
Serine Palmitoyltransferase (SPT)-related Neurodegenerative and Neurodevelopmental Disorders
Abstract
Motor neuron diseases and peripheral neuropathies are heterogeneous groups of neurodegenerative disorders that manifest with distinct symptoms due to progressive dysfunction or loss of specific neuronal subpopulations during different stages of development. A few monogenic, neurodegenerative diseases associated with primary metabolic disruptions of sphingolipid biosynthesis have been recently discovered. Sphingolipids are a subclass of lipids that form critical building blocks of all cellular and subcellular organelle membranes including the membrane components of the nervous system cells. They are especially abundant within the lipid portion of myelin. In this review, we will focus on our current understanding of disease phenotypes in three monogenic, neuromuscular diseases associated with pathogenic variants in components of serine palmitoyltransferase, the first step in sphingolipid biosynthesis. These include hereditary sensory and autonomic neuropathy type 1 (HSAN1), a sensory predominant peripheral neuropathy, and two neurodegenerative disorders: juvenile amyotrophic lateral sclerosis affecting the upper and lower motor neurons with sparing of sensory neurons, and a complicated form of hereditary spastic paraplegia with selective involvement of the upper motor neurons and more broad CNS neurodegeneration. We will also review our current understanding of disease pathomechanisms, therapeutic approaches, and the unanswered questions to explore in future studies.
OVERVIEW OF SPHINGOLIPID BIOSYNTHESIS PATHWAY
Eukaryotic cell membranes are comprised of lipids and proteins that support their structure and normal functions. Sphingolipids are principal components of myelin and highly enriched in the peripheral and central nervous system, where they regulate neuronal function, axonal growth, and myelination and thus contribute to neurodegenerative diseases [1]. Some sphingolipids can also serve as potent signaling molecules by activating specific cell surface receptors [2, 3].
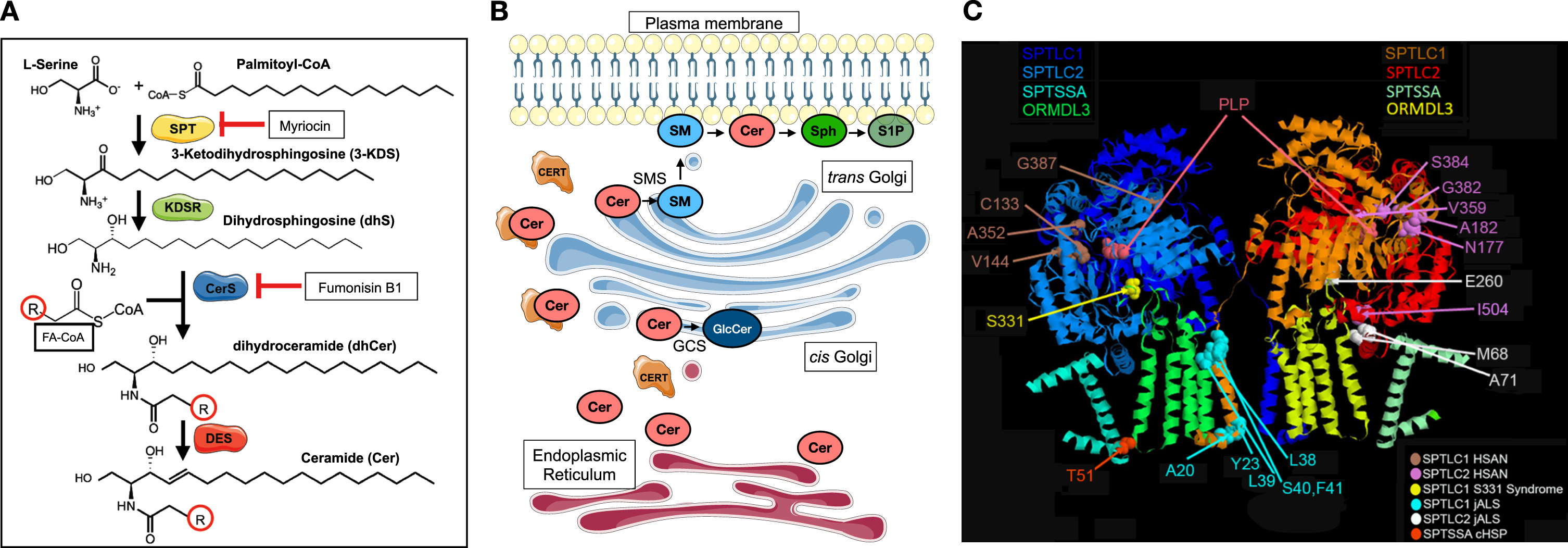
Sphingolipid biosynthesis involves a complex network of enzymes in different subcellular compartments. Serine palmitoyltransferase (SPT), localized to the endoplasmic reticulum (ER), is a multi-subunit enzyme that catalyzes the condensation of L-serine and fatty acyl-CoAs (commonly palmitoyl-CoA) to produce 3-ketodihydrosphingosine (3-KDS). This first step in sphingolipid biosynthesis is rate-limiting and subject to close homeostatic regulation [3]. 3-KDS is then reduced and acylated to form dihydroceramides (dhCer), which in turn are desaturated to form ceramides (Cer). These steps also predominantly take place in the ER, where the different enzyme complexes that catalyze each of these reactions are presumed to hand off each product to serve as the substrate for each subsequent reaction. Ceramide, which is the principal backbone substrate for higher order sphingolipids, is then transported to the Golgi apparatus, via the ceramide transport protein (CERT) or by vesicular transport, where it is converted to sphingomyelin and complex glycosphingolipids [4] (Fig. 1A-B).
Fig. 1
Serine palmitoyltransferase catalyzes the first and rate-limiting step in sphingolipid biosynthesis. (A) Condensation of L-Serine and fatty acyl-CoA (FA-CoA—commonly palmityol-CoA) forms 3-KDS, which is reduced to form dihydrosphingosine (dhS). N-acylation of dhS by different isoforms of ceramide synthases (CerS) forms dihydroceramides (dhCer), which are then desaturated to form ceramides (Cer). There is considerable diversity in the acyl chain length (R) of ceramides. Myriocin is a potent inhibitor of SPT. Fumonisin B1 inhibits CerS. DES = dihydroceramide desaturase (B) The multistep biosynthesis of ceramides from serine and fatty acyl-CoAs primarily occurs in the endoplasmic reticulum and its associated membranes (red). Ceramides are transported to the Golgi (blue) by ceramide transport protein (CERT) or by vesicular transport, where they serve as substrates for synthesis of higher order sphingolipids. Glucosylceramides (GlcCer) are synthesized by glucosylceramide synthase (GCS) and sphingomyelins (SMs) are synthesized by sphingomyelin synthases. Sph = sphingosine; S1P = sphingosine-1-phosphate. (C) The cryo-EM structure of SPT shows its organization into a dimer of heterotetramers (SPTLC1, SPTLC2, ORMDL3, and small subunit of SPT A are shown). The disease causing variants that lead to hereditary sensory and autonomic neuropathy type 1 (HSAN1), juvenile amyotrophic lateral sclerosis (jALS), or a complicated form of HSP (cHSP) are highlighted. In general, variants that cluster near the active site (PLP binding site), in SPTLC1 or SPTLC2, are usually associated with HSAN1 whereas those variants that affect the transmembrane domains of SPTLC1 or SPTSSA or intermembrane domain of SPTLC2 are more likely to cause jALS or cHSP. SPTLC1 S331 residue interacts both with ORMDLs and is near the active site. Substitutions at this residue typically cause a mixed phenotype of sensory neuropathy and motor neuron disease. Parts of the figure were drawn by using pictures from Servier Medical Art. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/).
Given the hydrophobicity of most sphingolipids, their proper catabolism and coordinated breakdown are essential to avoid their accumulation in subcellular compartments. Loss-of-function pathogenic variants in enzymes responsible for coordinated breakdown of complex sphingolipids, which are typically associated with membranes and lysosomes, underlie several lipid storage and lysosomal storage disorders with clinical features including neurodegeneration, leukodystrophy, neuropathy, and other broad systemic features such as hematologic abnormalities and renal disease. These complex disorders fall outside of the purview of this review; readers are encouraged to refer to other excellent manuscripts on this topic for a more in-depth understanding [5, 6]. In this review, we will focus on monogenic neurodegenerative diseases caused by primary pathogenic variants in components of the SPT enzyme complex and their differential effects on SPT function that can in turn inform rational therapeutic design for these diseases.
SERINE PALMITOYLTRANSFERASE (SPT) AND SPHINGOLIPID BIOSYNTHESIS
SPT is a pyridoxal 5-phosphate (PLP)-dependent enzyme of the α-oxoamine synthase family that resides in the ER membrane. SPT is highly evolutionarily conserved in all eukaryotes as well as some bacteria [7]. Structural studies of purified mammalian SPT have established its organization into a dimer of four subunits: SPTLC1, SPTLC2 (or SPTLC3), one small subunit of SPT (SPTSSA or SPTSSB), and one of ORMDL1, ORMDL2, or ORMDL3 proteins [8, 9]. The different SPT subunits determine its localization, function, catalytic activity, and homeostatic regulation (Fig. 1C). The interface between SPTLC1 and SPTLC2 forms the enzyme’s active site [10]. SPTLC3, which can substitute for SPTLC2, has limited tissue distribution in placenta and some endocrine glands, and confers distinct acyl-CoA substrate preferences [11, 12]. Small subunits of SPT dramatically enhance its catalytic activity [13]. Unlike the ubiquitously expressed SPTSSA, which is essential for normal embryonic development [14–16], SPTSSB has more limited tissue distribution, though it is expressed in the neurons of the brain and spinal cord [17]. Although SPTSSB is not essential for normal embryonic development, it confers preference for formation of longer chain-length sphingoid bases by SPT and activating pathogenic variants in this gene lead to neurodegeneration in a mouse model [13, 17].
While preliminary studies had reported no effect of ORMDL knockdown on SPT activity [18], more rigorous comparative studies confirm their inhibitory effect on SPT activity in cellular and animal models [19–21]. Homeostatic control of SPT activity is mediated through the ORMDL proteins in conjunction with ceramides [3, 20–22]. The separate effects of each individual ORMDL isoform on SPT activity are not fully understood. Together with studies of Ormdl knockout mice, recent studies demonstrate that while overlapping in function, ORMDL isoforms’ inhibitory effects on SPT are not fully redundant [21, 23]. However, further studies are needed to gain a more comprehensive understanding of different ORMDL isoforms in different tissues, specific cell types, and during development.
SPT ACTIVITY AND NEURODEVELOPMENT
SPT activity is essential for normal embryonic development. Sptlc1, Sptlc2, and Sptssa knockout mice do not advance beyond early developmental stages (adsorbed between E6.5 to E7.5) [14, 16]. Studies in conditional knockout animals also show an essential role for SPT activity in adipocyte development, myelopoiesis, and endothelial cell and vascular development [14, 15]. The specific role of SPT activity in development of the nervous systems has not been extensively studied. In a pioneering and detailed study of rat brain myelination during the early postnatal period, Davis et al have established the temporal course of sphingolipid biosynthesis in brain tissue and specifically oligodendrocytes [24]. These studies expose the previously unappreciated complexity of regulation of SPT activity and modulation of its subunit components to produce specific sphingolipid subspecies and meet cellular and tissue demands during critical periods of myelination, in particular the need to curtail sphingolipid synthesis following the peak of myelination. It remains unclear how sphingolipid biosynthesis is coordinated with synthesis of protein components of myelin, or with cholesterol synthesis, and how these processes converge to contribute to the complex process of myelination in the developing nervous system.
GAIN-OF-FUNCTION PATHOGENIC VARIANTS IN SPT COMPONENTS AND NEURODEGENERATIVE DISEASE
Several monogenic human diseases have been linked to pathogenic variants in SPT enzyme components (Table 1; Fig. 1C). Hereditary sensory and autonomic neuropathy type 1 (HSAN1) was the first disease linked to specific pathogenic variants in SPTLC1 and SPTLC2. More recently, we and others have reported a new class of variants in SPTLC1 and SPTLC2 that cause juvenile amyotrophic lateral sclerosis (ALS). Likewise, activating SPTSSA variants have also recently been reported to cause a related phenotype of complicated hereditary spastic paraplegia. In this section, we will focus on each of these diseases, the different consequences of their causative variants on SPT enzyme activity, our current understanding of disease mechanisms and disease models, and potential therapeutic approaches.
Table 1
Monogenic neurologic diseases associated with pathogenic variants in components of serine palmitoyltransferase
| Clinical findings | Genotypes | Biochemical mechanism | Treatment strategy | |
| Hereditary sensory and autonomic neuropathy type 1 (HSAN1) | Progressive sensory neuropathy Length dependent motor neuropathy Sensorineural hearing loss (some) Macular telangiectasia type 2 (some) Early cataracts (some) | SPTLC11: p.C133W, p.C133Y, p.V144D, p.A352 V, p.G387A SPTLC22: p. N177D, p.A182P, p.V359M, p.G382 V, p.S384F, p.I504F All dominantly inherited | Shift in SPT substrate preference from L-serine to L- alanine and glycine | Serine supplementation |
| SPTLC1 S331 syndrome | Progressive sensory neuropathy Motor neuron disease Early cataracts | SPTLC1: p.S331F, p.S331Y All dominantly inherited | Shift in SPT substrate preference from L-serine to L- alanine and glycine AND SPT overactivity due to loss of ORMDL inhibition | Avoid serine supplementation |
| SPT-related juvenile amyotrophic lateral sclerosis | Atrophy, weakness, fasciculations Spasticity, hyperreflexia Respiratory and bulbar dysfunction No sensory neuropathy Cognitive dysfunction (some) Early Cataracts (some) | SPTLC1: p.A20 S, p.Y23F, p.L38 R, p.L39del, p.F40_S41del SPTLC2: p.M68 R, p.A71 V p.E260K All dominantly inherited | SPT overactivity due to loss of ORMDL inhibition | Avoid serine supplementation; SPT inhibition |
| SPT-related complex hereditary spastic paraplegia (HSP) | Lower extremity spasticity Seizures Cognitive decline Sensorineural hearing loss | SPTSSA3: p.T51I (dominant) c.171_172del, p. Q58AfsTer10 (recessive) | SPT overactivity due to loss of ORMDL inhibition | Avoid serine supplementation; SPT inhibition |
1. NM_006415.4. 2. NM_004863.4. 3. NM_138288.3.
HEREDITARY SENSORY AND AUTONOMIC NEUROPATHY TYPE 1 (HSAN1)
HSAN1 is an autosomal dominant disease that primarily affects the peripheral nervous system. The classic phenotype includes early-onset and progressive sensory neuropathy with variable motor neuropathy; despite the name, there is usually little autonomic involvement [25]. Symptoms of HSAN1 typically start in the second or third decade of life though with notable variability even among affected individuals within the same family [26, 27]. Loss of sensation in the feet, dysesthesia, pain, traumatic perforating ulcers, and mutilating arthropathy are common manifestations. Electrodiagnostic studies typically show a symmetric sensory greater than motor neuropathy with mixed axonal and demyelinating features with nerve conduction velocities in the intermediate or demyelinating range [26]. Sural nerve biopsy findings corroborate the clinical findings, with severe loss of myelinated axons and the presence of flattened Schwann cells suggestive of unmyelinated axon loss. Teased nerve fiber studies of the few surviving myelinated axons typically demonstrate segmental demyelination [26]. Autopsy studies have confirmed this disease as a sensory predominant peripheral neuropathy, with relative preservation of ventral horn motor neurons and ventral roots, neuronal loss in the dorsal root ganglia, and loss of myelination in the dorsal columns [26, 28].
A few other systemic features have been associated with HSAN1. Hearing loss, which was reported in some of the originally described families, has not been a consistently reported clinical feature in all families. In some studies, a subset of HSAN1 patients are reported to develop macular telangiectasia, a progressive retinal disease linked to disruptions of glycine-serine metabolism [29, 30]. Chronic cough and gastroesophageal reflux have been reported in some patients, presumably in part due to the involvement of visceral afferents as part of the underlying neuropathy [31]. However, broader neurodegeneration, or other systemic features such as pulmonary, hematologic, bone, or cardiac disease are not known features of this disease.
Familial linkage studies over two decades ago identified the locus, and ultimately the causative variants in the majority of HSAN1 patients in SPTLC1, with two common, recurrent missense variants (p.C133W and p.C133Y) [32–35]. Subsequently, additional pathogenic missense variants in SPTLC1 as well as SPTLC2 were also linked to the same disease phenotype [36–39].
Initial studies of HSAN1 variants suggested a dominant-negative, inactivating effect on SPT [40–42]. Decreased SPT activity and reduced production of glycosphingolipids have also been proposed to contribute to the phenotype in a C elegans model of HSAN1 [43]. However, a large body of evidence have established that the HSAN1 variants cause a specific toxic gain-of-function in SPT activity [44, 45]. The mutant SPT enzyme has altered amino acid substrate preference: while it still maintains its affinity for L-serine, the HSAN1 variants increase its propensity to catalyze the condensation of L-alanine or glycine with palmitoyl-CoA to form 1-deoxysphingoid bases (DSBs) and ultimately lead to production of 1-deoxyceramides. 1-deoxysphingolipid metabolites lack the critical C1 hydroxyl group, which precludes their phosphorylation by sphingosine kinase and degradation by sphingosine-1-phosphate lyase [44]. 1-deoxysphingolipids can be metabolized in the liver by the cytochrome P450-4F enzymes [46]; however, their overproduction and insufficient degradation is thought to underlie the downstream neurotoxicity in HSAN1.
The precise underlying mechanisms of 1-deoxysphingolipid neurotoxicity are subjects of ongoing investigations. Studies using cell models have shown that exogenously added 1-deoxysphingolipids localize to the mitochondria and result in mitochondrial fractionation and dysfunction [47], increase autophagic flux [48], or impair plasma membrane endocytosis [49]. HSAN1 patient derived iPSCs and iPSC-derived sensory neurons also show disruption of axonal and myelin stability and abnormal glycosphingolipid metabolism [50].
Transgenic mice that overexpress the Sptlc1C133W variant recapitulate the biochemical findings of increased 1-deoxysphingolipids and demonstrate behavioral findings consistent with a mild sensory and motor neuropathy. The first manifestation is that of thermal but not mechanical hypersensitivity by 10 months of age [51], followed by small fiber neuropathy and discoordination when aged up to 14 months of age [52]. These behavioral findings are associated with loss of myelinated fibers and myelin thinning in dorsal root ganglia (DRG) as well as ventral roots at 10 months of age followed by loss of unmyelinated sensory axons in older mice (15 months) [51, 52]. In vitro studies of DRG cultures from the overexpressor Sptlc1C133W mouse show increased neurite growth and branching [53]. The aberrant excess outgrowth during development may not be sustainable over time and thus a setup for later vulnerability and degeneration.
In the absence of promoter-driven overexpression, the heterozygous knock-in mice with the Sptlc1C133 W variant do not show a disease-relevant phenotype, despite elevation of 1-deoxysphingolipids in plasma and nerve tissue [54]. Homozygous Sptlc1C133 W mice are not viable. These discrepancies add to the complexity of mechanistic studies in this mouse model. Relevant to these findings, elevation of 1-deoxysphingolipids has been reported in patients with diabetic neuropathy and animal models of this disease [30, 55, 56]. Interestingly, elevated 1-deoxysphingolipids in isolation do not seem to cause neuropathy but their elevation in conjunction with high fat dietary intake results in thermal hypopathia and decreased epidermal nerve fiber layer density, consistent with small fiber neuropathy [57]. Thus, future studies are needed to evaluate the effect of dietary and genetic modifiers that contribute to pathogenesis of diabetic peripheral neuropathy and to the phenotypic variability in HSAN1.
It remains unclear if elevated DSBs in neurons (or glial cells) vs serum result in the selective neurotoxicity in HSAN1. There are no known studies that have measured DSBs in the CNS or peripheral nerve tissues of HSAN1 patients. In studies of the Sptlc1C133W transgenic mice, DSBs were elevated in the sciatic nerve and serum but were not elevated in the brain or spinal cord tissues [52]. The underlying cause of this tissue specific increase in DSBs and its contribution to disease pathogenesis has not been explored but the apparent compartmentalization of increased DSBs can partially explain the selective vulnerability of DRG neurons in HSAN1, which are exposed to systemic circulation more than central nervous system neurons.
SPT-RELATED MOTOR NEURON DISEASES: JUVENILE AMYOTROPHIC LATERAL SCLEROSIS AND HEREDITARY SPASTIC PARAPLEGIA
Juvenile ALS is a neurodegenerative disorder of the upper and lower motor neurons. In contrast to the more common adult-onset ALS, the juvenile form of the disease typically manifests before 25 years of age. While both juvenile ALS and adult-onset ALS can be caused by Mendelian genetic variants, juvenile ALS is slower in progression and more likely to be genetic in etiology [58].
SPT-RELATED JUVENILE ALS
We and others have reported a number of patients with juvenile ALS caused by a novel class of gain-of-function SPTLC1 variants [59–63]. The disease initially presents with upper motor neuron dysfunction (e.g. spasticity) or lower motor neuron dysfunction (e.g. weakness, atrophy) without sensory symptoms. Initial symptoms were reported as early as 3–4 years of age but in some individuals the disease onset was in the second or third decade of life [62, 64]. The disease course has been universally and relentlessly progressive, albeit with variability in its rate, and includes involvement of the bulbar and respiratory function [64]. Sensory neuropathy is not a typical feature, even in late stages of the disease. Electrodiagnostic studies show normal sensory studies and diffuse acute and chronic denervation in multiple myotomes without demyelinating features [60]. Absence of sensory findings were confirmed on sural nerve biopsies of at least one patient (F3) [60] and by normal intraepidermal nerve fiber density (patient F7 II1 [60]—unpublished). Cognitive function has not been systematically assessed in SPTLC1-ALS patients but has not been reported including in individuals with this disease in their 50 s or 60 s [62, 64]. It is important to note that while sensory symptoms predominate in HSAN1, motor neuropathy with mixed demyelinating and axonal features is a common clinical and electrophysiologic finding in HSAN1 [26, 27]. However, unlike the SPT-associated motor neuron diseases, motor neuropathy in HSAN1 is typically length-dependent, always associated with sensory neuropathy and does not affect bulbar or respiratory function.
To date, five variants in SPTLC1 have been linked to the juvenile ALS phenotype (Table 1). The contrast between HSAN1 and juvenile ALS phenotypes suggested a dichotomy in their effect on SPT activity, leading to the pleiotropic disease phenotypes. In parallel, basic mechanistic studies of regulation of SPT activity had identified the N-terminal transmembrane domain of SPTLC1, the very domain where SPTLC1-ALS variants cluster, as a binding site for the negative regulators of SPT activity, ORMDL proteins [8, 9, 65]. Our untargeted sphingolipidomic studies of patient serum samples and patient derived fibroblasts revealed that in contrast with HSAN1-causing variants that confer catalytic substrate promiscuity to SPT, SPTLC1-ALS variants result in unrestrained SPT activity. Since they cluster in the first transmembrane domain of the protein, SPTLC1-ALS variants disrupt the interaction of SPTLC1 with ORMDLs, and result in excess biosynthesis of canonical SLs but not 1-deoxysphingolipids [60, 66].
In addition to SPTLC1, pathogenic variants in SPTLC2 (p.E260K, p.M68 R, and p.A71 V) that lead to SPT overactivity also cause a phenotype of juvenile ALS likely by disrupting the ORMDL regulation of the SPT complex [67–69]. Similar to SPTLC1-ALS, these patients also present with progressive, early childhood-onset upper and lower motor neuron signs without sensory neuropathy. Electrodiagnostic studies showed acute and chronic denervation in multiple myotomes, with normal sensory responses and no demyelinating features. Sphingolipidomic analysis of patient serum samples and patient derived cells also demonstrate unrestrained SPT activity and overproduction of canonical sphingolipid species instead of overproduction of 1-deoxysphingolipids. In contrast with SPTLC1-ALS, a few individuals with SPTLC2-related juvenile ALS are noted to develop cognitive dysfunction including frontotemporal dementia, likely due to broader neurodegeneration [67–69].
ACTIVATING SPTSSA VARIANTS AND HEREDITARY SPASTIC PARAPLEGIA
Recently, we also reported activating, pathogenic variants in SPTSSA, encoding the small subunit A of SPT, as a cause of a complex form of hereditary spastic paraplegia (cHSP) [70]. In this disease, symptoms emerge in early childhood predominantly with upper motor neuron dysfunction leading to lower extremity spasticity without lower motor neuron disease. Progressive cognitive decline and sensorineural hearing loss were also reported as clinical features. Similar to juvenile ALS variants, SPTSSA variants seem to interfere with ORMDL regulation of the SPT complex, thus resulting in unrestrained SPT activity. It is notable that unlike the SPTLC1 and SPTLC2-related ALS variants, the cHSP-related SPTSSA variants do not increase, and likely decrease (via ORMDL inhibition) the activity of SPTSSB-containing SPT. Despite the biochemical similarities and differences between SPT-related juvenile ALS and cHSP, the mechanisms underlying the phenotypic differences remain poorly understood.
Together, the recent discoveries of different motor neuron disease phenotypes caused by SPT-component variants establish excess sphingolipid biosynthesis as a novel metabolic mechanism for motor neuron disease and in some instances broader neurodegeneration. Studies of cell models (transfected HEK293 cells), patient-derived fibroblasts, and serum samples have been invaluable in understanding the primary effects of these mutations on SPT. Studies of iPSC-derived lower motor neuron-like cells harboring the SPTLC1-related ALS variant (p.F40_S41del) have shown elevated canonical sphingolipids without an effect on differentiation, morphology, and survival in these cells [60]; however, more detailed characterization of these cells has not been reported yet. Drosophila models of SPTSSA-related HSP variants show elevated sphingolipids, with motor deficits and shorter lifespans, consistent with motor neuron degeneration [70].
No murine models with SPT-related ALS or HSP variants have been reported so far. A few knock-in mouse models of SPT-related ALS and HSP are under development by different investigators. If successful in modelling the disease, they hold promise to facilitate the studies of the underlying mechanisms of neurodegeneration caused by SPT overactivity. Still, studies of other related mouse models confirm the importance of regulation of SPT for normal nervous system development and function. Although no human disease has been reported with variants in the SPTSSB gene, the “stellar” mouse model with the spontaneously occurring Sptssb H56 L substitution demonstrates a neurodegenerative phenotype with early-onset ataxia, low body weight, and premature death at around 10 weeks of age. Biochemical studies have shown accumulation of C20-sphingoid base containing sphingolipids in the eye and nervous system in this model [17]. In addition, knockout mice of all three Ormdl isoforms have been generated and loss of two Ormdl isoforms in the mouse results in neurodegeneration [21]. Similarly, conditional expression of a fusion-SPT transgene (fSPT) –a constitutively active enzyme with the human SPTLC2, SPTSSA, and SPTLC1 subunits fused into a single-chain SPT –under the control of Mx1-Cre, an interferon-inducible promoter in mice, results in progressive hindlimb paralysis and pathologic changes in the sciatic nerve in association with markedly increased sphingolipids [21].
THE COMPLEXITY OF SPT-RELATED NEURODEGENERATIVE DISEASES AND THEIR ASSOCIATED PHENOTYPES
SPT is a ubiquitous enzyme and yet distinct disruptions in its activity due to specific pathogenic variants underlie the sensory-predominant phenotype (HSAN1) or motor neuron degeneration (juvenile ALS or cHSP). While the dichotomy of the phenotypes seems to parallel the underlying dichotomy of the effect of the different classes of variants on SPT function, a few anomalies remain.
A few case reports have described a “hybrid” phenotype associated with specific SPTLC1 variants that alter the S331 residue [71–74]. The phenotypic manifestation of these patients includes features of both HSAN1 and ALS with early onset sensory neuropathy as well as motor neuron disease without broader neurodegeneration. In addition, early cataracts have been reported in several individuals with these specific variants. Sphingolipidomic evaluation of patient serum and patient-derived cells with SPTLC1 p.S331 substitutions have identified a concurrent increase in 1-deoxysphingolipis (a feature of HSAN1 variants) as well as an overproduction of canonical sphingolipids likely by disrupting ORMDL inhibition (a feature of ALS variants) [73, 74]. These findings suggest that SPTLC1-related HSAN1 and ALS phenotypes are not mutually exclusive, and aspects of both phenotypes may co-occur. Still, in these special cases, the correlation between the different phenotypes and specific changes in SPT biochemistry seems to be preserved.
We also have reported the phenotypic overlap between HSAN1 and the ALS phenotypes in the father in a family with the SPTLC1-ALS variant p.L39del (F7 I-1) [60]. This individual had elevation of 1-deoxysphingolipids as well as canonical sphingolipids, in association with otherwise unexplained elevated alanine/serine level ratio in his serum [66]. These findings suggested that availability of amino acid substrates may influence the disease phenotype in SPT-related diseases. However, elevated serum 1-deoxysphingolipids were also noted in an individual with the SPTLC2-ALS variant p.E260K [68]. In this individual, this finding was not associated with altered alanine/serine ratios or concurrent presence of sensory neuropathy. These findings highlight the presence of genetic, dietary, or environmental modifiers of the disease phenotype which add to the complexity of genotype phenotype correlations and warrant further investigation in future studies. It is also expected that new variants in SPT components will be identified that expand the phenotypic spectrum of the disease including in adult-onset ALS [59, 75].
THERAPEUTIC APPROACHES FOR SPT RELATED DISEASES
Mechanistic studies into the underlying biochemical abnormalities of SPT function due to specific pathogenic variants has informed rational therapeutic development. A few therapeutic approaches and potential use of RNA-and DNA-directed therapies for treatment of these diseases are reviewed in this section.
SERINE SUPPLEMENTATION FOR HSAN1, NOT SPT-RELATED MOTOR NEURON DISEASES
HSAN1-related SPTLC1 and SPTLC2 pathogenic variants shift the amino acid substrate preference of SPT to reduce specificity for serine and increase the usage of L-alanine and glycine. As a result, serine supplementation has been proposed to help return the incorporation of L-serine by SPT back to normal rates. When serine supplementation was investigated in cellular and mouse models of HSAN1, it mitigated the production of 1-deoxysphingolipids and demonstrated improvements in relevant cellular phenotypes [50, 53, 76]. In HSAN1 mouse models, a serine-enriched diet led to a rapid reduction in 1-deoxysphingolipid synthesis and short-term therapy in older, post-symptomatic mice also demonstrated improvements in disease relevant neurological function [76]. These findings paved the way for a randomized clinical trial of L-serine in patients with HSAN1, who showed improvements in intraepidermal nerve fiber layer density from serial skin biopsies, reduction of 1-deoxysphingolipids, and improvements in the clinical Charcot-Marie-Tooth neuropathy score (CMTNS) [77].
In SPT-related motor neuron diseases, which are caused by unrestrained SPT activity, serine supplementation would be predicted to exacerbate sphingolipid overproduction, with the potential to accelerate the disease progression; consistently, serine supplementation in iPSC-derived motor neurons with the SPTLC1del40 - 41 variant accentuated the consequences of SPT overactivity [60]. To counteract SPT overactivity, partial SPT inhibition provides a more rational therapeutic approach, though one must act with caution as complete inhibition of SPT is associated with systemic toxicity. Importantly, inhibition of SPT does not restore the dynamic regulatory function and range of the enzymatic activity.
SMALL MOLECULE INHIBITORS OF SPT
Myriocin (also known as thermozymocidin or ISP-10) was first isolated as a natural fungal product and exerts a potent inhibitory effect on SPT via a dual inhibitory mechanism. Studies of bacterial SPT showed it initially binds the active site PLP cofactor as an external aldimine with high affinity (dissociation constant [Kd] in nM range), and is subsequently cleaved to an aldehyde that covalently modifies the PLP-binding lysine 265 residue (K379 in human SPTLC2), resulting in irreversible/suicide inhibition of the enzyme [78]. While it has been used extensively in mechanistic studies of the enzyme in cellular and animal models, clinical development has been hampered by its toxicity [79, 80].
Stereoisomers L- and D-cycloserine also inhibit SPT both as competitive and covalent/irreversible inhibitors, with L-cycloserine showing much higher potency [81]. Other SPT inhibitors have also been developed (e.g. Imidazopyridine and Pyrazolopiperidine). All of these inhibitors show dose-related toxicity or off-target activity that have hampered their clinical development [79, 82]. The only exception is D-cycloserine (seromycin), which is FDA approved as a second-line anti-tubercular antibiotic [83] but its use as a therapeutic for SPT overactivity has not been systematically tested. It is important to note that while systemic SPT inhibition results in toxicity, specific inhibition of SPT in the nervous system (for example by intrathecal drug delivery) has not been evaluated. Likewise, while studies of knockout mice confirm that SPT activity is necessary for embryonic development and that of the nervous system especially during myelination, the role of SPT activity by cell or tissue-specific genetic manipulation of animal models in the post-natal nervous system has not been reported [14, 16]. Patients with primary pathogenic variants in the SPT components resulting in its overactivity may be less sensitive to toxicity associated with SPT inhibition. Thus, this approach remains a feasible therapeutic strategy, though in addition to assessment of efficacy, careful dose finding, assessment of optimal route of administration, and related toxicity in cellular and animal models are necessary.
SERINE DEPLETION
Since overproduction of canonical sphingolipids relies on availability of L-serine as a substrate, in addition to SPT inhibition, serine depletion is another theoretically feasible approach to ameliorate SPT overactivity. Serine depletion can be achieved by inhibition of the enzymes in the serine biosynthesis pathway (for example phosphoglycerate dehydrogenase inhibition) [84], or by dietary restriction of both L-serine and glycine (which can be readily converted to L-serine by serine hydroxymethyltransferase). This approach is only applicable to the SPT-related juvenile ALS and cHSP which are characterized by an overactive SPT complex. For HSAN1 patients, serine depletion would be predicted to have a detrimental effect by increasing 1-deoxysphingolipid production.
RNA- AND DNA-DIRECTED THERAPIES
In addition to supplements, dietary modifications, and small molecules, anti-sense oligonucleotides, RNAi, and other RNA- and DNA-directed therapies, hold great promise for precise targeting of the underlying genetic consequences of SPT related diseases. However, additional studies are necessary to evaluate their efficacy, mitigate toxicity, and circumvent target tissue delivery limitations of these approaches.
ALLELE-SPECIFIC KNOCKDOWN
Based on animal studies and frequency of loss of function variants in population databases, SPT genes are thought to be haplosufficient; that is, loss of function or deletion of one gene copy does not cause a phenotype. Thus, allele-specific knockdown is a feasible approach in correcting the dominant gain of function in all SPT-related diseases. In our original gene discovery study of SPTLC1-related ALS, siRNA mediated allele-specific knockdown of SPTLC1-related ALS variants rescued the enzyme overactivity in patient derived fibroblasts [60]. While effective, and imposing little chance for over-inhibition of SPT, this approach has limited generalizability and needs to be custom-designed and validated for each disease-causing SPT variant. Targeting of common SNVs that are in cis with the disease-causing variants in the SPT related genes can improve the generalizability of this approach in future studies for these dominantly inherited variants. Furthermore, siRNA delivery to the relevant cell types in SPT-related diseases remains a difficult challenge, requiring chemical modifications, conjugates, or novel packaging modalities. Allele specific knockdown is not restricted to siRNAs and can be pursued using other RNA-directed therapies.
NON-ALLELE SPECIFIC SPT COMPONENT KNOCKDOWN
Targeting SPT components without allele specificity, for example with anti-sense oligonucleotides (ASOs), improves on the generalizability limitations of the allele-specific approach and limitations of delivery to target cells and tissues. Certain ASO chemistries provide advantages over unmodified siRNAs, as they are endonuclease resistant and their biodistribution in the CNS tissue and cells, including motor neurons and glial cells are well characterized in animal models including nonhuman primates [85]. In addition, intrathecally administered gapmers as a class have already been used in clinical trials for genetic forms of ALS [86, 87] (NCT04768972) and one has been recently FDA-approved for treatment of SOD1-related ALS [88]. However, there remains a risk of over-inhibition of SPT activity depending on the SPT component target. As discussed above for small molecule inhibitors, the exact level of SPT activity necessary for normal cellular and organismal function, especially in the postnatal nervous system, has not been established. In this regard, knockdown of small subunits of SPT (SPTSSA or SPTSSB) instead of SPTLC1 or SPTLC2 provides theoretical advantages over small molecules or allele-specific knockdown approaches. First, it is a plausible and generalizable approach to treat all SPT-related variants or other diseases linked to gain of function variants in SPT components. Second, even with complete knockdown of target transcripts, which is unlikely to be achieved, SPT is predicted to maintain some level of basal activity without the presence of small subunits, thus providing a larger safety factor against over-inhibition.
CRISPR GENE TARGETING AND GENE EDITING
CRISPR-based approaches for allele specific inactivation [89], or CRISPR gene editing (e.g. prime editing) [90] are also theoretically feasible, though have yet to be tested in cellular and animal models of this disease. They provide a durable therapeutic approach, but are hampered by inefficient delivery of CRISPR machinery and gRNAs, immunogenicity, and their potential off target activity, which require further study and mitigation.
CONCLUSIONS
With recent discovery of new classes of variants in SPT components and the phenotypic pleiotropy in SPT-related monogenic diseases, the complexity of the role of sphingolipid biosynthesis pathway and its regulation and contribution to neurodegenerative disease has become more apparent. The study of these variants has informed biochemical and mechanistic studies that have in turn improved our understanding of SPT function and informed rational therapeutic design for SPT-related diseases. Nonetheless, future fundamental studies to establish the downstream cellular consequences of abnormal SPT activity, the selective vulnerability of sensory and motor neurons to distinct biochemical changes in SPT function, the important cells in the nervous system for each associated disease, the tolerated threshold for SPT inhibition, and optimal timing of interventions that mitigate abnormal SPT activity to ameliorate the disease manifestations and progression need to be more systematically studied.
ACKNOWLEDGMENTS
The authors would like to acknowledge Carsten G. Bönnemann (NIH/NINDS) for review of the manuscript and helpful discussions.
FUNDING
PM receives funding from the Packard Center for ALS, Live like Lou Foundation. TMD receives funding from the department of defense (DoD) (OSD(HA).2022IC D.WBH-1). FE is funded by the Connolly Family Charitable Fund, the NIH Common Fund, through the Office of Strategic Coordination/Office of the NIH Director under Award Number(s) [U01HG007672] and an R21 from the NINDS Subcontract Undiagnosed Disease Network, and NIH-NINDS U54NS115052. TMD and PM are also supported by a CDMRP Therapeutic Idea grant (AL190015) and by a CDMRP therapeutic development grant (AL220025). FE and TMD receive funding from the undiagnosed disease network (UDN: 5U01HG007530).
CONFLICT OF INTEREST
PM is a consultant for Leal Therapeutics LLC (Cash only, no stock options). PM is an Editorial Board Member of this journal but was not involved in the peer-review process nor had access to any information regarding its peer-review. FE has received consulting fees from UpToDate, Prime Medical Education, bluebird bio, Leal Therapeutics, Sanofi, Takeda and SwanBio Therapeutics. All other authors declare no competing interests.
REFERENCES
[1] | Alaamery M , Albesher N , Aljawini N , Alsuwailm M , Massadeh S , Wheeler MA , et al. Role of sphingolipid metabolism in neurodegeneration. J Neurochem. (2021) ;158: (1):25–35. |
[2] | Breslow DK , Weissman JS . Membranes in balance: Mechanisms of sphingolipid homeostasis. Mol Cell. (2010) ;40: (2):267–79. |
[3] | Breslow DK . Sphingolipid homeostasis in the endoplasmic reticulum and beyond. Cold Spring Harb Perspect Biol. (2013) ;5: (4):a013326. |
[4] | Hanada K . Intracellular trafficking of ceramide by ceramide transfer protein. Proc Jpn Acad Ser B Phys Biol Sci. (2010) ;86: (4):426–37. |
[5] | Arenz C . Recent advances and novel treatments for sphingolipidoses. Future Med Chem. (2017) ;9: (14):1687–700. |
[6] | Grassi S , Chiricozzi E , Mauri L , Sonnino S , Prinetti A . Sphingolipids and neuronal degeneration in lysosomal storage disorders. J Neurochem. (2019) ;148: (5):600–11. |
[7] | Lowther J , Naismith JH , Dunn TM , Campopiano DJ . Structural, mechanistic and regulatory studies of serine palmitoyltransferase. Biochem Soc Trans. (2012) ;40: (3):547–54. |
[8] | Li S , Xie T , Liu P , Wang L , Gong X . Structural insights into the assembly and substrate selectivity of human SPT-ORMDL3 complex. Nat Struct Mol Biol. (2021) ;28: (3):249–57. |
[9] | Wang Y , Niu Y , Zhang Z , Gable K , Gupta SD , Somashekarappa N , et al. Structural insights into the regulation of human serine palmitoyltransferase complexes. Nat Struct Mol Biol. (2021) ;28: (3):240–8. |
[10] | Yasuda S , Nishijima M , Hanada K . Localization, topology, and function of the LCB1 subunit of serine palmitoyltransferase in mammalian cells. J Biol Chem. (2003) ;278: (6):4176–83. |
[11] | Hornemann T , Penno A , Rutti MF , Ernst D , Kivrak-Pfiffner F , Rohrer L , et al. The SPTLC3 subunit of serine palmitoyltransferase generates short chain sphingoid bases. J Biol Chem. (2009) ;284: (39):26322–30. |
[12] | Hornemann T , Richard S , Rutti MF , Wei Y , von Eckardstein A . Cloning and initial characterization of a new subunit for mammalian serine-palmitoyltransferase. J Biol Chem. (2006) ;281: (49):37275–81. |
[13] | Han G , Gupta SD , Gable K , Niranjanakumari S , Moitra P , Eichler F , et al. Identification of small subunits of mammalian serine palmitoyltransferase that confer distinct acyl-CoA substrate specificities. Proc Natl Acad Sci U S A. (2009) ;106: (20):8186–91. |
[14] | Parthibane V , Lin J , Acharya D , Abimannan T , Srideshikan SM , Klarmann K , et al. SSSPTA is essential for serine palmitoyltransferase function during development and hematopoiesis. J Biol Chem. (2021) ;296: :100491. |
[15] | Parthibane V , Acharya D , Srideshikan SM , Lin J , Myerscough DG , Abimannan T , et al. Sptlc1 is essential for myeloid differentiation and hematopoietic homeostasis. Blood Adv. (2019) ;3: (22):3635–49. |
[16] | Hojjati MR , Li Z , Jiang XC . Serine palmitoyl-CoA transferase (SPT) deficiency and sphingolipid levels in mice. Biochim Biophys Acta. (2005) ;1737: (1):44–51. |
[17] | Zhao L , Spassieva S , Gable K , Gupta SD , Shi LY , Wang J , et al. Elevation of 20-carbon long chain bases due to a mutation in serine palmitoyltransferase small subunit b results in neurodegeneration. Proc Natl Acad Sci U S A. (2015) ;112: (42):12962–7. |
[18] | Zhakupova A , Debeuf N , Krols M , Toussaint W , Vanhoutte L , Alecu I , et al. ORMDL3 expression levels have no influence on the activity of serine palmitoyltransferase. FASEB J. (2016) ;30: (12):4289–300. |
[19] | Siow DL , Wattenberg BW . Mammalian ORMDL proteins mediate the feedback response in ceramide biosynthesis. J Biol Chem. (2012) ;287: (48):40198–204. |
[20] | Xie T , Liu P , Wu X , Dong F , Zhang Z , Yue J , et al. Ceramide sensing by human SPT-ORMDL complex for establishing sphingolipid homeostasis. Nat Commun. (2023) ;14: (1):3475. |
[21] | Clarke BA , Majumder S , Zhu H , Lee YT , Kono M , Li C , et al. The Ormdl genes regulate the sphingolipid synthesis pathway to ensure proper myelination and neurologic function in mice. Elife. (2019) ;8. |
[22] | Davis DL , Gable K , Suemitsu J , Dunn TM , Wattenberg BW . The ORMDL/Orm-serine palmitoyltransferase (SPT) complex is directly regulated by ceramide: Reconstitution of SPT regulation in isolated membranes. J Biol Chem. (2019) ;294: (13):5146–56. |
[23] | Green CD , Weigel C , Oyeniran C , James BN , Davis D , Mahawar U , et al. CRISPR/Cas9 deletion of ORMDLs reveals complexity in sphingolipid metabolism. J Lipid Res. (2021) ;62: :100082. |
[24] | Davis DL , Mahawar U , Pope VS , Allegood J , Sato-Bigbee C , Wattenberg BW . Dynamics of sphingolipids and the serine palmitoyltransferase complex in rat oligodendrocytes during myelination. J Lipid Res. (2020) ;61: (4):505–22. |
[25] | Hicks EP . Hereditary perforating ulcer of the foot. The Lancet. (1922) ;199: (5138):319–21. |
[26] | Houlden H , King R , Blake J , Groves M , Love S , Woodward C , et al. Clinical, pathological and genetic characterization of hereditary sensory and autonomic neuropathy type 1 (HSAN I). Brain. (2006) ;129: (Pt 2):411–25. |
[27] | Fridman V , Oaklander AL , David WS , Johnson EA , Pan J , Novak P , et al. Natural history and biomarkers in hereditary sensory neuropathy type 1. Muscle Nerve. (2015) ;51: (4):489–95. |
[28] | Lindahl AJ , Lhatoo SD , Campbell MJ , Nicholson G , Love S . Late-onset hereditary sensory neuropathy type I due to SPTLC1 mutation: Autopsy findings. Clin Neurol Neurosurg. (2006) ;108: (8):780–3. |
[29] | Rodrigues FG , Pipis M , Heeren TFC , Fruttiger M , Gantner M , Vermeirsch S , et al. Description of a patient cohort with Hereditary Sensory Neuropathy type 1 without retinal disease Macular Telangiectasia type 2 – implications for retinal screening in HSN1. J Peripher Nerv Syst. (2022) ;27: (3):215–24. |
[30] | Gantner ML , Eade K , Wallace M , Handzlik MK , Fallon R , Trombley J , et al. Serine and Lipid Metabolism in Macular Disease and Peripheral Neuropathy. N Engl J Med. (2019) ;381: (15):1422–33. |
[31] | Spring PJ , Kok C , Nicholson GA , Ing AJ , Spies JM , Bassett ML , et al. Autosomal dominant hereditary sensory neuropathy with chronic cough and gastro-oesophageal reflux: Clinical features in two families linked to chromosome 3p22-p24. Brain. (2005) ;128: (Pt 12):2797–810. |
[32] | Bejaoui K , Wu C , Scheffler MD , Haan G , Ashby P , Wu L , et al. SPTLC1 is mutated in hereditary sensory neuropathy, type 1. Nat Genet. (2001) ;27: (3):261–2. |
[33] | Dawkins JL , Hulme DJ , Brahmbhatt SB , Auer-Grumbach M , Nicholson GA . Mutations in SPTLC1, encoding serine palmitoyltransferase, long chain base subunit-1, cause hereditary sensory neuropathy type I. Nat Genet. (2001) ;27: (3):309–12. |
[34] | Nicholson GA , Dawkins JL , Blair IP , Kennerson ML , Gordon MJ , Cherryson AK , et al. The gene for hereditary sensory neuropathy type I (HSN-I) maps to chromosome 9q22.1-q22.3. Nat Genet. (1996) ;13: (1):101–4. |
[35] | Bejaoui K , McKenna-Yasek D , Hosler BA , Burns-Deater E , Deater LM , O’Neill G , et al. Confirmation of linkage of type 1 hereditary sensory neuropathy to human chromosome 9q22. Neurology. (1999) ;52: (3):510–5. |
[36] | Suriyanarayanan S , Othman A , Drager B , Schirmacher A , Young P , Mulahasanovic L , et al. A Novel Variant (Asn177Asp) in SPTLC2 Causing Hereditary Sensory Autonomic Neuropathy Type 1C. Neuromolecular Med. (2019) ;21: (2):182–91. |
[37] | Suriyanarayanan S , Auranen M , Toppila J , Paetau A , Shcherbii M , Palin E , et al. The Variant (Arg183Trp) in SPTLC2 Causes Late-Onset Hereditary Sensory Neuropathy. Neuromolecular Med. (2016) ;18: (1):81–90. |
[38] | Murphy SM , Ernst D , Wei Y , Laura M , Liu YT , Polke J , et al. Hereditary sensory and autonomic neuropathy type 1 (HSANI) caused by a novel mutation in SPTLC2. Neurology. (2013) ;80: (23):2106–11. |
[39] | Rotthier A , Auer-Grumbach M , Janssens K , Baets J , Penno A , Almeida-Souza L , et al. Mutations in the SPTLC2 subunit of serine palmitoyltransferase cause hereditary sensory and autonomic neuropathy type I. Am J Hum Genet. (2010) ;87: (4):513–22. |
[40] | Gable K , Han G , Monaghan E , Bacikova D , Natarajan M , Williams R , et al. Mutations in the yeast LCB1 and LCB2 genes, including those corresponding to the hereditary sensory neuropathy type I mutations, dominantly inactivate serine palmitoyltransferase. J Biol Chem. (2002) ;277: (12):10194–200. |
[41] | Bejaoui K , Uchida Y , Yasuda S , Ho M , Nishijima M , Brown RH, Jr., et al. Hereditary sensory neuropathy type 1 mutations confer dominant negative effects on serine palmitoyltransferase, critical for sphingolipid synthesis. J Clin Invest. (2002) ;110: (9):1301–8. |
[42] | Raman MCC , Johnson KA , Yard BA , Lowther J , Carter LG , Naismith JH , et al. The external aldimine form of serine palmitoyltransferase: Structural, kinetic, and spectroscopic analysis of the wild-type enzyme and HSAN1 mutant mimics. J Biol Chem. (2009) ;284: (25):17328–39. |
[43] | Cui M , Ying R , Jiang X , Li G , Zhang X , Zheng J , et al. A Model of Hereditary Sensory and Autonomic Neuropathy Type 1 Reveals a Role of Glycosphingolipids in Neuronal Polarity. J Neurosci. (2019) ;39: (29):5816–34. |
[44] | Penno A , Reilly MM , Houlden H , Laura M , Rentsch K , Niederkofler V , et al. Hereditary sensory neuropathy type 1 is caused by the accumulation of two neurotoxic sphingolipids. J Biol Chem. (2010) ;285: (15):11178–87. |
[45] | Gable K , Gupta SD , Han G , Niranjanakumari S , Harmon JM , Dunn TM . A disease-causing mutation in the active site of serine palmitoyltransferase causes catalytic promiscuity. J Biol Chem. (2010) ;285: (30):22846–52. |
[46] | Alecu I , Othman A , Penno A , Saied EM , Arenz C , von Eckardstein A , et al. Cytotoxic 1-deoxysphingolipids are metabolized by a cytochrome P450-dependent pathway. J Lipid Res. (2017) ;58: (1):60–71. |
[47] | Alecu I , Tedeschi A , Behler N , Wunderling K , Lamberz C , Lauterbach MA , et al. Localization of 1-deoxysphingolipids to mitochondria induces mitochondrial dysfunction. J Lipid Res. (2017) ;58: (1):42–59. |
[48] | Lauterbach MA , Saavedra V , Mangan MSJ , Penno A , Thiele C , Latz E , et al. 1-Deoxysphingolipids cause autophagosome and lysosome accumulation and trigger NLRP3 inflammasome activation. Autophagy. (2021) ;17: (8):1947–61. |
[49] | Cordes T , Kuna RS , McGregor GH , Khare SV , Gengatharan J , Muthusamy T , et al. 1-Deoxysphingolipid synthesis compromises anchorage-independent growth and plasma membrane endocytosis in cancer cells. J Lipid Res. (2022) ;63: (10):100281. |
[50] | Clark AJ , Kugathasan U , Baskozos G , Priestman DA , Fugger N , Lone MA , et al. An iPSC model of hereditary sensory neuropathy-1 reveals L-serine-responsive deficits in neuronal ganglioside composition and axoglial interactions. Cell Rep Med. (2021) ;2: (7):100345. |
[51] | McCampbell A , Truong D , Broom DC , Allchorne A , Gable K , Cutler RG , et al. Mutant SPTLC1 dominantly inhibits serine palmitoyltransferase activity in vivo and confers an age-dependent neuropathy. Hum Mol Genet. (2005) ;14: (22):3507–21. |
[52] | Eichler FS , Hornemann T , McCampbell A , Kuljis D , Penno A , Vardeh D , et al. Overexpression of the wild-type SPT1 subunit lowers desoxysphingolipid levels and rescues the phenotype of HSAN1. J Neurosci. (2009) ;29: (46):14646–51. |
[53] | Jun BK , Chandra A , Kuljis D , Schmidt BP , Eichler FS . Substrate Availability of Mutant SPT Alters Neuronal Branching and Growth Cone Dynamics in Dorsal Root Ganglia. J Neurosci. (2015) ;35: (40):13713–9. |
[54] | Hines TJ , Tadenev ALD , Lone MA , Hatton CL , Bagasrawala I , Stum MG , et al. Precision mouse models of Yars/dominant intermediate Charcot-Marie-Tooth disease type C and Sptlc1/hereditary sensory and autonomic neuropathy type 1. J Anat. (2022) ;241: (5):1169–85. |
[55] | Othman A , Bianchi R , Alecu I , Wei Y , Porretta-Serapiglia C , Lombardi R , et al. Lowering plasma 1-deoxysphingolipids improves neuropathy in diabetic rats. Diabetes. (2015) ;64: (3):1035–45. |
[56] | Bertea M , Rutti MF , Othman A , Marti-Jaun J , Hersberger M , von Eckardstein A , et al. Deoxysphingoid bases as plasma markers in diabetes mellitus. Lipids Health Dis. (2010) ;9: :84. |
[57] | Handzlik MK , Gengatharan JM , Frizzi KE , McGregor GH , Martino C , Rahman G , et al. Insulin-regulated serine and lipid metabolism drive peripheral neuropathy. Nature. (2023) ;614: (7946):118–24. |
[58] | Lehky T , Grunseich C . Juvenile Amyotrophic Lateral Sclerosis: A Review. Genes (Basel). (2021) ;12: (12) |
[59] | Johnson JO , Chia R , Miller DE , Li R , Kumaran R , Abramzon Y , et al. Association of Variants in the SPTLC1 Gene With Juvenile Amyotrophic Lateral Sclerosis. JAMA Neurol. (2021) ;78: (10):1236–48. |
[60] | Mohassel P , Donkervoort S , Lone MA , Nalls M , Gable K , Gupta SD , et al. Childhood amyotrophic lateral sclerosis caused by excess sphingolipid synthesis. Nat Med. (2021) ;27: (7):1197–204. |
[61] | Lone MA , Zeng S , Bourquin F , Wang M , Huang S , Lin Z , et al. SPTLC1 Leu38Arg, a novel mutation associated with childhood ALS. Biochim Biophys Acta Mol Cell Biol Lipids. (2023) ;1868: (9):159359. |
[62] | Ajjarapu A , Feely SME , Shy ME , Trout C , Zuchner S , Moore SA , et al. Thirty-Year Follow-Up of Early Onset Amyotrophic Lateral Sclerosis with a Pathogenic Variant in SPTLC1. Case Rep Neurol. (2023) ;15: (1):146–52. |
[63] | Liu X , He J , Yu W , Fan D . A de novo c.113 T>C: p.L38R mutation of SPTLC1: Case report of a girl with sporadic juvenile amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener.. (2022) ;23: (7-8):634–7. |
[64] | Guissart C , De la Cruz E , Flabeau O , Grapperon AM , Corazza G , Junilhon L , et al. Heterozygous SPTLC1 p.Leu39del is a major cause of slow-progressing juvenile ALS. J Neurol Neurosurg Psychiatry. 2023. |
[65] | Han G , Gupta SD , Gable K , Bacikova D , Sengupta N , Somashekarappa N , et al. The ORMs interact with transmembrane domain 1 of Lcb1 and regulate serine palmitoyltransferase oligomerization, activity and localization. Biochim Biophys Acta Mol Cell Biol Lipids. (2019) ;1864: :(3):245–59. |
[66] | Lone MA , Aaltonen MJ , Zidell A , Pedro HF , Morales Saute JA , Mathew S , et al. SPTLC1 variants associated with ALS produce distinct sphingolipid signatures through impaired interaction with ORMDL proteins. J Clin Invest. (2022) ;132: :(18). |
[67] | Dohrn MF , Beijer D , Lone MA , Bayraktar E , Oflazer P , Orbach R , et al. Recurrent de-novo gain-of-function mutation in SPTLC2 confirms dysregulated sphingolipid production to cause juvenile amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2023. |
[68] | Syeda SB , Lone MA , Mohassel P , Donkervoort S , Munot P , Franca MC,Jr. , et al. Recurrent de novo SPTLC2 variant causes childhood-onset amyotrophic lateral sclerosis (ALS) by excess sphingolipid synthesis. J Neurol Neurosurg Psychiatry. 2023. |
[69] | Naruse H , Ishiura H , Esaki K , Mitsui J , Satake W , Greimel P , et al. SPTLC2 variants are associated with early-onset ALS and FTD due to aberrant sphingolipid synthesis. Ann Clin Transl Neurol. 2024. |
[70] | Srivastava S , Shaked HM , Gable K , Gupta SD , Pan X , Somashekarappa N , et al. SPTSSA variants alter sphingolipid synthesis and cause a complex hereditary spastic paraplegia. Brain. 2023. |
[71] | Auer-Grumbach M , Bode H , Pieber TR , Schabhuttl M , Fischer D , Seidl R , et al. Mutations at Ser331 in the HSN type I gene SPTLC1 are associated with a distinct syndromic phenotype. Eur J Med Genet. (2013) ;56: (5):266–9. |
[72] | Rotthier A , Penno A , Rautenstrauss B , Auer-Grumbach M , Stettner GM , Asselbergh B , et al. Characterization of two mutations in the SPTLC1 subunit of serine palmitoyltransferase associated with hereditary sensory and autonomic neuropathy type I. Hum Mutat. (2011) ;32: (6):E2211–25. |
[73] | Bode H , Bourquin F , Suriyanarayanan S , Wei Y , Alecu I , Othman A , et al. HSAN1 mutations in serine palmitoyltransferase reveal a close structure-function-phenotype relationship. Hum Mol Genet ((2016) ;25: (5):853–65. |
[74] | Fiorillo C , Capodivento G , Geroldi A , Tozza S , Moroni I , Mohassel P , et al. The SPTLC1 p.S331 mutation bridges sensory neuropathy and motor neuron disease and has implications for treatment. Neuropathol Appl Neurobiol. 2022:e12842. |
[75] | Li C , Hou Y , Wei Q , Lin J , Jiang Z , Jiang Q , et al. Mutation screening of SPTLC1 and SPTLC2 in amyotrophic lateral sclerosis. Hum Genomics. (2023) ;17: (1):28. |
[76] | Garofalo K , Penno A , Schmidt BP , Lee HJ , Frosch MP , von Eckardstein A , et al. Oral L-serine supplementation reduces production of neurotoxic deoxysphingolipids in mice and humans with hereditary sensory autonomic neuropathy type 1. J Clin Invest. (2011) ;121: (12):4735–45. |
[77] | Fridman V , Suriyanarayanan S , Novak P , David W , Macklin EA , McKenna-Yasek D , et al. Randomized trial of l-serine in patients with hereditary sensory and autonomic neuropathy type 1. Neurology. (2019) ;92: (4):e359–e70. |
[78] | Wadsworth JM , Clarke DJ , McMahon SA , Lowther JP , Beattie AE , Langridge-Smith PR , et al. The chemical basis of serine palmitoyltransferase inhibition by myriocin. J Am Chem Soc. (2013) ;135: (38):14276–85. |
[79] | Genin MJ , Gonzalez Valcarcel IC , Holloway WG , Lamar J , Mosior M , Hawkins E , et al. Imidazopyridine and Pyrazolopiperidine Derivatives as Novel Inhibitors of Serine Palmitoyl Transferase. J Med Chem. (2016) ;59: (12):5904–10. |
[80] | Kluepfel D , Bagli J , Baker H , Charest MP , Kudelski A . Myriocin, a new antifungal antibiotic from Myriococcum albomyces. J Antibiot (Tokyo). (1972) ;25: (2):109–15. |
[81] | Lowther J , Yard BA , Johnson KA , Carter LG , Bhat VT , Raman MC , et al. Inhibition of the PLP-dependent enzyme serine palmitoyltransferase by cycloserine: Evidence for a novel decarboxylative mechanism of inactivation. Mol Biosyst. (2010) ;6: (9):1682–93. |
[82] | Polc P , Pieri L , Bonetti EP , Scherschlicht R , Moehler H , Kettler R , et al. L-cycloserine: Behavioural and biochemical effects after single and repeated administration to mice, rats and cats. Neuropharmacology. (1986) ;25: (4):411–8. |
[83] | Boyd LJ , Epstein IG , Nair KG . The treatment of human tuberculosis with cycloserine: A year’s progress. Antibiot Annu. (1955) ;3: :141–7. |
[84] | Pacold ME , Brimacombe KR , Chan SH , Rohde JM , Lewis CA , Swier LJ , et al. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat Chem Biol. (2016) ;12: (6):452–8. |
[85] | Jafar-Nejad P , Powers B , Soriano A , Zhao H , Norris DA , Matson J , et al. The atlas of RNase H antisense oligonucleotide distribution and activity in the CNS of rodents and non-human primates following central administration. Nucleic Acids Res. (2021) ;49: (2):657–73. |
[86] | Tran H , Moazami MP , Yang H , McKenna-Yasek D , Douthwright CL , Pinto C , et al. Suppression of mutant C9orf72 expression by a potent mixed backbone antisense oligonucleotide. Nat Med. (2022) ;28: (1):117–24. |
[87] | Korobeynikov VA , Lyashchenko AK , Blanco-Redondo B , Jafar-Nejad P , Shneider NA . Antisense oligonucleotide silencing of FUS expression as a therapeutic approach in amyotrophic lateral sclerosis. Nat Med. (2022) ;28: (1):104–16. |
[88] | Miller TM , Cudkowicz ME , Genge A , Shaw PJ , Sobue G , Bucelli RC , et al. Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N Engl J Med. (2022) ;387: (12):1099–110. |
[89] | Mandegar MA , Huebsch N , Frolov EB , Shin E , Truong A , Olvera MP , et al. CRISPR Interference Efficiently Induces Specific and Reversible Gene Silencing in Human iPSCs. Cell Stem Cell. (2016) ;18: (4):541–53. |
[90] | Chen PJ , Liu DR . Prime editing for precise and highly versatile genome manipulation. Nat Rev Genet. (2023) ;24: (3):161–77. |


