
The IAAM LTBP4 Haplotype is Protective Against Dystrophin-Deficient Cardiomyopathy
Abstract
Background:
Dilated cardiomyopathy (DCM) is a major complication of, and leading cause of mortality in Duchenne muscular dystrophy (DMD). Its severity, age at onset, and rate of progression display wide variability, whose molecular bases have been scarcely elucidated. Potential DCM-modifying factors include glucocorticoid (GC) and cardiological treatments, DMD mutation type and location, and variants in other genes.
Methods and Results:
We retrospectively collected 3138 echocardiographic measurements of left ventricular ejection fraction (EF), shortening fraction (SF), and end-diastolic volume (EDV) from 819 DMD participants, 541 from an Italian multicentric cohort and 278 from the Cooperative International Neuromuscular Group Duchenne Natural History Study (CINRG-DNHS). Using generalized estimating equation (GEE) models, we estimated the yearly rate of decrease of EF (–0.80%) and SF (–0.41%), while EDV increase was not significantly associated with age. Utilizing a multivariate generalized estimating equation (GEE) model we observed that mutations preserving the expression of the C-terminal Dp71 isoform of dystrophin were correlated with decreased EDV (–11.01 mL/m2, p = 0.03) while for dp116 were correlated with decreased EF (–4.14%, p = <0.001). The rs10880 genotype in the LTBP4 gene, previously shown to prolong ambulation, was also associated with increased EF and decreased EDV (+3.29%, p = 0.002, and –10.62 mL/m2, p = 0.008) with a recessive model.
Conclusions:
We quantitatively describe the progression of systolic dysfunction progression in DMD, confirm the effect of distal dystrophin isoform expression on the dystrophin-deficient heart, and identify a strong effect of LTBP4 genotype of DCM in DMD.
INTRODUCTION
Duchenne muscular dystrophy (DMD) is caused by out-of-frame mutations in DMD, leading to absence or near-absence of dystrophin in muscle fibers [1]. Over the last few decades, life expectancy for DMD patients has improved, due to the implementation of interventions like nocturnal ventilation, scoliosis correction, physical therapy, and glucocorticoid (GC) treatment [2, 3]. Nevertheless, prolonged survival is paralleled by an increased incidence of dilated cardiomyopathy (DCM) [4]. The absence of dystrophin is the determinant of chronic damage localized into cardiomyocytes, leading to fibrosis, reduced contractility, and eventually heart failure [5]. Onset of DCM starts from the age of 10 years, and prevalence increases until the age of 18, when almost all patients are affected to some extent. There is no obvious correlation between the severity of muscle weakness and that of cardiac manifestations; rather, some studies suppose that earlier muscle weakness, and subsequent reduced exertion, may protect from progression to heart failure [6].
Current standards of care for DMD patients recommend regular cardiac echo-color-Doppler studies, starting in young asymptomatic children, including evaluations of left ventricular systolic function (assessed as shortening fraction [SF] and/or ejection fraction [EF]) and eccentric remodelling (assessed as ventricular diameter or end-diastolic volume [EDV] indexed by body surface) [7]. Thresholds of EF (<50–55%), SF (<28%) and/or EDV (>70 mL/m2) are often adopted [22] to define the onset of DCM (hypokinetic and/or dilative), although these may vary according to evaluation methods (e.g. echography vs. magnetic resonance) and current guidelines.
The onset, rate of progression, and overall severity of DCM present substantial variability, whose molecular and clinical bases have not been clearly elucidated [8]. The effect of GC treatment DCM is still debated, despite prevailing reports of a beneficial effect [9–12]. Moreover, several studies have attempted to identify correlations between DMD mutation characteristics (size, location) and the severity of DCM. In the milder allelic Becker muscular dystrophy, in which measurable amounts of dystrophin are expressed, deletions in the N-terminal domain of dystrophin were associated with early DCM [13]. This was not confirmed in DMD; [11, 14] on the contrary, in a recent Japanese study, it was distal mutations, disrupting one of the shorter dystrophin isoforms Dp116, that were associated with improved cardiac function [15, 16]. DMD mutations may also be classified by their amenability to emerging molecular treatments, such as exon skipping [17], and some of these mutation classes have been associated with prolonged ambulation [18–20], and to have an effect on upper limb function [21]. However, this kind of association has not yet been tested with the DCM phenotype.
There is also room for the hypothesis that genetic modifiers, i.e. variants in loci different than DMD, may play a role in phenotypic variability of dystrophin-deficient DCM. This hypothesis was initially explored in a smaller Italian multicentric study of DCM onset [22]. Of particular interest are 5 single nucleotide polymorphisms (SNPs), situated in genes expressed in the myocardium, and known to be modifiers of the loss of ambulation in DMD population, because of their involvement in inflammatory and fibrotic pathways. These are: SPP1 rs28357094 [23], LTBP4 rs10880, rs2303729 and rs1131620 [24], CD40 rs1883832 [25]. Therefore, we aimed to retrospectively explore the effects of GC treatment, DMD genotype, and modifier genotypes on EF, SF, and EDV measurements, in a large multicentric, international cohort of DMD patients.
METHODS
Cohort and measures
We collected retrospective data regarding left ventricular EF (henceforth just “EF”), left ventricular EDV indexed by body surface (henceforth “EDV”), and left ventricular SF (henceforth “SF”) from 11 Italian Centers which are listed in the authors’ affiliations (all Centers contributed patient data). We also included EF and SF data available from the Cooperative International Neuromuscular Research Group Duchenne Natural History Study (CINRG-DNHS).
Ethics statement
All participants or their parents/guardians gave informed consent to study procedure, which were carried out in accordance with the Declaration of Helsinki.
Inclusion criteria
Inclusion criteria were consistent between Italian and CINRG Centers [26], and were as follows: 1) molecularly defined diagnosis of DMD, i.e. out-of-frame DMD mutation and/or absent or trace-level (<3%) dystrophin in skeletal muscle immunohistochemistry or immunoblot, in the presence of a compatible phenotype; 2) availability of at least one echocardiographic measurement of either left ventricular EF, SF, EDV; 3) availability of a DNA sample for genotyping (optional).
Echocardiographic evaluation
Echocardiographic data were derived from routine diagnostic and follow-up exams, obtained retrospectively from clinical records of patients. At all participating Centers, echocardiographic studies were performed with last generation echocardiographic machines, most frequently from Philips or General Electric. Two-dimensional images and M-mode echocardiograms of atrial and ventricular cavities were obtained in multiple cross-sectional planes, with the transducer in standard positions according to the recommendations of the American Society of Echocardiography. Left ventricular EF was calculated from two-dimensional images, with modified Simpson’s formula or area-length method [27].
DMD genotype
Information about pathogenetic DMD mutations were collected when available from clinical or genetic reports. We classified deletions based on amenability to molecular treatments, i.e. skipping of exons 8, 44, 45, 51, and 53 (henceforth: “skip 8”, “skip 44”, etc. [18, 19, 28]). Nonsense and splice site mutations were considered as separate groups. Moreover, all mutations were subdivided into “proximal” and “distal” to dystrophin Dp140, Dp116 and Dp71 isoforms (i.e. mutations situated 5’ of intron 44 are proximal to Dp140, 5’ of intron 55 are proximal to Dp116 and 5’ of intron 62 are proximal to Dp71). These isoforms were selected because they are known to be relevant for central nervous system [29], respiratory [30], and cardiac [15] phenotypes in DMD.
SNP genotypes
SNP genotypes were determined by TaqMan (Thermo Fisher Scientific) assays, at the known DMD modifier genes expressed in cardiac tissue: SPP1 rs28357094 [23], LTBP4 rs10880, rs2303729 and rs1131620 [24], CD40 rs1883832 [25]. For LTBP4, both the effect of the supposedly most functional rs10880 SNP and of the full VTTT/IAAM haplotype were considered. To test genotype/phenotype correlation, the published inheritance models (i.e. dominant, additive, or recessive) were applied. Genotypes at modifier SNPs included in this work respected the Hardy-Weinberg equilibrium.
Statistical analysis
Quantitative variables were summarized as mean±standard error (SE) and median (range), unless otherwise specified. Distributions of quantitative variables between groups were compared by Mann-Whitney U test. Generalized Estimating Equations (GEEs), with patient identifier as a clustering variable, and using the “exchangeable” correlation structure, were used to estimate effects of: age; GC treatment (on vs. off at each evaluation); DMD mutation (tested separately: each specified mutation group vs. “other” mutations; or “distal” vs. “proximal” relative to dystrophin isoforms of interes); and SNP genotypes. Covariates were first tested individually, adjusting for age; subsequently, significant covariates were combined in multivariate models. This stepwise approach was preferred to a multivariate model including all variables, in order to minimize loss of statistical power due to missing data. In the GEE analysis, covariates are assumed independent from each other, so rs10880 and IAAM haplotype were evaluated in separate analyses, as well as DMD mutation types vs. position. GEEs were preferred over linear mixed models because of our focus on “marginal” estimates of population-level, rather than individual-level effects of covariates. Statistical significance was set at p < 0.05 (nominal significance). Analyses were carried out with R v.4.2.2 implementing the “gee” and “dplyr” packages.
RESULTS
Cohorts and demographics
We recruited 819 participants, 541 from the Italian cohort and 278 from the CINRG-DNHS. Distribution by age at the first available echocardiographic evaluation is listed in Table 1. Age ranged from very young children to adults, with a mean of 12.57±11.59 years.
Table 1
Distribution by age (years) at the time of first evaluation
| Cohort | n | Mean | SD | Median | Min | Max |
| (years) | (years) | (years) | (years) | (years) | ||
| CINRG-DNHS | 278 | 13 | 12.02 | 5.8 | 2.49 | 30.5 |
| Italian cohort | 541 | 12.35 | 11.3 | 6,92 | 0.53 | 45.09 |
| Total | 819 | 12.57 | 11.59 | 6.56 | 0.53 | 45.09 |
CINRG-DNHS: Cooperative International Neuromuscular Research Group Duchenne Natural history Study. SD: standard deviation.
Evaluations and follow-up
We collected a total of 3138 echocardiographic evaluations (2104 from the Italian cohort, 1034 from the CINRG-DNHS), divided as follows: 2883 EF measures (2072 Italy in 512 patients, 811 CINRG in 203 patients); 1170 SF measures (334 Italy in 54 patients, 836 CINRG in 227 patients); and 346 EDV measures (in 129 patients, all from the Italian cohort). Participants were followed-up with 3.83±3.27 evaluations, for a duration of 3.46±3.42 years on average. Data were collected retrospectively in the Italian cohort from echocardiograms carried out from April 1984 through November 2018, average date being November 2012; while the CINRG-DNHS was carried out between 2006 and 2016 (see https://cinrgresearch.org/duchenne-natural-history/, last accessed August 2023).
GC treatment
Distribution by GC treatment showed that 420/819 (51.28%) participants were being treated with GCs at least at one evaluation, while 201 (24.54%) were off treatment during the whole follow-up period. For the remaining participants, GC treatment status was variable during follow-up or unknown; in particular, GC treatment could not be defined at the time of 572/3138 evaluations (18.22%).
Angiotensin-converting enzyme inhibitor (ACEi) and beta-blocker (BB) treatment
Due to the retrospective design of the study, data about ACEi and BB treatments were available in only a minority of participants. In particular, of 269 participants with available ACEi treatment data, 177 (65.8%) were on ACEi at least at one evaluation during follow-up, and of 266 with available BB data, 82 (30.83%) were on BBs at least at one evaluation. Detailed data with average age by treatment (which was older in treated participants) is shown in Supplementary Table 1.
DMD mutations
The distribution by DMD mutations in the studied population reflected the expected relative frequencies of different mutation types [31]. Table 2 provides a summary of the distribution by mutation, grouped by amenability to exon skipping treatments, and other relevant mutational categories, as explained in Methods. The disease-causing mutation was undefined in a conspicuous number of participants, 182 (22.22 %). This was partly due to missing data in a retrospective study design, and partly because inclusion criteria allowed the recruitment of participants with a diagnosis only based on muscle biopsy protein assays.
Table 2
Distribution by DMD mutation category
| Criterion | Mutation group | Italian cohort | CINRG-DNHS | Total |
| Molecular treatment amenability | “skip44” deletions | 24 (4.44 %) | 18 (6.47 %) | 42 (5.13 %) |
| “skip45” deletions | 25 (4.62 %) | 22 (7.91 %) | 47 (5.74 %) | |
| “skip51” deletions | 40 (7.39 %) | 32 (11.51 %) | 72 (8.79 %) | |
| “skip53” deletions | 46 (8.5 %) | 12 (4.32 %) | 58 (7.08 %) | |
| “skip8” deletions | 7 (1.29 %) | 6 (2.16 %) | 13 (1.59 %) | |
| Nonsense | 34 (6.28 %) | 14 (5.04 %) | 48 (5.86 %) | |
| Splice site mutations | 18 (3.33 %) | 0 (0 %) | 18 (2.2 %) | |
| Other* | 245 (45.29 %) | 94 (33.81 %) | 339 (41.39 %) | |
| Undefined† | 102 (18.85 %) | 80 (28.78 %) | 182 (22.22 %) | |
| Position of the mutation | Dp140- (3’ intron 44) | 265 (48.98 %) | 134 (48.2 %) | 399 (48.72 %) |
| Dp140+ (5’ intron 44) | 170 (31.42 %) | 67 (24.1 %) | 237 (28.94 %) | |
| Dp 140 undefined† | 106 (19.59 %) | 77 (27.7 %) | 183 (22.34 %) | |
| Dp116- (3’ intron 55) | 37 (6.84 %) | 10 (3.6 %) | 47 (5.74 %) | |
| Dp116+ (5’ intron 55) | 398 (73.57 %) | 163 (58.63%) | 561 (68.5 %) | |
| Dp116 undefined† | 106 (19.59 %) | 105 (37.77 %) | 211 (25.76 %) | |
| Dp71- (3’ intron 62) | 18 (3.33 %) | 6 (2.16 %) | 24 (2.93 %) | |
| Dp71+ (5’ intron 62) | 417 (77.08 %) | 167 (60.07 %) | 584 (71.31 %) | |
| Dp71 undefined† | 106 (19.59 %) | 105 (37.77 %) | 211 (25.76 %) | |
| Total | 541 | 278 | 819 |
*Other: non-skippable deletions, duplications, frameshift mutations. CINRG-DNHS: Cooperative International Neuromuscular Research Group Duchenne Natural history Study. †Undefined: the number of undefined mutations may differ between criteria (e.g. reported nonsense mutation without nucleotide number or exon position available).
Modifier SNP genotypes
Due to limited DNA availability, not all participants could be genotyped at all SNPs of interest: SPP1 rs28357094 and CD40 rs1883832 were genotyped in 570 (69.60%) and LTBP4 rs10880 in 558 (68.13%) of participants (full VTTT/IAAM haplotype in 526, 64.22%).
Age and GCs treatment effect
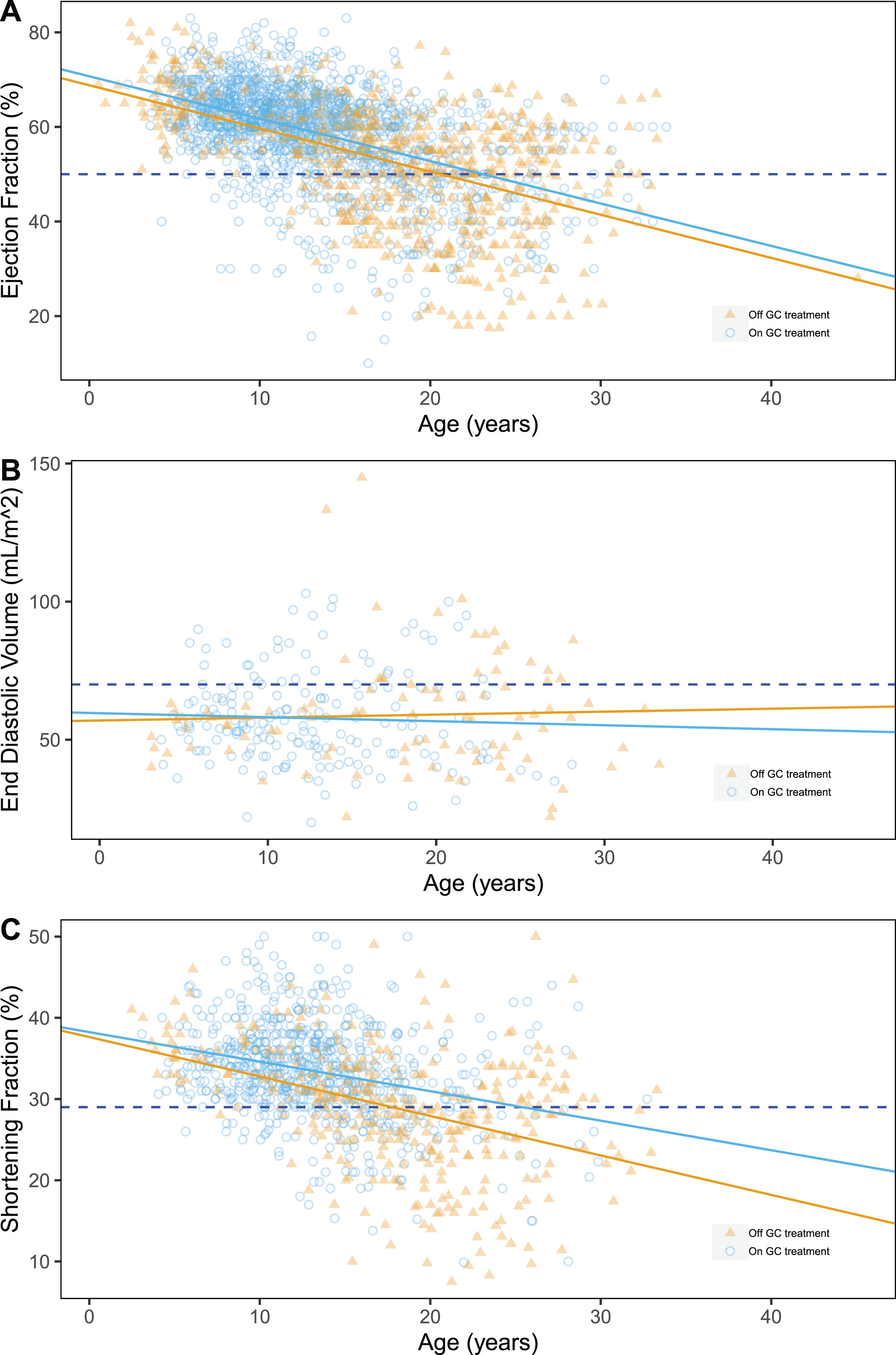
GEE models showed a significant correlation of both EF and SF with age. In particular, we were able to quantify the yearly decrease of EF and SF at the rate of –0.80±0.03% (p < 0.0001) (Fig. 1A,) and –0.41±0.04% per year (p < 0.0001) (Fig. 1B), respectively (results of the univariate analyses are summarized in Table S2-S3). On the contrary, the expected increase of EDV with age was not statistically significant, although a trend was observed (+0.42±0.16 mL/m2 per year, p = 0.060, Fig. 1C,). Concerning GC treatment, we detected a statistically significant protective effect on SF% (+1.69±0.49%, p = 0.00087, Fig. 1B), and EF% (+1.12±0.36% p = 0.048) but not on EDV.
Fig. 1
Scatter plots of ejection fraction (%) (A), shortening fraction (%) (B) and end-diastolic volume (mL/m2) (C) by age, grouped by GC treatment at evaluation time. Pathological thresholds are marked by dashed lines.
ACEi and BB effect
Data for ACEi and BBs were sparse, and the vast majority of treated participants among those with available data suggested bias in the data (i.e. those treated were more frequently reported). Therefore, we did not include these drugs as covariates in the GEE models. However, available data were sufficient to estimate yearly decrease of EF and SF by GEE in a subset of ACEi treated patients only: these amounted to –0.72±0.07% and –0.40±0.08%, respectively.
DMD mutation effects
Both “skip 44” and “skip 53” deletions appeared to have a significant effect on EF, with estimated GEE coefficients of +2.21±1.46%, p = 0.024, and –2.58±1.24%, p = 0.04 (Table S2), respectively. (Fig. 2A), i.e. protective for “skip 44” and detrimental for “skip 53”. A protective effect associated of “skip 44” deletions was detected also on EDV (–17.63±10.24 mL/m2, p = 0.0085), together with a detrimental effect of “skip 45” (+14.03±19 mL/m2, p = 0.00016). None of the DMD mutation groups have effect on SF (Fig. 2).
Fig. 2
Scatter plots of ejection fraction (%) (A), end diastolic volume (mL/m2) (B) by age, grouped by LTBP4 genotype. Pathological thresholds are marked by the dashed lines.
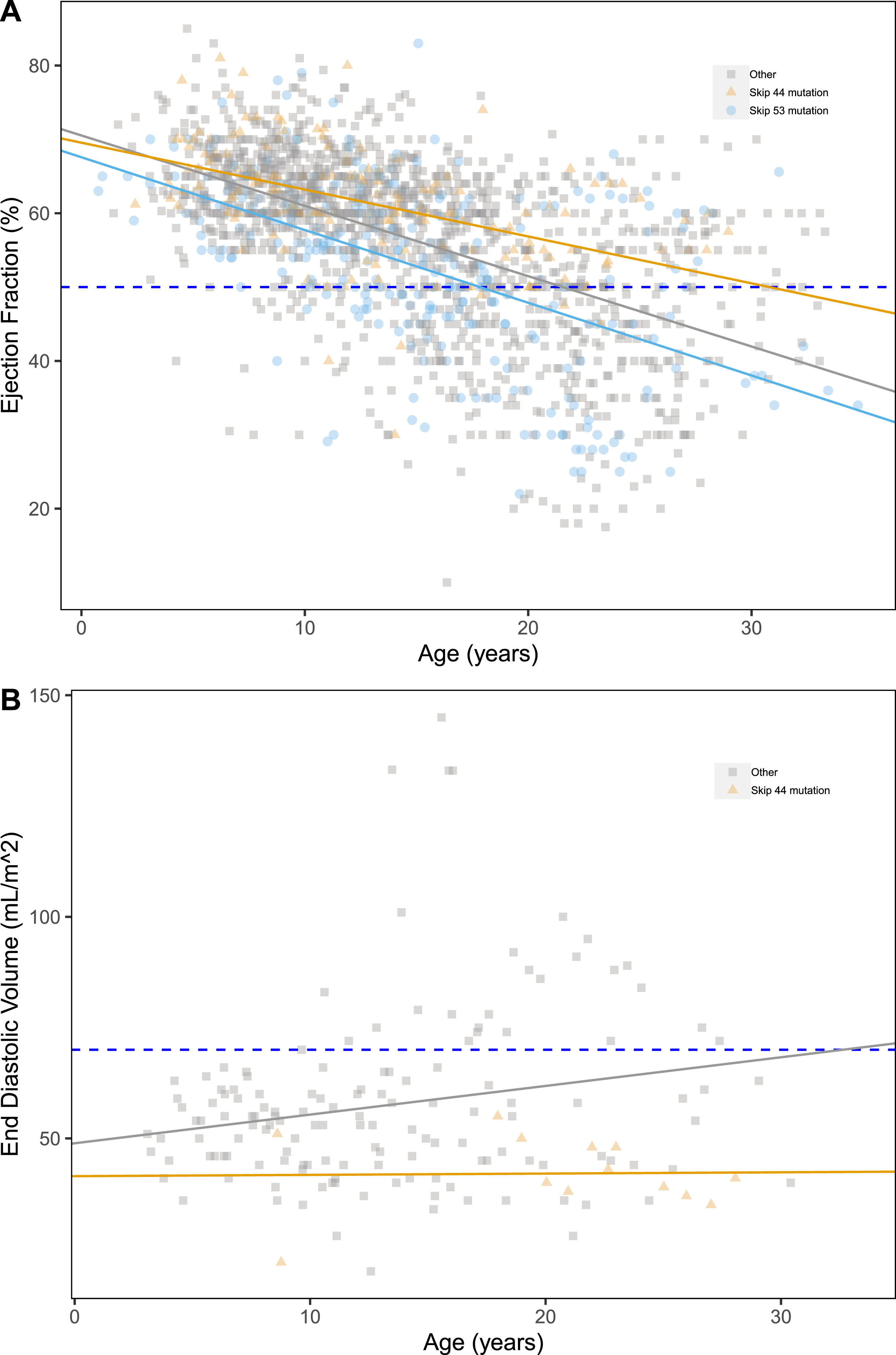
We detected a significant detrimental effect of mutations proximal to Dp116 on EDV (+11.88±8.42 mL/m2, p = 0.0063) and EF% (–3.52±1.93%, p = 0.019), but not on SF. Finally, we detected no significant effects of mutations proximal to Dp140 or Dp71 (Table S2).
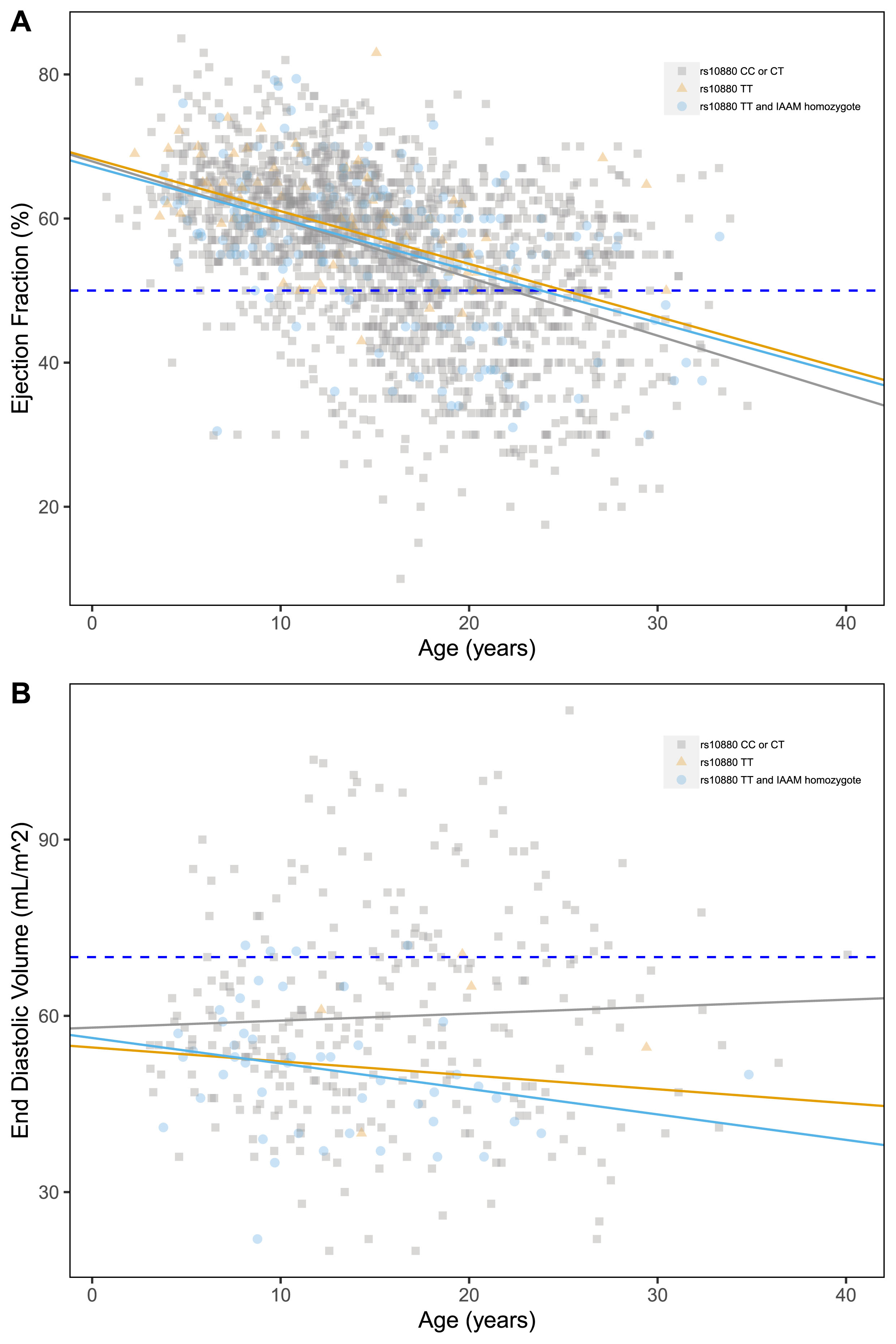
Modifier SNPs effects
Of the three genotyped modifier loci, only LTBP4 presented significant associations with the outcomes of interest. The IAAM haplotype appeared to have a protective effect on ventricular dilation (Fig. 3B), with a significant reduction in EDV associated with both rs10880 genotype and IAAM haplotype (–7.19±3.78 mL/m2, p = 0.016; and –8.61±4.21 mL/m2, p = 0.010, respectively). None of the tested SNPs was significantly correlated with EF% and SF% (Table S2). Despite this, in the multivariate model (described below), the effect of the LTBP4 genotype (both considering the single rs10880 SNP and the entire IAAM haplotype) was confirmed on EF.
Fig. 3
Scatter plots of ejection fraction (%) (A), end diastolic volume (mL/m2) (B) by age, grouped by relevant DMD mutation type. Pathological thresholds are marked by the dashed lines.
Multivariate models
To confirm the effect of each individual variable evaluated thus far, we utilized the GEE model for conducting a multivariate analysis (Table 3), enabling us to assess potential effects due to collinearity among the considered variables. First, we confirmed the detrimental effect of mutations proximal to the Dp116 isoform on EF (but not for EDV, although a trend is visible, Table 3), with an estimated coefficient of –4.14±2.59%, p < 0.001. Additionally, we confirmed the detrimental effect of “skip45” (+18.48±17.22 mL/m2 p < 0.001) and protective effect of “skip 44” (Table 3) solely on EDV (–15.24±9.49 mL/m2 p = 0.005), but not on EF. The multivariate model further corroborated the protective effect on EDV of both the rs10880 genotype (–9.2±6.05 mL/m2, p = 0.007) and the entire LTBP4 haplotype (–10.48±5.81 mL/m2, p = 0.0097), revealing a significant protective effect on EF as well (+3.26±1.44%, p = 0.003 and +2.89±1.69 mL/m2, p = 0.033). These results were confirmed in each individual cohort (Italy and CINRG), both considering the single SNP and the entire haplotype (Table S5). Moreover, we observed a detrimental effect of SPP1 rs28357094 on EDV (+6.5±4.26 mL/m2, p = 0.04). In addition to what was observed through univariate analyses, significant associations were found regarding splicing mutations and SF (–5.41±4.79%, p < 0.001) and the presence of mutations proximal to the Dp71 isoform and EDV (–11.01±13.71, p = 0.003). A beneficial effect of GC treatment could be confirmed only on SF (1.37±0.63%, p = 0.04).
Table 3
Coefficients of Multivariate Generalized Estimating Equation (GEE) analyses obtained (whole study)
| Covariate | Ejection Fraction (%) | Shortening Fraction (%) | End Diastolic Volume | |||||||
| indexed (mL/m2) | ||||||||||
| Estimate | SE | p-value | Estimate | SE | p-value | Estimate | SE | p-value | ||
| Intercept | 69.11 | 1.3 | 0 | 37.6 | 1.16 | <0.001 | 42.45 | 5.15 | <0.001 | |
| Age (years) | –0.87 | 0.05 | <0.001 | –0.41 | 0.05 | <0.001 | 0.54 | 0.18 | n.s. | |
| On glucocorticoids | 0.72 | 0.66 | n.s. | 1.37 | 0.63 | 0.04 | 2.51 | 2.58 | n.s. | |
| Mutation groups by molecular treatment amenability | “skip 8” deletions | 3.18 | 3.36 | n.s. | 0.9 | 2.16 | n.s. | 6.39 | 11.2 | n.s. |
| “skip 44” deletions | 1.8 | 1.93 | n.s. | –0.27 | 1.21 | n.s. | –15.24 | 9.49 | 0.005 | |
| “skip 45” deletions | 0.64 | 1.75 | n.s. | 0.05 | 1.14 | n.s. | 18.48 | 17.22 | <0.001 | |
| “skip 51” deletions | –0.09 | 1.43 | n.s. | –1.2 | 0.99 | n.s. | –2.62 | 6.59 | n.s. | |
| “skip 53” deletions | 0.38 | 1.86 | n.s. | –0.48 | 1.39 | n.s. | 6.57 | 6.57 | n.s. | |
| Nonsense | –0.62 | 1.8 | n.s. | –0.14 | 1.15 | n.s. | 5.41 | 6.23 | n.s. | |
| Splice site | 0.18 | 3.88 | n.s. | –5.41 | 4.79 | <0.001 | 0.52 | 7.65 | n.s. | |
| Modifier genotypes | SPP1 rs28357094 (dom) | –0.77 | 1.01 | n.s. | –0.05 | 0.71 | n.s. | 6.5 | 4.26 | 0.04 |
| CD40 rs1883832 (add) | –0.3 | 0.76 | n.s. | –0.34 | 0.53 | n.s. | 2.06 | 3.86 | n.s. | |
| LTBP4 rs10880 (rec) | 3.26 | 1.44 | 0.003 | 1.01 | 1.04 | n.s. | –9.2 | 6.05 | 0.007 | |
| IAAM haplotype (rec)* | 2.90 | 1.70 | 0.038 | 0.90 | 1.29 | n.s. | –10.48 | 5.81 | 0.0098 | |
*The values related to this parameter were calculated by substituting rs10880 in the multivariate model. EF: ejection fraction; EDV: end diastolic volume; SF: shortening fraction; SE: Standard Error; skip 8: mutations amenable to treatment with skipping of exon 8 (same for other exon numbers); n.s.: not significant; dom: dominant inheritance model; add: additive inheritance model; rec: recessive inheritance model. Significant p-values (p < 0.05) are bold.
DISCUSSION
In this retrospective study, we present data from the Italian and the CINRG-DNHS multicentric cohorts. In order to reach sufficient sample sizes, especially for genetic association tests, a large collaborative effort involved around 30 tertiary neuromuscular Centers across 4 continents, possibly raising doubts of excessive heterogeneity in the data. We acknowledge some differences in standards of care and follow-up duration between the two sub-cohorts. Indeed, in the CINRG-DNHS, patients underwent a longer follow-up than in the Italian cohort (approximately 4.0 vs. 3.12 years on average). On the other hand, individuals from the CINRG-DNHS were evaluated less frequently: approximately every 1.1 years on average, vs. approximately 0.8 year in the Italian cohort, which is closer to recommended standards of care in this age range [7]. In fact, a relatively insufficient application of cardiological standards of care in the CINRG-DNHS had been already commented on by CINRG investigators [12].
Furthermore, in the Italian cohort, patients were less frequently treated with GCs: approximately 69% of evaluations with available GC data, vs. 71% in the CINRG-DNHS. This difference in the standard of care is probably due to the older age in the Italian cohort, in which several patients born in the ‘80 s and even in the ‘70 s were included. At the time these patients were evaluated, it was not a common practice to treat DMD patients with GCs after loss of ambulation.
GEE models confirmed that EF and SF decrease significantly with age in the total study population, at the rate of –0.79±0.03% and –0.41±0.04% per year respectively. This EF decrease is in line with our knowledge on the progressive nature of the hypokinetic cardiomyopathy of DMD [4, 32]. However, such a quantitative estimation of the rate of EF decline, based on a long-term observation with serial echocardiographic measures, had not been provided in the literature. As for SF decrease, our data point to a smaller decrease than described in a recent longitudinal study of SF [33], which estimated an approximate –1.5% yearly decrease. This may be explained by the larger age range in our study population, which “distributes” the decrease over a wider time interval. The estimated values of age decrease of EF and SF may be used as a benchmark for powering clinical trials, as well as a reference for gauging the severity of individual disease progression in clinical practice.
Finally, it was not possible to detect a statistically significant effect of age on EDV measures, although a trend was observed (+0.42±0.16 mL/m2 per year, p = 0.060, Fig. 1C). With the limits of a smaller sample size, this suggests that ventricular dilation is not as constant and predictable a feature in dystrophin-deficient DCM as the loss of systolic function.
Concerning GC therapy, we identify a significant effect on EF (1.21±0.36%, p = 0.048) and SF (1.68±0.49, p = 0.00087) in the overall study. This was in part surprising, as we expected similar findings with EF and SF, which are both an expression of the same underlying physiological feature, i.e. ventricular contractile function. Furthermore, the multivariate model confirmed this finding only for SF.
It should also be noted that, while GCs are now generally considered to be beneficial also for DCM in dystrophinopathies, the subject has been debated in the literature with some contradictory findings. Their beneficial effects has been demonstrated in several studies [9, 10, 34], but put in doubt in others [11, 12]. Furthermore, prednisone exacerbated myocardial fibrosis in several murine muscular dystrophy models [35, 36], because of its agonist function at the mineralocorticoid receptor which activates the renin-angiotensin axis. In the overall population presented here, several patients were treated with deflazacort rather than prednisone, which may act differently at the level of the mineralocorticoid receptor, as shown by studies of dissociative steroids [37]. Unfortunately, we currently do not have sufficiently detailed data on deflazacort vs. prednisone treatments to perform a targeted analysis at this point. Differences between these two drugs have already been found in relation to ambulatory function and tolerability [38]. A large, long-term study comparing glucocorticoid regimens, FOR-DMD [39], did not show differences in cardiac outcomes between prednisone and deflazacort regimens, although in a young population recruited between the ages of 4 to 7 and followed up to 36 months [40].
Regarding the effects of different kinds of mutations, “skip 44” deletions seemed to be protective in EF (2.2±1.46%, p = 0.024) and EDV (–17.64±10.24 95 mL/m2, p = 0.0085), although the multivariate model only confirms the association regarding EDV (–15.24±9.49 mL/m2 p = 0.005).
These mutations were expected to be protective, probably due to a predominance of patients with an isolated exon 45 deletion, who present frequent spontaneous exon skipping events leading to low levels of an in-frame delta-44-45 transcript, resulting in in low amounts of an internally deleted dystrophin protein [41]. Our data suggest that this alternative splicing event may be ongoing in the heart as well as in skeletal muscle.
Our analysis demonstrated a decrease in systolic function (–3.52±1.92%, p = 0.019) and a significant increase of EDV (11.87±8.41, p = 0.0063) associated with mutations proximal to Dp116, compared to distal mutations. The multivariate analysis confirmed a significant decrease in systolic function (–4.14±2.59%, p = <0.001, Table 3) and also a trend in EDV (7.53±7.98, p = 0.08), confirming the observations of Yamamoto et al. [15]. The detrimental effect of this isoform on cardiac function has not been mechanistically elucidated, although Yamamoto et al. did identify Dp116 expression in the myocardium. A mechanism in which Dp116 competes with residual traces of Dp427 for the binding to the dystrophin-associated glycoprotein complex has been hypothesized [15, 42]. A novel finding in our data was an apparent effect, emerging from the multivariate analysis, of Dp71 on EDV, its absence aggravating ventricular dilation. This finding is based on a relatively low sample size, and should therefore be confirmed in independent cohorts. A role of this ubiquitous isoform, which is strongly expressed in blood vessels, on the cardiovascular system may be hypothesized.
Finally, considering genetic modifiers, of the 3 genotyped loci tested (SPP1 rs28357094, CD40 rs1883832 and LTBP4 rs2303729, rs1131620, rs10880, and IAAM haplotype), only LTBP4 rs10880, and the linked IAAM haplotype, presented significant associations with cardiac function.
The recessive rs10880 T allele and IAAM haplotype seemed to have a protective effect on EF and EDV, with estimated effect sizes of +3.26±1.44% (p = 0.003) and –9.2±6.05 mL/m2 (p = 0.007; +2.90% and –10.48 mL/m2 in the multivariate GEE model), respectively. The EF effect was independently significant in both the Italian and international CINRG-DNHS sub-cohorts, which is relevant because the effect of this genotype in prolonging ambulation was previously described as dependent on ancestry [43].
The protective effect of LTBP4 on cardiological outcomes may be interpreted thatthe IAAM isoprotein of LTBP4 is able to sequester TGF-β with a greater avidity and in a more stable latent complex. TGF-β is thus prevented from signalling at the surface of cells (cardiomyocytes, fibroblasts, immune cells) and promote pro-fibrotic changes in the myocardium, which in turn lead to reduce contractility and dilative remodelling [44].
We recognize several limitations to this study. First of all, due to the retrospective study design, several data points were missing, mostly regarding DMD and SNP genotype characterization, GC treatment history, but especially cardiological treatments. Data regarding the latter variable were at best sparse in the present study. Indeed, current guidelines [7] recommend to start ACEi (Angiotensin-Converting-Enzyme inhibitors) or ARBs (Angiotensin II Receptor Blockers) at a young age, with a later association of β-blockers according to symptoms and follow-up findings. These drugs have been found to be effective in preventing and delaying the progression of dystrophinopathic DCM [7, 45, 46]. The analysis of the limited data available suggested a slightly slower decrease of EF during ACEi treatment, and confirmed an additive effect of ACEi with LTBP4 genotype, probably due to concurrent effects on TGF-β activation downstream of mineralocorticoid receptor activation [44]. A further limitation is inherent to the limited accuracy and sensitivity of echocardiographic assessments, compared to more modern MRI-based measurements [16]. The construction and analyses of large datasets of cardiac MRI measurements in dystrophinopathies is warranted, in order to refine genotype-phenotype correlations and better delineate natural history trajectories. Some limitations were methodological in nature, e.g. the lack of MRI data, which is more sensitive than echocardiography especially in respect to the quantification of myocardial fibrosis [47]; and the fact that methods used to calculate cardiac outcomes (EF, SF, and EDV) might have slightly differed between sites. Also, the effects of studied variable on these three outcomes were not always concordant, as may be expected.
In conclusion, we describe a strong effect of LTBP4 genotype on dystrophin-deficient DCM. Detailed studies of the interaction of genotype with cardiological treatment efficacy are needed, however, before it can be implemented in DMD care as a prognostic biomarker. With its limitations, mainly due to a retrospective approach and a lack of MRI data, this study represents an initial characterization of variables that may be relevant for the design and interpretation of experimental studies for DCM in DMD.
ACKNOWLEDGMENTS
LB and EP are part of the European Reference Network for Neuromuscular diseases. The authors are grateful to all patients and their parents/caregivers.
FUNDING
EP acknowledges funding from the Cariparo Foundation (Progetto di Eccellenza “GenMod” 2017), and from Telethon Genetic BioBank (GTB12001D to EP and GTB12001 H to LP) and the Eurobiobank network. The CINRG-DNHS was funded by the Department of Education/NIDRR (#H133B031118, #H133B090001); U.S. Department of Defense (#W81XWH-12-1-0417); National Institutes of Health/NIAMS (#R01AR061875); Parent Project Muscular Dystrophy.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JND-230129.
REFERENCES
[1] | Hoffman EP , Brown RH Jr. , Kunkel LM Dystrophin: The protein product of the duchenne muscular dystrophy locus. Cell. (1987) ;51: :919–28. |
[2] | Eagle M , Baudouin SV , Chandler C , Giddings DR , Bullock R , Bushby K . Survival in Duchenne muscular dystrophy: Improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscular Disorders. (2002) ;12: :926–9. |
[3] | Klitzner TS , Beekman RH , Galioto FM , Jones TK , Manning PB , Morrow WR , Newburger J , Moore JWM , Cripe LH , Colegrove L . Cardiovascular health supervision for individuals affected by duchenne or becker muscular dystrophy. Pediatrics. (2005) ;116: :1569–73. |
[4] | Nigro G , Comi LI , Politano L , Bain RJI . The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int J Cardiol. (1990) ;26: :271–7. |
[5] | Melacini P Cardiac problems in DMD. In: Muscular Dystrophy: Causes and Management. Nova Science Publishers, Inc.; 2013. pp. 367-380. |
[6] | Spurney CF . Cardiomyopathy of duchenne muscular dystrophy: Current understanding and future directions. Muscle Nerve. (2011) ;44: :8–19. |
[7] | Birnkrant DJ , Bushby K , Bann CM , Alman BA , Apkon SD , Blackwell A , Case LE , Cripe L , Hadjiyannakis S , Olson AK , Sheehan DW , Bolen J , Weber DR , Ward LM . Diagnosis and management of Duchenne muscular dystrophy, part 2: Respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. (2018) ;17: :347–61. |
[8] | Birnkrant DJ , Bello L , Butterfield RJ , Carter JC , Cripe LH , Cripe TP , Mckim DA , Nandi D , Pegoraro E Cardiorespiratory management of Duchenne muscular dystrophy: Emerging therapies, neuromuscular genetics, and new clinical challenges [Internet]. Available from: www.thelancet.com/ |
[9] | Markham LW , Kinnett K , Wong BL , Woodrow Benson D , Cripe LH . Corticosteroid treatment retards development of ventricular dysfunction in Duchenne muscular dystrophy. Neuromuscular Disorders. (2008) ;18: :365–70. |
[10] | Silversides CK , Webb GD , Harris VA , Biggar DW . Effects ofdeflazacorton left ventricular function in patients with Duchenne muscular dystrophy. American Journal of Cardiology. (2003) ;91: :769–72. |
[11] | Ashwath ML , Jacobs IB , Crowe CA , Ashwath RC , Super DM , Bahler RC . Left ventricular dysfunction in Duchenne muscular dystrophy and genotype. American Journal of Cardiology. (2014) ;114: :284–9. |
[12] | Spurney C , Shimizu R , Morgenroth LP , Kolski H , Gordish-Dressman H , Clemens PR , Investigators the C Cooperative international neuromuscular research group duchenne natural history study demonstrates insufficient diagnosis and treatment of cardiomyopathy in duchenne muscular dystrophy. Muscle Nerve. (2014) ;50: :250–6. |
[13] | Kaspar RW , Hugh DA , Ray CW , Alvarez EC , Kissel TJ , Pestronk A , Weiss RB , Flanigan MK , Mendell JR , Montanaro F . Analysis of Dystrophin Deletion Mutations Predicts Age of Cardiomyopathy Onset in Becker Muscular Dystrophy. Circ Cardiovasc Genet. (2009) ;2: :544–51. |
[14] | Jefferies JL , Eidem BW , Belmont JW , Craigen WJ , Ware SM , Fernbach SD , Neish SR , Smith EO , Towbin JA . Genetic predictors and remodeling of dilated cardiomyopathy in muscular dystrophy. Circulation. (2005) ;112: :2799–804. |
[15] | Yamamoto T , Awano H , Zhang Z , Sakuma M , Kitaaki S , Matsumoto M , Nagai M , Sato I , Imanishi T , Hayashi N , Matsuo M , Iijima K , Saegusa J . Cardiac Dysfunction in Duchenne Muscular Dystrophy Is Less Frequent in Patients With Mutations in the Dystrophin Dp116 Coding Region Than in Other Regions. Circ Genom Precis Med. (2018) ;11: :e001782. |
[16] | Reza N , Owens AT . Decoding Dysfunction in Duchenne Muscular Dystrophy Cardiomyopathy. Circ Genom Precis Med. (2018) ;11: :e002051. |
[17] | Bello L , Pegoraro E . Genetic diagnosis as a tool for personalized treatment of Duchenne muscular dystrophy. Acta Myol. (2016) ;35: :122–7. |
[18] | Bello L , Morgenroth LP , Gordish-Dressman H , Hoffman EP , McDonald CM , Cirak S . DMD genotypes and loss of ambulation in the CINRG Duchenne Natural History Study. Neurology. (2016) ;87: :401 LP–409. |
[19] | Wang RT , Barthelemy F , Martin AS , Douine ED , Eskin A , Lucas A , Lavigne J , Peay H , Khanlou N , Sweeney L , Cantor RM , Miceli MC , Nelson SF . DMD genotype correlations from the Duchenne Registry: Endogenous exon skipping is a factor in prolonged ambulation for individuals with a defined mutation subtype. Hum Mutat. (2018) ;39: :1193–202. |
[20] | van den Bergen JC , Ginjaar HB , Niks EH , Aartsma-Rus A , Verschuuren JJGM . Prolonged ambulation in duchenne patients with a mutation amenable to exon 44 skipping. J Neuromuscul Dis. (2014) ;1: :91–4. |
[21] | Sabbatini D , Fusto A , Vianello S , Villa M , Janik J , D’Angelo G , Diella E , Magri F , Comi GP , Panicucci C , Bruno C , D’Amico A , Bertini E , Astrea G , Battini R , Politano L , Masson R , Baranello G , Previtali SC , Messina S , Vita G , Berardinelli A , Mongini T , Pini A , Pane M , Mercuri E , Hoffman EP , Morgenroth L , Gordish-Dressman H , Duong T , McDonald CM , Bello L , Pegoraro E Genetic modifiers of upper limb function in Duchenne muscular dystrophy. J Neurol [Internet]. 2022;Available from: https://doi.org/10.1007/s00415-022-11133-8 |
[22] | Barp A , Bello L , Politano L , Melacini P , Calore C , Polo A , Vianello S , Sorarú G , Semplicini C , Pantic B , Taglia A , Picillo E , Magri F , Gorni K , Messina S , Vita GL , Vita G , Comi GP , Ermani M , Calvo V , Angelini C , Hoffman EP , Pegoraro E . Genetic modifiers of duchenne muscular dystrophy and dilated cardiomyopathy. PLoS One. (2015) ;10: :1–14. |
[23] | Pegoraro E , Hoffman EP , Piva L , Gavassini BF , Cagnin S , Ermani M , Bello L , Soraru G , Pacchioni B , Bonifati MD , Lanfranchi G , Angelini C , Kesari A , Lee I , Gordish-Dressman H , Devaney JM , McDonald CM . SPP1 genotype is a determinant of disease severity in Duchenne muscular dystrophy. Neurology. (2011) ;76: :219 LP–226. |
[24] | Flanigan KM , Ceco E , Lamar KM , Kaminoh Y , Dunn DM , Mendell JR , King WM , Pestronk A , Florence JM , Mathews KD , Finkel RS , Swoboda KJ , Gappmaier E , Howard MT , Day JW , McDonald C , McNally EM , Weiss RB . LTBP4 genotype predicts age of ambulatory loss in duchenne muscular dystrophy. Ann Neurol. (2013) ;73: :481–8. |
[25] | Bello L , Flanigan KM , Weiss R , United Dystrophinopathy Project , Spitali P , Aartsma-Rus A , Muntoni F , Mercuri E , Tuffery-Giraud S , Claustres M , Straub V , Lochmuller H , Barp A , Vianello S , Pegoraro E , Punetha J , Gordish-Dressman H , Giri M , McDonald CM , Hoffman EP , Cooperative International Neuromuscular Research Group. Association Study of Exon Variants in the NF-κB and TGFβ Pathways Identifies CD40 as a Modifier of Duchenne Muscular Dystrophy. Am J Hum Genet. (2016) ;99: :1163–71. |
[26] | McDonald CM , Henricson EK , Abresch RT , Han JJ , Escolar DM , Florence JM , Duong T , Arrieta A , Clemens PR , Hoffman EP , Cnaan A , Investigators the C . The cooperative international neuromuscular research group duchenne natural history study—a longitudinal investigation in the era of glucocorticoid therapy: Design of protocol and the methods used. Muscle Nerve [Internet]. (2013) ;48: :32–54.Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/mus.23807 |
[27] | Schiller NB , Shah PM , Crawford M , DeMaria A , Devereux R , Feigenbaum H , Gutgesell H , Reichek N , Sahn D , Schnittger I , Silverman NH , Tajik AJ . Recommendations for Quantitation of the Left Ventricle by Two-Dimensional Echocardiography. Journal of the American Society of Echocardiography. (1989) ;2: :358–67. |
[28] | Brogna C , Coratti G , Pane M , Ricotti V , Messina S , D’Amico A , Bruno C , Vita G , Berardinelli A , Mazzone E , Magri F , Ricci F , Mongini T , Battini R , Bello L , Pegoraro E , Baranello G , Previtali SC , Politano L , Comi GP , Sansone VA , Donati A , Bertini E , Muntoni F , Goemans N , Mercuri E . Long-term natural history data in Duchenne muscular dystrophy ambulant patients with mutations amenable to skip exons 44, 45, 51 and 53. PLoS One. (2019) ;14: :e0218683. |
[29] | Doorenweerd N , Mahfouz A , van Putten M , Kaliyaperumal R , T’ Hoen PAC , Hendriksen JGM , Aartsma-Rus AM , Verschuuren JJGM , Niks EH , Reinders MJT , Kan HE , Lelieveldt BPF . Timing and localization of human dystrophin isoform expression provide insights into the cognitive phenotype of Duchenne muscular dystrophy. Sci Rep. (2017) ;7: :12575. |
[30] | Bello L , D’Angelo G , Villa M , Fusto A , Vianello S , Merlo B , Sabbatini D , Barp A , Gandossini S , Magri F , Comi GP , Pedemonte M , Tacchetti P , Lanzillotta V , Trucco F , D’Amico A , Bertini E , Astrea G , Politano L , Masson R , Baranello G , Albamonte E , De Mattia E , Rao F , Sansone VA , Previtali S , Messina S , Vita GL , Berardinelli A , Mongini T , Pini A , Pane M , Mercuri E , Vianello A , Bruno C , Hoffman EP , Morgenroth L , Gordish-Dressman H , McDonald CM , Investigators C-D , Pegoraro E Genetic modifiers of respiratory function in Duchenne muscular dystrophy. Ann Clin Transl Neurol. 2020;n/a. |
[31] | Aartsma-Rus A , Van Deutekom JCT , Fokkema IF , Van Ommen GJB , Den Dunnen JT . Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve. (2006) ;34: :135–44. |
[32] | Romfh A , McNally EM . Cardiac Assessment in Duchenne and Becker Muscular Dystrophies. Curr Heart Fail Rep. (2010) ;7: :212–8. |
[33] | Wang M , Birnkrant DJ , Super DM , Jacobs IB , Bahler RC . Progressive left ventricular dysfunction and long-term outcomes in patients with Duchenne muscular dystrophy receiving cardiopulmonary therapies. Open Heart. (2018) ;5: :e000783–e000783. |
[34] | Barber BJ , Andrews JG , Lu Z , West NA , Meaney FJ , Price ET , Gray A , Sheehan DW , Pandya S , Yang M , Cunniff C . Oral Corticosteroids and Onset of Cardiomyopathy in Duchenne Muscular Dystrophy. J Pediatr. (1084) ;163: :1080–1084.e1. |
[35] | Bauer R , MacGowan GA , Blain A , Bushby K , Straub V . Steroid treatment causes deterioration of myocardial function in the δ-sarcoglycan-deficient mouse model for dilated cardiomyopathy. Cardiovasc Res. (2008) ;79: :652–61. |
[36] | Sali A , Guerron AD , Gordish-Dressman H , Spurney CF , Iantorno M , Hoffman EP , Nagaraju K . Glucocorticoid-Treated Mice Are an Inappropriate Positive Control for Long-Term Preclinical Studies in the mdx Mouse. PLoS One. (2012) ;7: :e34204. |
[37] | Heier CR , Yu Q , Fiorillo AA , Tully CB , Tucker A , Mazala DA , Uaesoontrachoon K , Srinivassane S , Damsker JM , Hoffman EP , Nagaraju K , Spurney CF . Vamorolone targets dual nuclear receptors to treat inflammation and dystrophic cardiomyopathy. Life Sci Alliance. (2019) ;2: :e201800186. |
[38] | Bello L , Gordish-Dressman H , Morgenroth LP , Henricson EK , Duong T , Hoffman EP , Cnaan A , McDonald CM . Prednisone/prednisolone and deflazacort regimens in the CINRG Duchenne Natural History Study. Neurology. (2015) ;85: :1048 LP–1055. |
[39] | Crow RA , Hart KA , McDermott MP , Tawil R , Martens WB , Herr BE , McColl E , Wilkinson J , Kirschner J , King WM , Eagle M , Brown MW , Hirtz D , Lochmuller H , Straub V , Ciafaloni E , Shieh PB , Spinty S , Childs A-M , Manzur AY , Morandi L , Butterfield RJ , Horrocks I , Roper H , Flanigan KM , Kuntz NL , Mah JK , Morrison L , Darras BT , von der Hagen M , Schara U , Wilichowski E , Mongini T , McDonald CM , Vita G , Barohn RJ , Finkel RS , Wicklund M , McMillan HJ , Hughes I , Pegoraro E , Bryan Burnette W , Howard JF , Thangarajh M , Campbell C , Griggs RC , Bushby K , Guglieri M . A checklist for clinical trials in rare disease: Obstacles and anticipatory actions—lessons learned from the FOR-DMD trial. Trials. (2018) ;19: :291. |
[40] | Guglieri M , Bushby K , McDermott MP , Hart KA , Tawil R , Martens WB , Herr BE , McColl E , Speed C , Wilkinson J , Kirschner J , King WM , Eagle M , Brown MW , Willis T , Griggs RC , Straub V , Van Ruiten H , Childs AM , Ciafaloni E , Shieh PB , Spinty S , Maggi L , Baranello G , Butterfield RJ , Horrocks IA , Roper H , Alhaswani Z , Flanigan KM , Kuntz NL , Manzur A , Darras BT , Kang PB , Morrison L , Krzesniak-Swinarska M , Mah JK , Mongini TE , Ricci F , Von Der Hagen M , Finkel RS , O’Reardon K , Wicklund M , Kumar A , McDonald CM , Han JJ , Joyce N , Henricson EK , Schara-Schmidt U , Gangfuss A , Wilichowski E , Barohn RJ , Statland JM , Campbell C , Vita G , Vita GL , Howard JF , Hughes I , McMillan HJ , Pegoraro E , Bello L , Burnette WB , Thangarajh M , Chang T . Effect of Different Corticosteroid Dosing Regimens on Clinical Outcomes in Boys with Duchenne Muscular Dystrophy: A Randomized Clinical Trial. JAMA. (2022) ;327: :1456–68. |
[41] | Dwianingsih EK , Malueka RG , Nishida A , Itoh K , Lee T , Yagi M , Iijima K , Takeshima Y , Matsuo M . A novel splicing silencer generated by DMD exon 45 deletion junction could explain upstream exon 44 skipping that modifies dystrophinopathy. J Hum Genet. (2014) ;59: :423–9. |
[42] | Barnabei MS , Sjaastad F V , Townsend DW , Bedada FB , Metzger JM . Severe dystrophic cardiomyopathy caused by the enteroviral protease 2A-mediated C-terminal dystrophin cleavage fragment. Sci Transl Med. (2015) ;7: :294ra106–294ra106. |
[43] | Bello L , Kesari A , Gordish-Dressman H , Cnaan A , Morgenroth LP , Punetha J , Duong T , Henricson EK , Pegoraro E , McDonald CM , Hoffman EP , Investigators on behalf of the CINRG Genetic modifiers of ambulation in the cooperative international Neuromuscular research group Duchenne natural history study. Ann Neurol. (2015) ;77: :684–96. |
[44] | Dobaczewski M , Chen W , Frangogiannis NG . Transforming growth factor (TGF)-β signaling in cardiac remodeling. J Mol Cell Cardiol. (2011) ;51: :600–6. |
[45] | Duboc D , Meune C , Pierre B , Wahbi K , Eymard B , Toutain A , Berard C , Vaksmann G , Weber S , Bècane HM . Perindopril preventive treatment on mortality in Duchenne muscular dystrophy: 10 years’ follow-up. Am Heart J. (2007) ;154: :596–602. |
[46] | McNally EM , Kaltman JR , Woodrow Benson D , Canter CE , Cripe LH , Duan D , Finder JD , Hoffman EP , Judge DP , Kertesz N , Kinnett K , Kirsch R , Metzger JM , Pearson GD , Rafael-Fortney JA , Raman S V , Spurney CF , Targum SL , Wagner KR , Markham LW . Contemporary cardiac issues in Duchenne muscular dystrophy. Circulation. (2015) ;131: :1590–8. |
[47] | Tandon A , Villa CR , Hor KN , Jefferies JL , Gao Z , Towbin JA , Wong BL , Mazur W , Fleck RJ , Sticka JJ , Benson DW , Taylor MD . Myocardial fibrosis burden predicts left ventricular ejection fraction and is associated with age and steroid treatment duration in duchenne muscular dystrophy. J Am Heart Assoc [Internet]. 2015 [cited 2023 Aug 21];4. Available from: https://www.ahajournals.org/doi/abs/10.1161/JAHA.114.001338 |


