pathways are suppressed by the c-NHEJ core machinery proteins. B) HR-mediated DSB repair in post-mitotic neurons results in long ssDNA resection and the failure in subsequent engagement into homologous sequence invasion caused by cell cycle constraints. This results in the persistence of single-stranded repair intermediates and chronic activation of ATR, where the latter is a strong driver of cell cycle checkpoint and senescence.
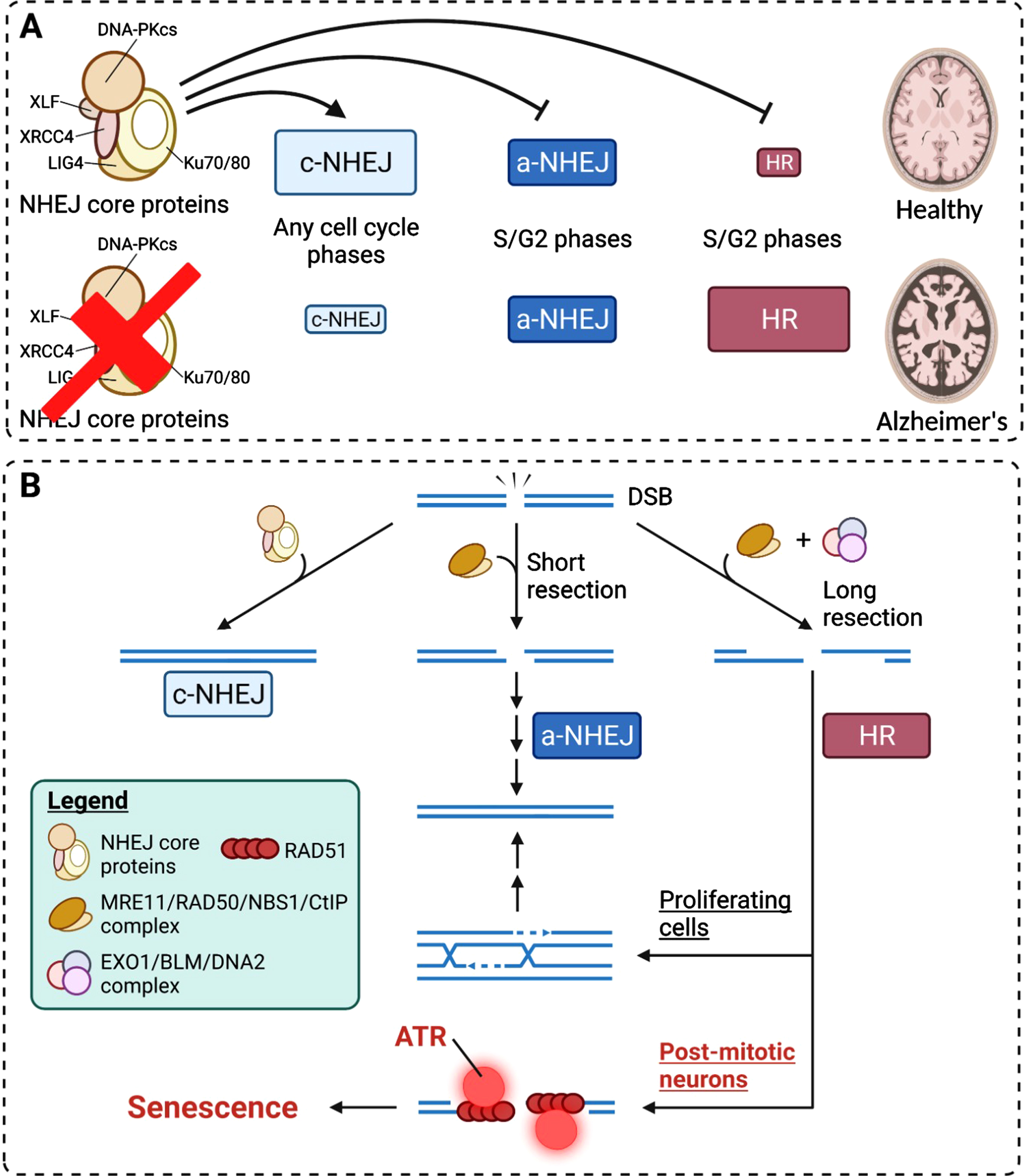
We speculate that the non-homeostatic fates manifest as either neuronal death or cellular senescence (Fig. 1) and these fates will emerge at times when the optimal functioning of NHEJ repair machinery is compromised, particularly in the context of AD, or on occasions when the primary lesions are left unattended (Fig. 3A). In the former scenario, neurons are likely reset back into the “cell cycling” status. Upon reaching the S or G2 phase, the DDR can then be facilitated by the HR and a-NHEJ, despite how the decision is made between the two remains unclear. The HR is likely by default a failure among these cells, as homologous sequencing invasion is unlikely to occur (Figs. 2 and 3B). This results in lingering ssDNA repair intermediates, which prompt the sustained activation of the ATR pathway to consolidate a cellular senescence response (Fig. 3B). For the unattended lesions, these may prime the occurrence of somatic mutations. Under most circumstances, these could be tolerated as long as they are located in regions that will not impede the expressions of essential genes; or else a detrimental death fate is assured. This proposed model tightly links how DNA damage response is closely associated with the cell cycle status, and how failure in the repair network results in neuronal senescence and cell death, contributing to the hallmarks of brain aging. Of note, this model bears emphasis on NHEJ and HR, yet they are not the exclusive mechanisms of DNA repair in neurons.
CURRENT APPROACHES FOR TARGETING SENESCENT CELLS
The idea of selectively targeting senescent cells has emerged since 2004 when an inverse relationship between senescence cell burden and health span was identified in mice [248]. Since then, multiple strategies have been identified, which can be classified into two major categories: senomorphics and senolytics [249]. While senomorphics are compounds that aim at neutralizing SASP components without the actual killing effect [250], senolytics are ones that aim at eliminating senescent cells directly [251]. The earliest senolytics were identified by a bioinformatics approach, targeted at disrupting the senescent cell anti-apoptotic and pro-survival network [251]. The ultimate effect is to render senescent cells that express SASP no longer “immuned” to the associated damaging effect, thereby killing themselves [251]. A famous example of this approach is the combined treatment of Src kinase inhibitor dasatinib and the flavonoid quercertin, so as the use of BCL2 family protein inhibitors and others [60, 251–254].
More recently, the application of the oxidized form of nicotinamide adenine dinucleotide (NAD+) has become one of the hotspots of anti-aging and longevity science [255]. Strong links between NAD+ depletion and hallmarks of brain aging have been found, so as between NAD+ and DDR at the molecular level [256, 257]. NAD+ is a substrate for DNA repair through the reaction called PARylation for “flagging” sites of lesions for subsequent processing, and this is mediated by DDR signaling enzymes PARP1, PARP2, and PARP3 [258]. In genetic diseases such as the XPA [259], Cockayne syndrome group B (CSB) [260] and A-T [261] where genes involved in certain DDR are mutated, unresolved and sustained activation of PARP1 is frequently observed, leading to uncontrolled PARylation and hence severe NAD+ depletion [259]. The latter situation consequently de-activates the NAD-dependent sirtuins [259], an anti-aging family of deacylases in the eukaryotic system. A similar phenomenon is also observed in AD and other age-related disorders [262]. Supplementation of NAD+ precursors, either in the form of nicotinamide riboside or nicotinamide monucleotide, is found to be beneficial in improving the cellular DNA repair capacities, as well as alleviating the clinical symptoms associated with these DNA-repair deficient [259, 263, 264] and age-related neurological diseases (i.e. AD, PD, Huntington’s disease, amyotrophic lateral sclerosis) [256].
FUTURE PERSPECTIVES
It is now encouraging to witness a rise in research interest in cellular senescence biology in the context of brain aging and neurodegenerative diseases. It is not difficult to realize that cellular senescence and the pathogenesis of AD, at some point, show reciprocal causality. Here in this review, the argument for how senescent cells may serve as a major contributor to tissue aging and pathogenesis is laid out. We argue that neuronal senescence is “a new face of an old acquaintance”. This phenomenon possibly explains how certain cell cycle re-engaged neurons could persist in the brain if they do not die instantly (Fig. 1). If the analysis is correct, this will extend our understanding of the influences of these “lingering” cells on the brain milieu.
With reference to both the classic and the latest evidence related to the subject of cellular senescence, it is logical to reason that unresolved DDR is likely the trigger of neuronal senescence, as this logically narrates our knowledge of the relationship between the DDR, cell cycle re-entry, and permanent checkpoint arrest that we have acquired in post-mitotic neurons over the past two decades (Fig. 2). The p53 dynamics is the key controlling element of the terminal fate of a cell after DDR [265], which the protein itself is a known downstream phosphorylation target of both ATM and ATR [266]. Therefore, it is crucial to understand how the resulting dynamics differ when triggered by different kinases, and that should help explain the differences in the terminal fates achieved.
Research into the possibilities of senescence cell-targeting senotherapies in reversing the effects of pathological aging has been surging in recent years. However, targeting senescent cells is not an easy task, not only due to the fact that the brain is difficult to access, but emerging evidence also indicates that there is a large heterogeneity in the molecular characteristics of senescent cells, depending on the inducing agent, cell type, and life stages [249, 267]. Therefore, the mechanisms that distinguish the beneficial from the deleterious senescence events and details of such heterogeneity are the critical knowledge gaps at present. With the recent advances in spatial and single-cell-based omics and multiplex-based imaging technologies, unique markers and maps that describe the evolutionary details of neuronal senescence at the molecular, cellular, morphological, and functional levels in a spatial-and-temporal framework can potentially be identified in the near future. Alongside with the traditional focus on the anti-amyloid and tauopathy drug development, senotherapeutic strategies may therefore hold great promises as adjuvants in sustaining brain cell resilience and tissue homeostasis in acting against the inevitable aging factor of life.
ACKNOWLEDGMENTS
The work was supported, in part, by grants from the following: The Hong Kong Research Grants Council (RGC)-General Research Fund (GRF) (PI: ECS24107121, GRF16100219 and GRF16100718); the National Natural Science Foundation-Excellent Young Scientists Fund 2020 (Ref: 32022087); Alzheimer’s Association Research Fellowship (PI: AARF-17-531566); CUHK-Improvement on Competitiveness in Hiring New Faculties Funding Scheme (PI: Ref. 133) and CUHK-School of Life Sciences Start-up funding to H.-M.C.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/22-0203r1).
REFERENCES
[1] | Ryan NS , Rossor MN , Fox NC ((2015) ) Alzheimer’s disease in the 100 years since Alzheimer’s death. Brain 138: , 3816–3821. |
[2] | Collaborators GBDDF ((2022) ) Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: An analysis for the Global Burden of Disease Study 2019. Lancet Public Health 7: , e105–e125. |
[3] | DeTure MA , Dickson DW ((2019) ) The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener 14: , 32. |
[4] | Grundke-Iqbal I , Iqbal K , Tung YC , Quinlan M , Wisniewski HM , Binder LI ((1986) ) Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A 83: , 4913–4917. |
[5] | Nukina N , Ihara Y ((1986) ) One of the antigenic determinants of paired helical filaments is related to tau protein. J Biochem 99: , 1541–1544. |
[6] | Glenner GG , Wong CW ((1984) ) Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 120: , 885–890. |
[7] | Glenner GG , Wong CW ((1984) ) Alzheimer’s disease and Down’s syndrome: Sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun 122: , 1131–1135. |
[8] | Hardy J , Allsop D ((1991) ) Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci 12: , 383–388. |
[9] | Selkoe DJ ((1991) ) The molecular pathology of Alzheimer’s disease. Neuron 6: , 487–498. |
[10] | Goedert M , Spillantini MG , Jakes R , Rutherford D , Crowther RA ((1989) ) Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 3: , 519–526. |
[11] | Goedert M ((1993) ) Tau protein and the neurofibrillary pathology of Alzheimer’s disease. Trends Neurosci 16: , 460–465. |
[12] | Goedert M ((1996) ) Tau protein and the neurofibrillary pathology of Alzheimer’s disease. Ann N Y Acad Sci 777: , 121–131. |
[13] | Selkoe DJ , Hardy J ((2016) ) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8: , 595–608. |
[14] | Mullane K , Williams M ((2020) ) Alzheimer’s disease beyond amyloid: Can the repetitive failures of amyloid-targeted therapeutics inform future approaches to dementia drug discovery? . Biochem Pharmacol 177: , 113945. |
[15] | Mullard A ((2021) ) Failure of first anti-tau antibody in Alzheimer disease highlights risks of history repeating. Nat Rev Drug Discov 20: , 3–5. |
[16] | Herrup K ((2015) ) The case for rejecting the amyloid cascade hypothesis. Nat Neurosci 18: , 794–799. |
[17] | Kametani F , Hasegawa M ((2018) ) Reconsideration of amyloid hypothesis and tau hypothesis in Alzheimer’s disease. Front Neurosci 12: , 25. |
[18] | Uddin MS , Kabir MT , Rahman MS , Behl T , Jeandet P , Ashraf GM , Najda A , Bin-Jumah MN , El-Seedi HR , Abdel-Daim MM ((2020) ) Revisiting the amyloid cascade hypothesis: From anti-Abeta therapeutics to auspicious new ways for Alzheimer’s disease. Int J Mol Sci 21: , 5858. |
[19] | Daviglus ML , Bell CC , Berrettini W , Bowen PE , Connolly ES Jr. , Cox NJ , Dunbar-Jacob JM , Granieri EC , Hunt G , McGarry K , Patel D , Potosky AL , Sanders-Bush E , Silberberg D , Trevisan M ((2010) ) National Institutes of Health State-of-the-Science Conference statement: Preventing Alzheimer disease and cognitive decline. Ann Intern Med 153: , 176–181. |
[20] | Kotter-Gruhn D , Kornadt AE , Stephan Y ((2015) ) Looking beyond chronological age: Current knowledge and future directions in the study of subjective age. Gerontology 62: , 86–93. |
[21] | Stephan Y , Sutin AR , Luchetti M , Terracciano A ((2018) ) Subjective age and risk of incident dementia: Evidence from the National Health and Aging Trends survey. J Psychiatr Res 100: , 1–4. |
[22] | Lopez-Otin C , Blasco MA , Partridge L , Serrano M , Kroemer G ((2013) ) The hallmarks of aging. Cell 153: , 1194–1217. |
[23] | Tan FC , Hutchison ER , Eitan E , Mattson MP ((2014) ) Are there roles for brain cell senescence in aging and neurodegenerative disorders? . Biogerontology 15: , 643–660. |
[24] | Stefanatos R , Sanz A ((2018) ) The role of mitochondrial ROS in the aging brain. FEBS Lett 592: , 743–758. |
[25] | Martinez-Cue C , Rueda N ((2020) ) Cellular senescence in neurodegenerative diseases. Front Cell Neurosci 14: , 16. |
[26] | Hu Y , Fryatt GL , Ghorbani M , Obst J , Menassa DA , Martin-Estebane M , Muntslag TAO , Olmos-Alonso A , Guerrero-Carrasco M , Thomas D , Cragg MS , Gomez-Nicola D ((2021) ) Replicative senescence dictates the emergence of disease-associated microglia and contributes to Abeta pathology. Cell Rep 35: , 109228. |
[27] | Martínez-González K , Islas-Hernández A , Martínez-Ezquerro JD , Bermúdez-Rattoni F , Garcia-delaTorre P ((2020) ) Telomere length and oxidative stress variations in a murine model of Alzheimer’s disease progression. Eur J Neurosci 52: , 4863–4874. |
[28] | Yoon YS , You JS , Kim TK , Ahn WJ , Kim MJ , Son KH , Ricarte D , Ortiz D , Lee SJ , Lee HJ ((2022) ) Senescence and impaired DNA damage responses in alpha-synucleinopathy models. Exp Mol Med 54: , 115–128. |
[29] | Walton CC , Begelman D , Nguyen W , Andersen JK ((2020) ) Senescence as an amyloid cascade: The amyloid senescence hypothesis. Front Cell Neurosci 14: , 129. |
[30] | Kang C , Xu Q , Martin TD , Li MZ , Demaria M , Aron L , Lu T , Yankner BA , Campisi J , Elledge SJ ((2015) ) The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 349: , aaa5612. |
[31] | Moreno-Blas D , Gorostieta-Salas E , Pommer-Alba A , Muciño-Hernández G , Gerónimo-Olvera C , Maciel-Barón LA , Konigsberg M , Massieu L , Castro-Obregón S ((2019) ) Cortical neurons develop a senescence-like phenotype promoted by dysfunctional autophagy. Aging (Albany NY) 11: , 6175–6198. |
[32] | Tasdemir N , Lowe SW ((2013) ) Senescent cells spread the word: Non-cell autonomous propagation of cellular senescence. EMBO J 32: , 1975–1976. |
[33] | Di Mitri D , Alimonti A ((2016) ) Non-cell-autonomous regulation of cellular senescence in cancer. Trends Cell Biol 26: , 215–226. |
[34] | Jurk D , Wang C , Miwa S , Maddick M , Korolchuk V , Tsolou A , Gonos ES , Thrasivoulou C , Saffrey MJ , Cameron K , von Zglinicki T ((2012) ) Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 11: , 996–1004. |
[35] | Chow HM , Shi M , Cheng A , Gao Y , Chen G , Song X , So RWL , Zhang J , Herrup K ((2019) ) Age-related hyperinsulinemia leads to insulin resistance in neurons and cell-cycle-induced senescence. Nat Neurosci 22: , 1806–1819. |
[36] | Dodig S , Cepelak I , Pavic I ((2019) ) Hallmarks of senescence and aging. Biochem Med (Zagreb) 29: , 030501. |
[37] | von Kobbe C ((2018) ) Cellular senescence: A view throughout organismal life. Cell Mol Life Sci 75: , 3553–3567. |
[38] | Munoz-Espin D , Canamero M , Maraver A , Gomez-Lopez G , Contreras J , Murillo-Cuesta S , Rodriguez-Baeza A , Varela-Nieto I , Ruberte J , Collado M , Serrano M ((2013) ) Programmed cell senescence during mammalian embryonic development. Cell 155: , 1104–1118. |
[39] | Hinds P , Pietruska J ((2017) ) Senescence and tumor suppression. F1000Res 6: , 2121. |
[40] | Antelo-Iglesias L , Picallos-Rabina P , Estevez-Souto V , Da Silva-Alvarez S , Collado M ((2021) ) The role of cellular senescence in tissue repair and regeneration. Mech Ageing Dev 198: , 111528. |
[41] | Helman A , Klochendler A , Azazmeh N , Gabai Y , Horwitz E , Anzi S , Swisa A , Condiotti R , Granit RZ , Nevo Y , Fixler Y , Shreibman D , Zamir A , Tornovsky-Babeay S , Dai C , Glaser B , Powers AC , Shapiro AM , Magnuson MA , Dor Y , Ben-Porath I ((2016) ) p16(Ink4a)-induced senescence of pancreatic beta cells enhances insulin secretion. Nat Med 22: , 412–420. |
[42] | Wyld L , Bellantuono I , Tchkonia T , Morgan J , Turner O , Foss F , George J , Danson S , Kirkland JL ((2020) ) Senescence and cancer: A review of clinical implications of senescence and senotherapies. Cancers (Basel) 12: , 2134. |
[43] | Shimizu I , Minamino T ((2019) ) Cellular senescence in cardiac diseases. J Cardiol 74: , 313–319. |
[44] | Palmer AK , Gustafson B , Kirkland JL , Smith U ((2019) ) Cellular senescence: At the nexus between ageing and diabetes. Diabetologia 62: , 1835–1841. |
[45] | Pignolo RJ , Law SF , Chandra A ((2021) ) Bone aging, cellular senescence, and osteoporosis. JBMR Plus 5: , e10488. |
[46] | Guerrero A , De Strooper B , Arancibia-Carcamo IL ((2021) ) Cellular senescence at the crossroads of inflammation and Alzheimer’s disease. Trends Neurosci 44: , 714–727. |
[47] | Paramos-de-Carvalho D , Jacinto A , Saude L ((2021) ) The right time for senescence. Elife 10: , e72449. |
[48] | van Deursen JM ((2014) ) The role of senescent cells in ageing. Nature 509: , 439–446. |
[49] | Xue W , Zender L , Miething C , Dickins RA , Hernando E , Krizhanovsky V , Cordon-Cardo C , Lowe SW ((2007) ) Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445: , 656–660. |
[50] | Kang TW , Yevsa T , Woller N , Hoenicke L , Wuestefeld T , Dauch D , Hohmeyer A , Gereke M , Rudalska R , Potapova A , Iken M , Vucur M , Weiss S , Heikenwalder M , Khan S , Gil J , Bruder D , Manns M , Schirmacher P , Tacke F , Ott M , Luedde T , Longerich T , Kubicka S , Zender L ((2011) ) Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 479: , 547–551. |
[51] | Krizhanovsky V , Yon M , Dickins RA , Hearn S , Simon J , Miething C , Yee H , Zender L , Lowe SW ((2008) ) Senescence of activated stellate cells limits liver fibrosis. Cell 134: , 657–667. |
[52] | Lujambio A , Akkari L , Simon J , Grace D , Tschaharganeh DF , Bolden JE , Zhao Z , Thapar V , Joyce JA , Krizhanovsky V , Lowe SW ((2013) ) Non-cell-autonomous tumor suppression by p53. Cell 153: , 449–460. |
[53] | Sagiv A , Krizhanovsky V ((2013) ) Immunosurveillance of senescent cells: The bright side of the senescence program. Biogerontology 14: , 617–628. |
[54] | Karin O , Alon U ((2021) ) Senescent cell accumulation mechanisms inferred from parabiosis. Geroscience 43: , 329–341. |
[55] | Ovadya Y , Landsberger T , Leins H , Vadai E , Gal H , Biran A , Yosef R , Sagiv A , Agrawal A , Shapira A , Windheim J , Tsoory M , Schirmbeck R , Amit I , Geiger H , Krizhanovsky V ((2018) ) Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat Commun 9: , 5435. |
[56] | Perez-Lanzon M , Zitvogel L , Kroemer G ((2019) ) Failure of immunosurveillance accelerates aging. Oncoimmunology 8: , e1575117. |
[57] | Soto-Gamez A , Quax WJ , Demaria M ((2019) ) Regulation of survival networks in senescent cells: From mechanisms to interventions. J Mol Biol 431: , 2629–2643. |
[58] | Pahl HL ((1999) ) Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 18: , 6853–6866. |
[59] | Catz SD , Johnson JL ((2001) ) Transcriptional regulation of bcl-2 by nuclear factor kappa B and its significance in prostate cancer. Oncogene 20: , 7342–7351. |
[60] | Yosef R , Pilpel N , Tokarsky-Amiel R , Biran A , Ovadya Y , Cohen S , Vadai E , Dassa L , Shahar E , Condiotti R , Ben-Porath I , Krizhanovsky V ((2016) ) Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun 7: , 11190. |
[61] | Wang E ((1995) ) Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved. Cancer Res 55: , 2284–2292. |
[62] | Yosef R , Pilpel N , Papismadov N , Gal H , Ovadya Y , Vadai E , Miller S , Porat Z , Ben-Dor S , Krizhanovsky V ((2017) ) p21 maintains senescent cell viability under persistent DNA damage response by restraining JNK and caspase signaling. EMBO J 36: , 2280–2295. |
[63] | Strzeszewska A , Alster O , Mosieniak G , Ciolko A , Sikora E ((2018) ) Insight into the role of PIKK family members and NF-small ka, Cyrillic B in DNA damage-induced senescence and senescence-associated secretory phenotype of colon cancer cells. Cell Death Dis 9: , 44. |
[64] | Rodier F , Coppe JP , Patil CK , Hoeijmakers WA , Munoz DP , Raza SR , Freund A , Campeau E , Davalos AR , Campisi J ((2009) ) Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol 11: , 973–979. |
[65] | Rea IM , Gibson DS , McGilligan V , McNerlan SE , Alexander HD , Ross OA ((2018) ) Age and Age-related diseases: Role of inflammation triggers and cytokines. Front Immunol 9: , 586. |
[66] | Geldmacher DS , Levin BE , Wright CB ((2012) ) Characterizing healthy samples for studies of human cognitive aging. Front Aging Neurosci 4: , 23. |
[67] | Dykiert D , Der G , Starr JM , Deary IJ ((2012) ) Age differences in intra-individual variability in simple and choice reaction time: Systematic review and meta-analysis. , e. PLoS One 7: , 45759. |
[68] | Mattson MP , Arumugam TV ((2018) ) Hallmarks of brain aging: Adaptive and pathological modification by metabolic states. Cell Metab 27: , 1176–1199. |
[69] | Sikora E , Bielak-Zmijewska A , Dudkowska M , Krzystyniak A , Mosieniak G , Wesierska M , Wlodarczyk J ((2021) ) Cellular senescence in brain aging. Front Aging Neurosci 13: , 646924. |
[70] | Niccoli T , Partridge L ((2012) ) Ageing as a risk factor for disease. , R. Curr Biol 22: , 741–752. |
[71] | Ishikawa S , Ishikawa F ((2020) ) Proteostasis failure and cellular senescence in long-term cultured postmitotic rat neurons. Aging Cell 19: , e13071. |
[72] | Ogrodnik M , Evans SA , Fielder E , Victorelli S , Kruger P , Salmonowicz H , Weigand BM , Patel AD , Pirtskhalava T , Inman CL , Johnson KO , Dickinson SL , Rocha A , Schafer MJ , Zhu Y , Allison DB , von Zglinicki T , LeBrasseur NK , Tchkonia T , Neretti N , Passos JF , Kirkland JL , Jurk D ((2021) ) Whole-body senescent cell clearance alleviates age-related brain inflammation and cognitive impairment in mice. Aging Cell 20: , e13296. |
[73] | Ippati S , Deng Y , van der Hoven J , Heu C , van Hummel A , Chua SW , Paric E , Chan G , Feiten A , Fath T , Ke YD , Haass NK , Ittner LM ((2021) ) Rapid initiation of cell cycle reentry processes protects neurons from amyloid-beta toxicity. Proc Natl Acad Sci U S A 118: , e2011876118. |
[74] | He N , Jin WL , Lok KH , Wang Y , Yin M , Wang ZJ ((2013) ) Amyloid-beta(1-42) oligomer accelerates senescence in adult hippocampal neural stem/progenitor cells via formylpeptide receptor 2. Cell Death Dis 4: , e924. |
[75] | Riessland M , Kolisnyk B , Kim TW , Cheng J , Ni J , Pearson JA , Park EJ , Dam K , Acehan D , Ramos-Espiritu LS , Wang W , Zhang J , Shim JW , Ciceri G , Brichta L , Studer L , Greengard P ((2019) ) Loss of SATB1 induces p21-dependent cellular senescence in post-mitotic dopaminergic neurons. Cell Stem Cell 25: , 514–530e518. |
[76] | Verma DK , Seo BA , Ghosh A , Ma SX , Hernandez-Quijada K , Andersen JK , Ko HS , Kim YH ((2021) ) Alpha-synuclein preformed fibrils induce cellular senescence in Parkinson’s disease models. Cells 10: , 1694. |
[77] | Bigan E , Sasidharan Nair S , Lejeune FX , Fragnaud H , Parmentier F , Mégret L , Verny M , Aaronson J , Rosinski J , Neri C ((2020) ) Genetic cooperativity in multi-layer networks implicates cell survival and senescence in the striatum of Huntington’s disease mice synchronous to symptoms. Bioinformatics 36: , 186–196. |
[78] | Birger A , Ben-Dor I , Ottolenghi M , Turetsky T , Gil Y , Sweetat S , Perez L , Belzer V , Casden N , Steiner D , Izrael M , Galun E , Feldman E , Behar O , Reubinoff B ((2019) ) Human iPSC-derived astrocytes from ALS patients with mutated C9ORF72 show increased oxidative stress and neurotoxicity. E Bio Medicine 50: , 274–289. |
[79] | Vazquez-Villaseñor I , Garwood CJ , Heath PR , Simpson JE , Ince PG , Wharton SB ((2020) ) Expression of p16 and p21 in the frontal association cortex of ALS/MND brains suggests neuronal cell cycle dysregulation and astrocyte senescence in early stages of the disease. Neuropathol Appl Neurobiol 46: , 171–185. |
[80] | Si Z , Sun L , Wang X ((2021) ) Evidence and perspectives of cell senescence in neurodegenerative diseases. Biomed Pharmacother 137: , 111327. |
[81] | Bhat R , Crowe EP , Bitto A , Moh M , Katsetos CD , Garcia FU , Johnson FB , Trojanowski JQ , Sell C , Torres C ((2012) ) Astrocyte senescence as a component of Alzheimer’s disease. PLoS One 7: , e45069. |
[82] | Limbad C , Oron TR , Alimirah F , Davalos AR , Tracy TE , Gan L , Desprez PY , Campisi J ((2020) ) Astrocyte senescence promotes glutamate toxicity in cortical neurons. PLoS One 15: , e0227887. |
[83] | Bryant AG , Hu M , Carlyle BC , Arnold SE , Frosch MP , Das S , Hyman BT , Bennett RE ((2020) ) Cerebrovascular senescence is associated with tau pathology in Alzheimer’s disease. Front Neurol 11: , 575953. |
[84] | von Zglinicki T , Wan T , Miwa S ((2021) ) Senescence in post-mitotic cells: A driver of aging? . Antioxid Redox Signal 34: , 308–323. |
[85] | Eckermann M , Schmitzer B , van der Meer F , Franz J , Hansen O , Stadelmann C , Salditt T ((2021) ) Three-dimensional virtual histology of the human hippocampus based on phase-contrast computed tomography. Proc Natl Acad Sci U S A 118: , e2113835118. |
[86] | Dehkordi SK , Walker J , Sah E , Bennett E , Atrian F , Frost B , Woost B , Bennett RE , Orr TC , Zhou Y , Andhey PS , Colonna M , Sudmant PH , Xu P , Wang M , Zhang B , Zare H , Orr ME ((2021) ) Profiling senescent cells in human brains reveals neurons with CDKN2D/p19 and tau neuropathology. Nat Aging 1: , 1107–1116. |
[87] | Arendt T , Holzer M , Gartner U ((1998) ) Neuronal expression of cycline dependent kinase inhibitors of the INK4 family in Alzheimer’s disease. J Neural Transm (Vienna) 105: , 949–960. |
[88] | McShea A , Harris PL , Webster KR , Wahl AF , Smith MA ((1997) ) Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer’s disease. Am J Pathol 150: , 1933–1939. |
[89] | Sun A , Liu M , Nguyen XV , Bing G ((2003) ) P38 MAP kinase is activated at early stages in Alzheimer’s disease brain. Exp Neurol 183: , 394–405. |
[90] | Hayflick L , Moorhead PS ((1961) ) The serial cultivation of human diploid cell strains. Exp Cell Res 25: , 585–621. |
[91] | de Magalhaes JP , Passos JF ((2018) ) Stress, cell senescence and organismal ageing. Mech Ageing Dev 170: , 2–9. |
[92] | Storer M , Mas A , Robert-Moreno A , Pecoraro M , Ortells MC , Di Giacomo V , Yosef R , Pilpel N , Krizhanovsky V , Sharpe J , Keyes WM ((2013) ) Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 155: , 1119–1130. |
[93] | Jia G , Aroor AR , Jia C , Sowers JR ((2019) ) Endothelial cell senescence in aging-related vascular dysfunction. Biochim Biophys Acta Mol Basis Dis 1865: , 1802–1809. |
[94] | Simpson RJ , Cosgrove C , Ingram LA , Florida-James GD , Whyte GP , Pircher H , Guy K ((2008) ) Senescent T-lymphocytes are mobilised into the peripheral blood compartment in young and older humans after exhaustive exercise. Brain Behav Immun 22: , 544–551. |
[95] | Wang J , Uryga AK , Reinhold J , Figg N , Baker L , Finigan A , Gray K , Kumar S , Clarke M , Bennett M ((2015) ) Vascular smooth muscle cell senescence promotes atherosclerosis and features of plaque vulnerability. Circulation 132: , 1909–1919. |
[96] | Shay JW , Wright WE ((2000) ) Hayflick, his limit, and cellular ageing. Nat Rev Mol Cell Biol 1: , 72–76. |
[97] | Gillooly JF , Hayward A , Hou C , Burleigh JG ((2012) ) Explaining differences in the lifespan and replicative capacity of cells: A general model and comparative analysis of vertebrates. Proc Biol Sci 279: , 3976–3980. |
[98] | Harley CB , Futcher AB , Greider CW ((1990) ) Telomeres shorten during ageing of human fibroblasts. Nature 345: , 458–460. |
[99] | Aikata H , Takaishi H , Kawakami Y , Takahashi S , Kitamoto M , Nakanishi T , Nakamura Y , Shimamoto F , Kajiyama G , Ide T ((2000) ) Telomere reduction in human liver tissues with age and chronic inflammation. Exp Cell Res 256: , 578–582. |
[100] | Effros RB ((2009) ) Kleemeier Award Lecture 2008–the canary in the coal mine: Telomeres and human healthspan. J Gerontol A Biol Sci Med Sci 64: , 511–515. |
[101] | Pan C , Xue BH , Ellis TM , Peace DJ , Diaz MO ((1997) ) Changes in telomerase activity and telomere length during human T lymphocyte senescence. Exp Cell Res 231: , 346–353. |
[102] | Hastie ND , Dempster M , Dunlop MG , Thompson AM , Green DK , Allshire RC ((1990) ) Telomere reduction in human colorectal carcinoma and with ageing. Nature 346: , 866–868. |
[103] | Allsopp RC , Vaziri H , Patterson C , Goldstein S , Younglai EV , Futcher AB , Greider CW , Harley CB ((1992) ) Telomere length predicts replicative capacity of human fibroblasts. Proc Natl Acad Sci U S A 89: , 10114–10118. |
[104] | Kammori M , Nakamura K , Kawahara M , Mimura Y , Kaminishi M , Takubo K ((2002) ) Telomere shortening with aging in human thyroid and parathyroid tissue. Exp Gerontol 37: , 513–521. |
[105] | Njajou OT , Cawthon RM , Damcott CM , Wu SH , Ott S , Garant MJ , Blackburn EH , Mitchell BD , Shuldiner AR , Hsueh WC ((2007) ) Telomere length is paternally inherited and is associated with parental lifespan. Proc Natl Acad Sci U S A 104: , 12135–12139. |
[106] | Mondello C , Petropoulou C , Monti D , Gonos ES , Franceschi C , Nuzzo F ((1999) ) Telomere length in fibroblasts and blood cells from healthy centenarians. Exp Cell Res 248: , 234–242. |
[107] | Nwosu BU , Nilsson O , Mitchum RD Jr , Coco M , Barnes KM , Baron J ((2005) ) Lack of telomere shortening with age in mouse resting zone chondrocytes. Horm Res 63: , 125–128. |
[108] | Renault V , Thornell LE , Eriksson PO , Butler-Browne G , Mouly V ((2002) ) Regenerative potential of human skeletal muscle during aging. Aging Cell 1: , 132–139. |
[109] | Serra V , von Zglinicki T ((2002) ) Human fibroblasts senesce with a donor-specific telomere length. FEBS Lett 516: , 71–74. |
[110] | Takubo K , Izumiyama-Shimomura N , Honma N , Sawabe M , Arai T , Kato M , Oshimura M , Nakamura K ((2002) ) Telomere lengths are characteristic in each human individual. Exp Gerontol 37: , 523–531. |
[111] | Gomes NM , Ryder OA , Houck ML , Charter SJ , Walker W , Forsyth NR , Austad SN , Venditti C , Pagel M , Shay JW , Wright WE ((2011) ) Comparative biology of mammalian telomeres: Hypotheses on ancestral states and the roles of telomeres in longevity determination. Aging Cell 10: , 761–768. |
[112] | de Lange T ((2005) ) Shelter: Therotein complex that shapes and safeguards human telomeres . Genes Dev 19: , 2100–2110. |
[113] | Griffith JD , Comeau L , Rosenfield S , Stansel RM , Bianchi A , Moss H , de Lange T ((1999) ) Mammalian telomeres end in a large duplex loop. Cell 97: , 503–514. |
[114] | Jacobs JJ ((2013) ) Loss of telomere protection: Consequences and opportunities. Front Oncol 3: , 88. |
[115] | Takai H , Smogorzewska A , de Lange T ((2003) ) DNA damage foci at dysfunctional telomeres. Curr Biol 13: , 1549–1556. |
[116] | Artandi SE , Attardi LD ((2005) ) Pathways connecting telomeres and p53 in senescence, apoptosis, and cancer. Biochem Biophys Res Commun 331: , 881–890. |
[117] | Jackson SP , Bartek J ((2009) ) The DNA-damage response in human biology and disease. Nature 461: , 1071–1078. |
[118] | Ciccia A , Elledge SJ ((2010) ) The DNA damage response: Making it safe to play with knives. Mol Cell 40: , 179–204. |
[119] | Giglia-Mari G , Zotter A , Vermeulen W ((2011) ) DNA damage response. Cold Spring Harb Perspect Biol 3: , a000745. |
[120] | Chatterjee N , Walker GC ((2017) ) Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen 58: , 235–263. |
[121] | Nussenzweig A , Nussenzweig MC ((2007) ) A backup DNA repair pathway moves to the forefront. Cell 131: , 223–225. |
[122] | Wu J , Starr S ((2014) ) Low-fidelity compensatory backup alternative DNA repair pathways may unify current carcinogenesis theories. Future Oncol 10: , 1239–1253. |
[123] | Hewitt G , Jurk D , Marques FD , Correia-Melo C , Hardy T , Gackowska A , Anderson R , Taschuk M , Mann J , Passos JF ((2012) ) Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat Commun 3: , 708. |
[124] | Petersen S , Saretzki G , von Zglinicki T ((1998) ) Preferential accumulation of single-stranded regions in telomeres of human fibroblasts. Exp Cell Res 239: , 152–160. |
[125] | Chan SW , Blackburn EH ((2002) ) New ways not to make ends meet: Telomerase, DNA damage proteins and heterochromatin. Oncogene 21: , 553–563. |
[126] | Marcand S , Pardo B , Gratias A , Cahun S , Callebaut I ((2008) ) Multiple pathways inhibit NHEJ at telomeres. Genes Dev 22: , 1153–1158. |
[127] | Mao P , Liu J , Zhang Z , Zhang H , Liu H , Gao S , Rong YS , Zhao Y ((2016) ) Homologous recombination-dependent repair of telomeric DSBs in proliferating human cells. Nat Commun 7: , 12154. |
[128] | Verdun RE , Karlseder J ((2006) ) The DNA damage machinery and homologous recombination pathway act consecutively to protect human telomeres. Cell 127: , 709–720. |
[129] | Ghadaouia S , Olivier MA , Martinez A , Kientega T , Qin J , Lambert-Lanteigne P , Cardin GB , Autexier C , Malaquin N , Rodier F ((2021) ) Homologous recombination-mediated irreversible genome damage underlies telomere-induced senescence. Nucleic Acids Res 49: , 11690–11707. |
[130] | Fumagalli M , Rossiello F , Mondello C , d’Adda di Fagagna F ((2014) ) Stable cellular senescence is associated with persistent DDR activation. PLoS One 9: , e110969. |
[131] | Wengerodt D , Schmeer C , Witte OW , Kretz A ((2019) ) Amitosenescence and pseudomitosenescence: Putative new players in the aging process. Cells 8: , 1546. |
[132] | Feringa FM , Raaijmakers JA , Hadders MA , Vaarting C , Macurek L , Heitink L , Krenning L , Medema RH ((2018) ) Persistent repair intermediates induce senescence. Nat Commun 9: , 3923. |
[133] | Brack C , Lithgow G , Osiewacz H , Toussaint O ((2000) ) EMBO WORKSHOP REPORT: Molecular and cellular gerontology Serpiano, Switzerland, September 18-22, 1999. EMBO J 19: , 1929–1934. |
[134] | Bielak-Zmijewska A , Mosieniak G , Sikora E ((2018) ) Is DNA damage indispensable for stress-induced senescence? . Mech Ageing Dev 170: , 13–21. |
[135] | Illis LS ((2012) ) Central nervous system regeneration does not occur. Spinal Cord 50: , 259–263. |
[136] | Chow HM , Herrup K ((2015) ) Genomic integrity and the ageing brain. Nat Rev Neurosci 16: , 672–684. |
[137] | Levy WB , Calvert VG ((2021) ) Communication consumes 35 times more energy than computation in the human cortex, but both costs are needed to predict synapse number. Proc Natl Acad Sci U S A 118: , e2008173118. |
[138] | Kann O , Kovacs R ((2007) ) Mitochondria and neuronal activity. , C. Am J Physiol Cell Physiol 292: , 641–657. |
[139] | Wang X , Michaelis EK ((2010) ) Selective neuronal vulnerability to oxidative stress in the brain. Front Aging Neurosci 2: , 12. |
[140] | Beckhauser TF , Francis-Oliveira J , De Pasquale R ((2016) ) Reactive oxygen species: Physiological and physiopathological effects on synaptic plasticity. J Exp Neurosci 10: , 23–48. |
[141] | Dhawan S , Myers P , Bailey DMD , Ostrovsky AD , Evers JF , Landgraf M ((2021) ) Reactive oxygen species mediate activity-regulated dendritic plasticity through NADPH oxidase and aquaporin regulation. Front Cell Neurosci 15: , 641802. |
[142] | Loebrich S , Nedivi E ((2009) ) The function of activity-regulated genes in the nervous system. Physiol Rev 89: , 1079–1103. |
[143] | Jauregui-Lozano J , Escobedo S , Easton A , Lanman NA , Weake VM , Hall H ((2022) ) Proper control of R-loop homeostasis is required for maintenance of gene expression and neuronal function during aging. Aging Cell 21: , e13554. |
[144] | Belotserkovskii BP , Tornaletti S , D’Souza AD , Hanawalt PC ((2018) ) R-loop generation during transcription: Formation, processing and cellular outcomes. DNA Repair (Amst) 71: , 69–81. |
[145] | McKinnon PJ ((2016) ) Topoisomerases and the regulation of neural function. Nat Rev Neurosci 17: , 673–679. |
[146] | Minatohara K , Akiyoshi M , Okuno H ((2015) ) Role of immediate-early genes in synaptic plasticity and neuronal ensembles underlying the memory trace. Front Mol Neurosci 8: , 78. |
[147] | Sun X , Lin Y ((2016) ) Npas4: Linking neuronal activity to memory. Trends Neurosci 39: , 264–275. |
[148] | Gallo FT , Katche C , Morici JF , Medina JH , Weisstaub NV ((2018) ) Immediate early genes, memory and psychiatric disorders: Focus on c-Fos, Egr1 and Arc. Front Behav Neurosci 12: , 79. |
[149] | Baker-Andresen D , Ratnu VS , Bredy TW ((2013) ) Dynamic DNA methylation: A prime candidate for genomic metaplasticity and behavioral adaptation. Trends Neurosci 36: , 3–13. |
[150] | Li X , Wei W , Zhao QY , Widagdo J , Baker-Andresen D , Flavell CR , D’Alessio A , Zhang Y , Bredy TW ((2014) ) Neocortical Tet3-mediated accumulation of 5-hydroxymethylcytosine promotes rapid behavioral adaptation. Proc Natl Acad Sci U S A 111: , 7120–7125. |
[151] | Li X , Marshall PR , Leighton LJ , Zajaczkowski EL , Wang Z , Madugalle SU , Yin J , Bredy TW , Wei W ((2019) ) The DNA repair-associated protein Gadd45gamma regulates the temporal coding of immediate early gene expression within the prelimbic prefrontal cortex and is required for the consolidation of associative fear memory. J Neurosci 39: , 970–983. |
[152] | Barreto G , Schafer A , Marhold J , Stach D , Swaminathan SK , Handa V , Doderlein G , Maltry N , Wu W , Lyko F , Niehrs C ((2007) ) Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature 445: , 671–675. |
[153] | Rai K , Huggins IJ , James SR , Karpf AR , Jones DA , Cairns BR ((2008) ) DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and gadd45. Cell 135: , 1201–1212. |
[154] | Ma DK , Guo JU , Ming GL , Song H ((2009) ) DNA excision repair proteins and Gadd45 as molecular players for active DNA demethylation. Cell Cycle 8: , 1526–1531. |
[155] | Madabhushi R , Gao F , Pfenning AR , Pan L , Yamakawa S , Seo J , Rueda R , Phan TX , Yamakawa H , Pao PC , Stott RT , Gjoneska E , Nott A , Cho S , Kellis M , Tsai LH ((2015) ) Activity-induced DNA breaks govern the expression of neuronal early-response genes. Cell 161: , 1592–1605. |
[156] | Suberbielle E , Sanchez PE , Kravitz AV , Wang X , Ho K , Eilertson K , Devidze N , Kreitzer AC , Mucke L ((2013) ) Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-beta. Nat Neurosci 16: , 613–621. |
[157] | Suberbielle E , Djukic B , Evans M , Kim DH , Taneja P , Wang X , Finucane M , Knox J , Ho K , Devidze N , Masliah E , Mucke L ((2015) ) DNA repair factor BRCA1 depletion occurs in Alzheimer brains and impairs cognitive function in mice. Nat Commun 6: , 8897. |
[158] | Peze-Heidsieck E , Bonnifet T , Znaidi R , Ravel-Godreuil C , Massiani-Beaudoin O , Joshi RL , Fuchs J , Retrotransposons as a source of DNA damage in neurodegeneration. Front Aging Neurosci 13: , 786897. |
[159] | Martinez-Vicente M , Sovak G , Cuervo AM ((2005) ) Protein degradation and aging. Exp Gerontol 40: , 622–633. |
[160] | Nixon RA ((2013) ) The role of autophagy in neurodegenerative disease. Nat Med 19: , 983–997. |
[161] | Nixon RA , Yang DS , Lee JH ((2008) ) Neurodegenerative lysosomal disorders: A continuum from development to late age. Autophagy 4: , 590–599. |
[162] | Nixon RA ((2020) ) The aging lysosome: An essential catalyst for late-onset neurodegenerative diseases. Biochim Biophys Acta Proteins Proteom 1868: , 140443. |
[163] | Rodrigue KM , Kennedy KM , Park DC ((2009) ) Beta-amyloid deposition and the aging brain. Neuropsychol Rev 19: , 436–450. |
[164] | Lan YY , Londono D , Bouley R , Rooney MS , Hacohen N ((2014) ) Dnase2a deficiency uncovers lysosomal clearance of damaged nuclear DNA via autophagy. Cell Rep 9: , 180–192. |
[165] | Lan YY , Heather JM , Eisenhaure T , Garris CS , Lieb D , Raychowdhury R , Hacohen N ((2019) ) Extranuclear DNA accumulates in aged cells and contributes to senescence and inflammation. Aging Cell 18: , e12901. |
[166] | Rodier F , Munoz DP , Teachenor R , Chu V , Le O , Bhaumik D , Coppe JP , Campeau E , Beausejour CM , Kim SH , Davalos AR , Campisi J ((2011) ) DNA-SCARS: Distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. J Cell Sci 124: , 68–81. |
[167] | Ivanov A , Pawlikowski J , Manoharan I , van Tuyn J , Nelson DM , Rai TS , Shah PP , Hewitt G , Korolchuk VI , Passos JF , Wu H , Berger SL , Adams PD ((2013) ) Lysosome-mediated processing of chromatin in senescence. J Cell Biol 202: , 129–143. |
[168] | Lu T , Pan Y , Kao SY , Li C , Kohane I , Chan J , Yankner BA ((2004) ) Gene regulation and DNA damage in the ageing human brain. Nature 429: , 883–891. |
[169] | Wu W , Hill SE , Nathan WJ , Paiano J , Callen E , Wang D , Shinoda K , van Wietmarschen N , Colon-Mercado JM , Zong D , De Pace R , Shih HY , Coon S , Parsadanian M , Pavani R , Hanzlikova H , Park S , Jung SK , McHugh PJ , Canela A , Chen C , Casellas R , Caldecott KW , Ward ME , Nussenzweig A ((2021) ) Neuronal enhancers are hotspots for DNA single-strand break repair. Nature 593: , 440–444. |
[170] | Lodato MA , Rodin RE , Bohrson CL , Coulter ME , Barton AR , Kwon M , Sherman MA , Vitzthum CM , Luquette LJ , Yandava CN , Yang P , Chittenden TW , Hatem NE , Ryu SC , Woodworth MB , Park PJ , Walsh CA ((2018) ) Aging and neurodegeneration are associated with increased mutations in single human neurons. Science 359: , 555–559. |
[171] | Bae T , Tomasini L , Mariani J , Zhou B , Roychowdhury T , Franjic D , Pletikos M , Pattni R , Chen BJ , Venturini E , Riley-Gillis B , Sestan N , Urban AE , Abyzov A , Vaccarino FM ((2018) ) Different mutational rates and mechanisms in human cells at pregastrulation and neurogenesis. Science 359: , 550–555. |
[172] | Ataian Y , Krebs JE ((2006) ) Five repair pathways in one context: Chromatin modification during DNA repair. Biochem Cell Biol 84: , 490–504. |
[173] | Iyama T , Wilson DM 3rd ((2013) ) DNA repair mechanisms in dividing and non-dividing cells. DNA Repair (Amst) 12: , 620–636. |
[174] | Welty S , Teng Y , Liang Z , Zhao W , Sanders LH , Greenamyre JT , Rubio ME , Thathiah A , Kodali R , Wetzel R , Levine AS , Lan L ((2018) ) RAD52 is required for RNA-templated recombination repair in post-mitotic neurons. J Biol Chem 293: , 1353–1362. |
[175] | Brasnjevic I , Hof PR , Steinbusch HW , Schmitz C ((2008) ) Accumulation of nuclear DNA damage or neuron loss: Molecular basis for a new approach to understanding selective neuronal vulnerability in neurodegenerative diseases. DNA Repair (Amst) 7: , 1087–1097. |
[176] | Huang K ((2010) ) DNA repair deficiency in a newly identified neurological disease. Clin Genet 78: , 418–419. |
[177] | McKinnon PJ ((2009) ) DNA repair deficiency and neurological disease. Nat Rev Neurosci 10: , 100–112. |
[178] | Andrews AD , Barrett SF , Robbins JH ((1978) ) Xeroderma pigmentosum neurological abnormalities correlate with colony-forming ability after ultraviolet radiation. Proc Natl Acad Sci U S A 75: , 1984–1988. |
[179] | Uribe-Bojanini E , Hernandez-Quiceno S , Cock-Rada AM ((2017) ) Xeroderma pigmentosum with severe neurological manifestations/De Sanctis-Cacchione syndrome and a novel XPC mutation. Case Rep Med 2017: , 7162737. |
[180] | van Os NJH , van Deuren M , Weemaes CMR , van Gaalen J , Hijdra H , Taylor AMR , van de Warrenburg BPC , Willemsen M ((2020) ) Classic ataxia-telangiectasia: The phenotype of long-term survivors. J Neurol 267: , 830–837. |
[181] | Wang H , Lautrup S , Caponio D , Zhang J , Fang EF ((2021) ) DNA damage-induced neurodegeneration in accelerated ageing and Alzheimer’s disease. Int J Mol Sci 22: , 6748. |
[182] | Nouspikel T , Hanawalt PC ((2000) ) Terminally differentiated human neurons repair transcribed genes but display attenuated global DNA repair and modulation of repair gene expression. Mol Cell Biol 20: , 1562–1570. |
[183] | Starling S ((2018) ) Ageing: Somatic mutations accumulate in ageing and diseased neurons. Nat Rev Neurol 14: , 64. |
[184] | Herrup K , Neve R , Ackerman SL , Copani A ((2004) ) Divide and die: Cell cycle events as triggers of nerve cell death. J Neurosci 24: , 9232–9239. |
[185] | Martin LJ , Liu Z , Pipino J , Chestnut B , Landek MA ((2009) ) Molecular regulation of DNA damage-induced apoptosis in neurons of cerebral cortex. Cereb Cortex 19: , 1273–1293. |
[186] | Tuxworth RI , Taylor MJ , Martin Anduaga A , Hussien-Ali A , Chatzimatthaiou S , Longland J , Thompson AM , Almutiri S , Alifragis P , Kyriacou CP , Kysela B , Ahmed Z ((2019) ) Attenuating the DNA damage response to double-strand breaks restores function in models of CNS neurodegeneration. Brain Commun 1: , fcz005. |
[187] | Kruman II , Wersto RP , Cardozo-Pelaez F , Smilenov L , Chan SL , Chrest FJ , Emokpae R Jr , Gorospe M , Mattson MP ((2004) ) Cell cycle activation linked to neuronal cell death initiated by DNA damage. Neuron 41: , 549–561. |
[188] | Gupta S , You P , SenGupta T , Nilsen H , Sharma K ((2021) ) Crosstalk between different DNA repair pathways contributes to neurodegenerative diseases. Biology (Basel) 10: . |
[189] | Shen X , Chen J , Li J , Kofler J , Herrup K ((2016) ) Neurons in vulnerable regions of the Alzheimer’s disease brain display reduced ATM signaling. eNeuro 3: , 163. |
[190] | Kanungo J ((2016) ) DNA-PK deficiency in Alzheimer’s disease. J Neurol Neuromedicine 1: , 17–22. |
[191] | Jacobsen E , Beach T , Shen Y , Li R , Chang Y ((2004) ) Deficiency of the Mre11 DNA repair complex in Alzheimer’s disease brains. Brain Res Mol Brain Res 128: , 1–7. |
[192] | Lovell MA , Xie C , Markesbery WR ((2000) ) Decreased base excision repair and increased helicase activity in Alzheimer’s disease brain. Brain Res 855: , 116–123. |
[193] | Weissman L , Jo DG , Sorensen MM , de Souza-Pinto NC , Markesbery WR , Mattson MP , Bohr VA ((2007) ) Defective DNA base excision repair in brain from individuals with Alzheimer’s disease and amnestic mild cognitive impairment. Nucleic Acids Res 35: , 5545–5555. |
[194] | Sfeir A , Symington LS ((2015) ) Microhomology-mediated end joining: A back-up survival mechanism or dedicated pathway? . Trends Biochem Sci 40: , 701–714. |
[195] | Herrup K , Li J , Chen J ((2013) ) The role of ATM and DNA damage in neurons: Upstream and downstream connections. DNA Repair (Amst) 12: , 600–604. |
[196] | Iijima-Ando K , Zhao L , Gatt A , Shenton C , Iijima K ((2010) ) A DNA damage-activated checkpoint kinase phosphorylates tau and enhances tau-induced neurodegeneration. Hum Mol Genet 19: , 1930–1938. |
[197] | Guo Z , Tian Y , Guo Y , Li B , Liu X , Xie K , Song Y , Wang D ((2019) ) RAD6B plays a critical role in neuronal DNA damage response to resist neurodegeneration. Front Cell Neurosci 13: , 392. |
[198] | Shanbhag NM , Evans MD , Mao W , Nana AL , Seeley WW , Adame A , Rissman RA , Masliah E , Mucke L ((2019) ) Early neuronal accumulation of DNA double strand breaks in Alzheimer’s disease. Acta Neuropathol Commun 7: , 77. |
[199] | Yang J , Xu ZP , Huang Y , Hamrick HE , Duerksen-Hughes PJ , Yu YN ((2004) ) ATM and ATR: Sensing DNA damage. World J Gastroenterol 10: , 155–160. |
[200] | Marechal A , Zou L ((2013) ) DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol 5: , a012716. |
[201] | Schwartz EI , Smilenov LB , Price MA , Osredkar T , Baker RA , Ghosh S , Shi FD , Vollmer TL , Lencinas A , Stearns DM , Gorospe M , Kruman II ((2007) ) Cell cycle activation in postmitotic neurons is essential for DNA repair. Cell Cycle 6: , 318–329. |
[202] | Tomashevski A , Webster DR , Grammas P , Gorospe M , Kruman II ((2010) ) Cyclin-C-dependent cell-cycle entry is required for activation of non-homologous end joining DNA repair in postmitotic neurons. Cell Death Differ 17: , 1189–1198. |
[203] | Davydov V , Hansen LA , Shackelford DA ((2003) ) Is DNA repair compromised in Alzheimer’s disease? . Neurobiol Aging 24: , 953–968. |
[204] | Decottignies A ((2013) ) Alternative end-joining mechanisms: A historical perspective. Front Genet 4: , 48. |
[205] | Shibata A , Jeggo PA ((2021) ) ATM’s role in the repair of DNA double-strand breaks. Genes (Basel) 12: , 1370. |
[206] | Ottaviani D , LeCain M , Sheer D ((2014) ) The role of microhomology in genomic structural variation. Trends Genet 30: , 85–94. |
[207] | Densham RM , Morris JR ((2019) ) Moving mountains-the BRCA1 promotion of DNA resection. Front Mol Biosci 6: , 79–79. |
[208] | Truong LN , Li Y , Shi LZ , Hwang PY , He J , Wang H , Razavian N , Berns MW , Wu X ((2013) ) Microhomology-mediated end joining and homologous recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc Natl Acad Sci U S A 110: , 7720–7725. |
[209] | Liu S , Kong D ((2021) ) End resection: A key step in homologous recombination and DNA double-strand break repair. Genome Instab Dis 2: , 39–50. |
[210] | Cortez D , Guntuku S , Qin J , Elledge SJ ((2001) ) ATR and ATRIP: Partners in checkpoint signaling. Science 294: , 1713–1716. |
[211] | Zou L , Elledge SJ ((2003) ) Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300: , 1542–1548. |
[212] | Baumann P , Benson FE , West SC ((1996) ) Human Rad51 protein promotes ATP-dependent homologous pairing and strand transfer reactions. Cell 87: , 757–766. |
[213] | Ward IM , Chen J ((2001) ) Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem 276: , 47759–47762. |
[214] | Petermann E , Caldecott KW ((2006) ) Evidence that the ATR/Chk1 pathway maintains normal replication fork progression during unperturbed S phase. Cell Cycle 5: , 2203–2209. |
[215] | Yurov YB , Vorsanova SG , Iourov IY ((2011) ) The DNA replication stress hypothesis of Alzheimer’s disease. ScientificWorldJournal 11: , 2602–2612. |
[216] | Haigis MC , Yankner BA ((2010) ) The aging stress response. Mol Cell 40: , 333–344. |
[217] | Liu S , Bekker-Jensen S , Mailand N , Lukas C , Bartek J , Lukas J ((2006) ) Claspin operates downstream of TopBP1 to direct ATR signaling towards Chk1 activation. Mol Cell Biol 26: , 6056–6064. |
[218] | Brown AD , Sager BW , Gorthi A , Tonapi SS , Brown EJ , Bishop AJ ((2014) ) ATR suppresses endogenous DNA damage and allows completion of homologous recombination repair. PLoS One 9: , e91222. |
[219] | Bonda DJ , Evans TA , Santocanale C , Llosa JC , Vina J , Bajic V , Castellani RJ , Siedlak SL , Perry G , Smith MA , Lee HG ((2009) ) Evidence for the progression through S-phase in the ectopic cell cycle re-entry of neurons in Alzheimer disease. Aging (Albany NY) 1: , 382–388. |
[220] | Barrio-Alonso E , Hernandez-Vivanco A , Walton CC , Perea G , Frade JM ((2018) ) Cell cycle reentry triggers hyperploidization and synaptic dysfunction followed by delayed cell death in differentiated cortical neurons. Sci Rep 8: , 14316. |
[221] | van Leeuwen LA , Hoozemans JJ ((2015) ) Physiological and pathophysiological functions of cell cycle proteins in post-mitotic neurons: Implications for Alzheimer’s disease. Acta Neuropathol 129: , 511–525. |
[222] | Moh C , Kubiak JZ , Bajic VP , Zhu X , Smith MA , Lee HG ((2011) ) Cell cycle deregulation in the neurons of Alzheimer’s disease. Results Probl Cell Differ 53: , 565–576. |
[223] | Kumar P , Raman T , Swain MM , Mishra R , Pal A ((2017) ) Hyperglycemia-induced oxidative-nitrosative stress induces inflammation and neurodegeneration via augmented tuberous sclerosis complex-2 (TSC-2) activation in neuronal cells. Mol Neurobiol 54: , 238–254. |
[224] | Torres-Querol C , Torres P , Vidal N , Portero-Otin M , Arque G , Purroy F ((2021) ) Acute ischemic stroke triggers a cellular senescence-associated secretory phenotype. Sci Rep 11: , 15752. |
[225] | Rashidian J , Iyirhiaro GO , Park DS ((2007) ) Cell cycle machinery and stroke. Biochim Biophys Acta 1772: , 484–493. |
[226] | Aubrecht TG , Faden AI , Sabirzhanov B , Glaser EP , Roelofs BA , Polster BM , Makarevich O , Stoica BA ((2018) ) Comparing effects of CDK inhibition and E2F1/2 ablation on neuronal cell death pathways and after traumatic brain injury. Cell Death Dis 9: , 1121. |
[227] | Nandakumar S , Rozich E , Buttitta L ((2021) ) Cell cycle re-entry in the nervous system: From polyploidy to neurodegeneration. Front Cell Dev Biol 9: , 698661. |
[228] | Frade JM , Ovejero-Benito MC ((2015) ) Neuronal cell cycle: The neuron itself and its circumstances. Cell Cycle 14: , 712–720. |
[229] | Zhang XP , Liu F , Cheng Z , Wang W ((2009) ) Cell fate decision mediated by p53 pulses. Proc Natl Acad Sci U S A 106: , 12245–12250. |
[230] | Shadfar S , Brocardo M , Atkin JD ((2022) ) The complex mechanisms by which neurons die following DNA damage in neurodegenerative diseases. Int J Mol Sci 23: , 2484. |
[231] | Vaughn AE , Deshmukh M ((2007) ) Essential postmitochondrial function of p53 uncovered in DNA damage-induced apoptosis in neurons. Cell Death Differ 14: , 973–981. |
[232] | Son JH , Shim JH , Kim KH , Ha JY , Han JY ((2012) ) Neuronal autophagy and neurodegenerative diseases. Exp Mol Med 44: , 89–98. |
[233] | Eliopoulos AG , Havaki S , Gorgoulis VG ((2016) ) DNA damage response and autophagy: A meaningful partnership. Front Genet 7: , 204. |
[234] | Gomes LR , Menck CFM , Leandro GS ((2017) ) Autophagy roles in the modulation of DNA repair pathways. Int J Mol Sci 18: , 2351. |
[235] | Liu Q , Qiu J , Liang M , Golinski J , van Leyen K , Jung JE , You Z , Lo EH , Degterev A , Whalen MJ ((2014) ) Akt and mTOR mediate programmed necrosis in neurons. Cell Death Dis 5: , e1084. |
[236] | Xu C , Wu J , Wu Y , Ren Z , Yao Y , Chen G , Fang EF , Noh JH , Liu YU , Wei L , Chen X , Sima J ((2021) ) TNF-alpha-dependent neuronal necroptosis regulated in Alzheimer’s disease by coordination of RIPK1-p62 complex with autophagic UVRAG. Theranostics 11: , 9452–9469. |
[237] | Lim J , Park H , Heisler J , Maculins T , Roose-Girma M , Xu M , McKenzie B , van Lookeren Campagne M , Newton K , Murthy A ((2019) ) Autophagy regulates inflammatory programmed cell death via turnover of RHIM-domain proteins. Elife 8: , e44452. |
[238] | Herrup K , Yang Y ((2007) ) Cell cycle regulation in the postmitotic neuron: Oxymoron or new biology? . Nat Rev Neurosci 8: , 368–378. |
[239] | Sah E , Krishnamurthy S , Ahmidouch MY , Gillispie GJ , Milligan C , Orr ME ((2021) ) The cellular senescence stress response in post-mitotic brain cells: Cell survival at the expense of tissue degeneration.. Life (Basel) 11: , 229. |
[240] | Magrassi L , Leto K , Rossi F ((2013) ) Lifespan of neurons is uncoupled from organismal lifespan. Proc Natl Acad Sci U S A 110: , 4374–4379. |
[241] | Krenning L , Feringa FM , Shaltiel IA , van den Berg J , Medema RH ((2014) ) Transient activation of p53 in G2 phase is sufficient to induce senescence. Mol Cell 55: , 59–72. |
[242] | Mullers E , Silva Cascales H , Jaiswal H , Saurin AT , Lindqvist A ((2014) ) Nuclear translocation of Cyclin B1 marks the restriction point for terminal cell cycle exit in G2 phase. Cell Cycle 13: , 2733–2743. |
[243] | Johmura Y , Shimada M , Misaki T , Naiki-Ito A , Miyoshi H , Motoyama N , Ohtani N , Hara E , Nakamura M , Morita A , Takahashi S , Nakanishi M ((2014) ) Necessary and sufficient role for a mitosis skip in senescence induction. Mol Cell 55: , 73–84. |
[244] | Li F , Lo TY , Miles L , Wang Q , Noristani HN , Li D , Niu J , Trombley S , Goldshteyn JI , Wang C , Wang S , Qiu J , Pogoda K , Mandal K , Brewster M , Rompolas P , He Y , Janmey PA , Thomas GM , Li S , Song Y ((2021) ) The Atr-Chek1 pathway inhibits axon regeneration in response to Piezo-dependent mechanosensation. Nat Commun 12: , 3845. |
[245] | Toledo LI , Murga M , Gutierrez-Martinez P , Soria R , Fernandez-Capetillo O ((2008) ) ATR signaling can drive cells into senescence in the absence of DNA breaks. Genes Dev 22: , 297–302. |
[246] | Ye W , Blain SW ((2010) ) S phase entry causes homocysteine-induced death while ataxia telangiectasia and Rad3 related protein functions anti-apoptotically to protect neurons. Brain 133: , 2295–2312. |
[247] | Vazquez-Villasenor I , Garwood CJ , Simpson JE , Heath PR , Mortiboys H , Wharton SB ((2021) ) Persistent DNA damage alters the neuronal transcriptome suggesting cell cycle dysregulation and altered mitochondrial function. Eur J Neurosci 54: , 6987–7005. |
[248] | Krishnamurthy J , Torrice C , Ramsey MR , Kovalev GI , Al-Regaiey K , Su L , Sharpless NE ((2004) ) Ink4a/Arf expression is a biomarker of aging. J Clin Invest 114: , 1299–1307. |
[249] | Gasek NS , Kuchel GA , Kirkland JL , Xu M ((2021) ) Strategies for targeting senescent cells in human disease. Nat Aging 1: , 870–879. |
[250] | Tchkonia T , Zhu Y , van Deursen J , Campisi J , Kirkland JL ((2013) ) Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J Clin Invest 123: , 966–972. |
[251] | Zhu Y , Tchkonia T , Pirtskhalava T , Gower AC , Ding H , Giorgadze N , Palmer AK , Ikeno Y , Hubbard GB , Lenburg M , O’Hara SP , LaRusso NF , Miller JD , Roos CM , Verzosa GC , LeBrasseur NK , Wren JD , Farr JN , Khosla S , Stout MB , McGowan SJ , Fuhrmann-Stroissnigg H , Gurkar AU , Zhao J , Colangelo D , Dorronsoro A , Ling YY , Barghouthy AS , Navarro DC , Sano T , Robbins PD , Niedernhofer LJ , Kirkland JL ((2015) ) The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 14: , 644–658. |
[252] | Zhu Y , Tchkonia T , Fuhrmann-Stroissnigg H , Dai HM , Ling YY , Stout MB , Pirtskhalava T , Giorgadze N , Johnson KO , Giles CB , Wren JD , Niedernhofer LJ , Robbins PD , Kirkland JL ((2016) ) Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 15: , 428–435. |
[253] | Tchkonia T , Palmer AK , Kirkland JL ((2021) ) New horizons: Novel approaches to enhance healthspan through targeting cellular senescence and related aging mechanisms. J Clin Endocrinol Metab 106: , e1481–e1487. |
[254] | Robbins PD , Jurk D , Khosla S , Kirkland JL , LeBrasseur NK , Miller JD , Passos JF , Pignolo RJ , Tchkonia T , Niedernhofer LJ ((2021) ) Senolytic drugs: Reducing senescent cell viability to extend health span. Annu Rev Pharmacol Toxicol 61: , 779–803. |
[255] | Zhou FQ ((2021) ) NAD(+), senolytics, or pyruvate for healthy aging? . Nutr Metab Insights 14: , 11786388211053407. |
[256] | Lautrup S , Sinclair DA , Mattson MP , Fang EF ((2019) ) NAD(+) in brain aging and neurodegenerative disorders. Cell Metab 30: , 630–655. |
[257] | Wilk A , Hayat F , Cunningham R , Li J , Garavaglia S , Zamani L , Ferraris DM , Sykora P , Andrews J , Clark J , Davis A , Chaloin L , Rizzi M , Migaud M , Sobol RW ((2020) ) Extracellular NAD(+) enhances PARP-dependent DNA repair capacity independently of CD73 activity. Sci Rep 10: , 651. |
[258] | Hassa PO , Hottiger MO ((2008) ) The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADP-ribose polymerases. Front Biosci 13: , 3046–3082. |
[259] | Fang EF , Scheibye-Knudsen M , Brace LE , Kassahun H , SenGupta T , Nilsen H , Mitchell JR , Croteau DL , Bohr VA ((2014) ) Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell 157: , 882–896. |
[260] | Boetefuer EL , Lake RJ , Dreval K , Fan HY ((2018) ) Poly(ADP-ribose) polymerase 1 (PARP1) promotes oxidative stress-induced association of Cockayne syndrome group B protein with chromatin. J Biol Chem 293: , 17863–17874. |
[261] | Haince JF , Kozlov S , Dawson VL , Dawson TM , Hendzel MJ , Lavin MF , Poirier GG ((2007) ) Ataxia telangiectasia mutated (ATM) signaling network is modulated by a novel poly(ADP-ribose)-dependent pathway in the early response to DNA-damaging agents. J Biol Chem 282: , 16441–16453. |
[262] | Braidy N , Berg J , Clement J , Khorshidi F , Poljak A , Jayasena T , Grant R , Sachdev P ((2019) ) Role of nicotinamide adenine dinucleotide and related precursors as therapeutic targets for age-related degenerative diseases: Rationale, biochemistry, pharmacokinetics, and outcomes. Antioxid Redox Signal 30: , 251–294. |
[263] | Fang EF , Kassahun H , Croteau DL , Scheibye-Knudsen M , Marosi K , Lu H , Shamanna RA , Kalyanasundaram S , Bollineni RC , Wilson MA , Iser WB , Wollman BN , Morevati M , Li J , Kerr JS , Lu Q , Waltz TB , Tian J , Sinclair DA , Mattson MP , Nilsen H , Bohr VA ((2016) ) NAD(+) replenishment improves lifespan and healthspan in ataxia telangiectasia models via mitophagy and DNA repair. Cell Metab 24: , 566–581. |
[264] | Scheibye-Knudsen M , Mitchell SJ , Fang EF , Iyama T , Ward T , Wang J , Dunn CA , Singh N , Veith S , Hasan-Olive MM , Mangerich A , Wilson MA , Mattson MP , Bergersen LH , Cogger VC , Warren A , Le Couteur DG , Moaddel R , Wilson DM , 3rd , Croteau DL , de Cabo R , Bohr VA ((2014) ) A high-fat diet and NAD(+) activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab 20: , 840–855. |
[265] | Purvis JE , Karhohs KW , Mock C , Batchelor E , Loewer A , Lahav G ((2012) ) p53 dynamics control cell fate. Science 336: , 1440–1444. |
[266] | Fedak EA , Adler FR , Abegglen LM , Schiffman JD ((2021) ) ATM and ATR activation through crosstalk between DNA damage response pathways. Bull Math Biol 83: , 38. |
[267] | Kirschner K , Rattanavirotkul N , Quince MF , Chandra T ((2020) ) Functional heterogeneity in senescence. Biochem Soc Trans 48: , 765–773. |


