
Hidden Gems in Neurology: The Syndrome of Hemiparkinsonism-Hemiatrophy
This article will shed some light on “hidden gems” in the neurological literature of Parkinson disease regarding a rare but distinctive parkinsonian syndrome associated with hemiparkinsonism and somatic/cerebral hemiatrophy (HP-HA). This phenomenon was brought to the medical world’s attention in a 1981 report of four patients by Harold Klawans.1 Klawans, one of the pioneers of movement disorders and a keen observer of clinical phenomenology,2 reported instances of skeletal hemiatrophy on the same side in which these patients manifested parkinsonism. On examination, they exhibited unilaterally smaller extremities (smaller hands and/or feet). On the smaller side, the parkinsonism developed in their fourth to fifth decades of life, which is younger than the typical age of onset for sporadic PD of 58.3 Although for these patients the origin of their hemiatrophy was not determined, two of them had a history of perinatal injury. Klawans and colleagues noted that, for all patients, parkinsonian features remained unilateral, were slowly progressive, and tended to respond poorly to levodopa. One of the patients showed dystonia on his parkinsonian side (described as left foot inversion during exercise). Klawans and colleagues postulated that, in this novel syndrome, hemiparkinsonism might have been a delayed consequence of developmental or acquired hemiatrophy. Given the poor response to levodopa therapy, the authors also suggested that the parkinsonism might have a component of postsynaptic striatal dysfunction.
Seven years later, Buchman and colleagues published another series of 15 patients with HP-HA (adding 11 new cases to those described previously).4 For all patients, their mean age of onset for parkinsonian features was relatively young (mean age: 43.7 years, range: 31–61). Ten of the 15 patients were noted to have dystonia ipsilateral with the hemiparkinsonism and somatic hemiatrophy. In each instance, the limb dystonic symptoms arose prior to the start of levodopa treatment. Approximately half of these patients reported some aspect of an abnormal birth history, such as prolonged labor or delivery, traumatic delivery, breech presentation, premature delivery, or neonatal hypoxia. This background was in support of a hypothesis that HP-HA might arise from perinatal cerebral insults. In their report, Klawans and colleagues reiterated key differences between HP-HA and typical presentations of idiopathic PD. Although HP-HA tended to be highly or fully asymmetric, more than half of the patients eventually developed bilateral parkinsonian features.
Another series of 11 HP-HA patients were described in 1990 by Giladi and colleagues.5 In this “hidden gem,” the authors defined hemiatrophy not only on the basis of somatic asymmetry but also recognizing a structural cerebral change contralateral to the hemiatrophic side on which parkinsonian features were manifested. Ten of the 11 patients reported had body hemiatrophy, while 9 manifested neuroradiologic findings of contralateral cerebral hemiatrophy—either an enlarged lateral ventricle, cortical atrophy, or basal ganglia atrophy. To explain the subsequent bilateral parkinsonian involvement observed in some of the patients, these authors postulated that relative brain atrophy can be appreciated only in the most significantly affected area, and that the seemingly non-atrophic side might have been damaged to a lesser extent. In this series, the mean onset age of parkinsonian symptoms was 38.1 years (range: 18–54).
Though some degree of skeletal asymmetry can be normal, grossly observable asymmetry is rare and implies some mechanisms of abnormal development.6 In patients with HP-HA, asymmetry of limbs was generally recognized in childhood, requiring, in some instances, altered shoe or glove sizes.1,7 Hence, the young age of onset for asymmetry reflects developmental growth failure rather than loss of body mass (as might be implied by the term hemiatrophy). In general, asymmetry of the cerebral hemispheres is a common physiological finding that is determined by genetic (such as handedness) and various environmental factors.8,9 Marked brain hemiatrophy is rare and arises as a complication of perinatal or early childhood brain insults, such as in hypoxia or hypotension in infants, or due to secondary cell loss, demyelination, or gliosis.10 Penfield and Robertson were among the first neuroscientists to report this phenomenon. In their 1943 study, they recognized that the patients who demonstrated asymmetry of the body (relative smallness on one side) had focal lesions in the contralateral postcentral gyrus before age 3, whereas lesions restricted to frontal, occipital, or temporal cortices were not associated with hemiatrophy.11 In a 17-patient study evaluating PET scans of infants following asphyxia, one of the cases showed an asymmetric decrease in cerebral blood flow in the atrophic hemisphere.12
Another series of six HP-HA patients was described in a 1994 report—the first to compare HP-HA patients to groups of healthy controls and to idiopathic PD patients.13 In this study, Przedborski and colleagues compared the HP-HA patients (defined by either somatic or contralateral cerebral atrophy).
The HP-HA patients were contrasted to ten healthy individuals and ten others with a unilateral presentation of PD but lacking brain or body hemiatrophy. 18F-fluorodeoxyglucose tracer PET images taken at the level of the basal ganglia demonstrated a metabolic reduction in the contralateral caudate and putamen compared to the normal control and unilateral PD groups. The HP-HA cohort had reduced glucose utilization in the contralateral medial frontal cortex as compared to normal controls, although not relative to the unilateral PD group. Two patients were also studied with 18F-fluorodopa tracer PET; in both instances, the contralateral striatal update was significantly reduced compared to normal controls. The authors concluded that the findings of nigral and extranigral dysfunction suggested both pre- and post-synaptic nigrostriatal dopaminergic dysfunction in HP-HA.
In 2004, Wijemanne and colleagues added 30 HP-HA cases to the medical literature, detailing this disorder’s clinical and radiographic characteristics.7 The mean age of onset in this series was 44.2 years (range: 15–63). Half of them manifested dystonia before the onset of hemiparkinsonism. Brain asymmetry was noted in 30%, and 47% of those whose birth histories were available were reported to have difficult birth or severe febrile illness in the first few months of life. This publication highlighted that brain injuries resulting in HP-PA might occur not only in the perinatal period, but also in early childhood. Tables 1 and 2 summarize previously reported case series and reports on HP-HA.
Table 1
Summary of Previously Published Case Series of Hemiparkinsonism-Hemiatrophy
| Reference | Number of patients | Mean age of onset, y | Male: Female ratio | Body hemiatrophy, % | Cerebral hemiatrophy, % | Dystonia, % | Abnormal birth or perinatal history∧, % | Response to levodopa, % | Progression to bilateral symptoms, % | Mean length of follow up, y |
| 4 | 15 | 43.7 (31–61) | 11 : 4 | 100 | 8 | 67 | 47 | 78 | 53 | 3.4 (1–35) |
| 5 | 11 | 38.1 (18–54) | 9 : 2 | 91 | 55 | 27 | 0 | 64 | 46 | 1.7 (0.33–5) |
| 13 | 6 | 35.1 (23–59) | 1 : 1 | 100 | 67 | 83 | 17 | 33 | 0 | 3.3 (1–7) |
| 16 | 2 | 47.5 (44–51) | 1 : 1 | 100 | 0 | 50 | 0 | 50 | 0 | N/A |
| 7 | 30 | 44.2 (15–63) | 2 : 3 | 100 | 30 | 70 | 47 | 80 | 23 | 4.2 (1–9) |
| 17 | 2 | 41.5 (34–49) | 2 : 0 | 100 | 50 | 100 | 0 | 50 | 0 | 5.8 (5.5–6) |
| 18 | 4 | 26.3 (2–50) | 1 : 1 | 100 | 50 | 50 | 75 | 50 | 25 | 2 (1–3) |
∧Abnormal birth, perinatal, early childhood, or head trauma history. N/A, not available. This table includes all published case series indexed in PubMed and written in English as of June 6, 2024.
Table 2
Summary of Previously Published Case Reports of Hemiparkinsonism-Hemiatrophy
| Reference | Age of onset, y | Sex | Body hemiatrophy | Cerebral hemiatrophy seen on imaging | Dystonia | Abnormal birth or perinatal history∧ | Response to levodopa | Progression to bilateral symptoms | Length of follow up, y | Unique findings |
| 15 | 19 | F | Yes | No | Yes | No | N/A | No | 0.75 | Responsive to subthalamic nucleus stimulator |
| 19 | 35 | M | Yes | No | Yes | N/A | No | No | 4 | Spontaneous limb elevation on same side as parkinsonism symptoms |
| 20 | 52 | F | Yes | Yes | No | Yes | Minimal | No | 1.67 | – |
| 21 | 45 | F | Yes | Yes | No | No | No | No | 1 | – |
| 22 | 36 | M | Yes | No | No | No | Yes | No | 3 | Abnormal signal in the midbrain on the side contralateral to symptoms |
| 23 | 27 | F | Yes | No | Yes | Yes | Yes | No | 11 | Exertional alternating leg paresis, dystonia, and hypotonia; abnormal signal in the substantia nigra on the contralateral side to symptoms |
| 24 | 22 | M | No | Yes | Yes | No | N/A | No | N/A | Bilateral asymmetric cerebral atrophy |
| 25 | 43 | M | Yes | Yes | No | No | Slight and transient | Yes | 5 | Brain hemihypoplasia, abnormal eye movements |
| 26 | 30 | F | Yes | No | Yes | N/A | Yes | No | 5 | Parkin gene mutations–missense mutation (Arg275Trp) and a duplication of exon 7 of parkin |
| 27 | 42 | M | Yes | No | Yes | N/A | N/A | No | N/A | Unilateral freezing of gait |
| 28 | 55 | F | Yes | Yes | No | No | Yes | Yes | 9 | – |
| 29 | 50 | F | Yes | No | Yes | No | Yes | No | 1 | – |
| 30 | 46 | M | Yes | No | Yes | No | Yes | No | N/A | Mirror movements |
| 31 | 16 | F | Yes | No | Yes | N/A | No | No | N/A | – |
| 32 | 26 | M | Yes | Yes | Yes | No | Yes | No | 1 | – |
| 33 | 36 | M | Yes | No | No | N/A | Yes | No | N/A | Concomitant Alzheimer disease |
| 34 | 46 | M | Yes | Yes | No | No | Yes | No | N/A | Midbrain atrophy and substantia nigra rarefaction contralateral to parkinsonism symptoms |
| 35 | 39 | M | Yes | No | No | No | No | No | 3 | SPECT with 99mTc TRODAT-1 imaging showed decreased 99mTc TRODAT-1 binding in the right basal ganglia |
| 36 | 65 | M | Yes | Yes | Yes | No | Yes | No | 3 | Schizencephaly |
| 37 | 20 | M | No | Yes | Yes | No | No | No | 1 | Iron deposition in globus pallidus and substantia nigra on the same side as cerebral hemiatrophy |
| 38 | 14 | M | Yes | No | Yes | Yes | Yes | No | 18 | Long-term (15 year) follow up brain MRI remained unremarkable and DaT scan was unchanged |
∧Abnormal birth, perinatal, early childhood, or head trauma history. N/A, not available. This table includes all published case reports indexed in PubMed and written in English as of June 6, 2024.
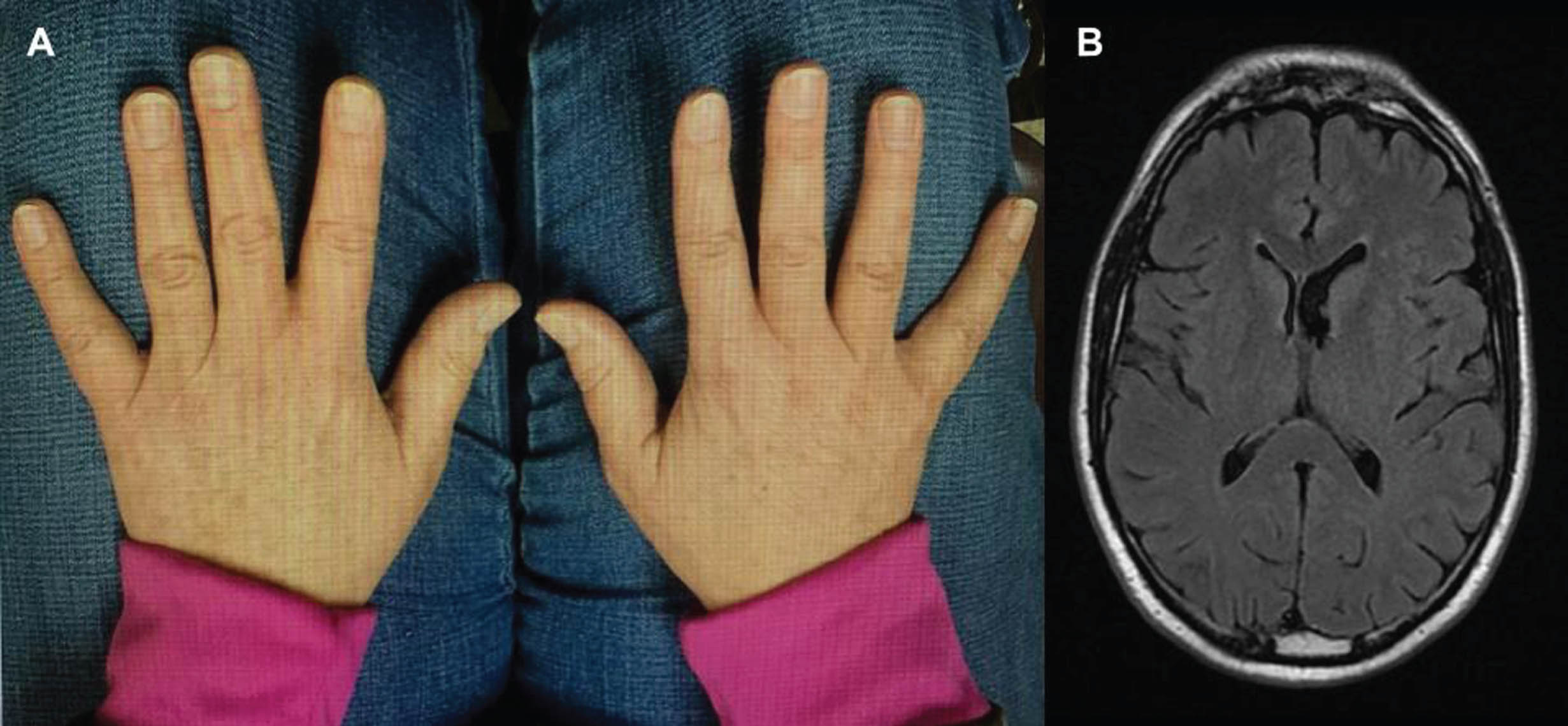
In Fig. 1, typical features of the HP-HA syndrome are illustrated in the cerebrum and hand size images. This 61-year-old right-handed man gradually developed resting hand tremor, small and slow handwriting, decreased dexterity and decrementing at repetitive tapping only on the right hand and foot. No features of dystonia, acting out dreams, impaired olfaction, or other neurological problems were reported. The patient reported that his right foot was one-half American shoe size smaller than the left and his right hand revealed slightly shorter digits than the left (Fig. 1A). In addition to features described above, his examination revealed mild right arm cogwheel rigidity, and decreased arm swing while walking. A brain MRI image demonstrated enlargement of the left lateral ventricle (Fig. 1B). Over 6 years of follow-up, this patient has responded well to carbidopa-levodopa 25–100 mg three times daily. He has not developed left-sided symptoms, dyskinesia or dystonia, or motor fluctuations.
Fig. 1
In panel A, the smaller sizes of right-hand digits are evident. The axial FLAIR brain MRI image in panel B shows enlargement of the left lateral ventricle.
In summary, HP-HA is a rare and unique clinical pattern of parkinsonism and likely to be under-recognized, especially since asymmetrical parkinsonism is a common feature of sporadic PD. Similarly, many cases of otherwise typical PD have the preceding onset of limb dystonia.14 While the phenomenology of HP-PA is well characterized by unilateral symptoms on the side affected by skeletal hemiatrophy (contralateral to cerebral hemiatrophy), its neuropathology has not yet been reported. Histopathological examination of cases is needed to explore the cause of the bilaterality of this syndrome.
HP-HA presents in adulthood, though typically at an age younger than that of sporadic PD. Abnormal birth history or other early childhood cerebral insults appear to be major risk factors, and it may be illuminating to inquire about perinatal history especially if a patient presents with symptoms concerning for HP-HA. Awareness of this unique syndrome makes it informative to inquire about birth injury. Clinicians may choose to pursue brain imaging in patients with parkinsonism features who are found to have gross somatic asymmetry although, in general, we do not feel the need to image patients with bilateral features of parkinsonism. As mentioned previously, grossly observable asymmetry that is at once noticeable to the naked eye is documented to be rare in literature.6 We must remain vigilant of HP-HA, especially in patients who present with parkinsonism symptoms and are noted to have gross somatic asymmetry on physical examination. Given the early life origin of some cases of HP-HA, its etiology merits further study in experimental models ofparkinsonism.
Recognizing the diagnosis of HP-HA carries relevant prognostic information, especially since its course tends to be slower and with symptoms remaining unilateral for longer than typical PD. Responsiveness to levodopa is variable amongst patients. To the best of our knowledge, there has only been one reported case of deep brain stimulation and the target was subthalamic nucleus.15 This patient had dystonia and was reported to have had a dramatic response after subthalamic stimulation while anticholinergic and dopamine agonist therapy resulted in poor improvement. We would postulate the globus pallidus internus to be a reasonable target as well given that dystonia is often involved. Further studies are needed to confirm the efficacy of different stimulation targets in HP-HA.
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
Dr. Park has no conflict of interest to report. Dr. LeWitt is an Editorial Board Member of this journal but was not involved in the peer-review process nor had access to any information regarding its peer-review.
REFERENCES
1. | Klawans HL . Hemiparkinsonism as a late complication of Hemiatrophy. Neurology (1981) ; 31: : 625–625. |
2. | Goetz CG . In Memoriam: Harold L. Klawans, MD. Mov Disord (2004) ; 13: : 625. |
3. | Pagano G , Ferrara N , Brooks DJ , et al. Age at onset and Parkinson disease phenotype. Neurology (2016) ; 86: : 1400–1407. |
4. | Buchman AS , Goetz CG and Klawans HL. Hemiparkinsonism with hemiatrophy. Neurology (1988) ; 38: : 527–527. |
5. | Giladi N , Burke RE , Kostic V , et al. Hemiparkinsonism-Hemiatrophy syndrome. Neurology (1990) ; 40: : 1731–1731. |
6. | Halperin G . Normal asymmetry and unilateral hypertrophy. Arch Intern Med (1931) ; 48: : 676. |
7. | Wijemanne S , Jankovic J . Hemiparkinsonism-hemiatrophy syndrome. Neurology (2007) ; 69: : 1585–1594. |
8. | Amunts K , Schlaug G , Schleicher A , et al. Asymmetry in the human motor cortex and handedness. Neuroimage (1996) ; 4: : 216–222. |
9. | Corballis MC . Evolution of cerebral asymmetry. Prog Brain Res (2019) ; 250: : 153–178. |
10. | Alpers BJ and Dear RB. Hemiatrophy of the brain. J Nerv Ment Dis (1939) ; 89: : 653–671. |
11. | Penfield W and Robertson JSM. Growth asymmetry due to lesion of the post central cortex. Arch Neurol Psychiatry (1943) ; 50: : 405–430. |
12. | Volpe JJ , Herscovitch P , Perlman JM , et al. Positron emission tomography in the asphyxiated term newborn: parasagittal impairment of cerebral blood flow. Ann Neurol (1985) ; 17: : 287–296. |
13. | Przedborski S , Giladi N , Takikawa S , et al. Metabolic topography of the hemiparkinsonism-hemiatrophy syndrome. Neurology (1994) ; 44: : 1622–1628. |
14. | LeWitt PA , Burns RS and Newman RP. Dystonia in untreated parkinsonism. Clin Neuropharmacol (1986) ; 9: : 293–297. |
15. | Jenkins M , Mendonca D , Parrent A , et al. Hemiparkinsonism-somatic hemiatrophy syndrome. Can J Neurol Sci (2002) ; 29: : 184–187. |
16. | Nardone R , Lochner P and Tezzon F. Hemiparkinson-hemiatrophy syndrome: a transcranial magnetic stimulation study. Electromyogr Clin Neurophysiol (2003) ; 43: : 235–240. |
17. | Dziadkiewicz A , Białecka M and Janik P. Hemiparkinsonism-hemiatrophy syndrome – report on two cases and review of the literature. Neurol Neurochir Pol (2013) ; 47: : 387–392. |
18. | He C , Zhang P , Li Y , et al. Hemiparkinsonism or hemidystonia with hemiatrophy syndrome: a case series with follow-up. Front Neurosci (2020) ; 14: : 64. |
19. | Giladi N and Fahn S. Hemiparkinsonism-hemiatrophy syndrome may mimic early-stage cortical-basal ganglionic degeneration. Mov Disord (1992) ; 7: : 384–385. |
20. | Przedborski S , Goldman S , Levivier M , et al. Brain glucose metabolism and dopamine D2 receptor analysis in a patient with hemiparkinsonism-hemiatrophy syndrome. Mov Disord (1993) ; 8: : 391–395. |
21. | Przedborski S , Goldman S , Giladi N , et al. Positron emission tomography in hemiparkinsonism-hemiatrophy syndrome. Adv Neurol (1993) ; 60: : 501–505. |
22. | Costa B , Zanette G , Bertolasi L , et al. Hemiparkinsonism-hemiatrophy syndrome: neuroradiological and neurophysiological findings. Eur Neurol (1994) ; 34: : 107–109. |
23. | Lang AE . Hemiatrophy, juvenile-onset exertional alternating leg paresis, hypotonia, and hemidystonia and adult-onset hemiparkinsonism: the spectrum of hemiparkinsonism-hemiatrophy syndrome. Mov Disord (1995) ; 10: : 489–495. |
24. | Sharma S , Prabhakar S and Parihar PS. Hemiparkinsonism-hemiatrophy syndrome (HHS): a clinical dilemma. Neurol India (1996) ; 44: : 231. |
25. | Marchioni E , Soragna D , Versino M , et al. Hemiparkinsonism-hemiatrophy with brain hemihypoplasia. Mov Disord (1999) ; 14: : 359–364. |
26. | Pramstaller PP , Künig G , Leenders K , et al. Parkin mutations in a patient with hemiparkinsonism-hemiatrophy: a clinical-genetic and PET study. Neurology (2002) ; 58: : 808–810. |
27. | Lee PH , Joo US , Yong SW , et al. Asymmetric freezing of gait in hemiparkinsonism-hemiatrophy. Neurology (2004) ; 63: : E7. |
28. | Martinelli P , Scaglione C , Capocasa M , et al. Hemiparkinsonism-hemiatrophy: a new observation. J Neurol (1998) ; 245: : 180–182. |
29. | Tessitore A , Russo A , Cirillo M , et al. Hemiparkinsonism and hemiatrophy syndrome: a rare observation. Clin Neurol Neurosurg (2010) ; 112: : 524–526. |
30. | Silvers DS and Menkes DL. Hemibody mirror movements in hemiparkinsonism-hemiatrophy syndrome. J Neurol Sci (2009) ; 287: : 260–263. |
31. | Pisani V , Nicoletti A , Meschini A , et al. Putaminal, but not nigral alterations, characterize hemiparkinsonism-hemiatrophy syndrome: a case report. Mov Disord (2011) ; 26: : 352–354. |
32. | Ayromlou H , Najmi S and Arami MA. Hemiparkinsonism-hemiatrophy syndrome. Arch Iran Med (2011) ; 14: : 152–154. |
33. | Yang Y and Kwak YT. Hemiparkinsonism-hemiatrophy syndrome with Alzheimer’s disease. Geriatr Gerontol Int (2015) ; 15: : 122–123. |
34. | Barbagallo G , Arabia G , Lupo A , et al. Midbrain hemiatrophy and nigral rarefaction in a patient with hemiparkinsonism-hemiatrophy syndrome. Mov Disord (2016) ; 31: : 1756–1757. |
35. | Vale TC , De Lima Pinto FC , Pedroso JL , et al. Case Hemiparkinsonism-hemiatrophy-SPECT with 99mTc TRODAT-1 and muscle MR imaging abnormalities. Radiology (2017) ; 283: : 613–619. |
36. | Shin C , Kwon YN , Lee D , et al. Incongruent hemiatrophy and hemiparkinsonism in a patient with schizencephaly. Clin Neurol Neurosurg (2018) ; 175: : 106–107. |
37. | Chung SJ . Subcortical and brainstem hemiatrophy accompanied by iron deposition in a patient with hemiparkinsonism-hemiatrophy syndrome: a case report. BMC Neurol (2021) ; 21: : 51. |
38. | Pascarella A , Manzo L and Estraneo A. Hemiparkinsonism-hemiatrophy syndrome: a long-term clinical, neurophysiological, and neuroimaging follow-up study. Neurol Sci (2023) ; 44: : 731–734. |


