
Methodological Quality of Clinical Trials in Amyotrophic Lateral Sclerosis: A Systematic Review
Abstract
Background:
More than 200 clinical trials have been performed worldwide in ALS so far, but no agents with substantial efficacy on disease progression have been found.
Objective:
To describe the methodological quality of all clinical trials performed in ALS and published before December 31, 2022.
Methods:
We conducted a systematic review following the Preferred Reporting Items for Systematic Reviews and Meta Analyses.
Results:
213 trials were included. 47.4% manuscripts described preclinical study evaluation, with a positive effect in all. 67.6% of trials were conducted with a parallel-arm design, while 12.7% were cross-over studies; 77% were randomized, while in 5.6% historical-controls were used for comparison. 70% of trials were double blind. Participant inclusion allowed forced vital capacity (or corresponding slow vital capacity)<50% in 15% cases, between 55–65% in 21.6%, between 70–80% in 14.1% reports, and 49.3% of the evaluated manuscripts did not provide a minimum value for respiratory capacity at inclusion. Disease duration was < 6-months in 6 studies, 7–36 months in 68, 37–60 months in 24, 8 trials requested more than 1-month of disease duration, while in 107 reports a disease duration was not described. Dropout rate was ≥20% in 30.5% trials, while it was not reported for 8.5%.
Conclusion:
The methodological quality of the included studies was highly variable. Major issues to be addressed in future ALS clinical trials include: the requirement for standard animal toxicology and phase I studies, the resource-intensive nature of phase II-III studies, adequate study methodology and design, a good results reporting.
INTRODUCTION
Amyotrophic Lateral Sclerosis (ALS) is a neurodegenerative disease predominantly affecting upper and lower motor neurons, with several genetic and environmental risk factors [1]. The disease is progressive, leading to death in 3–5 years after onset of symptoms. Notwithstanding some recent findings in uncovering etiological clues [2], the causes of the disease remain mostly unknown. Because of a lack of validated biomarkers and the absence of distinguishing pathognomonic clinical features at the start, the early stage of the disease is not easily discernible.
More than 200 clinical trials have been performed worldwide so far, but no agents with substantial efficacy on disease progression have been found. All drugs currently available for the treatment of ALS have modest effects, extending the lifespan only for a few months, thus making ALS a disease with a clear unmet therapeutic need. Only riluzole (Rilutek®) has been approved as a disease modifier in all countries, and some additional treatments like edaravone (Radicava®), sodium phenylbutyrate and tauroursodeoxycholic acid (Relyvrio®) and tofersen (Qalsody®) are available only in a few selected countries.
Previous ALS clinical trials might have had poor methodology or approach, leading to possible inconclusive or false-negative results. We therefore conducted a systematic review to evaluate the methodological quality of all clinical trials performed in ALS (methodology, statistical analyses, reporting) and published before December 31, 2022.
Our specific objective was to answer the following research questions: 1.What is the methodological quality of trials in ALS? 2.What study designs are used in ALS trials? 3.Is the study rationale (including pre-clinical assessment) adequate?
METHODS
Search strategy
An expert reference librarian and a study author with expertise in conducting systematic reviews and in ALS clinical trial design and management (EP), developed the search strategy from the time of database inception until December 2022. We searched PubMed, EMBASE, and the Cochrane Library. The detailed search strategy is reported in Appendix 1. The study followed the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) reporting guidelines in terms of study selection, data collection and synthesis, and assessment of bias and quality[3]. We checked the reference list of the retrieved eligible studies to find relevant articles missed from database searching. We also searched for additional references from recent systematic reviews to avoid any potentially missed papers.
The following databases were searched only with the purpose of evaluating publication bias: The International Clinical Trials Registry Platform, the European Union Clinical Trials Register, and clinicaltrials.gov datasets.
Eligibility criteria
We included all original full-text reports related to clinical trials (any type: open label, double-blind, randomized, not-randomized, cross-over, adaptative) performed on people with ALS/MND aged 18 + years, and conducted in hospitals, nursing homes, outpatient clinics, or at community level, published in the English language before December 31, 2022. We included studies of disease progression or symptomatic pharmaceutical intervention, with efficacy as primary or secondary endpoint. We included studies of phase I/II, II and III, according to the Authors’ definition. When the phase was not stated, we categorized the study depending on the study characteristics and aims. Excluded were phase I trials, reports describing a study protocol or interim analyses or post-hoc analyses, reports, letters to the editor, book chapters, conference proceedings, dissertations, theses, and animal studies.
We considered placebo, any active drug, standard practice “for comparative studies” and “none, with before-after comparison” for non-comparative studies.
Study selection
We exported all the references retrieved from the searches into the Rayyan online tool [4]. Duplicated records were manually removed by one author (EP) prior to screening. After that, four pairs of reviewers working independently, blindly assessed each article’s title and abstract for eligibility. When each pair completed all the evaluations, the blind was open and disagreements were solved by discussion and consensus during a direct meeting between the two members of the pair. In a few cases, when the reviewers did not reach an agreement, a third evaluator (EP, ML) reviewed the study and made the final decision. The reasons for the exclusion were listed for all excluded documents. After the title and abstract screening, four pairs of independent reviewers read the full articles of the remaining studies and completed a full-text review.
Reviewers extracted study details from the full text articles using a pilot-tested form defined by three senior epidemiologists and two junior investigators. Variables were selected according to Sackett [5]. All variables were pre-defined and categorized. The main domains of data extraction were: general information, demographic characteristics (number of participants recruited and withdrawn), comorbidities, investigational medication(s) used, diagnostic criteria, inclusion/exclusion criteria, primary objective, study design, primary (with effect sizes) and secondary outcomes, statistical aspects, risk of bias. Data were extracted directly from published reports and no additional information was requested from the corresponding authors nor collected by external sources.
To ensure accurate data extraction, a training meeting was performed and any question was solved before starting the extraction. Data were extracted independently by 5 reviewers in double. After the evaluation of 3% of the eligible reports, a second meeting was arranged and the data extracted were compared to discuss any discrepancy and to ensure consistency between reviewers. A third meeting was planned and carried out after the evaluation of 30% of the full texts. After the third meeting, data were extracted by a single reviewer for the remaining publications.
The risk of bias was also assessed using the appropriate Cochrane tools for clinical trials (ROB2 for randomized trial, ROB2 for cross over trial) structured into a fixed set of domains describing different sources of bias, focusing on different aspects of trial design, conduct and reporting. The risk of bias for non-randomized trials was not assessed. The risk of bias was classified into three categories including “low”, “high” or “some concerns” according to the Cochrane manual [6]. The risk of bias was assessed by the same pairs of reviewers and was performed in double for 36% of studies (during the 3 meetings performed by reviewers, the risk of bias was also discussed). The Risk-of-bias visualization tool (ROBVIS) [7] was used to produce the plots that summarize the risk of bias assessment.
Systematic review methods were established before conducting the review by determining the search strategy, article inclusion criteria, quality assessment methods, data extraction methods, and statistical analysis plans. No protocol deviations were made.
The study protocol was published in the PROSPERO database to minimize reporting bias (CRD42022381689).
RESULTS
From the first literature search, a total of 287 full-text documents were evaluated. After abstract reading, 60 reports were excluded, leaving a total of 227 full texts. In March 2023 a search update was performed to include reports published from May 2022 to December 2022 and 8 additional full texts were added. Also, we checked references of systematic reviews and all eligible studies to find articles that met the inclusion criteria but which had been missed from database search; 31 additional full texts were included.
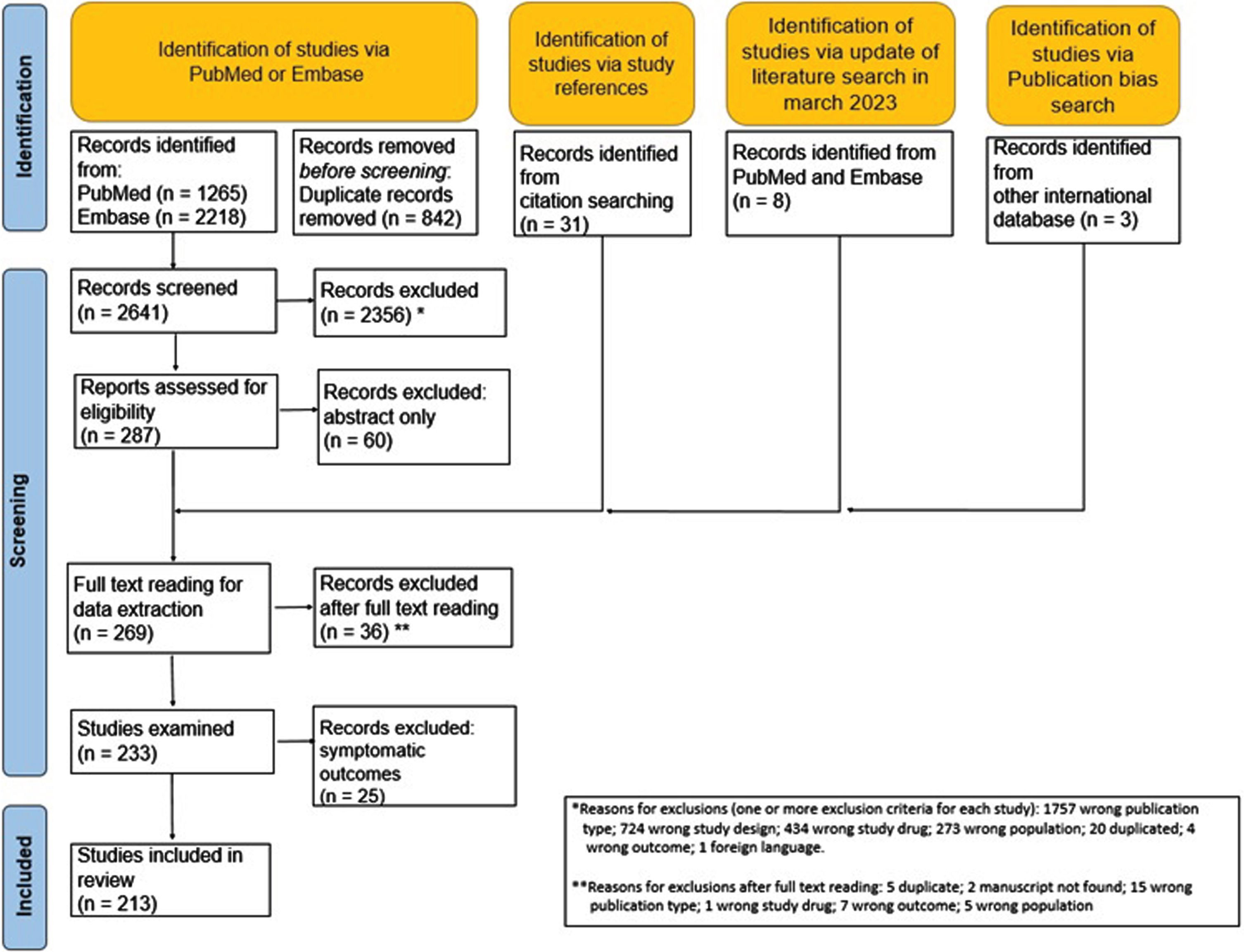
After reading 269 full texts, a total of 36 reports were excluded for various reasons, leaving 233 documents for data extraction. Five manuscripts reported 2 trials, each performed in 2 different steps, with data reported separately for each step. Thus, the total number of accountable trials was 238. After full-text reading and data extraction, we decided to exclude 25 trials because their objective was to evaluate the effect of treatment on symptoms only, both in primary and secondary endpoints, without any evaluation of the effect on disease progression. Overall, the total number of trials included in the present review is 213. The list of all documents after full-text reading is available in supplementary material (eTable 1, included, and eTable 2, excluded). Figure 1 shows the flow diagram of documents included and excluded at different stages of the review based on the PRISMA statements.
Fig. 1
PRISM study flow chart.
The number of trials increased from an average of 1/year in the 70s up to 10/year, with a steady increase of phase II and parallel arms designs after 1995 (Fig. 2a and 2b). Most trials (eFigure 1) were from the USA (N = 104), followed by Italy [36], Canada, Germany and France (21–25 in each country). The main characteristics of studies, study participants, inclusion and exclusion criteria are described in Table 1 : 41.8% of the articles were published before 2003 and 30.5% in 2013 or after.
Fig. 2
A. Number of trials by year and phase; B. Number of trials by year and design.
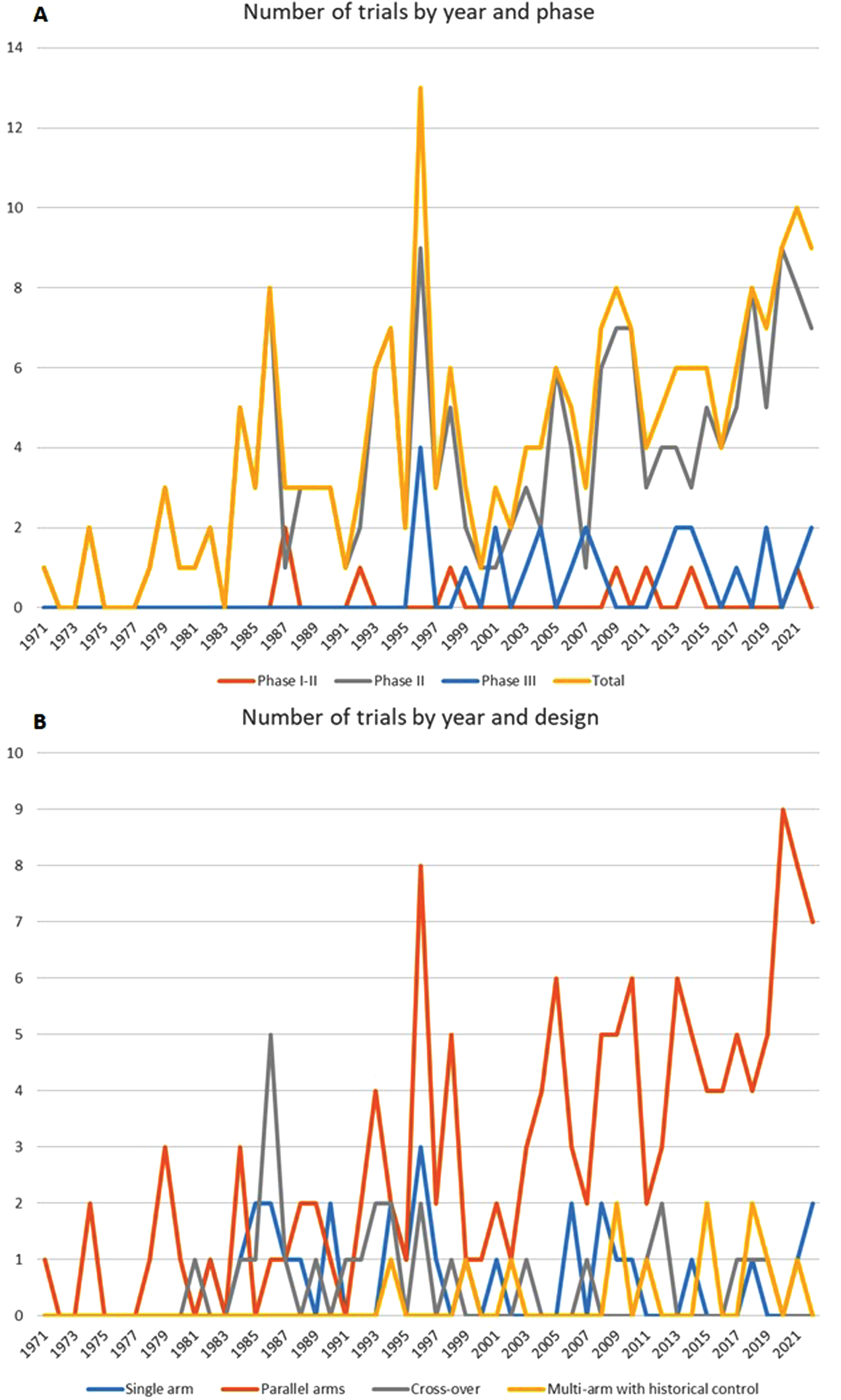
Table 1
Main characteristics of studies included in the review, characteristics of study participants, inclusion and exclusion criteria
| Main characteristics of studies included in the review | ||
| n or median | % or range | |
| Year of publication | ||
| Before 2003 | 89 | 41.8 |
| Between 2003 and 2013 | 59 | 27.7 |
| After 2013 | 65 | 30.5 |
| Phase | ||
| I/II | 8 | 3.8 |
| II | 179 | 84 |
| III | 26 | 12.2 |
| Centers involved | ||
| Monocentric | 85 | 39.9 |
| Multicentric | 98 | 46 |
| Not specified | 30 | 14.1 |
| Number of centers (in multicenter studies) | 11 | 2 - 674 |
| Number of subjects screened | 141* | 4–83 |
| Number of subjects included | 210** | 4–83 |
| Total follow-up duration (weeks) | 36 | 1–182 |
| Treatment duration (weeks) | 24 | 1–182 |
| Characteristics of study participants, inclusion and exclusion criteria | ||
| n or median | % or range | |
| Mean age of included participants*** | 57 | 44 - 66 |
| Males % *** | 63 | 0.43 - 100 |
| Females % *** | 36 | 0 - 83 |
| ALS type | ||
| Sporadic ALS | 35 | 16.4 |
| Familial ALS | 3 | 1.4 |
| Both | 41 | 19.3 |
| Not specified | 134 | 62.9 |
| Included both bulbar and spinal onset ALS | ||
| Yes | 113 | 53.1 |
| No | 5 | 2.4 |
| Not specified | 95 | 44.6 |
| Included participants with comorbidities | ||
| Yes | 0 | 0 |
| No | 36 | 16.9 |
| Not specified | 177 | 83.1 |
| Included smokers | ||
| Yes | 3 | 1.4 |
| No | 2 | 0.9 |
| Not specified | 208 | 97.7 |
| Included participants drinking alcohol | ||
| Yes | 0 | 0 |
| No | 9 | 4.2 |
| Not specified | 204 | 95.8 |
| Included participants taking concomitant medications | ||
| Yes | 105 | 49.3 |
| No | 20 | 9.4 |
| Not specified | 88 | 41.3 |
| Included participants taking riluzole (only studies performed after 1994) | ||
| No | 1 | 0.6 |
| Add-on | 53 | 33.6 |
| As per clinical practice | 44 | 27.8 |
| Not specified | 60 | 38 |
| Minimum FVC/SVC level allowed in inclusion criteria | ||
| 30–50 | 32 | 15 |
| 55–65 | 46 | 21.6 |
| 70–80 | 30 | 14.1 |
| Not specified | 105 | 49.3 |
| < 2 | 1 | 0.5 |
| < 3 | 2 | 0.9 |
| < 6 | 3 | 1.4 |
| < 12 | 4 | 1.9 |
| < 18 | 6 | 2.8 |
| < 24 | 16 | 7.5 |
| < 30 | 3 | 1.4 |
| < 36 | 39 | 18.3 |
| < 48 | 4 | 1.9 |
| < 60 | 20 | 9.4 |
| > 1 | 1 | 0.5 |
| > 6 | 3 | 1.4 |
| > 12 | 1 | 0.5 |
| > 24 | 1 | 0.5 |
| > 36 | 1 | 0.5 |
| > 60 | 1 | 0.5 |
| No restrictions | 1 | 0.5 |
| Not specified | 106 | 49.8 |
| Diagnostic criteria | ||
| El Escorial revised | 44 | 20.7 |
| Awaji | 3 | 1.4 |
| El Escorial | 87 | 40.9 |
| World Federation of Neurology | 4 | 1.9 |
| Clinical evaluation | 26 | 12.2 |
| Not specified | 49 | 23 |
*Missing data in 72 cases. **Missing data in 3 cases. ***Not specified in 29 studies.
In the post-riluzole era (after 1994), 18 studies (11.5%) did not use controls, 45 (28.7%) trials used placebo.
Respiratory function was not used (or not specified) as an inclusion criterion in 49.3% of the studies (79.8% of them were published before 2003). When respiratory function was reported as an inclusion criterion, in 15% of trials participants were enrolled with Forced Vital Capacity (FVC) (or the corresponding value of Slow Vital Capacity (SVC))<50% of the predicted value (23.7% of them published between 2003 and 2013 and 18.5% after 2013).
Eight studies were phase I/II. Phase II trials were the most represented (179, 84%), with a wide range of enrolled participants, from 4 to 605. Among the 179 phase II trials, 105 enrolled less than 50 participants, thirty-one 51–100 participants and thirty-eight 101–500 participants. There were 26 Phase III trials and the number of enrolled participants was higher than 501 in twelve of them, while in 11 trials 101–500 participants were enrolled.
Characteristics of the planning stage (study rationale, design, sample size calculation and statistical analysis) are reported in Table 2. Two thirds of studies used a parallel arms design, followed by single arm and cross over. Adaptative study design was used in 6 trials only, always as a group sequential design. Power calculation was not provided in 56.3% cases (75.3%, 47.5% and 38.5% of studies published, respectively, before 2003, between 2003 and 2013 or after 2013).
Table 2
Planning phase: study rationale, design, sample size calculation and statistical analysis
| n or median | % or range | |
| Pre-clinical studies performed | ||
| Yes, with positive results | 101 | 47.4 |
| Yes, with negative results | 0 | 0.0 |
| No | 16 | 7.5 |
| Not specified | 96 | 45.1 |
| Toxicity studies performed | ||
| Yes | 53 | 24.9 |
| No | 10 | 4.7 |
| Not specified | 150 | 70.4 |
| Study design | ||
| Single arm | 30 | 14.1 |
| Parallel arms | 144 | 67.6 |
| Cross-over | 27 | 12.7 |
| Multi-arm with historical controls | 12 | 5.6 |
| Number of experimental groups | ||
| 1 | 30 | 14.1 |
| 2 | 149 | 70.0 |
| 3 | 22 | 10.3 |
| 4 | 9 | 4.2 |
| 5 | 2 | 0.9 |
| 6 | 1 | 0.5 |
| Type of control group | ||
| Not present | 30 | 14.1 |
| Not specified | 3 | 1.4 |
| Not randomized | 3 | 1.4 |
| Randomized | 164 | 77.0 |
| Randomized+historical | 1 | 0.5 |
| Historical control group | 12 | 5.6 |
| Randomized study | ||
| No | 48 | 22.5 |
| Yes | 165 | 77.5 |
| Centralized randomization | ||
| Yes | 105 | 49.3 |
| No | 3 | 1.4 |
| Not specified | 57 | 26.8 |
| Not randomized | 48 | 22.5 |
| Blindness | ||
| Not blinded | 50 | 23.5 |
| Single blind | 6 | 2.8 |
| Double blind | 149 | 70.0 |
| Not specified | 8 | 3.8 |
| Adaptive design | ||
| No | 207 | 97.2 |
| Yes | 6 | 2.8 |
| Type of adaptive design | ||
| Multi-stage | 2 | 33.3 |
| Group sequential | 4 | 66.7 |
| Sample size calculation performed | ||
| Yes, with power < 80% | 7 | 3.3 |
| Yes, with power = 80% | 55 | 25.8 |
| Yes, with power > 80% | 28 | 13.2 |
| Yes, with power not specified | 3 | 1.4 |
| Not performed | 120 | 56.3 |
| Level of significance | ||
| < 5% | 10 | 4.7 |
| 5% | 105 | 49.3 |
| Not specified | 98 | 46.0 |
| Primary hypothesis type | ||
| Superiority | 150 | 70.4 |
| Non inferiority | 2 | 0.9 |
| Not specified | 60 | 28.2 |
| Futility | 1 | 0.5 |
Inclusion criteria were not described in 23.9% of the studies, almost half of them published before the year 2003 (41 documents, 46.1%). Similarly, 69.5% of studies did not report exclusion criteria, and about half of them were published before the year 2003 (N = 46, 51.7%). In 183 studies (85.1%) a control group was present. Among them, 70 (38.2%) were published before 2003, 53 (29%) between 2003 and 2013 and 60 (32.8%) after 2013. The use of historical controls increased from 6 trials (4%) before 2013, to 6 studies (9%) in 2013 or thereafter.
Considering only studies performed after 1994 (year of riluzole approval) (N = 157), riluzole was described as concomitant medication in 97 trials (61.8%); among these, in 53 cases (54.6%) it was an add-on with the tested treatment, while in 44 (45.4%) it was used or not as per clinical practice.
Subgroup analysis was performed in 53 studies (24.5%), in 19 studies with less than 50 participants, in 8 with 51–100, in 19 with 101–500, and in 7 with more than 501 participants. The most frequent subgroup analyses were performed for site of onset (N = 22), sex (N = 13), and use of riluzole (N = 10 among those that accepted riluzole as per clinical practice).
Additional data on study conduct and results reporting are available in Table 3. A list of all outcome measures is shown in Table 4.
Table 3
Study conduction and reporting of results
| n or median | % or range | |
| Study rationale reported | 196 | 92.0 |
| Inclusion criteria reported | 162 | 76.1 |
| Exclusion criteria reported | 148 | 69.5 |
| Sources for participants recruitment, setting of the study, characteristics participants properly described | 164 | 77.0 |
| Treatment allocation properly described | ||
| Described | 80 | 37.6 |
| Not described | 48 | 22.5 |
| Not Applicable | 85 | 39.9 |
| Allocation concealment described | ||
| Yes | 63 | 29.6 |
| No | 91 | 42.7 |
| Not applicable | 59 | 27.7 |
| Outcome measures declared | 207 | 97.2 |
| Primary outcome described | 207 | 97.2 |
| The primary outcome was clinically relevant | 204 | 95.8 |
| Study flow-chart reported | 94 | 44.1 |
| Recruitment stopped | 9 | 4.2 |
| Included participants representative of general ALS patients population | ||
| Yes | 100 | 47.0 |
| No | 30 | 14.1 |
| Not specified | 83 | 39.0 |
| Characteristics of included participants comparable between treatment groups | ||
| Yes | 115 | 54.0 |
| No | 23 | 10.8 |
| Not specified | 75 | 35.2 |
| Not applicable | ||
| Study procedures comparable between treatment groups | ||
| Yes | 128 | 60.1 |
| No | 1 | 0.5 |
| Not specified | 36 | 16.9 |
| Not applicable | 48 | 22.5 |
| Allocation respected | ||
| Yes | 136 | 63.9 |
| No | 1 | 0.5 |
| Not specified | 28 | 13.2 |
| Not available | 48 | 22.5 |
| Planned sample size reached | ||
| Yes | 17 | 8.4 |
| Higher sample size | 40 | 19.7 |
| Lower sample size | 26 | 12.8 |
| Sample size calculation not reported | 120 | 59.1 |
| Missing | 10 | |
| Withdrawal reasons reported | ||
| No withdrawals | 8 | 3.8 |
| No | 86 | 40.4 |
| Yes | 119 | 55.9 |
| Number of withdrawals reported | ||
| No | 83 | 39.0 |
| Yes | 130 | 61.0 |
| Drop-out % | ||
| < 20% | 130 | 61.0 |
| ≥20% | 65 | 30.5 |
| Not specified | 18 | 8.5 |
| Population analyzed | ||
| Intention to treat | 120 | 56.3 |
| Per protocol | 22 | 10.3 |
| Both | 26 | 12.2 |
| Not specified | 45 | 21.1 |
| Percentage of patients with adverse events | ||
| < 10% | 76 | 35.7 |
| ≥10% | 111 | 52.1 |
| Not specified | 26 | 12.2 |
| Percentage of patients with adverse events leading to treatment discontinuation | ||
| < 10% | 147 | 69.0 |
| ≥10% | 44 | 20.7 |
| Not specified | 22 | 10.3 |
| Statistical plan reported | ||
| Interim analysis performed | ||
| Yes | 24 | 11.3 |
| No | 123 | 57.8 |
| Not specified | 66 | 31.0 |
| Decision rule | ||
| Stop for efficacy | 6 | 2.8 |
| Stop for futility | 9 | 4.2 |
| Sample size re-estimation | 3 | 1.4 |
| Other | 2 | 0.9 |
| Not specified | 74 | 34.7 |
| Not applicable | 119 | 55.9 |
| Subgroup analyses performed | ||
| Yes | 53 | 24.9 |
| No/Not specified | 160 | 75.1 |
| Study results | ||
| Positive | 68 | 31.9 |
| Negative | 145 | 68.1 |
Table 4
Primary Outcomes by study phase
| Test | N in phase 1–2 | N in phase 2 | N in phase 3 | Total |
| ALSFRS-R | 1 | 46 | 10 | 57 |
| Muscles parameters | 1 | 31 | 4 | 36 |
| Survival | 2 | 22 | 9 | 33 |
| Adverse events (AE) | 3 | 24 | 1 | 28 |
| Norris scale | 2 | 24 | 0 | 26 |
| Laboratory tests | 0 | 24 | 0 | 24 |
| Forced vital capacity (FVC) | 0 | 16 | 2 | 18 |
| Medical Research Council (MRC) | 1 | 16 | 0 | 17 |
| Slow vital capacity (SVC) | 0 | 4 | 3 | 7 |
| Appel scale | 0 | 5 | 1 | 6 |
| Manual Muscle Testing (MMT) | 0 | 4 | 1 | 5 |
| Bulbar scale | 0 | 4 | 0 | 4 |
| Biomarkers | 0 | 1 | 0 | 1 |
| MITOS | 0 | 1 | 0 | 1 |
| Other | 1 | 55 | 1 | 57 |
Sixty-eight trials (31.9%) had positive results on the primary endpoint. Among phase II trials, 121 (67.6%) did not reach statistical significance thresholds, while in phase III studies, 20 (76.9%) did not detect any difference between groups. Final results were comparable in randomized and non-randomized trials (no differences observed in 68.7% of the randomized reports and 50% of the non-randomized).
Figure 3A–C represent the risk of bias assessment divided per study design.
Fig. 3
Risk of bias evaluation: A. ROB 2 for parallel arm studies and ITT (N = 137); B. ROB 2 for parallel arm studies and PPT (N = 6); C. ROB 2 for cross over studies (N = 24).
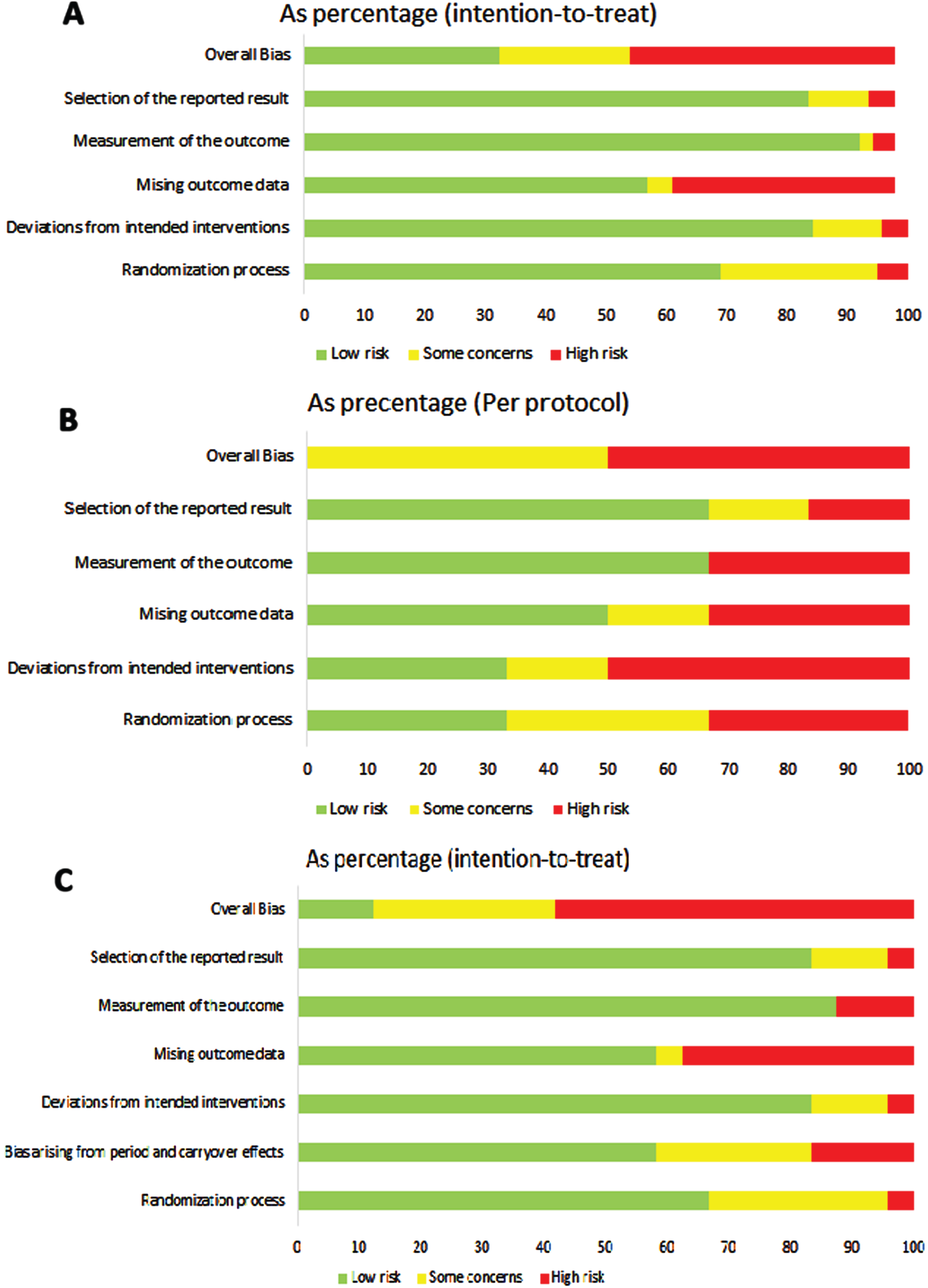
The risk of bias assessment was performed for parallel arms and cross over trials separately. In the 139 trials with parallel arms and ITT analysis, overall bias was rated as high, some concerns or low in 43.9%, 21.6% and 32.4%, respectively. Of the six parallel arms with Per Protocol (PP) analysis, the overall risk of bias was judged as some concerns in half and with high risk of bias in the other half. In cross over studies (N = 24) only the Intention To Treat (ITT) analysis was reported. The overall ROB2 was high in 58.3%, some concern in 29.2% and low in 12.5%. Missing outcome data was always in the domains with the highest risk in all study designs.
Overall, in 77.9% of the trials, no information was provided about the management of missing data, while in 15.0% cases an imputation procedure was described. Four trials referred to had no missing data.
The risk of bias did not change significantly by publication period nor by study phase (Supplementary Figure 2).
To evaluate publication bias, we searched predefined databases (The International Clinical Trials Registry Platform, the European Union Clinical Trials Register and clinicaltrials.gov) for trials fulfilling all the inclusion criteria of this review, but that were not identified in PubMed or Embase. After excluding duplicate records found in 1 or more of the 3 databases, a total of 206 clinical trials fulfilling inclusion criteria for the present review were detected (Fig. 1). The contact person of each trial was contacted: in 72 cases no further information was obtained (34 did not indicated the contact person and 38 did not answer); in 24 cases the trial was still ongoing; in 13 cases the contact person answered that a publication of the results was planned and still in progress; in 97 cases the publication status was obtained. Of the 97 clinical trials in which information about publication was obtained, 81 (83.5%) had a corresponding published report (3 of them were not previously detected and were subsequently added to the review), while 16 did not. Of these, 10 were prematurely interrupted (10.3%) and 6 were completed trials (6.2%). One of the 6 completed trials without published reports had negative results, as explicitly stated by the contact person.
Among the 72 trials in which no further information was obtained from contacts, the status reported in the corresponding database was: completed in 33, interrupted in 5, ongoing in 18 and not reported in 16.
The list of all active substances used in clinical trials in ALS by results is reported in eTable 3. The most tested substances were thyrotropin releasing hormone (N = 10), riluzole (N = 7) and lithium carbonate (N = 6). eTable 4 reports the mechanism of action of all tested drugs and results.
DISCUSSION
Our systematic review describes the quality of published phase I/II, II, and III clinical trials in which active substances were tested in people living with ALS to detect an effect on disease progression. This is the most comprehensive systematic review of ALS clinical trials carried out, with no restriction of time, outcome, or sample size. Two hundred-thirty-four trials, published from 1971 to December 2022, were evaluated, and more than 130 different active substances with different mechanism of action were considered.
Although some of these agents exhibited a good profile of safety and tolerability in animal models, they were unable to reproduce these benefits in the clinical trials. A wide range of factors, such as the poor methodological quality with high risk of bias of many studies, the late disease stage in which the clinical trials were initiated, and the heterogeneity of pathogenic mechanism occurring in ALS, could explain these failures.
Methodological flaws could lead to over-emphasis of the results of a study as “positive drug effect” on one side, and on the other side to rejection of potentially useful therapies because previous pivotal trials were inadequately designed. It is therefore crucial to consider the challenges in designing, delivering and conducting trials in ALS, and to review the possible causes of lack of confirmed treatment effect in so many studies.
Study rationale
Potential active substances should be identified in the laboratory by several preclinical studies and their results should be fully described in the introduction of all reports. Consideration should be given to which animal models may be appropriate, whether they provide sufficient information to advance a clinical program, if doses can be extrapolated from these models, and the need to test comedications [8]. FDA recommends that a pragmatic trial should be initiated only after an appropriate preclinical study is performed in specific disease models [9].
Although rationale was declared in most trials, less than 50% reported preclinical studies, in all cases positive. Unfortunately, almost all drugs showing positive effects in animals failed in subsequent human trials, but some of these negative trials might have more properly been evaluated as inconclusive for multiple reasons, including poor quality design, underpowering and excessive quantities of missing data [10].
Choice of controls
The choice of controls in ALS trials cannot disregard the use of riluzole, since this is the gold standard therapy, available since 1994. The use of riluzole implies knowledge of its possible interaction with the tested drug. For this reason, preclinical animal studies should assess the potential conse-quences of possible drug-drug interaction [11] and the safety profile when used in add-on. This could be one reason for negative results in human studies. The risk in the use of riluzole as add-on therapy in ALS is exemplified by the results of the xaliproden study in which the potential efficacy of a new drug may have been masked or even negatively affected when Xaliproden was combined with riluzole [12].
Ideally, the use of placebo arms would be avoided in ALS because of the ethics of placebo use in a terminal illness, but there are significant challenges in such an approach.
In the present review, 5.5% of the trials used historical controls for comparison. The alternative use of an historical control group remains controversial and problematic [13], and is discouraged by FDA, because several controlled trials have demonstrated differences in rates of progression and survival among placebo cohorts [8, 13].
In line with previous observations [14], we support the idea that it is not appropriate to use historical controls for comparisons, due to a large degree of variability in disease course among individual patients, the evolution of standards of patient care (such as the availability of new treatments or changes in the frequency of use for some treatments) during the course of the year and by sites. Historical controls might be of value in the design of future trials as we develop a more comprehensive and reliable characterization of the disease course [9].
The use of matched historical controls could not be fully justified even if they share comparable standards of care and inclusion criteria as the participants. In particular, matching by propensity score allows one to obtain comparable arms. Propensity score is defined as the conditional probability of receiving a treatment given a set of patient-specific covariates [15]. In the presence of a non-randomized historical arm the propensity score is not known, but could be estimated using a known set of covariates. Propensity score matched groups will then be comparable in terms of this set of covariates [15]. This method sometimes is taken into account because it allows one to avoid the use of placebo. However, this is methodology less robust than a parallel-arm and randomized design [16].
Disease heterogeneity and inclusion criteria
The heterogeneity of ALS means, despite all the best stratification strategies, the risk of having groups that are not perfectly comparable. This challenges trialists to categorize patients in homogeneous subgroups based on phenotype characteristics, such as age, clinical type, comorbidities, respiratory function, stage of disease progression, disease management [8], and even genotype [17], all possible predictors of disease course also related to aging [18–20]. Furthermore, the stage of neurodegeneration is not the same in all neurons, and different biological manifestations might be active in different regional groups of motor neurons. Diagnostic delay is another concern, since it correlates with rate of disease progression [21].
Researchers can minimize the effects of this variability with different strategies at different stages of study design: the first is at inclusion level. Selection of the right patients for participation in clinical trials is key to the potential success of the study, and necessitates recruitment of sufficient numbers of participants and the exclusion of those not fulfilling the study aims.
The importance of the definition of eligibility criteria in the design of ALS clinical trials was observed by Torrieri et al. [22]. Researchers could minimize the effect of disease heterogeneity by enrolling for example only people with a given diagnostic delay, or with a defined progression rate (fast progressors), this strategy could markedly reduce trial heterogeneity and boost statistical power [22, 23]). The observation that only specific subsets of patients responded to experimental drugs has highlighted the importance of considering patient subgroups in designing clinical trials [22, 24, 25]. On the other hand, the probability of detecting a meaningful effect size by minimizing heterogeneity should balance with the need to ensure that any finding can be generalized to the real ALS population (external validity).
The other two strategies to account for heterogeneity are to stratify randomization based on predictors or to perform subgroup analysis.
Half the trials we reviewed did not specify in their inclusion criteria any limits for FVC/SVC, diagnostic delay, or disease duration, nor did they state whether site of onset or familiarity were considered for inclusion. The other half used extremely different inclusion criteria. For example, FVC ranged from < 50% to > 80% and disease duration at enrolment ranged from < 2 months to > 60 months, determining a large heterogeneity as regard to progression rate, including participants with different stage of disease and a diverse disease course.
Choosing to enroll patients with different levels of prognostic factors in the same trial helps the generalizability of results and allows for a more rapid enrolment but requires the analysis of data in subgroups to evaluate the treatment effect for the different categories of predictors. This requires a larger number of enrolled participants to allow for an adequate power per group. For example, in studies allowing the inclusion of participants with more than 24 months’ disease duration, the median number of enrolled participants in our review was only 44.5, thus probably not permitting subgroup analyses with adequate power.
To overcome these flaws, several solutions are possible. Based on the supposed mechanism of action of a specific drug and the trial aim, the sponsor should consider the most adequate disease durations as inclusion criteria. Patients at a relatively early disease stage should be included [14]. Given that rarely enough power is reached in post-hoc subgroup analyses, these should be considered only as exploratory analyses used to provide some additional indication that, however, must be confirmed in specific subsequent studies. Subgroups, based on genotypes, should also be considered as recommended by the FDA [9].
Study phase and related aims
As previously described [26], the choice of trial design depends on the stage of development of a given drug and which study objective trialists are interested in. Briefly, the goal of phase II studies is to evaluate the safety, tolerability, dose finding, and the effect of a treatment on a specific disease, using a superiority design. Preliminary efficacy, but not definitive testing for efficacy, could also be tested in phase II trials [9, 23, 27–29].
As described in the recommendations from FDA guidelines, clinical trialists should consider including the evaluation of appropriate biomarkers or biological targets in the design of a phase II study, to demonstrate that an investigational therapy achieves its anticipated function or biological target [9].
We identified 203 phase II studies. Of these, only 79 (38.91%) were conducted with safety, tolerability, dose finding or effect as main objective, in line with study phase requirements.
The results from phase II studies should help trialists to better design subsequent efficacy phase III studies [9]. In this context, out of 26 phase III trials only 11 had at least one previous positive phase II study investigating the same active principle. Methodological characteristics of Phase II trials that proceeded to phase III were not different from those that did not, suggesting that commercial strategies or scientific interest of the single researchers impact this decision more than methodological quality, rationale and previous trial results.
Sample size
Trials must be designed specifying a pre-defined effect size that should be detected with adequate power at a pre-specified level of significance. If the initial hypothesis on the effect size is not well set, the sample size required and defined in the design phase may not be sufficient to detect differences between treatment groups.
A small sample size can be reached in a pilot study to confirm the results coming from preclinical study and to provide preliminary data for a next confirmatory study. When conducting phase III study, a formal sample size calculation must be provided based on previous observation and the expected clinical relevant benefit.
In our review, the planned sample size was not reached in 12.8% of the studies and a sample size calculation was not reported in 59.1%. In phase III studies, 92.3% provided a sample size calculation (power≥80%).
Dropouts
Disease progression, travel difficulties and caregiver burden represent common reasons for high attrition rates [30]. Since participants who drop out are usually unrepresentative of those who are randomized at the beginning of the trial, the credibility of the result is impacted in the presence of missing data [31], especially if this differentially affects study arms, leading to biased estimates. Participants who discontinue the study are those with a higher risk of not reaching the end of the study and at higher risk of experiencing more adverse events, which will affect the results of the per protocol analyses and could undermine the interpretation of the final results.
In our review, a drop-out rate higher than 20% was detected in 30.5% of trials. Also, one trial evaluated per protocol population only and the same study had a dropout rate higher than 20%.
Under the principle of intention-to-treat, in order to have an unbiased and statistically valid analysis, it is necessary to have a complete dataset to be used for the primary analysis. It is therefore necessary to use appropriate methods to manage missing data. The most efficient solution is the ability of the Sponsor to keep missing data as low as possible, acting on more appropriate inclusion criteria and study procedures.
Provided their validity and reproducibility is evaluated, several alternatives may be considered to carry on a patient’s evaluation when they cannot reach the study site with the aim to lower the dropout rate and missing data. These include the use of telemedicine technology, even if in-person evaluation is to be preferred [32–34], home self-administration of the Revised Amyotrophic Lateral Sclerosis Functional Rating Scale (ALSFRS-R) [35–38], remote assessment of FVC and of maximum inspiratory pressure (MIP) through smart phone-based technology with real time transmission of the data [39]. During the COVID-19 pandemic restriction, this strategy was the basis for a new trial approach.
Outcomes
A primary end point should be clearly stated and easily verifiable at the end of the study in order to be able to provide a clinical interpretation of the obtained results. We found a large variety of primary end points in our review: clinical, instrumental parameters and functional scales, with different degrees of accuracy and precision over the decades.
The choice of the most appropriate primary outcome is crucial, given that it is used to calculate the sample size needed for the trial to detect a treatment effect. There is need for outcomes sufficiently sensible to changes induced by treatments, even during the restricted time limits of a clinical trial.
Phase III trials used the ALSFRS-R and survival as the most common endpoints, whereas phase II studies used a larger variety of endpoints, including also muscle parameters, adverse events, other scales and respiratory evaluation. The time of death is strongly influenced by the use of devices and palliative care available to manage disease symptoms. For this reason, the assessment of survival should be combined with an evaluation of the need for full-time (or nearly full-time) respiratory support. Thus, survival is not the best option for early phase trials to be conducted in a short timeframe. Surrogate outcomes are better suited in this context.
Notably, the primary outcome of a trial, is used to calculate the sample size of both phase II and III. In this context, survival is an inefficient primary outcome measure for Phase II trials due to the large sample size and long trial duration needed to detect adequate numbers of events [11]. If patient function is intended to be assessed by the primary outcome, mortality should be integrated as a secondary endpoint [9]. It should be noted however, that the only disease-modifying therapy approved worldwide is Riluzole, and those trials used survival as an endpoint.
The ALSFRS-R can be used instead of mortality, because it strongly predicts survival, is easy and inexpensive to administer, and minimizes dropout rates. However, the ALSFRS-R varies with time and requires moderately large sample sizes with at least 6 months duration of the trial. Combining different outcomes (eg, functioning, muscle strength, lung function, and survival) can considerably increase the efficiency of clinical trials, providing a more accurate measure of drug efficacy than the use of a single end point [11].
Although phase II clinical trials are not designed to determine clinical efficacy [9], they often include several clinical outcome measures to evaluate unexpected or large clinical impact (positive or negative). However, the inclusion of clinical outcome measures in phase II trials creates a paradox: phase II clinical trials do not have enough power to detect clinical effect [9], lacking adequate sample size but a long follow up period is required to identify small effects of the treatment [40].
Furthermore, we found that many phase II studies used more than one measure as a primary endpoint, thus providing conflicting data.
Since phase II studies are still expected to be the prominent type in the future, but they require large cohorts of patients and a long follow up period to identify effects of the treatment [40], new surrogate measures, including biomarkers, should be used in phase II studies to screen and help to select the most promising compounds to be brought into phase III clinical trials.
Huge strides in identifying biomarkers for ALS progression could lead to the optimization of already available outcomes and provide the possibility to better investigate potential therapies for ALS.
Study design
Most of the clinical trials analyzed were carried out using two study models: parallel arms, in which, after randomization, one group of patients receives the drug and the other the placebo for the entire duration of the trial, and a crossover design, in which the same person is exposed at different times, to one or more treatments, according to a random sequence. In this way all participants receive all the tested treatments.
A disease with rapid progression such as ALS, with currently available outcomes that make it difficult to detect clinically relevant changes in the earliest phases of a study, does not lend itself to a crossover study design. The rapid evolution that characterizes neurodegeneration in ALS constitutes a bias in the choice of crossover designs as evaluations made on the same person at two different consecutive times are not comparable in terms of disease progression.
Risk of bias
The reliability of the results of a randomized trial depends on the extent to which potential sources of bias have been avoided. A systematic error may lead to underestimation or overestimation of an effect. The risk-of-bias analysis carried out in this review has revealed that most published studies in ALS have biases that negatively affect the veracity of the results obtained. The most prevalent bias is represented by the presence of missing data caused by the high number of drop-outs in clinical trials on ALS. Thus, the risk of bias is expected to be directly correlated with the final results of a trial.
Publication bias
Publication bias is not only a scientific problem, but also an ethical wrongdoing. People who participate in a clinical trial will expect that the research will add information to the present knowledge. Failing to report research is a waste of time and resources and it undermines the ability to make truly evidence-informed decisions about health care.
In order to give a complete picture of clinical trials in ALS, we performed a publication bias evaluation by consulting additional sources. Only 6/92 trials (6.2%) were completed but not published and 10 were prematurely interrupted without any published report (10.3%). In addition, 33 trials were marked as completed in registers but no publications were available. Thus, the publication bias is present but arguably is not a major problem in ALS.
Final considerations
Our review shows that many substances have been evaluated in ALS, usually with phase II trials, using a large number of outcomes, but without identifying a sufficient signal for progression in most cases. Although a third of the revised trials claimed to be “positive”, only riluzole has been approved in Europe; in North America, riluzole, sodium phenylbutyrate with taurursodiol and edaravone have been approved by the FDA, all with marginal efficacy. This astounding record of failures can be partly attributed to flaws in trial design which can lead to false negative and false positive results.
Major issues to be addressed in the design and conduct of clinical trials in ALS include the requirement for standard animal toxicology and phase I safety studies, the resource-intensive nature of phase II and III studies and the always challenging need to balance homogeneity of included patients and external validity. This and previous reviews of clinical trials for ALS identify a number of issues possibly contributing to their failure. It is important that these are considered in the design and implementation of future trials in this therapeutic area [8].
The primary limitation of the current study was that we included only articles in English, possibly excluding relevant trials in other languages. We also excluded literature such as theses, websites and similar documents that may or may not have been through peer review, or published elsewhere, but our analysis of publication bias makes us quite confident that our review was comprehensive.
This review has some strengths. First, it is the first review that investigated the methodological quality of all clinical trial performed in ALS. Second, most of the activities were performed in double and in blind, allowing authors to provide a robust final evaluation. Third, the assessment of the publication bias was performed using different sources and with the involvement of the contact point of each trial. Future studies should expand upon the promising preliminary results from phase II to Phase III studies by conducting large, multicentre, randomized controlled trials that examine the impact of various treatments over a longer period to assist in elucidating superior regimens and optimal dosage parameters in this vulnerable patient population.
It is crucial to develop alternative clinical trial models or any strategy to make investigators able to move through to precision medicine in such a heterogeneous disease. For example, umbrella trials, adaptative models [41], testing different drugs, possibly with multiple subgroups, and the use of biomarkers both for characterizing phenotypes and to serve as surrogate outcomes may be used. Composite outcomes, such as progression-free survival may also be used to increase the power of the study [42].
ACKNOWLEDGMENTS
We thanks Dr. Luca Porcu for technical support on the selection of variables to be evaluate in the present review, Dr. Veronica Andrea Fittipaldo for technical support in the literature search and Dr. Caterina Bendotti for biological support in the definition of mechanisms of action of the evaluated drugs.
FUNDING
No grant available for the present review. The study was supported by the Istituto di Ricerche Farmacologiche Mario Negri IRCCS. AAC is an NIHR Senior Investigator (NIHR202421) and supported through the Motor Neurone Disease Association, My Name’5 Doddie Foundation, and Alan Davidson Foundation. This study represents independent research part funded by the National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London.
DECLARATION OF INTEREST
No conflict of interest to be declared for the present review.
DATA AVAILABILITY STATEMENT
Data are available from the corresponding author upon request.
AUTHOR CONTRIBUTIONS
EP: study conceptualization (lead), developed the search strategy, performed the check of duplicated records to be manually removed, blindly assessed each article’s title and abstract for eligibility, reviewed the study in case of disagreement and made the final decision, defined the data to be extracted by each included manuscript, read the full articles to perform data extraction process, published the study protocol in the PROSPERO database, writing –Original Draft Preparation (lead).
SS: blindly assessed each article’s title and abstract for eligibility, defined the data to be extracted by each included manuscript, read the full articles to perform data extraction process, writing – Original Draft Preparation (equal).
EA: blindly assessed each article’s title and abstract for eligibility, defined the data to be extracted by each included manuscript, read the full articles to perform data extraction process, writing – Original Draft Preparation (equal).
LT: blindly assessed each article’s title and abstract for eligibility, read the full articles to perform data extraction process, writing – Original Draft Preparation (equal).
AA: study conceptualization (equal), writing – Original Draft Preparation and editing (lead).
ML: study conceptualization (lead), reviewed the study in case of disagreement and made the final decision, defined the data to be extracted by each included manuscript, read the full articles to perform data extraction process, writing – Original Draft Preparation (lead).
MC: writing – Original Draft Preparation (equal).
EB: study conceptualization (lead), defined the data to be extracted by each included manuscript, statistical analysis plans and results, writing – Original Draft Preparation (lead).
EV: blindly assessed each article’s title and abstract for eligibility, writing – Original Draft Preparation (equal).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JND-230217.
REFERENCES
[1] | Goutman SA , Hardiman O , Al-Chalabi A , Chió A , Savelieff MG , Kiernan MC , et al. Recent advances in the diagnosis and prognosis ofamyotrophic lateral sclerosis. Lancet Neurol. (2022) ;21: (5):480–93. |
[2] | Goutman SA , Hardiman O , Al-Chalabi A , Chió A , Savelieff MG , Kiernan MC , et al. Emerging insights into the complex genetics andpathophysiology of amyotrophic lateral sclerosis. Lancet Neurol. (2022) ;21: (5):465–79. |
[3] | Moher D , Liberati A , Tetzlaff J , Altman D , PRISMA Group. Preferred reporting items for systematic reviews and metaanalyses: the PRISMA statement. 2010. |
[4] | Ouzzani M , Hammady H , Fedorowicz Z , Elmagarmid A . Rayyan-a web andmobile app for systematic reviews. Syst Rev. (2016) ;5: (1):210. |
[5] | Sackett D , Straus S , Richardson S , Rosenberg W , Haynes R . Evidence-based medicine: how to practice and teach EBM. 2000. |
[6] | Higgins J , Savović J , Page M , Elbers RG , Sterne JAC . Cochrane Handbook for Systematic Reviews of Interventions version 6.3 (updated February 2022). Cochrane, 2022. Vol. Chapter 8: Assessing risk of bias in a randomized trial. 2022. |
[7] | McGuinness LA , Higgins JPT . Risk-of-bias VISualization (robvis): AnR package and Shiny web app for visualizing risk-of-biasassessments. Res Synth Methods. (2021) ;12: (1):55–61. |
[8] | Leigh PN , Swash M , Iwasaki Y , Ludolph A , Meininger V , Miller RG , et al. Amyotrophic lateral sclerosis: A consensus viewpoint ondesigning and implementing a clinical trial. Amyotroph Lateral SclerOther Motor Neuron Disord. (2004) ;5: (2):84–98. |
[9] | Food and Drug Administration. Amyotrophic Lateral Sclerosis: Developing Drugs for Treatment Guidance for Industry. 2019. |
[10] | Bhatt JM , Gordon PH . Current clinical trials in amyotrophic lateralsclerosis. Expert Opin Investig Drugs. (2007) ;16: (8):1197–207. |
[11] | Gordon PH , Meininger V . How can we improve clinical trials inamyotrophic lateral sclerosis? Nat Rev Neurol. ((2011) ;7: (11):650–4. |
[12] | Meininger V , Bensimon G , Bradley WR , Brooks B , Douillet P , Eisen AA , et al. Efficacy and safety of xaliproden in amyotrophic lateralsclerosis: results of two phase III trials. Amyotroph Lateral SclerMot Neuron Disord Off Publ World Fed Neurol Res Group Mot NeuronDis. (2004) ;5: (2):107–17. |
[13] | Simmons Z . Can we eliminate placebo in ALS clinical trials? MuscleNerve. (2009) ;39: (6):861–5. |
[14] | Leigh PN , Swash M , Iwasaki Y , Ludolph A , Meininger V , Miller RG , et al. Amyotrophic lateral sclerosis: a consensus viewpoint ondesigning and implementing a clinical trial. Amyotroph Lateral SclerMot Neuron Disord Off Publ World Fed Neurol Res Group Mot NeuronDis. (2004) ;5: (2):84–98. |
[15] | Rosenbaum PR , Rubin DB . The Central Role of the Propensity Score inObservational Studies for Causal Effects. Biometrika. (1983) ;70: (1):41–55. |
[16] | Paganoni S , Quintana M , Sherman AV , Vestrucci M , Wu Y , Timmons J , Cudkowicz M . Pooled Resource Open-Access ALS Clinical TrialsConsortium. Analysis of sodium phenylbutyrate and taurursodiolsurvival effect in ALS using external controls. Ann Clin TranslNeurol. (2023) ;10: (12):2297–2304. |
[17] | Su WM , Gu XJ , Duan QQ , Jiang Z , Gao X , Shang HF , et al. Geneticfactors for survival in amyotrophic lateral sclerosis: an integratedapproach combining a systematic review, pairwise and networkmeta-analysis. BMC Med. (2022) ;20: (1):209. |
[18] | Georges M , Attali V , Golmard JL , Morélot-Panzini C , Crevier-Buchman L , Collet JM , et al. Reduced survival in patientswith ALS with upper airway obstructive events on non-invasiveventilation. J Neurol Neurosurg Psychiatry. (2016) ;87: (10):1045–50. |
[19] | Körner S , Kollewe K , Ilsemann J , Müller-Heine A , Dengler R , Krampfl K , et al. Prevalence and prognostic impact of comorbiditiesin amyotrophic lateral sclerosis. Eur J Neurol. (2013) ;20: (4):647–54. |
[20] | Moglia C , Calvo A , Canosa A , Bertuzzo D , Cugnasco P , Solero L , et al. Influence of arterial hypertension, type 2 diabetes andcardiovascular risk factors on ALS outcome: a population-basedstudy. Amyotroph Lateral Scler Front Degener. (2017) ;18: (7-8):590–7. |
[21] | Zoccolella S , Beghi E , Palagano G , Fraddosio A , Guerra V , Samarelli V , et al. Predictors of long survival in amyotrophic lateralsclerosis: a population-based study. J Neurol Sci. (2008) ;268: (1-2):28–32. |
[22] | Torrieri MC , Manera U , Mora G , Canosa A , Vasta R , Fuda G , et al. Tailoring patients’ enrollment in ALS clinical trials: the effect ofdisease duration and vital capacity cutoffs. Amyotroph Lateral SclerFront Degener. (2022) ;23: (1-2):108–15. |
[23] | Berry JD , Cudkowicz ME , Shefner JM . Predicting success: Optimizingphase II ALS trials for the transition to phase III. AmyotrophLateral Scler Front Degener. (2014) ;15: (1-2):1–8. |
[24] | Abe K , Aoki M , Tsuji S , Itoyama Y , Sobue G , Togo M , et al. Safetyand efficacy of edaravone in well defined patients with amyotrophiclateral sclerosis: a randomised, double-blind, placebo-controlledtrial. Lancet Neurol. (2017) ;16: (7):505–12. |
[25] | Mora JS , Genge A , Chio A , Estol CJ , Chaverri D , Hernández M , et al. Masitinib as an add-on therapy to riluzole in patients withamyotrophic lateral sclerosis: a randomized clinical trial. Amyotroph Lateral Scler Front Degener. (2020) ;21: (1-2):5–14. |
[26] | Shefner JM . Designing Clinical Trials in Amyotrophic LateralSclerosis. Phys Med Rehabil Clin N Am. (2008) ;19: (3):495–508. |
[27] | Cudkowicz ME , Katz J , Moore DH , O’Neill G , Glass JD , Mitsumoto H , et al. Toward more efficient clinical trials for amyotrophic lateralsclerosis. Amyotroph Lateral Scler. (2010) ;11: (3):259–65. |
[28] | Schoenfeld DA , Cudkowicz M . Design of phase II ALS clinical trials. Amyotroph Lateral Scler. (2008) ;9: (1):16–23. |
[29] | Simon NG , Turner MR , Vucic S , Al-Chalabi A , Shefner J , Lomen-Hoerth C , et al. Quantifying disease progression in amyotrophic lateralsclerosis. Ann Neurol. (2014) ;76: (5):643–57. |
[30] | Atassi N , Yerramilli-Rao P , Szymonifka J , Yu H , Kearney M , Grasso D , et al. Analysis of start-up, retention, and adherence in ALSclinical trials. Neurology. (2013) ;81: (15):1350–5. |
[31] | Thompson JLP , Levy G . ALS issues in clinical trials. Missing data.Amyotroph Lateral Scler Mot Neuron Disord Off Publ World Fed NeurolRes Group Mot Neuron Dis. (2004) ;5: (Suppl 1):48–51. |
[32] | Ghasemi M , Poulliot K , Daniello KM , Silver B . Experience withtelemedicine in neuromuscular clinic during COVID-19 pandemic. ActaMyol Myopathies Cardiomyopathies Off J Mediterr Soc Myol. (2023) ;42: (1):14–23. |
[33] | Grogan J , Walsh S , Haulman A , Yazgi H , Geronimo A , Mamarabadi M , et al. Rapid Conversion to a Completely Virtual Multidisciplinary ALSClinic in Response to the COVID-19 Pandemic: Implications for FutureCare Delivery. J Clin Neuromuscul Dis. (2023) ;24: (4):207–13. |
[34] | Vasta R , Moglia C , D’Ovidio F , Di Pede F , De Mattei F , Cabras S , et al. Telemedicine for patients with amyotrophic lateral sclerosisduring COVID-19 pandemic: an Italian ALS referral center experience. Amyotroph Lateral Scler Front Degener. (2021) ;22: (3-4):308–11. |
[35] | Bakker LA , Schröder CD , Tan HHG , Vugts SMAG , van Eijk RPA , vanEs MA , et al. Development and assessment of the inter-rater andintra-rater reproducibility of a self-administration version of theALSFRS-R. J Neurol Neurosurg Psychiatry. (2020) ;91: (1):75–81. |
[36] | Berry JD , Paganoni S , Carlson K , Burke K , Weber H , Staples P , et al. Design and results of a smartphone-based digital phenotyping studyto quantify ALS progression. Ann Clin Transl Neurol. (2019) ;6: (5):873–81. |
[37] | Felgoise SH , Feinberg R , Stephens HE , Barkhaus P , Boylan K , Caress J , et al. Amyotrophic lateral sclerosis-specific quality oflife-short form (ALSSQOL-SF): A brief, reliable, and valid versionof the ALSSQOL-R. Muscle Nerve. (2018) ;58: (5):646–54. |
[38] | Jenkinson C , Fitzpatrick R , Brennan C , Swash M . Evidence for thevalidity and reliability of the ALS assessment questionnaire: theALSAQ-40. Amyotroph Lateral Scler Mot Neuron Disord Off Publ WorldFed Neurol Res Group Mot Neuron Dis. (1999) ;1: (1):33–40. |
[39] | Geronimo A , Simmons Z . Evaluation of remote pulmonary functiontesting in motor neuron disease. Amyotroph Lateral Scler FrontDegener. (2019) ;20: (5-6):348–55. |
[40] | Paganoni S , Cudkowicz M , Berry JD . Outcome measures in amyotrophiclateral sclerosis clinical trials. Clin Investig. (2014) ;4: (7):605–18. |
[41] | Park JJH , Hsu G , Siden EG , Thorlund K , Mills EJ . An overview ofprecision oncology basket and umbrella trials for clinicians. CACancer J Clin. (2020) ;70: (2):125–37. |
[42] | Fallowfield LJ , Fleissig A . The value of progression-free survivalto patients with advanced-stage cancer. Nat Rev Clin Oncol. (2011) ;9: (1):41–7. |


