
More Bang for Your Buck: “Off-The-Shelf” Solutions for Cell Replacement Therapy
Abstract
The immune barrier to transplantation has widely been recognized as the ultimate hurdle to the translation of stem cell-based therapies. In particular the polymorphic nature of the human leucocyte antigens (HLA) poses an imminent barrier to the successful engraftment of cells from other than autologous sources. To make stem cell therapies available to a larger pool of patients and a commercially viable option several groups have attempted to create hypoimmunogenic “universal” donor stem cells that evaded immune detection.
The goal of this commentary is to give a brief overview of the current approaches taken and discuss challenges that need to be addressed to turn such cells into a viable commercial option.
INTRODUCTION - THE HOOK
The importance of induced pluripotent stem cells (iPSC) as an unlimited, renewable source of cellular building blocks for cell replacement therapy (CRT) cannot be overstated. Right from the start, they have captured the imagination of investigators and investors alike as iPSC seemed to allow stem cell-based treatments in a personalized manner. Unfortunately, it has become increasingly clear, that the concept of personalized cellular medicine is flawed for a variety of reasons: the process of generating iPSC takes too long, it is highly variable with regards to the quality of the cell lines generated and all together is too expensive. Based on the first clinical trials with autologous iPSC in Japan, it has been estimated that the costs for a personalized stem cell treatment amount to more than $1M, which will limit the number of patients that will have access to this form of treatment [1]. Moreover, even autologous iPSC have been found to be the subject of immune rejection most likely due to incomplete reprogramming, the untimely expression of fetal antigens, or acquisition of neoantigens that can prompt an immune response [2, 3]
Progress in the field of genome and cell engineering inspired efforts to alter the immunogenicity of stem cells in order to hide them from the immune system and create universal allogeneic “off-the-shelf” cell products that can be administrated on-demand to any given patient at any given time (Fig. 1). The prospects of such universal cell products are imminent: they would not only reach a much larger pool of patients when compared to a personalized approach, but also significantly drive down the cost of goods, that arise from manufacturing, quality control and cell banking, and thereby democratize access to cellular medicines.
Fig.1
The Promise of Regenerative Medicine. The goal of cell therapies is to replace diseased or missing cells with new healthy cells. Using universal donor cells as an unlimited, “off-the-shelf”, quality-controlled source will enable a much larger pool of patients to benefit from these emerging treatments.
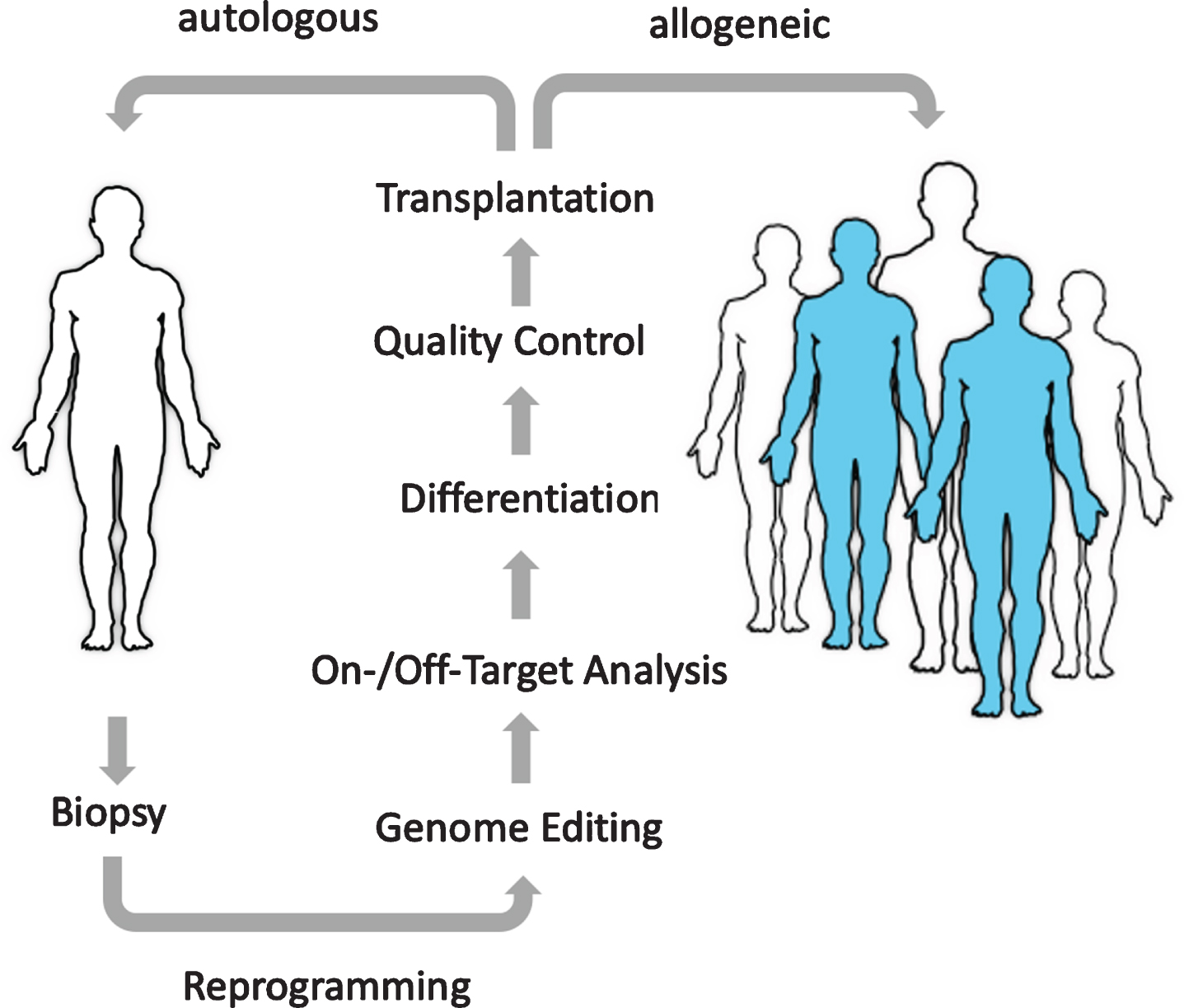
IMMUNE EVASION – AN HLA-CENTRIC TAKE
To address the problem of immune rejection, we and others have turned to genome engineering to introduce genetic modifications to hide allogeneic cells from the immune system. It is well known that hyperacute, as well as acute graft rejection are triggered predominantly by the expression of human leukocyte antigen (HLA) molecules, a family of highly polymorphic surface proteins, that provide each individual with a unique molecular barcode that allows the immune system to distinguish self from non-self. HLA come in two flavors, HLA class I and HLA class II molecules. While the expression of HLA class II molecules can be prevented by targeting the transcriptional master regulator CIITA [4, 5], targeting HLA class I molecules is more complex. The easiest way to prevent HLA class I expression is to target the accessory chain beta2microglobulin (B2M), which is required for the surface trafficking of all HLA class I proteins. This has been accomplished in a variety of cell types including T cells, ES and iPSC [5–7]. However, the downside to this approach is that it renders HLA-deficient cells prone to attack by natural killer (NK) cells, which patrol the body to eliminate virus-infected or cancerous cells that have lost HLA expression. This has motivated us to specifically ablate the expression of the polymorphic HLA molecules, HLA-A, -B, and -C, using a dual sgRNA strategy (Fig. 2), while leaving the expression of the invariant, tolerogenic HLA molecules HLA-E and -G intact [4].
Fig.2
Dual Guide Strategy for Selective Deletion of Polymorphic HLA Alleles. (A) Schematic depiction of the human HLA locus on chromosome 6. (B) The highly polymorphic HLA molecules HLA-A and HLA-B/-C can be excised using a dual CRISPR sgRNA approach without affecting other genes in the HLA locus. Red arrows indicate the position of the sgRNAs that will direct the Cas9 endonuclease to the desired target site. Since the B2M gene is still intact, expression of the tolerogenic HLA molecules HLA-E and -G is unhampered when using this approach [4].
![Dual Guide Strategy for Selective Deletion of Polymorphic HLA Alleles. (A) Schematic depiction of the human HLA locus on chromosome 6. (B) The highly polymorphic HLA molecules HLA-A and HLA-B/-C can be excised using a dual CRISPR sgRNA approach without affecting other genes in the HLA locus. Red arrows indicate the position of the sgRNAs that will direct the Cas9 endonuclease to the desired target site. Since the B2M gene is still intact, expression of the tolerogenic HLA molecules HLA-E and -G is unhampered when using this approach [4].](https://ip.ios.semcs.net:443/media/stj/2020/2-1/stj-2-1-stj200002/stj-2-stj200002-g002.jpg)
Cell banking of HLA-homozygous allogeneic iPSC has been suggested as an alternative approach [8], yet even HLA-matched cells have been shown to be rejected over time presumably due to the presence of other polymorphic proteins that can be detected by the immune system, so called minor histocompatibility antigens (mHAgs) [9]. An interesting hybrid approach combining genome engineering and cell banking of HLA-matched cell lines has recently been taken by the group of Akitsu Hotta at Kyoto University. The authors suggest that HLA KO cells engineered to express only one HLA-C allele could evade T and NK cell responses and will greatly reduce the number of cell lines required to match a large part of the human population [10]. One downside to this approach, however, is that HLA-C can still present foreign peptides derived from the allogeneic donor cells, and also the direct recognition of mHAgs as well as antibody-mediated chronic graft rejection are not accounted for. It is thus unclear as of yet, whether any of those HLA-engineered ‘immuno-compatible’ cell lines will persist long-term in the absence of immunosuppressive drugs or further genetic intervention.
Universal cells 2.0 – Managing immune tolerance
Recognizing the limitations of a mere immune evasive strategy, in a second phase of cell-engineering, academic labs have started to incorporate immunomodulatory molecules into their design process (Table 1). The general idea is to manage immune rejection by creating a local immune privileged niche allowing the engraftment of allogeneic cells, while maintaining systemic immune competence. This mirrors essentially what happens during pregnancy where the fetus – a semi allograft only 50% identical with the mother - is recognized by the maternal immune system yet effector cells are being muted in their response by the local immunosuppressive environment provided by the placenta. At the heart of this immune camouflage is the unique protein make-up of trophoblast cells which are of fetal origin and in direct contact with maternal blood. Trophoblasts are devoid of HLA-A and -B expression, and in addition to low levels of HLA-C, uniquely express the tolerogenic HLA molecule HLA-G [11]. Moreover, trophoblasts display high expression of the checkpoint inhibitor PD-L1 and incorporating PD-L1 and HLA-G into human pluripotent stem cells and their derivatives protected them from T and NK cell responses [4, 12].
Table 1
Expression of immunomodulatory factors in pluripotent stem cells
| Factor | PSC | Target Cell | Reference |
| HLA-E | hESC | NK | Gornalusse et al.[7] |
| HLA-G | hESC | NK, T cells | Han et al. [4] |
| PD-L1 | hESC, mESC | T cells, MF | Han et al. [4], Rong et al. [12], Harding et al. [15] |
| CTLA4-IG | hESC | T cells | Rong et al. [12] |
| CD47 | mESC, miPSC, hESC | MF, NK | Han et al. [4], Deuse et al. [5], Harding et al. [15] |
| CD200 | mESC | MF | Harding et al. [15] |
| FASL | mESC | T cells | Harding et al. [15] |
| SERPINB9 | mESC | T cells, NK | Harding et al. [15] |
| MFG-E8 | mESC | MF | Harding et al. [15] |
| CCL21 | mESC | T cells | Harding et al. [15] |
PSC = pluripotent stem cell; ESC = embryonic stem cell; iPSC = induced pluripotent stem cell; h = human; m = murine; NK = natural killer cells; MF = macrophages.
Another shift in design thinking has been to also target other immune cells, in particular innate immune cells, such as macrophages, NK cells, NKT, and neutrophils, whose contribution to allograft rejection is less defined. We have recently shown that incorporating CD47, a macrophage “don’t-eat-me” signal into human pluripotent stem cells can protect stem cell-derived vascular smooth muscle cells from excessive phagocytic activity by macrophages [4], a finding that has been extended to NK cells, as CD47 overexpression unexpectedly seems to protect HLA-deficient cells from NK cell lysis [5]. More recently, both cell types have been implicated in immune memory function, which could complicate engraftment of a secondary transplant from the same donor [13]. It is therefore important that additional steps are being taken to manage the activity of immune cells - beyond T cells - to allow for long-term graft survival [14].
Other factors that have been explored to target different arms of the immune system include SERPINB9 which inhibits Granzyme B, an effector molecule of cytotoxic T and NK cells, CD200 which binds to CD200R, an inhibitory receptor expressed in lymphoid and myeloid cells, and MFGE8 a proangiogenic factor. A recent report described that a combination of 8 factors may allow to overcome the allobarrier even in the presence of HLA, at least in the mouse models studied [15].
OUTLOOK
It is now widely accepted that CRT will change the way we treat degenerative and chronic diseases. With over 200 investigational new drug applications expected at the FDA in 2020 for cell-based therapies [16], the commercial value of these emerging therapies as well as the interest from the side of investors appears to be only increasing. Yet, while the tremendous progress in genome and cell engineering as outlined above has fueled efforts targeting the immune barrier, the jury is still out there and proof of a truly universal cell product that will withstand immune rejection upon transplantation into any patient long-term is missing.
One particular challenge that needs to be addressed is chronic graft rejection. While initial efforts have predominantly focused on HLA, which are drivers of hyperacute and acute graft rejection, antibodies reactive to mHAgs or neoantigens expressed by the engineered cell lines may precipitate activation of the complement cascade or antibody-dependent cellular cytotoxicity (ADCC) by NK cells. While serum of the recipient patients can be prescreened for anti-HLA antibodies, whole genome sequencing (WGS) can be indicative of the immunocompatibility between donor cell product and patient. Unfortunately, in most cases it is not clear, how preclinical findings - usually obtained in small animal models - translate to the human setting? There is no one humanized mouse model yet that fully recapitulates a human immune response to an allotransplant. While new mouse models are constantly being developed [17], the question rather seems to be, will an animal model ever predict faithfully an immune response against a transplanted human cell product?
With this in mind, a dialogue with regulators needs to be opened as to how clinical trials involving genetically modified cells will be conducted. On the road to the clinic there have to be clear cut quality control measures in place with regards to genomic stability, off-target effects as a result of genetic manipulations, the potential for tumorigenicity and of course the immunogenicity of the cell product if it is meant to last long-term. Moreover, standardized GMP manufacturing protocols and release criteria for the specific cell products have to be developed. There is a consensus that a therapeutic cell product should ideally be of clonal origin, and it seems desirable to keep the number of modifications as low as possible as quality control is already hard enough for a living cellular product alone, let alone a genome-edited one.
Despite the aforementioned challenges, there has been remarkable activity in the biotech space around the generation of “universal donor cells”. In 2018 Astellas bought Universal Cells – a small biotech company based in Seattle - for $102.5 M, and more recently, a series A funding to the cell therapy company Sana Biotechnology for $700 M is testimony of the significant enthusiasm from the side of investors and big pharma about this technology. Moreover, 2019 has seen the first clinical trials with genome edited hematopoietic stem and progenitor cells (HSPC) by CRISPR Therapeutics together with Vertex and Sangamo Therapeutics/Sanofi as a potential cure for sickle cell disease. Just this year the results of the first clinical trial using genome-edited CAR T cells have been reported by Carl June’s group at the University of Pennsylvania [18], and another clinical trial with NK cells derived from genome-edited iPSC has begun in the US conducted by Fate Therapeutics together with the University of Minnesota Medical Center to treat acute myeloid and B cell lymphoma. These are certainly exciting times for everyone working in the CRT space and it will take intimate collaborations between academic labs, biotech and large pharma companies as well as clinical centers and regulators to make the concept of engineered “cells as medicine” a cost-effective reality for all patients in need.
Conflict of interest statement
C.A.C is a founder of CRISPR Therapeutics and Sana Biotechnology. Both, C.A.C. and T.B.M. hold patents licensed by Harvard University to Sana Biotechnology.
REFERENCES
[1] | Mandai M , Watanabe A , Kurimoto Y , et al. Autologous Induced Stem-Cell-Derived Retinal Cells for Macular Degeneration. N Engl J Med. (2017) ;376: (11):1038–46. |
[2] | Zhao T , et al. Humanized Mice Reveal Differential Immunogenicity of Cells Derived from Autologous Induced Pluripotent Stem Cells. Cell Stem Cell. (2015) ;17: :353–9. |
[3] | Deuse T , et al. De novo mutations in mitochondrial DNA of iPSCs produce immunogenic neoepitopes in mice and humans. Nat Biotechnol. (2019) ;37: :1137–44. |
[4] | Han X , et al. Generation of hypoimmunogenic human pluripotent stem cells. P Natl Acad Sci Usa. (2019) ;116: :10441–6. |
[5] | Deuse T , et al. Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat Biotechnol. (2019) ;37: :252–58. |
[6] | Mandal PK , Ferreira LM , Collins R , et al. Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell. (2014) ;15: (5):643–52. |
[7] | Gornalusse GG , et al. HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nature Biotechnology. (2017) ;35: :765–72. |
[8] | Turner M , Leslie S , Martin NG , et al. Toward the development of a global induced pluripotent stem cell library. Cell Stem Cell. (2013) ;13: (4):382–4. |
[9] | Aron Badin R , Bugi A , Williams S , et al. MHC matching fails to prevent long-term rejection of iPSC-derived neurons in non-human primates. Nat Commun. (2019) ;10: (1):4357. |
[10] | Xu H , Wang B , Ono M , et al. Targeted Disruption of HLA Genes via CRISPR-Cas9 Generates iPSCs with Enhanced Immune Compatibility. Cell Stem Cell. (2019) ;24: (4):566–578.e7. |
[11] | Ferreira LMR , Meissner TB , Tilburgs T , Strominger JL . HLA-G: At the Interface of Maternal-Fetal Tolerance. Trends Immunol. (2017) ;38: (4):272–86. |
[12] | Rong Z , et al. An effective approach to prevent immune rejection of human ESC-derived allografts. Cell Stem Cell. (2014) ;14: :121–30. |
[13] | Dai H , Lan P , Zhao D , et al. PIRs mediate innate myeloid cell memory to nonself MHC molecules. Science. (2020) ;368: (6495):1122–7. |
[14] | Zhao D , Abou-Daya KI , Dai H , Oberbarnscheidt MH , Li XC , Lakkis FG . Innate Allorecognition and Memory in Transplantation. Front Immunol. (2020) ;11: :918. |
[15] | Harding J , et al. Induction of long-term allogeneic cell acceptance and formation of immune privileged tissue in immunocompetent hosts. Biorxiv 716571 (2019) . doi: 10.1101/716571 |
[16] | U.S. Food and Drug Administration. [homepage on the internet]. Statement from FDA Commissioner Scott Gottlieb, M.D. and Peter Marks, M.D., Ph.D., Director of the Center for Biologics Evaluation and Research on new policies to advance development of safe and effective cell and gene therapies. January 15, 2019. Available from: https://www.fda.gov/news-events/press-announcements/statement-fda-commissioner-scott-gottlieb-md-and-peter-marks-md-phd-director-center-biologics |
[17] | Allen TM , Brehm MA , Bridges S , et al. Humanized immune system mouse models: Progress, challenges and opportunities. Nat Immunol. (2019) ;20: (7):770–4. |
[18] | Stadtmauer EA , Fraietta JA , Davis MM , et al. CRISPR-engineered T cells in patients with refractory cancer. Science. (2020) ;367: (6481):eaba7365. |


