
Integrating molecular interactions and gene expression to identify biomarkers and network modules of chronic obstructive pulmonary disease
Abstract
BACKGROUND:
Chronic obstructive pulmonary disease (COPD) causes chronic obstructive conditions, chronic bronchitis, and emphysema, and is a major cause of death worldwide. Although several efforts for identifying biomarkers and pathways have been made, specific causal COPD mechanism remains unknown.
OBJECTIVE:
This study combined biological interaction data with gene expression data for a better understanding of the biological process and network module for COPD.
METHODS:
Using a sparse network-based method, we selected 49 genes from peripheral blood mononuclear cell expression data of 136 subjects, including 42 ex-smoking controls and 94 subjects with COPD.
RESULTS:
These 49 genes might influence biological processes and molecular functions related to COPD. For example, our result suggests that FoxO signaling may contribute to the atrophy of COPD peripheral muscle tissues via oxidative stress.
CONCLUSIONS:
Our approach enhances the existing understanding of COPD disease pathogenesis and predicts new genetic markers and pathways that may influence COPD pathogenesis.
1.Introduction
Chronic obstructive pulmonary disease (COPD) is the third leading cause of death globally. According to recent estimates, from 2010 to 2030, the number of COPD cases in developed countries will increase by over 150% [1]. COPD is a complex disease, so determining the genetic risk factors for the disease has been challenging. Several studies have been conducted to meet this challenge. Qiu et al. [2] examined the gene expression in COPD patients with related diseases by genome-wide association studies (GWAS) to determine the functional effects of known susceptible genes and find new genes associated with the disease. Sakornsakolpat et al. [3] identified 82 loci that may be associated with either COPD or population-based measures of lung function. However, the GWAS method often generates vast genome-wide “hits” and the cost to examine these “hits” is high. In another study, Bahr et al. [4] used multiple linear regression to adjust covariates to identify COPD candidate genes and pathways. They showed that differentially expressed selected pathways in COPD subjects included those that have a role in the inflammatory response of the immune system. According to Huang et al. [5], the secondary metabolic pathway, the xenobiotic metabolic pathway, and the cellular response to xenobiotic stimuli possibly contribute to the development and advancement of COPD.
Although various efforts have been made for identifying biomarkers and pathways, the specific causal COPD mechanism still remains unraveled. Most of the information generated in these studies is based on single-group data, wherein, the relationships among biological regulatory networks, protein-protein interactions, signaling pathways, and well-known genes in the statistical framework on which they are based have not been considered. In such a framework, the lack of biological information leads to the stability of prediction factors and reduces the predictive ability of the model [6]. Therefore, prior biological knowledge should be incorporated into the model to acquire more reliable and biologically significant results [7, 8, 9].
A few such studies include those of Li and Li [10], Chen et al. [11], and Wang et al. [12] who attempted to use biological knowledge. In their model, genome knowledge was encoded by a network, and its graph structure determined the specific relationship (edge) between genes (nodes). They then integrated the Laplace matrix into the penalty in linear, logistics, and Cox regression models. The network particularly represented many types of biological information such as the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways [7], the correlation between genes [9, 13], functional interaction network [14], or protein-protein interactions (PPIs) [15]. These models which utilize a priori biological knowledge are often called “network-based” methods.
The hypothesis that complex diseases such as COPD arise and develop due to interactions between several interrelated pathogenic genes, is supported by a growing body of evidence, indicating that the evaluation of the influence of any single variant is complicated [16]. In this study, we hypothesize that combining biological interaction information with gene expression data would provide better insight into biological processes and network modules for COPD. Therefore, we tried to unravel the biological process involved in COPD using a network-based method and the results were promising. This study enhanced our understanding of the network module of COPD and predicted the associated new candidate genes and pathways affecting its pathogenesis.
2.Method
We applied a Laplacian constraint approach [10] to integrate the biological network for the analysis of the gene expression data. Let a network
where
(1)
As shown in Eq. (1), the first term represents the loss function. The second term is a network-based penalty, which forms as a standard Laplacian matrix that captures the biological interaction knowledge.
Equation (1) is ill-posed in high-dimensional applications when the number of genes
(2)
where
(3)
The SCAD approach has several advantages over Lasso, such as better estimation accuracy and the oracle property. Consequently, we penalized the network-based method using the SCAD approach as we proposed earlier [14]. Finally, the method we used in this study was:
(4)
Where
(5)
and
We used the coordinate descent algorithm to solve Eq. (4). For more detailed information, please refer to our earlier publication [14].
| Algorithm: |
|---|
| Input: Training dataset |
| Output: Model parameter |
| Step 1: Update |
| Step 2: Let |
3.Results
3.1Data description
To identify the key biological process for COPD, we included 136 subjects with COPD (42 ex-smoking controls and 94 COPD subjects), in the study. Expression data from peripheral blood mononuclear cells from these subjects were collected [4].
We mapped the dataset to an official gene symbol, and calculated average expression levels for multiple probe-sets mapping to the same gene. BioGrid provides the biological interaction network
3.2Construction of the model and selection of biomarkers
We used tenfold cross-validation on multiple dimensions to find optimal regularization parameters of the model. With the estimated tuning parameters and all the training data, a classifier having 49 genes was constructed (Table 1) with a training classification error of 4.26%. Among all the cutoff points, the one with the highest sum of sensitivity and specificity was chosen.
Table 1
The selected 49 genes from chronic obstructive pulmonary disease gene expression data
| Genes | |||
|---|---|---|---|
| MMP1 | PAK2 | SLC25A38 | NSUN5 |
| TGFB2 | SOD2 | BACH2 | PMS2P1 |
| CTC1 | FAM13A | NUDT16L1 | CYFIP2 |
| FCMR | EFHC2 | SNRPN | THAP7 |
| GPCPD1 | NUMA1 | HMCES | TMEM134 |
| FOXP1 | VAPB | ITPKB | ZNF775 |
| OFD1 | PTEN | NMT2 | TSTD1 |
| USP20 | BCL6 | FBXO46 | DNLZ |
| RPARP-AS1 | PPARD | LONP1 | UBIAD1 |
| RAB43 | MTERF4 | PATZ1 | IL21R |
| MMP12 | AQP9 | HIVEP2 | RASGRP2 |
| FIP1L1 | NOSIP | SFMBT1 | SDR39U1 |
| IRAK3 | |||
Among these 49 genes, we observed some interesting findings. For example, smoke-induced emphysema has been implicated with matrix metalloproteinase (MMP)-1, a collagen degrader, and MMP-12, an elastin degrader, at least in animal models [25]. However, robust epidemiological data about serum MMPs in COPD are scarce, and there is a massive gap between experimental research and clinical epidemiology. MMPs genes may play a significant role in COPD, so with this perspective, we believe that our study could be a step towards bridge this gap. FoxP1 is a transcription factor important for the development of lung epithelial tissue. Recent data from the UK Biobank, ECLIPSE, and COPD Gene cohorts implicate genetic variants in the FOXP1 gene as important predictors of airflow limitation. Moreover, loss of FoxP1 protein increases endoplasmic reticulum stress in lung epithelial cells that may contribute to COPD development [26]. While the biological function of the FAM13A gene product is poorly understood. genetic variants in the FAM13A gene might determine susceptibility to COPD and lung cancer [27]. Iwona et al. [28] confirmed that the FAM13A variants are predisposed to increased susceptibility to COPD.
These findings imply that the selected genes may contribute to or act as a marker for the pathophysiology of COPD.
3.3Brief biological analysis
We then examined the 49 genes by GO and KEGG enrichment analyses (Figs 1 and 2). The results of GO analysis show that the selected 49 genes are involved in 17 significant pathways (
The enriched pathways may have possible roles in COPD. For example, the combination of chronic bronchiolitis and emphysema leading to COPD causes chronic airflow limitation and their ratio varies from patient to patient. B cell-related genes have been identified as being more prevalent in COPD with emphysema than in bronchiolitis, according to Faner et al. [29]. Likewise, Tang et al. [30] found an increased number of
This information can help us understand COPD pathobiology better, leading to new therapeutic possibilities for COPD.
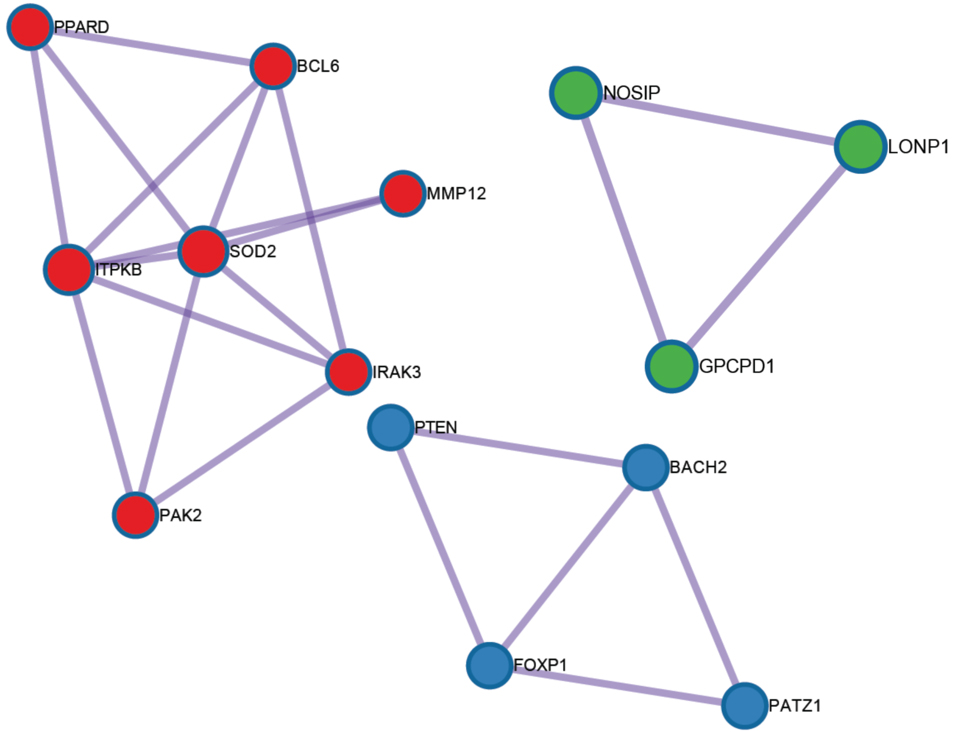
We conducted a PPI enrichment analysis of the selected genes and also performed the Molecular Complex Detection (MCODE) algorithm to identify densely connected network components from the constructed PPI network. The MCODE network result is presented in Fig. 3.
We then performed pathway analysis of the MCODE network, and the result is shown in Table 2.
Table 2
Pathway analysis of the molecular complex detection network
| GOID | Description | Log10 ( |
|---|---|---|
| hsa04068 | FoxO signaling pathway | |
| hsa05200 | Pathways in cancer | |
| hsa05206 | MicroRNAs in cancer |
Figure 1.
The gene ontology (GO) enrichment analysis.
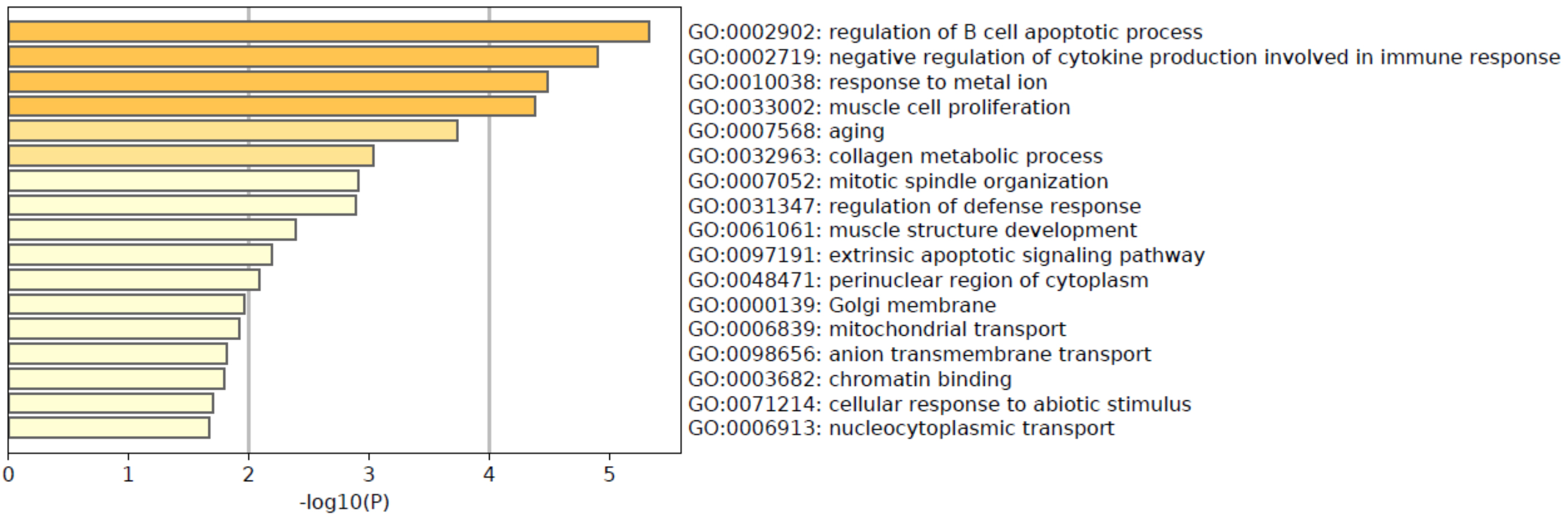
Figure 2.
The Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses.
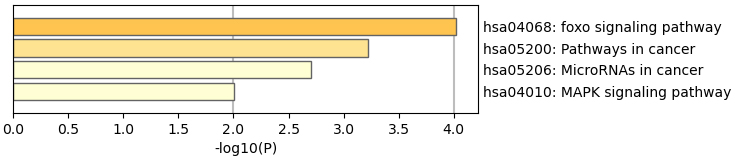
Figure 3.
The molecular complex detection algorithm resulted in the construction of protein-protein interaction (PPI) network from the selected 49 genes.
Atrophic muscle from COPD patients is shown to have increased expression of proteolysis pathway markers, such as transcription factors FoxO1 and FoxO3 [31]. Evidently, skeletal muscle atrophy involves increased expression of two E3 ubiquitin ligases, MuRF1 and atrogin-1, under the control of the FoxO family of transcription factors, particularly, FoxO1, activating protein degradation via the ubiquitin/proteasome system [32]. This information suggests that FoxO signaling may contribute to the atrophy of COPD peripheral muscle tissues via oxidative stress.
Data in Tables 1 and 2 and Figs 1–3 suggest that the selected 49 genes in this study might influence some biological processes and molecular functions related to COPD.
4.Discussion
Known for its chronic obstructive conditions, chronic bronchitis, and emphysema, COPD is the leading cause of death for the largest number of people worldwide. Although many efforts to identify biomarkers and pathways have been made, specific causal COPD mechanism remains unknown. This study combines biological interaction data with gene expression data for an improved understanding of the biological process and network module in COPD. We used a sparse network-based method and selected 49 genes from peripheral blood mononuclear cell expression data of 136 subjects, including 42 ex-smoking controls and 94 subjects with COPD. The results show that these 49 genes possibly influence biological processes and molecular functions related to COPD. For example, one of these genes coding for FoxO signaling may contribute to the atrophy of COPD peripheral muscle tissues via oxidative stress.
5.Conclusion
To conclude, our approach advances understanding of COPD disease pathogenesis and predicts new potential genetic markers and pathways that influence COPD pathogenesis. One limitation of this study was the lack of an in-depth verification of the selected genes and network modules.
Acknowledgments
This work was partially funded by the National Natural Science Foundation of China (62102261, 62006155, 6201101081), the Science and Technology Development Fund, Macau SAR (0002/2019/APD, 0056/2020/AFJ, 0158/2019/A3), and the Science and Technology Project of Shaoguan City (2008111045 31028).
Conflict of interest
None to report.
References
[1] | Iyer AS, Curtis JR, Meier DE. Proactive Integration of Geriatrics and Palliative Care Principles Into Practice for Chronic Obstructive Pulmonary Disease. JAMA Intern Med. (2020) ; 180: : 815. doi: 10.1001/jamainternmed.2020.1088. |
[2] | Qiu W, Cho MH, Riley JH, Anderson WH, Singh D, Bakke P, et al. Genetics of Sputum Gene Expression in Chronic Obstructive Pulmonary Disease. PLoS One. (2011) ; 6: : e24395. doi: 10.1371/journal.pone.0024395. |
[3] | Sakornsakolpat P, Prokopenko D, Lamontagne M, et al. Genetic landscape of chronic obstructive pulmonary disease identifies heterogeneous cell-type and phenotype associations. Nat Genet. (2019) ; 51: (3): 494–505. doi: 10.1038/s41588-018-0342-2. |
[4] | Bahr TM, Hughes GJ, Armstrong M, et al. Peripheral blood mononuclear cell gene expression in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. (2013) ; 49: (2): 316–323. doi: 10.1165/rcmb.2012-0230OC. |
[5] | Huang X, Lv D, Yang X, Li M, Zhang H. m6A RNA methylation regulators could contribute to the occurrence of chronic obstructive pulmonary disease. J Cell Mol Med. (2020) ; 24: (21): 12706–12715. doi: 10.1111/jcmm.15848. |
[6] | Huang H-H, Liang Y. A novel Cox proportional hazards model for high – dimensional genomic data in cancer prognosis. IEEE/ACM Trans Comput Biol Bioinforma. Published online (2019) : 1–1. doi: 10.1109/TCBB.2019.2961667. |
[7] | Huang HH, Liu XY, Li HM, Liang Y. Molecular pathway identification using a new L1/2 solver and biological network-constrained mode. Int J Data Min Bioinform. (2017) ; 17: (3): 189. doi: 10.1504/IJDMB.2017.085277. |
[8] | Huang H-H, Liang Y. Clinical drug response prediction by using a lq penalized network-constrained logistic regression method. Cell Physiol Biochem. (2018) ; 51: (5): 2073–2084. doi: 10.1159/000495826. |
[9] | Huang H-H, Liang Y, Liu X-Y. Network-based logistic classification with an enhanced L1/2 solver reveals biomarker and subnetwork signatures for diagnosing lung cancer. Biomed Res Int. (2015) ; 2015: : 713953. doi: 10.1155/2015/713953. |
[10] | Li C, Li H. Network-constrained regularization and variable selection for analysis of genomic data. Bioinformatics. (2008) ; 24: (9): 1175–1182. doi: 10.1093/bioinformatics/btn081. |
[11] | Chen J, Zhang S, C. S. Integrative analysis for identifying joint modular patterns of gene-expression and drug-response data. Bioinformatics. (2016) ; 32: (11): 1724–1732. doi: 10.1093/bioinformatics/btw059. |
[12] | Wang R, Su C, Wang X, et al. Global gene expression analysis combined with a genomics approach for the identification of signal transduction networks involved in postnatal mouse myocardial proliferation and development. Int J Mol Med. (2018) ; 41: (1): 311–321. doi: 10.3892/ijmm.2017.3234. |
[13] | Zhou Z, Huang H, Liang Y. Cancer classification and biomarker selection via a penalized logsum network-based logistic regression model. Technol Heal Care. (2021) ; 29: (S1): 287–295. doi: 10.3233/THC-218026. |
[14] | Huang H-H, Liang Y. An integrative analysis system of gene expression using self-paced learning and SCAD-Net. Expert Syst Appl. (2019) ; 135: : 102–112. doi: 10.1016/J.ESWA.2019.06.016. |
[15] | Zhang W, Wan YW, Allen GI, Pang K, Anderson ML, Liu Z. Molecular pathway identification using biological network-regularized logistic models. BMC Genomics. (2013) ; 14: (Suppl 8): S7-2164-14-S8-S7. Epub 2013 Dec 9. doi: 10.1186/1471-2164-14-S8-S7. |
[16] | Sharma A, Kitsak M, Cho MH, et al. Integration of molecular interactome and targeted interaction analysis to identify a COPD disease network module. Sci Rep. (2018) ; 8: (1): 14439. doi: 10.1038/s41598-018-32173-z. |
[17] | Liang Y, Chai H, Liu X-Y, Xu Z-B, Zhang H, Leung K-S. Cancer survival analysis using semi-supervised learning method based on Cox and AFT models with L1/2 regularization. BMC Med Genomics. (2016) ; 9: (1): 11. doi: 10.1186/s12920-016-0169-6. |
[18] | Liang Y, Liu C, Luan XZ, et al. Sparse logistic regression with a L1/2 penalty for gene selection in cancer classification. BMC Bioinformatics. (2013) ; 14: : 198. doi: 10.1186/1471-2105-14-198. |
[19] | Huang H, Peng X, Liang Y. SPLSN: An efficient tool for survival analysis and biomarker selection. Int J Intell Syst. Published online June 13, (2021) :int.22532. doi: 10.1002/int.22532. |
[20] | Huang H-H, Liang Y. Hybrid L1/2+2 method for gene selection in the Cox proportional hazards model. Comput Methods Programs Biomed. (2018) ; 164: : 65–73. doi: 10.1016/j.cmpb.2018.06.004. |
[21] | Huang H-H, Liu X-Y, Liang Y. Feature selection and cancer classification via sparse logistic regression with the hybrid L1/2+2 regularization. PLoS One. (2016) ; 11: (5): e0149675. doi: 10.1371/journal.pone.0149675. |
[22] | Huang H-H, Liu X-Y, Liang Y, Chai H, Xia L-Y. Identification of 13 blood-based gene expression signatures to accurately distinguish tuberculosis from other pulmonary diseases and healthy controls. Biomed Mater Eng. (2015) ; 26: (Suppl 1): S1837–43. doi: 10.3233/BME-151486. |
[23] | Liang Y, Leung K-S. Genetic Algorithm with adaptive elitist-population strategies for multimodal function optimization. Appl Soft Comput. (2011) ; 11: (2): 2017–2034. doi: 10.1016/J.ASOC.2010.06.017. |
[24] | Fan J, Li R. Variable selection via nonconcave penalized likelihood and its oracle properties. J Am Stat Assoc. (2001) ; 96: (456): 1348–1360. doi: 10.1198/016214501753382273. |
[25] | D’Armiento JM, Goldklang MP, Hardigan AA, et al. Increased Matrix Metalloproteinase (MMPs) Levels Do Not Predict Disease Severity or Progression in Emphysema. Lenburg M, ed. PLoS One. (2013) ; 8: (2): e56352. doi: 10.1371/journal.. |
[26] | Morrow JD, Cho MH, Hersh CP, et al. DNA methylation profiling in human lung tissue identifies genes associated with COPD. Epigenetics. (2016) ; 11: (10): 730–739. doi: 10.1080/15592294.2016.1226451. |
[27] | Young RP, Hopkins RJ, Hay BA, Whittington CF, Epton MJ, Gamble GD. FAM13A locus in COPD is independently associated with lung cancer – evidence of a molecular genetic link between COPD and lung cancer. Appl Clin Genet. (2011) ; 4: : 1–10. doi: 10.2147/TACG.S15758. |
[28] | Ziółkowska-Suchanek I, Mosor M, Gabryel P, et al. Susceptibility loci in lung cancer and COPD: Association of IREB2 and FAM13A with pulmonary diseases. Sci Rep. (2015) ; 5: (1): 13502. doi: 10.1038/srep13502. |
[29] | Faner R, Cruz T, Casserras T, et al. Network analysis of lung transcriptomics reveals a distinct b-cell signature in emphysema. Am J Respir Crit Care Med. (2016) ; 193: (11): 1242–1253. doi: 10.1164/rccm.201507-1311OC. |
[30] | Tang Y, Guan Y, Liu Y, Sun J, Xu L, Jiang Y. The Role of the Serum IL-33/sST2 Axis and Inflammatory Cytokines in Chronic Obstructive Pulmonary Disease. J Interf Cytokine Res. (2014) ; 34: (3): 162–168. doi: 10.1089/jir.2013.0063. |
[31] | Fermoselle C, Rabinovich R, Ausín P, et al. Does oxidative stress modulate limb muscle atrophy in severe COPD patients? Eur Respir J. (2012) ; 40: (4): 851–862. doi: 10.1183/09031936.00137211. |
[32] | Sandri M, Sandri C, Gilbert A, et al. Foxo Transcription Factors Induce the Atrophy-Related Ubiquitin Ligase Atrogin-1 and Cause Skeletal Muscle Atrophy. Cell. (2004) ; 117: (3): 399–412. doi: 10.1016/S0092-8674(04)00400-3. |


