
Antigenic targets in clear cell renal cell carcinoma
Abstract
Immune checkpoint inhibitors (ICIs) have transformed the management of advanced renal cell carcinoma (RCC), but most patients still do not receive a long-term benefit from these therapies, and many experience off-target, immune-related adverse effects. RCC is also different from many other ICI-responsive tumors, as it has only a modest mutation burden, and total neoantigen load does not correlate with ICI response. In order to improve the efficacy and safety of immunotherapies for RCC, it is therefore critical to identify the antigens that are targeted in effective anti-tumor immunity. In this review, we describe the potential classes of target antigens, and provide examples of previous and ongoing efforts to investigate and target antigens in RCC, with a focus on clear cell histology. Ultimately, we believe that a concerted antigen discovery effort in RCC will enable an improved understanding of response and resistance to current therapies, and lay a foundation for the future development of “precision” antigen-directed immunotherapies.
INTRODUCTION
The advent of immune checkpoint inhibitors (ICIs) brought a marked improvement in overall survival to patients with metastatic clear cell renal cell carcinoma (ccRCC) [1]. In fact, the current standard-of-care for ccRCC is dominated by combination therapies of ICIs that primarily target the PD-1—PD-L1 axis and tyrosine kinase inhibitors (TKIs). Since the approval of the anti-PD-1 antibody nivolumab and the anti-CTLA-4 antibody ipilimumab in 2018 for RCC [2], four other ICI-based combination therapies (together with anti-angiogenic TKIs) have demonstrated superior clinical efficacy to TKI therapy alone, and have subsequently been FDA-approved [3–6]. Although these new therapies represent substantial improvements over prior treatments, with median overall survival improving from just approximately 1 year in the 1990 s [7] to nearly 5 years today [8], these therapeutic regimens still do not lead to durable clinical responses for most patients. Improving the long-term clinical outcomes for patients with ccRCC will require a deeper understanding of the underlying immunobiology of ccRCC. Specifically, we must learn the biology of antigen-specific, immune-mediated tumor killing and understand how best to steer the immune response towards tumor cells.
Fig. 1
Overview of the cancer immunity cycle (as described by Chen and Mellman). The seven steps of the cancer immunity cycle are displayed in a cyclic format. In the first step, tumor cells undergo immunogenic cell death. Death of the malignant cells allows antigens to be released into the microenvironment. These antigens are taken up by antigen presenting cells, such as naïve dendritic cells; trafficked to lymph tissue; and processed with the internal antigen processing machinery. This involves proteosomes cleaving antigens into smaller peptides and presenting them on HLA complexes. Activated antigen presenting cells display antigens to prime and activate naïve T cells. The activated T cells differentiate into specific classes depending on factors such as co-stimulatory signals. Activated T cells migrate back to the site of the tumor via trafficking signals and infiltrate the tumor microenvironment. Once inside the tumor, T cells may recognize and kill malignant cells, thereby restarting the cycle.
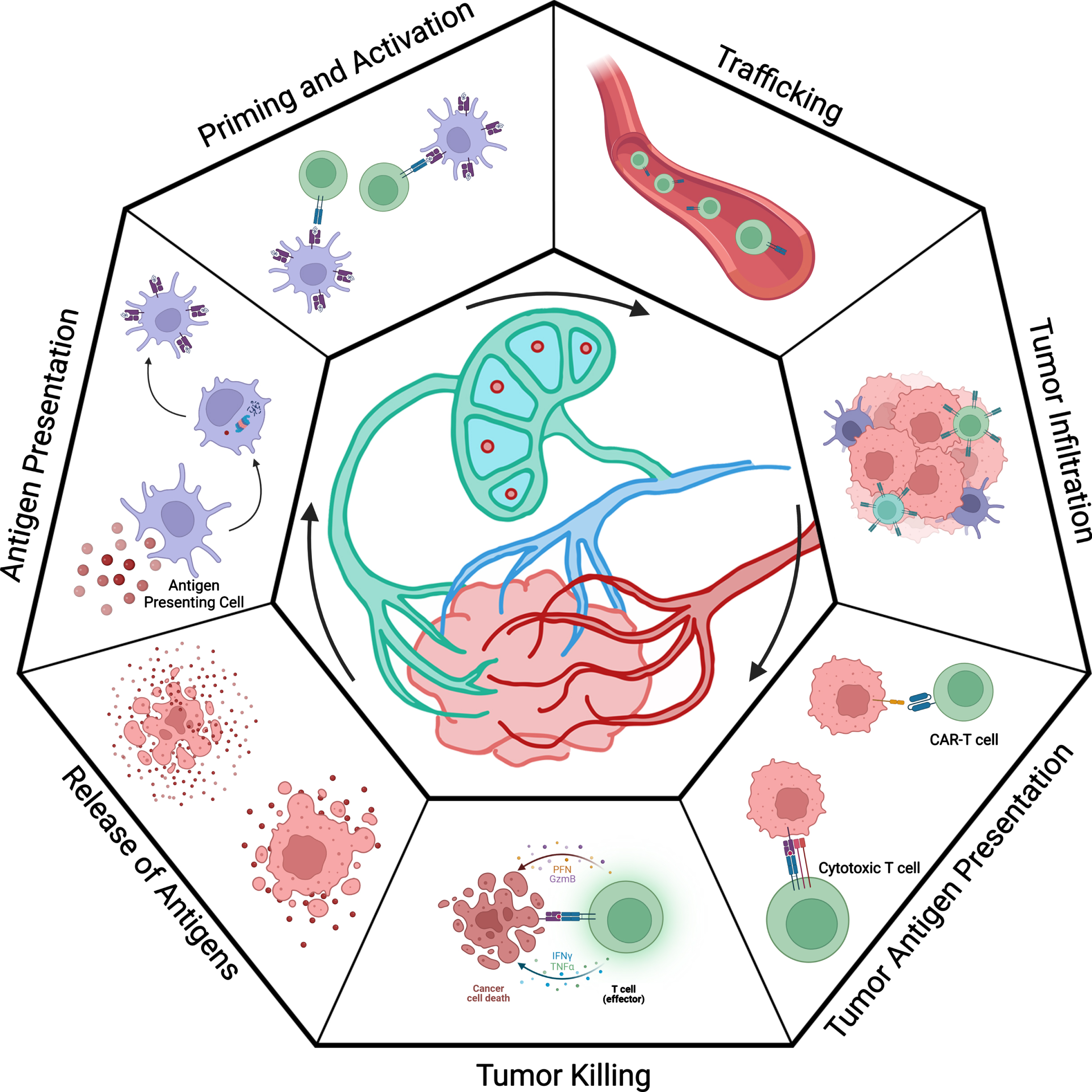
A critical framework for understanding why most patients do not experience long-term benefits from ICI-based therapy is described through the cancer-immunity cycle from Chen and Mellman (Fig. 1) [9, 10]. The cycle begins when a tumor cell dies and releases antigens to its surroundings. Tumor antigens are molecules (typically peptides) that can be presented to immune cells, potentially leading to an immune response that can target and kill malignant cells. These antigens are taken up by immature dendritic cells (DCs) or other antigen presenting cells (APCs) and then processed by antigen presenting machinery in these cells. The DCs migrate to draining lymph nodes or aggregate in antigen-presenting niches or tertiary lymphoid structures within the tumor [11]. At these sites, they mature and present the antigen on major histocompatibility complexes (MHCs, which are also known as human leukocyte antigens [HLAs] in humans) and express co-stimulatory markers. Circulating naïve T cells enter the lymph nodes through the high endothelial venules (HEV) and sample DCs with their T cell receptors (TCRs) in the cortical region. Upon recognition of an antigen, the bond between the DC and T cell strengthens, and an immunological synapse forms. With the correct co-stimulatory signals, T cells proliferate and differentiate into effector T cells. The HLA class and co-stimulatory signals will dictate which the lineage of the naïve T cell. Each cell lineage is specialized for distinct functions and canonically plays a role in handling different types of infections. Without the correct co-stimulatory markers, the T cells will not be activated and may in fact become tolerized to the target antigen.
The activated T cells are trafficked out of the lymph nodes via the efferent venule and then to the sites of tumors. Once they have infiltrated the tumor, they may recognize and kill cancer cells. However, cancers including RCC can adapt their tumor microenvironment to inhibit several steps of the cancer-immunity cycle. For example, they may hinder the infiltration of T cells or limit the activation of T cells by expressing inhibitory markers such as PD-L1. The PD-1/PD-L1 pathway inhibits the activation, proliferation, and cytolytic activity of T cells in the tumor microenvironment [12]. ICIs aim to block inhibitory signals, such as the PD-1/PD-L1 pathway, hijacked by the tumor cells and essentially “release the brakes” on the immune response.
Although this general framework for tumor killing is helpful, there are still many unknowns. When tumor antigens are released by apoptotic tumor cells, we generally do not know which antigens are processed and presented by APCs. Whereas many tumor antigens have been elucidated in other cancer types, such as melanoma [13], the landscape of antigens in RCC remains poorly characterized. Additionally, in the case where tumor antigens are presented by APCs, we do not know which ones initiate an effective immune response. Whereas the specific APC subsets effectively prime T cells against RCC antigens are not fully elucidated, there is a clear role for CD11c + dendritic cells in intratumoral antigen-presenting niches [14] and likely additional presentation in tertiary lymphoid structures [11]. Knowing which antigens (and which APCs) trigger an immune response would benefit new, precision immunotherapy approaches, including cancer vaccines and T cell therapies. Cancer vaccines are designed to use a select number of tumor antigens or a complete tumor antigen library to train the patients’ immune systems to recognize and kill cancer cells. Furthermore, adoptive T cell transfer aims to engineer T cells with specific TCRs that recognize tumor antigens presented on the surface of cancer cells. Understanding which antigens are effectively recognized by tumor-reactive T cells would be highly beneficial and would allow these therapies to take advantage of the whole cancer-immunity cycle to create a deep response to disease.
TUMOR ANTIGENS
Immune-mediated, antigen-specific tumor killing relies on the recognition of specific tumor antigens by the immune system (Fig. 2). Recognition occurs on the surface of the cancer cells in one of two ways. Antigen presenting machinery in a cancer cell can process intracellular proteins and present peptides on HLA molecules. Alternatively, the cancer cells can be recognized through surface antigen markers, typically engineered therapeutic antibodies, though there is evidence in other cancer types of endogenous antibody responses to surface antigens playing a role [15]. Aberrant proteins presented on HLA molecules are recognized by T cell-mediated immunity. Intracellular proteins are cleaved into smaller fragments within the tumor cells and presented on either HLA class I (for CD8 T cells) or, less commonly, HLA class II (for CD4 T cells). Once recognized, the cytotoxic T cells bind to the tumor cells through the TCR, and deliver cytotoxic effector proteins, including perforin, granzymes, and granulysin. These proteins perforate the tumor cell and trigger apoptosis. Additional mechanisms of T cell-mediated cytotoxicity include expression of cell death ligands (such as FasL and TRAIL) and production of effector cytokines (such as IFNγ and TNFα) [16].
Fig. 2
Antigen Classes. Antigens may be categorized as HLA-restricted or surface antigens. HLA-restricted antigens include all proteins generated within the cell that are processed by antigen presenting machinery. Surface antigens are a subset of proteins that are presented on the surface of the cell. Each class of antigen has been targeted by antigen-directed therapies in a personalized or universal basis. Personalized therapies are unique to each patient and have the opportunity to create a deeper immune response to malignancy. Universal therapies are not unique to an individual patient, but are cost effective and available immediately as an “off-the-shelf” therapy.
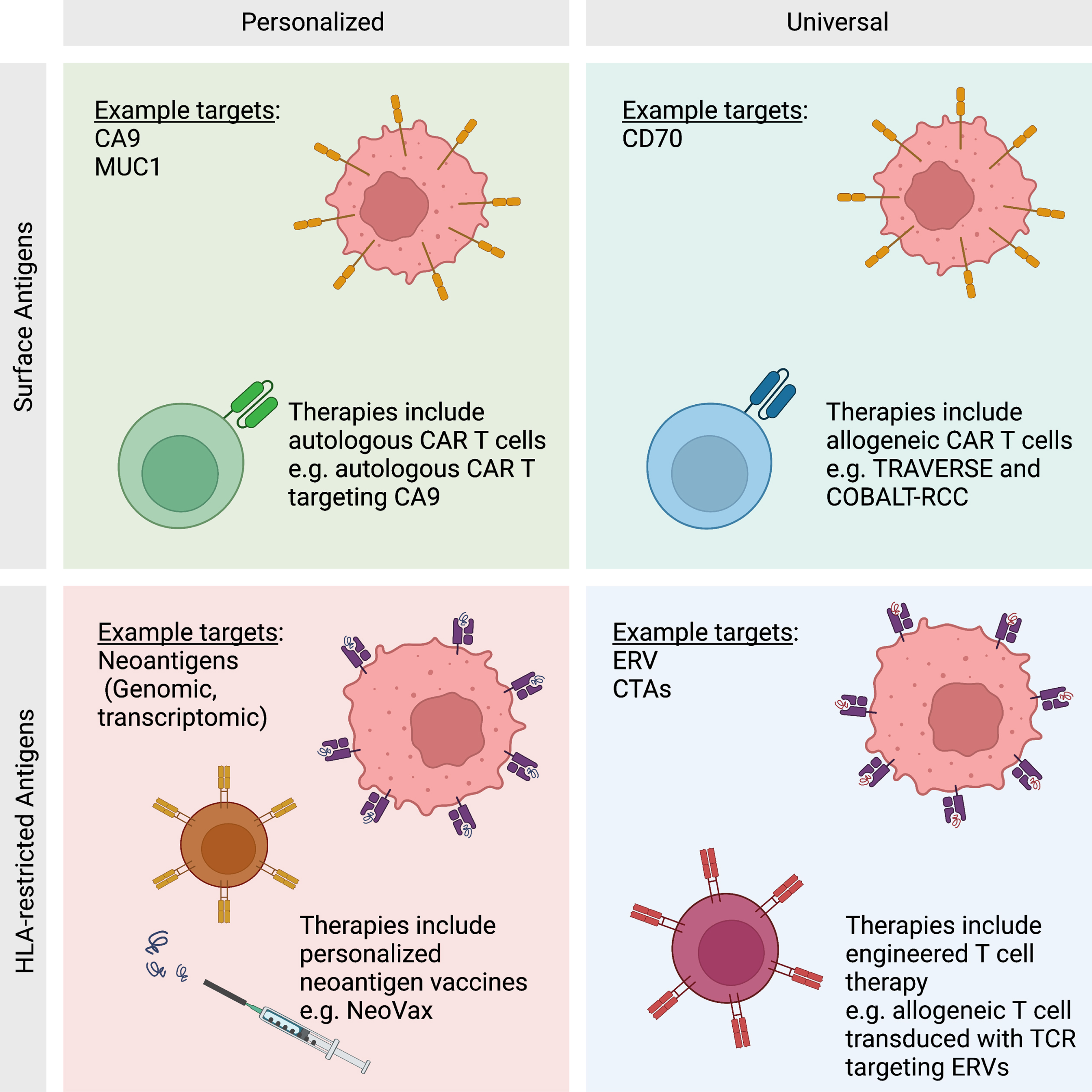
Tumor antigens presented in the context of HLA class II molecules are recognized by activated CD4 T cells, also known as helper T cells. These cells come in a range of classes, and each class is specialized for different purposes. Classes of CD4 T cells include the helper T cells TH1, TH2, TH17, and TFH. These helper T cells increase the ability of APCs to effectively present antigens and provide co-stimulation. Another class of CD4 T cells are regulatory T cells (Treg), which have an inhibitory function on other immune cells. Given the diversity of CD4 T cells phenotypes, it is expected that they would play myriad roles (and sometimes opposing roles) in the tumor microenvironment (TME). Beyond the traditional role of CD4 T cells in “helping” with antigen presentation to CD8 T cells [17], CD4 T cells can also have a direct cytotoxic effect on tumors [18]. Conversely, Treg cells can inhibit effective immune responses within the TME [19]. Overall, the roles of CD4 T cells in the TME are complex and require further study and clarification.
Distinct from HLA-restricted antigens, immunogenic tumor antigens on the surface of a cell may be recognized by antibody-mediated immunity. Antibody-mediated immunity begins when B cells recognize a chemical structure from the surface of a tumor cell with their B cell receptor (BCR). The activated B cells then internalize these structures and migrate to the peripheral lymphoid tissue or tertiary lymphoid structures (TLSs) in situ. Activated B cells will process the proteins and display them in shorter peptides on HLA class II complexes. The presented peptides may then be recognized by helper TFH cells which activate the B cells. Activated B cells proliferate and differentiate into plasma cells that produce antibodies against the target [11]. Currently, the precise role of antibody-mediated immunity in ccRCC is largely unknown. However, there is emerging evidence that TLSs within the tumor may be associated with improved clinical outcomes with ICI therapies and that these TLSs lead to the production of tumor-reactive auto-antibodies [11]. While much of the existing focus in RCC (and other solid tumors) has been centered on T cell-mediated immunity, there is clearly an emerging role for B-cell-mediated immunity as well.
With multiple ways to target and destroy tumor cells, the origin and presentation of tumor antigens is an important consideration when designing a therapy that leverages this machinery. Tumor antigens may be broadly categorized as tumor-associated antigens (TAAs) or tumor-specific antigens (TSAs). Tumor-associated antigens are derived from unaltered genes that are overexpressed in tumor cells, differentiation antigens specific to the local tissue, cancer germline/cancer testis antigens (CTAs), or human endogenous retroviruses (hERVs). On the other hand, tumor-specific antigens arise from genomic, transcriptomic, or proteomic alterations in the genome or oncoviruses that infect the genome. RCC is not known to be caused by an oncovirus, but examples of virally-driven cancers include human papillomavirus (HPV)-driven cervical, anal, head and neck cancers, and Merkel cell carcinoma (driven by Merkel cell polyomavirus). The modifications created by the viruses allow TSAs to be distinguishable from self-antigens. The extent to which distinct classes of TAAs and TSAs play a role in anti-tumor immunity in ccRCC is still largely unknown.
Tumor-associated antigens
Traditionally, antigens are thought to originate from a foreign source or from somatic mutations. However, unaltered genes expressed in tumor cells may be effectively targeted by the immune system and by immunotherapies. These antigens may be distinguishable from normal tissue because they are: (1) genes that are overexpressed in tumor cells, (2) genes that are differentiation markers that only arise in a specific local tissue, or (3) genes that have “leaked” expression in tumor cells because of demethylation or other epigenetic factors. For many of these tumor-associated antigens, they may arise from genes that are expressed in tumor cells because they support the cells’ survival and are therefore integral in the malignant cell.
In ccRCC, there are several genes that have been characterized as TAAs based on their overexpression. One example of a TAA was first discovered in 1996: receptor for advanced glycation end products (RAGE-1) [20]. RAGE-1 binds to free floating high mobility group box 1 (HMGB1). HMGB1 is a nuclear protein secreted by certain immune cells or released by apoptotic tumor cells. The interaction of RAGE-1 and HMGB1 promotes tumor cell survival, progression, and metastasis by its upregulation of the ERK/MAPK pathway [21, 22]. RAGE1 has been found to be expressed in the retina in normal tissue. A peptide derived from RAGE-1 has been shown to be an HLA-restricted antigen in ccRCC, presented on HLA-B*07.
Targeting overexpressed, HLA-restricted TAAs has been explored in RCC. In IMPRINT trial, investigated tested the multipeptide cancer vaccine IMA901 (targeting numerous overexpressed TAAs) in combination with the TKI sunitinib compared to sunitinib alone. The vaccine was composed of nine tumor-associated peptides predicted to present on HLA class I and one tumor-associated peptide predicted to present on HLA class II. The peptides were selected based on specific criteria, including overexpression in malignant RCC tissue compared with normal tissue and peptides that were predicted to be processed and presented by antigen processing machinery. Despite an initial signal in earlier phase trials, this multipeptide TAA vaccine did not improve overall survival in combination with sunitinib over sunitinib alone [23–25].
Beyond HLA-restricted antigens, surface antigens represent additional potential immune targets. An important example is carbonic anhydrase 9 (CA9), which is a classic ccRCC surface marker [26]. CA9 is important to the tumor cells’ survival by maintaining a neutral pH in the acidic tumor microenvironment. This cell surface enzyme catalyzes the reversible hydration of carbon dioxide into bicarbonate and a proton [27]. CA9 is also a hypoxia inducible gene, meaning its expression is increased in hypoxic environments. CA9 had been the focus of a major clinical trial targeted by chimeric antigen receptor (CAR) T cells [28], which are T cells engineered to recognize surface antigens through a fragment of an antibody linked to components of the endogenous TCR (and, more recently, also includes co-stimulatory machinery) [29]. Because CA9 is a cell surface protein, the molecule may be recognized by humoral (antibody-mediated) immunity. CA9 has been therapeutically targeted with the monoclonal antibody girentuximab, which was design to target RCC cells for NK cell-mediated killing through antibody-dependent cellular cytotoxicity. Unfortunately, girentuximab did not show clinical benefit in the adjuvant setting [30], but CA9 has remained a viable candidate target for CAR-T cells. Despite the importance of CA9 in ccRCC, the clinical trial was initially unsuccessful due to on-target, off-tumor interaction with cells in the liver inducing liver toxicity in patients [28]. In another cohort, patients were pre-treated with CA9 monoclonal antibody (mAb) which prevented this toxicity; however, no clinical responses were recorded. Importantly, that initial trial utilized a less effective earlier generation of CAR-T cells which lacked co-stimulatory machinery, so CA9 remains a target of potential interest. This trial, however, highlights a potential challenge to using TAAs as therapeutic targets: it is difficult to definitively know a priori the antigen’s expression levels in all normal tissues.
CD70 is an example of an overexpressed TAA that has more recently been targeted in RCC clinical trials. CD70 is a potent co-stimulatory marker typically expressed on DCs and binds to its receptor CD27 on naïve T cells. In addition, CD70 may also be found on activated T and B cells and several other cell types such as macrophages [31]. However, CD70 is commonly overexpressed in ccRCC [32], and therefore is a potential target for an antibody-mediated therapies, including CAR T cells. In fact, two clinical trials, COBALT-RCC (NCT04438083) and (NCT04696731), utilized allogeneic CAR T cells targeting CD70 on ccRCC. While substantial efforts are needed to further improve response rates, these studies provide an initial proof-of-concept that CAR T cell therapies can have clinical activity in ccRCC.
The second category of TAAs includes normal proteins expressed in the local tissue of the tumor because of its differentiation lineage [33]. Differentiation antigens were first investigated in melanoma in 1993 with the identification of tyrosinase, Melan-A/MART-1, and gp100/Pmel17 [34, 35]. In ccRCC, one well-characterized differentiation antigen, Mucin-1 (MUC1), is a transmembrane glycoprotein on endothelial cells and in cells related to kidney development. The protein may be recognized by the immune system through cell-mediated or humoral immunity. At least ten clinical trials have been designed to target MUC1 for a variety of cancer types. Examples of these trials include subunit vaccines, DNA vaccines, viral vectored vaccines, dendritic cell vaccines, and glycopeptide vaccines [36]. Multiple HLA-A*02-restricted peptides derived from MUC1 have been identified and have been shown to elicit in vitro T cell responses capable of killing MUC1 expressing cells (including RCC cell lines) [37]. In RCC specifically, numerous therapeutic approaches to target MUC1 have been investigated, including vaccination with a modified vaccinia virus expressing IL-2 and MUC1 [38] and a TAA peptide vaccine including a MUC1-targeting peptides [25, 36]. However, despite encouraging early clinical trial results, a phase III trial of TAA vaccine (including MUC1) did not improve overall survival [24].
The final category of TAAs includes genes that are typically silenced in the normal tissue of the malignancy but have “leaked” expression, typically due to epigenetic mechanisms such as DNA demethylation or histone modification [39]. One example of this category is cancer testis antigens (CTAs), which are typically expressed in normal tissue in immune privileged areas such as the placenta or testis. Immune privileged areas evade typical immune responses through several mechanisms, including downregulation of HLA molecules (to decrease antigen presentation) and the production of immunosuppressive cytokines. Therefore, immune privileged areas are able to avoid targeting by T cells [40]. The lack of presentation on normal tissue makes CTAs a good target for cancer therapies. In ccRCC, a targeted modified vaccinia Ankara (MVA) therapy was developed for the oncofetal 5T4 antigen, but it induced only low-level cellular immune responses against the CTA [41]. Additionally, a monoclonal antibody was developed to target 5T4, but also did not improve survival in patients [42].
Another example of “leaked” gene expression is aberrantly expressed endogenous retroviruses (ERVs). ERVs are defined as fragments of genomic DNA derived from the integration of retroviruses that infected the germ line cells of humans’ ancestors. They are estimated to make up approximately 1 to 8 percent of the human genome. However, they are incompetent in producing replicative viral particles, as they have accumulated mutations over time [43]. ERVs are epigenetically silenced through methods such as methylation, but they are de-repressed in certain malignancies, including ccRCC, making them a possible therapeutic target [44, 45]. ERVs are theorized to potentially make up a substantial fraction of the antigen burden for ccRCC. However, they are less well studied than other variants [46]. HERV-E (also known as ERVE-4) is particularly well characterized in ccRCC. In a ccRCC patient with a long-term complete response to allogeneic stem cell transplantation, the dominant circulating T cell population was found to recognize an HERV-E-derived peptide presented on HLA-A*11 [47]. Importantly, HERV-E expression was found to be directly regulated by HIF2α, which is typically constitutively active in ccRCCs [48, 49]. An engineered T cell therapy with a transduced TCR targeting the HERV-E-derived peptide is being tested in a clinical trial (NCT03354390).
Historically, many attempts to target TAAs have been unsuccessful due to on-target, off-tumor toxicity or poor clinical responses in clinical trials. The toxicity can be explained by a lack of understanding of where the self-antigen may be present. The lack of immune response may be due in large part to central tolerance mechanisms, where the TAAs are essentially considered as self-antigens, preventing a durable immune response from being developed [50, 51]. This is an inherent limitation of TAAs as a target for antigen-directed therapies –either the magnitude of the immune response will be low (and ineffective) because central immune tolerance is specifically designed to eliminate T cells capable of responding to such self-antigens, or the therapy would overcome central tolerance to elicit a high magnitude immune response, which would likely then also impact normal cells with low-level expression of the antigens. These limitations could potentially be overcome by targeting tumor-specific antigens.
Tumor-specific antigens
Tumor-specific antigens can be broken down into two major categories: neoantigens and oncoviral antigens. Neoantigens are antigens that arise in tumors from various mechanisms such as genomic mutations, aberrant transcriptomic variants, post-translational modifications, and viral open reading frames (ORFs). They represent classes of antigens that are unique to tumor cells and not otherwise native to normal cells in the body, and are therefore “new” and not subject to mechanisms of central immune tolerance [52]. ccRCC has only a moderate mutational burden, making other classes of neoantigens highly relevant [53]. Oncoviral antigens are created by oncogenic viruses, such as the Epstein-Barr virus or hepatitis B virus, which are not known to be relevant to ccRCC. However, the potential role of microbial-derived antigens has not been fully explored in ccRCC. In other tumor types, such as melanoma, bacteria have been seen to enter cells and serve as potential tumor antigens. Given the relatively modest number of mutation-derived neoantigens, this area is certainly worthy of further investigation in ccRCC.
Genomic
Genomic alterations in tumor cells are caused by non-synonymous single-nucleotide variants (SNVs), frameshifts due to insertions/deletions (INDELs), fusion, and chromosomal rearrangements [52]. Non-synonymous SNVs arise from a single nucleotide mutation to another nucleotide, leading to a change in the downstream amino acid. INDELs arise from the insertion or deletion of one or more nucleotides. INDELs could potentially lead to a frameshift resulting in all downstream amino acids being altered. While such frameshift neoantigens represent a high proportion of total mutations in ccRCC [54, 55], they are not associated with improved response to ICI [53]. While SNVs and INDELs are certainly relevant in ccRCC, fusion mutations and chromosomal rearrangements are not currently known to make up a significant portion of the genomic mutational burden [46]. Of note, though, in translocation RCC (tRCC), researchers have hypothesized that the TFE3 fusion event could potentially be immunogenic [56].
SNV and INDEL alterations lead to neoantigens that have an innate ability to avoid central tolerance [52, 57, 58]. T cells specific for neoantigens avoid central tolerance due to their highly antigenic properties and their absence in normal tissues [46, 59, 60]. However, because each tumor may contain dozens or even hundreds of potential neoantigens, and these antigens are patient-specific (i.e. “personal”), it is logistically challenging to generate effective personalized antigen-specific therapies for each patient [33, 61, 62]. The main logistical challenge in developing these personalized neoantigen therapies is the time and resources required to determine and choose which mutations are immunogenic and viable to include in a therapy. In other cancer settings, it often takes at least 8-12 weeks to develop a personalized cancer vaccine [63], which could be feasible in an adjuvant setting but limiting in a metastatic setting. There are ongoing efforts to target neoantigens directly in ccRCC using personalized vaccine approaches (NCT02950766), where tumor mutations are identified through whole-exome sequencing of tumor and normal tissue, expression of the variant is confirmed through RNA-sequencing, and antigenic peptides containing the mutation are predicted using advanced computational tools. In this trial, synthetic long peptides are synthesized and administered together with the immune adjuvant polyIC:LC in ccRCC patients with high-risk, resected disease (stage III or IV after surgical resection of all visible disease) [64].
Transcriptomic
Transcriptomic alterations may arise from RNA splicing, RNA editing, and allegedly non-coding regions [52]. Of these types of variants, RNA splicing has been most extensively examined in ccRCC [46]. Several types of RNA splicing events may lead to neoantigens in ccRCC, including exon skipping/inclusion, alternative 5’ splice sites, alternative 3’ splice sites, intron retention, mutually exclusive exons, and exitrons [52]. Excluding RNA splicing, many of these types of events are not well characterized in ccRCC, so their relevance as tumor antigens remains largely unknown.
ANTIGEN SPECIFICITY OF TUMOR-INFILTRATING T CELLS
First-line treatments for late-stage ccRCC involve immunotherapies that target immune checkpoints, such as PD-1/PD-L1 and CTLA-4. Anti-PD-1 therapies, in particular, are conventionally thought to function by revitalizing tumor-reactive T cells that have developed an exhausted phenotype in the tumor microenvironment. Exhaustion is a form of T cell dysfunction, and exhausted T cells are generally described to be hypofunctional cells that have upregulated expression of inhibitory receptors and reduced ability to secrete cytokines, proliferate, or exert cytolytic effector functions on target cells [65]. In ccRCC, T cells become progressively more exhausted with advancing disease stage [66]. Tumor-reactive T cells may target any of the types of tumor antigens discussed above [67]. In other types of cancers, such as melanoma (which has a high mutational burden), many of these tumor-specific antigens have been identified. However, in ccRCC, the specific tumor antigens and general classes of antigens that are most commonly recognized by these tumor-infiltrating lymphocytes (TILs) are not well understood.
The T cell infiltrate in ccRCC tumors is traditionally higher compared to most other solid tumors, and often experiences a highly immunosuppressive environment [68]. TILs have also been shown to harbor specificity for tumor antigens. In one study, investigators determined that up to 86% of TILs in 24 RCC patients were tumor-reactive [69]. They also showed that these tumor-reactive TILs had lower functional capabilities with mostly mono- or oligofunctional CD8 T cell responses [69], whereas polyfunctionality is typically considered important for optimal CD8 T cell responses [70–72]. In addition, two other studies additionally demonstrate that TILs in the tumor microenvironment of RCC are tumor-reactive [73, 74]. Of note, however, the immune cell infiltrate in advanced ccRCC develops an exhaustive phenotype, hindering their ability to control the disease. These terminally exhausted TILs may express multiple inhibitory immune checkpoints, including such markers as PD-1, TIM-3, LAG-4, and TIGIT, among others [66].
Until recently, there had been relatively few antigens identified to be recognized by TILs in ccRCC. An example of these antigens includes a HIF-1α-derived peptide [75]. In one immunogenomic analysis of the antigen specificity of T cells in six ccRCC patients, investigators performed whole exome sequencing (WES) of tumor samples, and then used neoantigen prediction software to predict which peptides may be presented on HLA class I molecules. With these predictions, they created multimers of peptide-MHC complexes that were used to probe the specificity of TILs. From this experiment, they identified 52 neoantigen-specific CD8 T cell responses in ccRCC TILs [76]. This important study demonstrated a potential role for targeting neoantigens in ccRCC, but only examined genomic-based neoantigens derived from SNVs and INDELs. Further work is needed to determine the contribution of other tumor-specific antigens to T cell immunity in ccRCC.
FUTURE DIRECTIONS
Despite multiple studies showing that the tumor microenvironment of RCC contains T cells that are tumor-reactive, little is known about their antigenic targets. To develop improved antigen-specific immunotherapies, the relevant classes of tumor antigens in ccRCC must be investigated more thoroughly. This may enable a next generation of off-the-shelf and personalized immunotherapies, including cell-based therapies and neoantigen vaccines.
Numerous cell-based therapy approach that have been brought into clinical trials, including dendritic cell-based vaccines [77, 78], engineered T cells [79], and autologous TILs [80]. In ccRCC, an initial clinical trial of an autologous dendritic cell vaccine where DCs were induced to present peptides from whole tumor samples demonstrated the feasibility of this approach, but did not yield positive clinical results [81]. Prior approaches have largely focused on TAAs, and their inherent limitations related to central tolerance mechanisms likely explain, at least in part, their lack of clinical efficacy. Engineered T cell approaches include transduced TCRs and chimeric antigen receptor (CAR) T cell therapies. Both of these approaches have been attempted in RCC and are being actively explored in clinical trials. Finally, autologous TIL therapy has also been explored in ccRCC, though has shown less promise than in other immunogenic tumors like melanoma [82]. Neoantigen vaccines are another strategy for personalized cancer treatment that has grown in interest in recent years and is actively being explored in RCC (NCT02950766). However, it is worth noting the potential challenges facing the effective development and implementation of antigen-directed therapies. Discovering antigens, particularly personalized tumor-specific antigens, requires access to clinical-grade sequencing services paired with advanced bioinformatic methods to successfully identify tumor-specific variants and predict antigenicity. Beyond merely identifying antigens, such therapies will require further refinement in delivery technologies and implementation. Current delivery methods, including peptides, nucleic acids (particularly messenger RNA), and engineered immune cell therapies all have demonstrated feasibility, but are costly and time-consuming (particularly with respect to personalized therapies). Further, even with a comprehensive understanding of tumor antigens, there are still many barriers to clinical activity, including the need to overcome immunosuppressive mechanisms in the tumor and beyond. It appears likely then that antigen-directed therapies could emerge as an important component of a combination immunotherapy strategy, adding the “steering wheel” to an immune response already released through immune cell activators or checkpoints inhibitors.
All of these antigen-directed therapeutic approaches would greatly benefit from understanding the antigen specificity of TILs. A deeper understanding of the classes of tumor antigens in RCC would provide important biological insight into this disease, with potential implications for understanding the high T cell infiltration in these tumors and contribute to an improved understanding of at least one potential mechanism resistant to current immunotherapies (i.e. lack of tumor antigens). However, knowledge of the exact tumor antigens in this disease would provide a foundation for the development an antigen-directed therapies, with this potential to further improve our immunotherapeutic armamentarium in RCC. Specifically, this knowledge would allow vaccines or cell-based therapies to be designed with a greater degree of accuracy by understanding which peptides elicit optimal immune responses. Therefore, a more concerted effort for antigen discovery is needed for ccRCC.
ACKNOWLEDGMENTS
We thank members of the Braun Laboratory for their input. Figures created using BioRender.com.
FUNDING
D.A.B. acknowledges support from the Dept of Defense Early Career Investigator grant (KCRP AKCI-ECI, W81XWH-20-1-0882), the Louis Goodman and Alfred Gilman Yale Scholar Fund, and the Yale Cancer Center (supported by NIH/NCI research grant P30CA016359).
AUTHOR CONTRIBUTIONS
D.A.B. contributed to the conception and writing of author. N.R.S. contributed to the conception and writing of article.
CONFLICT OF INTEREST
D.A.B. reports honoraria from LM Education/Exchange Services, advisory board fees from Exelixis and AVEO, equity in Fortress Biotech (subsidiary), personal fees from Schlesinger Associates, Cancer Expert Now, Adnovate Strategies, MDedge, CancerNetwork, Catenion, OncLive, Cello Health BioConsulting, PWW Consulting, Haymarket Medical Network, Aptitude Health, ASCO Post/Harborside, Targeted Oncology, AbbVie, and research support from Exelixis and AstraZeneca, outside of the submitted work. N.R.S has no conflict of interest to report.
REFERENCES
[1] | Demasure S , et al. Overall survival improvement in patients with metastatic clear-cell renal cell carcinoma between 2000 and 2020: a retrospective cohort study. Acta Oncol. (2022) ;61: (1):22–9. |
[2] | Motzer RJ , et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N Engl J Med. (2018) ;378: (14):1277–90. |
[3] | Motzer RJ , et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med. (2019) ;380: (12):1103–15. |
[4] | Rini BI , et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med. (2019) ;380: (12):1116–27. |
[5] | Motzer R , et al. Lenvatinib plus Pembrolizumab or Everolimus for Advanced Renal Cell Carcinoma. N Engl J Med. (2021) ;384: (14):1289–300. |
[6] | Choueiri TK , et al. Nivolumab plus Cabozantinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med. (2021) ;384: (9):829–41. |
[7] | Atkins MB , et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. (1999) ;17: (7):2105–16. |
[8] | Motzer RJ , et al. Conditional survival and long-term efficacy with nivolumab plus ipilimumab versus sunitinib in patients with advanced renal cell carcinoma. Cancer. (2022) ;128: (11):2085–97. |
[9] | Chen DS , Mellman I . Oncology meets immunology: the cancer-immunity cycle. Immunity. (2013) ;39: (1):1–10. |
[10] | Braun DA , et al. Beyond conventional immune-checkpoint inhibition –novel immunotherapies for renal cell carcinoma. Nat Rev Clin Oncol. (2021) ;18: (4):199–214. |
[11] | Meylan M , et al. Tertiary lymphoid structures generate and propagate anti-tumor antibody-producing plasma cells in renal cell cancer. Immunity. (2022) ;55: (3):527–41 e5. |
[12] | Han Y , Liu D , Li L . PD-1/PD-L1 pathway: current researches in cancer. Am J Cancer Res. (2020) ;10: (3):727–42. |
[13] | Oliveira G , et al. Phenotype, specificity and avidity of antitumour CD8(+) T cells in melanoma. Nature. (2021) ;596: (7870):119–25. |
[14] | Jansen CS , et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature. (2019) ;576: (7787):465–70. |
[15] | Ng KW , et al. Antibodies against endogenous retroviruses promote lung cancer immunotherapy. Nature. (2023) ;616: (7957):563–73. |
[16] | Raskov H , et al. Cytotoxic CD8(+) T cells in cancer and cancer immunotherapy. Br J Cancer. (2021) ;124: (2):359–67. |
[17] | Ahrends T , Borst J . The opposing roles of CD ((2018) ;4: (+) T cells in anti-tumour immunity. Immunology. 154(4):582–92. |
[18] | Oh DY , Fong L . Cytotoxic CD8(+) T cells in cancer: Expanding the immune effector toolbox. Immunity. (2021) ;54: (12):2701–11. |
[19] | Plitas GR , Alexander . Regulatory T Cells in Cancer. Annual Review of Cancer Biology. (2020) ;4: :459–77. |
[20] | Gaugler B , et al. A new gene coding for an antigen recognized by autologous cytolytic T lymphocytes on a human renal carcinoma. Immunogenetics. (1996) ;44: (5):323–30. |
[21] | Lin L , et al. Receptor for advanced glycation end products (RAGE) partially mediates HMGB1-ERKs activation in clear cell renal cell carcinoma. J Cancer Res Clin Oncol. (2012) ;138: (1):11–22. |
[22] | El-Far AH , et al. Role and Mechanisms of RAGE-Ligand Complexes and RAGE-Inhibitors in Cancer Progression. Int J Mol Sci. (2020) ;21: (10). |
[23] | Kirner A , Mayer-Mokler A , Reinhardt C . IMA a multi-peptide cancer vaccine for treatment of renal cell cancer. Hum Vaccin Immunother. (2014) ;10: (11):3179–89. |
[24] | Rini BI , et al. IMA901, a multipeptide cancer vaccine, plus sunitinib versus sunitinib alone, as first-line therapy for advanced or metastatic renal cell carcinoma (IMPRINT): a multicentre, open-label, randomised, controlled, phase 3 trial. Lancet Oncol. (2016) ;17: (11):1599–611. |
[25] | Walter S , et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat Med. (2012) ;18: (8):1254–61. |
[26] | Courcier J , et al. Carbonic Anhydrase IX in Renal Cell Carcinoma, Implications for Disease Management. Int J Mol Sci. (2020) ;21: (19). |
[27] | Tostain J , et al. Carbonic anhydrase 9 in clear cell renal cell carcinoma: a marker for diagnosis, prognosis and treatment. Eur J Cancer. (2010) ;46: (18):3141–8. |
[28] | Lamers CH , et al. Treatment of metastatic renal cell carcinoma (mRCC) with CAIX CAR-engineered T-cells-a completed study overview. Biochem Soc Trans. (2016) ;44: (3):951–9. |
[29] | Sterner RC , Sterner RM . CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. (2021) ;11: (4):69. |
[30] | Chamie K , et al. Adjuvant Weekly Girentuximab Following Nephrectomy for High-Risk Renal Cell Carcinoma: The ARISER Randomized Clinical Trial. JAMA Oncol. (2017) ;3: (7):913–20. |
[31] | Flieswasser T , et al. The CD70-CD27 axis in oncology: the new kids on the block. J Exp Clin Cancer Res. (2022) ;41: (1):12. |
[32] | Jilaveanu LB , et al. CD70 expression patterns in renal cell carcinoma. Hum Pathol. (2012) ;43: (9):1394–9. |
[33] | Feola S , et al. Uncovering the Tumor Antigen Landscape: What to Know about the Discovery Process. Cancers (Basel). (2020) ;12: (6). |
[34] | Brichard V , et al. The tyrosinase gene codes for an antigen recognized by autologous cytolytic T lymphocytes on HLA-A2 melanomas. J Exp Med. (1993) ;178: (2):489–95. |
[35] | Kawakami Y , et al. Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proc Natl Acad Sci U S A. (1994) ;91: (14):6458–62. |
[36] | Gao T , Cen Q , Lei H . A review on development of MUC1-based cancer vaccine. Biomed Pharmacother. (2020) ;132: :110888. |
[37] | Brossart P , et al. Identification of HLA-A2-restricted T-cellepitopes derived from the MUC1 tumor antigen for broadly applicablevaccine therapies. Blood. (1999) ;93: (12):4309–17. |
[38] | Oudard S , et al. A phase II study of the cancer vaccine TGalone and in combination with cytokines in patients with metastatic renal clear-cell carcinoma: clinical and immunological findings. Cancer Immunol Immunother. (2011) ;60: (2):261–71. |
[39] | Fratta E , et al. The biology of cancer testis antigens: putative function, regulation and therapeutic potential. Mol Oncol. (2011) ;5: (2):164–82. |
[40] | Gjerstorff MF , Andersen MH , Ditzel HJ . Oncogenic cancer/testis antigens: prime candidates for immunotherapy. Oncotarget. (2015) ;6: (18):15772–87. |
[41] | Kaufman HL , et al. Phase II trial of Modified Vaccinia Ankara (MVA) virus expressing 5T4 and high dose Interleukin-2 (IL-2) in patients with metastatic renal cell carcinoma. J Transl Med. (2009) ;7: :2. |
[42] | Stern PL , Harrop R . 5T4 oncofoetal antigen: an attractive target for immune intervention in cancer. Cancer Immunol Immunother. (2017) ;66: (4):415–26. |
[43] | Bannert N , et al. HERVs New Role in Cancer: From Accused Perpetrators to Cheerful Protectors. Front Microbiol. (2018) ;9: :178. |
[44] | Kassiotis G , Stoye JP . Immune responses to endogenous retroelements: taking the bad with the good. Nat Rev Immunol. (2016) ;16: (4):207–19. |
[45] | Attermann AS , et al. Human endogenous retroviruses and their implication for immunotherapeutics of cancer. Ann Oncol. (2018) ;29: (11):2183–91. |
[46] | Smith CC , et al. Alternative tumour-specific antigens. Nat Rev Cancer. (2019) ;19: (8):465–78. |
[47] | Takahashi Y , et al. Regression of human kidney cancer following allogeneic stem cell transplantation is associated with recognition of an HERV-E antigen by T cells. J Clin Invest. (2008) ;118: (3):1099–109. |
[48] | Cherkasova E , et al. Detection of an Immunogenic HERV-E Envelope with Selective Expression in Clear Cell Kidney Cancer. Cancer Res. (2016) ;76: (8):2177–85. |
[49] | Cherkasova E , et al. Inactivation of the von Hippel-Lindau tumor suppressor leads to selective expression of a human endogenous retrovirus in kidney cancer. Oncogene. (2011) ;30: (47):4697–706. |
[50] | Sahin U , et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature. (2020) ;585: (7823):107–12. |
[51] | Leko V , Rosenberg SA . Identifying and Targeting Human Tumor Antigens for T Cell-Based Immunotherapy of Solid Tumors. Cancer Cell. (2020) ;38: (4):454–72. |
[52] | Xie N , et al. Neoantigens: promising targets for cancer therapy. Signal Transduct Target Ther. (2023) ;8: (1):9. |
[53] | Braun DA , et al. Interplay of somatic alterations and immune infiltration modulates response to PD-1 blockade in advanced clear cell renal cell carcinoma. Nat Med. (2020) ;26: (6):909–18. |
[54] | Turajlic S , et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol. (2017) ;18: (8):1009–21. |
[55] | Alexandrov LB , et al. Signatures of mutational processes in human cancer. Nature. (2013) ;500: (7463):415–21. |
[56] | Marcon J , et al. Comprehensive Genomic Analysis of Translocation Renal Cell Carcinoma Reveals Copy-Number Variations as Drivers of Disease Progression. Clin Cancer Res. (2020) ;26: (14):3629–40. |
[57] | Schumacher TN , Schreiber RD . Neoantigens in cancer immunotherapy. Science. (2015) ;348: (6230):69–74. |
[58] | Schumacher TN , Scheper W , Kvistborg P . Cancer Neoantigens. Annu Rev Immunol. (2019) ;37: :173–200. |
[59] | Jiang T , et al. Tumor neoantigens: from basic research to clinical applications. J Hematol Oncol. (2019) ;12: (1):93. |
[60] | Richters MM , et al. Best practices for bioinformatic characterization of neoantigens for clinical utility. Genome Med. (2019) ;11: (1):56. |
[61] | Ilyas S , Yang JC . Landscape of Tumor Antigens in T Cell Immunotherapy. J Immunol. (2015) ;195: (11):5117–22. |
[62] | Vigneron N . Human Tumor Antigens and Cancer Immunotherapy. Biomed Res Int. (2015) ;2015: :948501. |
[63] | Ott PA , et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. (2017) ;547: (7662):217–21. |
[64] | Cristescu R , et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science. (2018) ;362: (6411). |
[65] | Blank CU , et al. Defining ‘T cell exhaustion’. Nat Rev Immunol. (2019) ;19: (11):665–74. |
[66] | Braun DA , et al. Progressive immune dysfunction with advancing disease stage in renal cell carcinoma. Cancer Cell. (2021) ;39: (5):632–48 e8. |
[67] | Tumeh PC , et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. (2014) ;515: (7528):568–71. |
[68] | Matsushita H , et al. Neoantigen Load, Antigen Presentation Machinery, and Immune Signatures Determine Prognosis in Clear Cell Renal Cell Carcinoma. Cancer Immunol Res. (2016) ;4: (5):463–71. |
[69] | Andersen R , et al. T-cell Responses in the Microenvironment of Primary Renal Cell Carcinoma-Implications for Adoptive Cell Therapy. Cancer Immunol Res. (2018) ;6: (2):222–35. |
[70] | Seder RA , Darrah PA , Roederer M . T-cell quality in memory and protection: implications for vaccine design. Nat Rev Immunol. (2008) ;8: (4):247–58. |
[71] | Donia M , et al. Aberrant Expression of MHC Class II in Melanoma Attracts Inflammatory Tumor-Specific CD4+T-Cells, Which Dampen CD8+T-cell Antitumor Reactivity. Cancer Res. (2015) ;75: (18):3747–59. |
[72] | Berinstein NL , et al. Survivin-targeted immunotherapy drives robust polyfunctional T cell generation and differentiation in advanced ovarian cancer patients. OncoImmunology. (2015) ;4: (8):e1026529. |
[73] | Markel G , et al. Preclinical evaluation of adoptive cell therapy for patients with metastatic renal cell carcinoma. Anti Cancer Res. (2009) ;29: (1):145–54. |
[74] | Baldan V , et al. Efficient and reproducible generation of tumour-infiltrating lymphocytes for renal cell carcinoma. Br J Cancer. (2015) ;112: (9):1510–8. |
[75] | Minami T , et al. Hypoxia-inducing factor (HIF)-1alpha-derived peptide capable of inducing cancer-reactive cytotoxic T lymphocytes from HLA-A24(+) patients with renal cell carcinoma. Int Immunopharmacol. (2017) ;44: :197–202. |
[76] | Hansen UK , et al. Tumor-Infiltrating T Cells From Clear Cell Renal Cell Carcinoma Patients Recognize Neoepitopes Derived From Point and Frameshift Mutations. Front Immunol. (2020) ;11: :373. |
[77] | Mastelic-Gavillet B , et al. Personalized Dendritic Cell Vaccines-Recent Breakthroughs and Encouraging Clinical Results. Front Immunol. (2019) ;10: :766. |
[78] | Carreno BM , et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. (2015) ;348: (6236):803–8. |
[79] | Yang JC , Rosenberg SA . Adoptive T-Cell Therapy for Cancer. Adv Immunol. (2016) ;130: :279–94. |
[80] | Hulen TMC C.A. , Svane IM , Met Ö . ACT Up TIL Now: The Evolution of Tumor-Infiltrating Lymphocytes in Adoptive Cell Therapy for the Treatment of Solid Tumors. Immuno. (2021) ;1: :194–211. |
[81] | Figlin RA , et al. Results of the ADAPT Phase 3 Study of Rocapuldencel-T in Combination with Sunitinib as First-Line Therapy in Patients with Metastatic Renal Cell Carcinoma. Clin Cancer Res. (2020) ;26: (10):2327–36. |
[82] | Andersen R , et al. Tumor infiltrating lymphocyte therapy for ovarian cancer and renal cell carcinoma. Hum Vaccin Immunother. (2015) ;11: (12):2790–5. |


