
Phenotype-Genotype Correlation of a Cohort of Patients with Congenital Myopathy: A Single Centre Experience from India
Abstract
Background:
Congenital myopathies (CMs) are a diverse group of inherited muscle disorders with broad genotypic and phenotypic heterogeneity. While the literature on CM is available from European countries, comprehensive data from the Indian subcontinent is lacking.
Objectives:
This study aims to describe the clinical and histopathological characteristics of a cohort of genetically confirmed CMs from India and attempts to do phenotype-genotype correlation.
Methods:
A retrospective chart review of genetically confirmed CMs was evaluated between January 2016 and December 2020 at the neuromuscular clinic. The clinical, genetic, and follow-up data were recorded in a pre-structured proforma as per the medical records, and the data was analyzed.
Results:
A total of 31(M: F = 14 : 17) unrelated patients were included. The median age at onset and duration of illness are 2.0(IQR:1–8) years and 6.0(IQR:3–10) years respectively. Clinical features observed were proximodistal weakness (54.8%), facial weakness (64.5%), and myopathic facies (54.8%), followed by ptosis (33.3%), and ophthalmoplegia (19.4%). Muscle histopathology was available in 38.7% of patients, and centronuclear myopathy was the most common histopathology finding. The pathogenic genetic variants were identified in RYR1 (29.0%), DNM2 (19.4%), SELENON (12.9%), KBTBD13 (9.7%), NEB (6.5%), and MYPN (6.5%) genes. Novel mutations were observed in 30.3% of the cohort. Follow-up details were available in 77.4% of children, and the median duration of follow-up and age at last follow-up was 4.5 (Range 0.5–11) years and 13 (Range 3–35) years, respectively. The majority were ambulant with minimal assistance at the last follow-up. Mortality was noted in 8.3% due to respiratory failure in Centronuclear myopathy 1 and congenital myopathy 3 with rigid spines (SELENON).
Conclusion:
This study highlights the various phenotypes and patterns of genetic mutations in a cohort of pediatric patients with congenital myopathy from India. Centronuclear myopathy was the most common histological classification and the mutations in RYR1 followed by DNM2 gene were the common pathogenic variants identified. The majority were independent in their activities of daily living during the last follow-up, highlighting the fact that the disease has slow progression irrespective of the genotype.
INTRODUCTION
Congenital myopathies (CM) are clinically and genetically heterogeneous groups of inherited muscle disorders characterized by distinct histopathological features and, in general, having a relatively stable or slowly progressive clinical course [1, 2]. Though the exact prevalence of CMs is not known, the recent systematic review reports the pooled prevalence of CM in children to be 2.73 (95% CI, 1.34 – 4.12) per 1,00,000 [3, 4]. Since the initial description of CM in 1956 by Shy and Magee, the diagnosis was largely based on the muscle biopsy findings through which they were classified into core myopathies, nemaline, centronuclear, myosin storage, and congenital fiber type disproportion myopathy [5–7].
With recent advances in gene panel testing and next-generation sequencing platforms, the diagnostic modality of choice is drifting away from muscle biopsy, which was standard clinical practice till a decade back [8]. Though a large number of genes are being described, a unique challenge in CMs is the wide heterogeneity between the clinical, histopathological, and genetic variants, even among children with features of myopathy belonging to the same family. This demands analysis of CMs from a comprehensively maintained database, which is available from European registries, however was limited from the Indian subcontinent [9–11]. This is all the more pertinent as the Indian subcontinent has a more ethnically and genetically diverse gene pool, which, in general, can have wide heterogeneity [3–6].
With the availability of Next Generation Sequencing (NGS) in our country since the last decade, increasing accessibility across the income range, availability of neuromuscular specialists, dedicated research centers, continuation of symptomatic care for floppy neonates, more and more children with CM are able to get a genetic diagnosis. The correlation of this genotypic information with the clinical and histological data provides crucial information for patient care, prognostication, and genetic counseling, and it would be the first step in exploring newer arenas for the treatment of CM. In India, with the advent of genetic testing, the knowledge about CMs’ clinical and genetic spectrum has been expanding. In this background, we undertook the present study to assess the clinical profile, histopathology, and mutational analysis in a cohort of pediatric patients with genetically confirmed CM and to determine their genotype-phenotype correlation.
MATERIALS AND METHODS
Study design
This is a retrospective study done from a quaternary care center for neurological disorders in South India (National Institute of Mental Health and Neurosciences, Bengaluru, India). A detailed chart review was done to identify and include the patients who (i) attended the neuromuscular clinic in the Neurology department between January 2016 and December 2020 (ii) were genetically confirmed to have congenital myopathy as per the “Gene table of monogenic neuromuscular disorders” guidelines [8] and (iii) were under the primary care of the authors. Patients were excluded from the analysis if (i) the diagnosis of CM was based on muscle biopsy, and genetic analysis was not available, and (ii) if the variants detected were benign or likely benign as per American College of Medical Genetics and Genomics (ACMG) guidelines. This study was approved by the Institute Ethics Committee (NIMHANS/IEC/2020-21). A prior informed consent was obtained from the participants at the time they underwent genetic testing. Written informed consent was obtained from parents/guardians for unmasking of the faces.
Patient cohort
Clinical data collection
The following data was recorded in a pre-structured proforma in an electronic database. Clinical details from their first visit to the hospital till their last clinical follow-up, either physical or tele-consultation, were noted. Clinical parameters included the demographic details, age at onset of symptoms, age at presentation, pattern of weakness-proximal/ distal/ axial/ combined, ocular-facial-bulbar, respiratory involvement, presence of ptosis, ophthalmoplegia, contractures, cardiac abnormalities, ambulatory status, and family history were noted. Creatine kinase (CK) levels and details of muscle biopsy were recorded wherever available. Follow-up details, including duration of illness, age at last follow-up, and clinical status, were recorded.
Genetic analysis methodology
All the study subjects who underwent genetic testing on a clinical basis were enrolled in the study at our center. Genomic DNA was extracted using standard procedures from peripheral blood samples. The libraries were sequenced as paired-end reads to mean > 80-100X coverage on Illumina sequencing platform (Illumina, CA). The sequences obtained were aligned to the human reference genome (GRCh37/hg19) using the BWA program, and gene annotation of the variants was performed using the VEP program [12–15]. The variant annotation was done using published literature and the following databases: ClinVar, Online Mendelian Inheritance in Man (OMIM), Genome-Wide Association Study (GWAS), Human Gene Mutation Database (HGMD), and SwissVar. Variants were classified according to the principles outlined in the American College of Medical Genetics and Genomics (ACMG) and Association for Molecular Pathology standards for interpretation of sequence variants. The clinically relevant variants were considered known if reported either in literature, ClinVar, or HGMD. Benign and likely benign variants were excluded from the study.
Statistical analysis
Data were analyzed using Statistical Package for Social Sciences (SPSS) software version 22.0 (Chicago, Illinois) using descriptive statistics for continuous variables like mean, median, interquartile range (IQR), and standard deviation for continuous variables. Frequency and percentages were used for categorical variables. For analysis of continuous variables among various subgroups, non-parametric tests were employed.
Results
During the study period, a total of 31 (M: F- 14 : 17) patients with genetically confirmed congenital myopathy (CM) fulfilling the eligibility criteria were recruited from 31 unrelated families. The majority of the patients in our cohort were from South India (70.9%), followed by East India (22.6%) and North India (3.1%). Referral diagnoses were myopathy (25.8%), congenital myopathy (22.5%), limb-girdle muscular dystrophy (16.1%), Duchenne/Becker’s muscular dystrophy (9.6%), spinal muscular atrophy (3.2%), cerebral palsy, developmental delay, hereditary motor neuropathy, myotonic dystrophy, neuropathy, congenital myasthenic syndrome and brachial plexopathy (3% each). The clinical profile, histopathology, mutational analysis, and follow-up details of patients with genetically confirmed CM are summarized in Table 1.
Table 1
Table summarizing the clinical profile, histopathology, mutational analysis and follow up of patients with congenital myopathy of our cohort
| P. number | Gender | Age at onset (Years) | Age at presentation (Years) | Pattern of weakness | Axial weakness | Facial | Ptosis | Myopathic facies | Extraocular movements | Ankle contractures | Consanguinity | CPK (U/L) | Histopathology | Loci of genetic mutation | Pattern of Inheritance | Age, at last, follow up (years) | Current clinical status |
| P- 1 | Male | 1 | 0.7 | Proximal and distal | No | No | No | Yes | Restricted | No | No | 103 | NA | RYR1 | AR | 6 | Assisted walking |
| P- 2 | Male | 2 | 9 | Proximal and distal | No | Yes | Yes | Yes | Restricted | No | No | 52 | Centronuclear myopathy | RYR1 | AR | 10 | Assisted walking |
| P- 3 | Female | 1 | 16 | Proximal | No | No | Yes | Yes | Restricted | No | No | 125 | Centronuclear myopathy | RYR1 | AR | 22 | Assistance to get up from floor |
| P- 4 | Female | 10 | 13 | Proximal | No | No | No | No | Normal | No | Yes | 925 | NA | RYR1 | AR | 16 | Assistance to get up from floor |
| P- 5 | Male | 1 | 5 | Proximal | No | No | Yes | No | Normal | No | No | 126 | NA | RYR1 | AR | 8 | Assisted walking |
| P- 6 | Female | 1 | 7 | Proximal and distal | Yes | No | No | Yes | Normal | Yes | Yes | 132 | NA | RYR1 | AD | 11 | Assisted walking |
| P- 7 | Female | 2 | 16 | Proximal | No | Yes | No | No | Restricted | No | Yes | 45 | NA | RYR1 | AR | 19 | Assistance to get up from floor |
| P- 8 | Male | 1 | 6 | Proximal and distal | No | Yes | No | Yes | Normal | No | Yes | 132 | Centronuclear myopathy | RYR1 | AR | 12 | Assisted walking |
| P- 9 | Female | 2 | 17 | Proximal | No | Yes | No | Yes | Normal | No | No | 67 | NA | RYR1 | AD | 20 | Independent walking |
| P- 10 | Female | 2 | 12 | Proximal and distal | No | Yes | No | Yes | Normal | Yes | No | 36 | Centronuclear>Core> Nemaline rods | NEB | AR | 18 | Assisted walking |
| P- 11 | Female | 1 | 4 | Proximal and distal | No | Yes | No | Yes | Normal | Yes | No | 128 | NA | NEB | AR | 7 | Assisted walking |
| P- 12 | Male | 14 | 16 | Proximal | No | No | No | No | Normal | No | Yes | 2077 | NA | DNM2 | AD | NA | NA |
| P- 13 | Female | 2 | 18 | Proximal and distal | No | Yes | Yes | No | Restricted | Yes | No | 19 | Centronuclear myopathy | DNM2 | AD | 25 | Assisted walking |
| P- 14 | Female | 8 | 9 | Proximal and distal | No | Yes | Yes | Yes | Normal | Yes | No | 103 | Centronuclear myopathy | DNM2 | AD | 16 | Walks independently |
| P- 15 | Male | 34 | 34 | Proximal and distal | No | Yes | Yes | No | Normal | No | No | 49 | Centronuclear myopathy | DNM2 | AR | 35 | Walks independently |
| P- 16 | Female | 11 | 13 | Proximal | No | No | No | No | Normal | No | No | 372 | NA | DNM2 | AD | NA | NA |
| P- 17 | Male | 11 | 13 | Proximal | Yes | No | Yes | No | Normal | No | No | 9082 | Centronuclear myopathy | DNM2 | AD | 20 | Died at 20 years of age |
| P- 18 | Female | 2 | 8 | Proximal | No | Yes | No | Yes | Normal | No | No | 90 | NA | SELENON | AR | 13 | Died at 12 years due to respiratory failure |
| P- 19 | Male | 5 | 22 | Proximal and distal | No | Yes | Yes | Yes | Normal | Yes | No | 1516 | NA | SELENON | AR | NA | NA |
| P- 20 | Female | 8 | 15 | Proximal and distal | No | No | No | No | Normal | No | Yes | 50 | NA | SELENON | AR | NA | NA |
| P- 21 | Female | 1 | 8 | Proximal | NA | NA | Yes | Yes | Normal | Yes | No | 191 | NA | SELENON | AR | 12 | Need support for climbing stairs |
| P- 22 | Male | 15 | 28 | Proximal and distal | No | Yes | No | Yes | Normal | Yes | No | 110 | NA | MYPN | AR | NA | NA |
| P- 23 | Male | 13 | 23 | Proximal and distal | No | No | No | No | Normal | No | Yes | 33 | NA | MYPN | AR | 32 | Assisted walking |
| P- 24 | Female | 1.5 | 8 | Proximal | No | Yes | No | Yes | Normal | No | Yes | 226 | Nemaline Rods> Centronuclear myopathy> Central Cores | TPM3 | AR | 9 | Assisted walking |
| P- 25 | Female | 1 | 5 | Distal | No | Yes | No | Yes | Normal | No | No | 154 | Nemaline rod myopathy | TPM3 | AR | 13 | Needs assistance to get up from floor |
| P- 26 | Male | 1 | 6 | Proximal and distal | Yes | Yes | No | No | Normal | Yes | No | 49 | NA | TPM2 | AD | 9 | Assisted walking |
| P- 27 | Female | 2 | 20 | Proximal and distal | No | Yes | Yes | Yes | Normal | Yes | No | 140 | Central Core> centronuclear myopathy | MTM1 | AR | 25 | Walks independently |
| P- 28 | Male | 1 | 15 | Proximal and distal | No | Yes | Yes | No | Restricted | No | No | 41 | Central Core> centronuclear myopathy | KBTBD13 | AD | 7 | Walks independently |
| P- 29 | Female | 7 | 14 | Distal | No | Yes | No | No | Normal | No | No | 57 | NA | KBTBD13 | AD | NA | NA |
| P- 30 | Male | 1.5 | 10 | Distal | No | Yes | No | No | Normal | Yes | Yes | 193 | NA | KBTBD13 | AD | NA | NA |
| P- 31 | Male | 0.5 | 5 | Proximal and distal | No | Yes | No | Yes | Normal | No | No | 118 | NA | ACTA1 | AD | 3 | Assisted walking |
Abbreviations: AD- Autosomal Dominant; AR- Autosomal recessive; CPK – Creatine phosphokinase; DNM2 – Dynamin 2; KBTBD13 – Kelch repeat and BTB domain containing13; MTM1 – Myotubularin; MYPN – Myopalladin; NA – Not Available; NEB – Nebulin; RYR1 – Ryanodine receptor type 1; SELENON – Selenoprotein N; TPM2 – b-Tropomyosin; TPM3 – Tropomyosin 3;U/L – Units per liter.
Demographic and clinical profile:
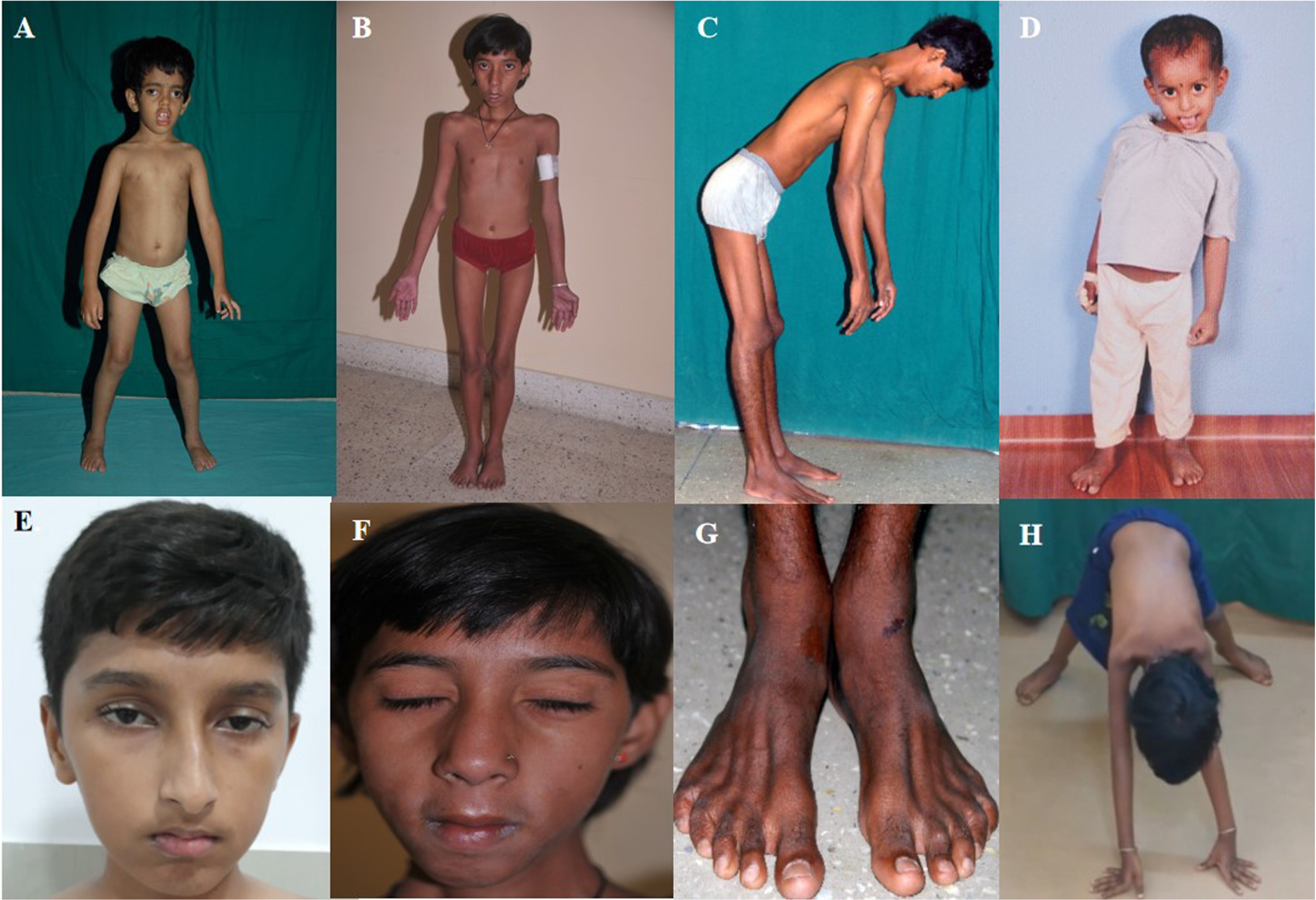
The median age at onset and duration of illness were 2.0 (IQR:1–8) years and 6.0 (IQR:3–10) years, respectively. The median age at presentation was 12.5 (IQR: 7–16) years. At the time of presentation to us, 54.8% had features of both proximal and distal muscle weakness, 35.4% had a limb-girdle pattern of proximal muscle weakness, and 10% had isolated distal muscle weakness. Around 10% of children had predominant weakness of axial musculature. Clinical features of motor predominant delay were present in 64.5% of children, 19.4% had a skeletal deformity in the form of scoliosis, 35.5% had ankle contractures, and only 3.2% of children had features of bulbar weakness. Facial weakness was observed in 64.5% and ptosis in 33.3% of children. Myopathic facies and features of external ophthalmoplegia were noted in 54.8% and 19.4% of children, respectively. Consanguinity was observed in 29% and siblings of the patients were affected in 10%. The mean CK level was 547.4±1650 U/L. Representative clinical images are shown in Fig. 1.
Fig. 1
Representative photographs of children with congenital myopathy showing features of – A. dental mal-occlusion and bilateral ptosis (P-14), B: myopathic facies as elongated face and sunken cheeks (P-18), C. rigid spine (P-19), D. skeletal deformity- scoliosis (P-8), E bilateral ptosis (P-28), F. bifacial weakness(P-18), G. wasting of extensor digitorum brevis in both legs (P-19), H. features suggesting proximal lower limb weakness on trying to get up from floor (P-20). (Consent obtained)
Characterization of histopathological profile:
Muscle biopsy was available in 12/31 (38.7%) patients and their median age at the time of biopsy was 12.5 (IQR 8.6–16.5) years. Biopsy was obtained either from the biceps or quadriceps on the non-dominant side. About eighty percent of the patients had central nuclei (muscle fibre with central nuclei constituting > 30% of muscle fibre population) [Fig. 2 A, B, C, H, I] and more than half of these patients exhibiting multiple internalised nuclei. Rod bodies were detected on Modified Gomori Trichrome (MGT) stain [Fig. 2 D, E] and features of core myopathy on oxidative enzyme staining (NADH, SDH) [Fig. 2 F, G] in three patients (25%) each, of which two of them had both rods and cores (NEB, TPM3) in their muscle tissue. The biopsy in all the twelve patients showed variation in size comprising of variable density and distribution of both hypertrophic and atrophic fibers, and two-thirds of the biopsies exhibited well-preserved fascicular architecture in paraffin and cryosections. The rest showed focal/ partial effacement in the form of adipocytic infiltration and focal/ early fibrosis. Two patients (DNM2, MTM1) also exhibited non specific neurogenic changes in muscle biopsy, who had clinical phenotype of myopathy. In one of the patient’s biopsies (NEB), a few fibers displayed a rimmed vacuole on the MGT stain. All RYR1 and Centronuclear myopathy 1 myopathy patients had centronuclear myopathy in muscle biopsy. However, rods were predominantly noticed in patients with TPM3 mutation and cores in MTM1 and KBTBD13 myopathies. The details of histopathology are mentioned in Table 2, and representative images are shown in Fig. 2.
Fig. 2
Microphotograph showing transverse section of P-2, centronuclear myopathy with many muscle fibres (>30%) possessing central nuclei (H & E x100) [A, B] and radial staining pattern (NADH × 200) [C]. Microphotograph showing transverse section in P-25, of Nemaline myopathy with many muscle fibres displaying Nemaline rods of variable density, configuration and distribution (MGT × 200) [D]displaying Nemaline rods (black arrow) of variable density, configuration and distribution (MGT × 400) [E]. Microphotograph showing transverse section of P-27, central core myopathy with many muscle fibres displaying central areas (Cores) of absence of oxidative activity (SDH × 200) [F] and (NADH × 200) [G]. Note the type I fibre predominance. Microphotograph showing transverse section in P-15 of centronuclear myopathy with many muscle fibres (>30%) possessing central nuclei. Also observed is the adipocytic infiltration and fibrosis (H & E ×100) [H]. Microphotograph showing transverse section in P-17 of centronuclear myopathy with many muscle fibres (>30%) possessing central nuclei (H & E × 200) [I].
![Microphotograph showing transverse section of P-2, centronuclear myopathy with many muscle fibres (>30%) possessing central nuclei (H & E x100) [A, B] and radial staining pattern (NADH × 200) [C]. Microphotograph showing transverse section in P-25, of Nemaline myopathy with many muscle fibres displaying Nemaline rods of variable density, configuration and distribution (MGT × 200) [D]displaying Nemaline rods (black arrow) of variable density, configuration and distribution (MGT × 400) [E]. Microphotograph showing transverse section of P-27, central core myopathy with many muscle fibres displaying central areas (Cores) of absence of oxidative activity (SDH × 200) [F] and (NADH × 200) [G]. Note the type I fibre predominance. Microphotograph showing transverse section in P-15 of centronuclear myopathy with many muscle fibres (>30%) possessing central nuclei. Also observed is the adipocytic infiltration and fibrosis (H & E ×100) [H]. Microphotograph showing transverse section in P-17 of centronuclear myopathy with many muscle fibres (>30%) possessing central nuclei (H & E × 200) [I].](https://ip.ios.semcs.net:443/media/jnd/2024/11-5/jnd-11-5-jnd230021/jnd-11-jnd230021-g002.jpg)
Table 2
Table describing the characteristic histopathological features in our cohort of patients with genetically confirmed congenital myopathy
| Patient number | Histology | Enzyme Histochemistry | Rods | Cores | Others | Final interpretation | Gene mutation | ||||||||
| Fascicular archicture | Hypertrophic fibres | Atrophic fibres | Location of nuclei | Single nuclei | Multiple scattered nuclei | Inflammation | MGT | NADH/SDH | ATPase | ||||||
| P-2 | Relatively PreservedEarly fibrosis+ | Present | Present | Central | Absent | Present | Absent | No new changes | Subsarcolemmal staining | Mosaic pattern | Absent | Absent | CNM | RYR1 | |
| P-3 | Preserved | Present | Present | Central | Present | Absent | Absent | No new changes | Absent | NA | Absent | Absent | CNM | RYR1 | |
| P-8 | Preserved | Present | Present | Central | Present | Absent | Absent | No new changes | No cores | Type 1 fibre hypoplasiaType 2 fibre hypertrophy | Absent | Absent | Coexisting CFTD | CNM | RYR1 |
| P-10 | Preserved | Present | Present | Central | Present (40%) | Absent | Absent | Rods present- subsarcolemmal region | Type 1 fibre predominateFew unstained areas suggestive of cores | Nil | Present | Present | One myofiber with vacuole rimmed by basophilic granular material | CNM>Cores> Rods | NEB |
| P-13 | Preserved | Present | Present | Central | Present | Present | Absent | Perinuclear staining | Type 1 fibre predominateType 2 fibre hypertrophy | Type 1 fibre predominateType 2 fibre hypertrophy | Absent | Absent | Coexisting CFTD | CNM | DNM2 |
| P-14 | Preserved | Present | Present | Central | Present | Absent | Absent | No inclusions | Type I fibre predominance | NA | Absent | Absent | CNM | DNM2 | |
| P-15 | Partly effaced with adipocytic infiltration and focal fibrosis | Present | Present | Central | Present | Present | Absent | No new changes | No ragged blue fibres | Type 1 predominanceMosaic pattern maintained | Absent | Absent | Coexisting neurogenic changes | CNM | DNM2 |
| P-17 | Preserved | Present | Present | Central | Absent | Present | Absent | NA | NA | NA | Absent | Absent | CNM | DNM2 | |
| P-24 | Partly effaced with adipocytic infiltration | Present | Present | Central | Present | Present | Absent | Numerous rod bodies in majority of fibres | Multiple cores notedType 1 fibrepredominace | Nil | Present | Present | Rods> CNM> Cores | TPM3 | |
| P-25 | Effaced | Present | Present | Central | Present | PresentOccasional fibre with peripheral ring of nuclear arrangement seen, (necklace fibres) | Absent | Rods present | NA | NA | Present | Absent | Occasional Necklace fibre | Rods>CNM | TPM3 |
| P-27 | Effaced | Present | Present | Central | Present (20%) | Present (minimal) | Absent | No inclusions | No cores | Nil | Absent | Present | Coexisting neurogenic changes | Cores>CNM | MTM1 |
| P-28 | Effaced | Present | Present | Central | Present (40%) | Present (minimal) | Absent | No inclusions | Single, multicores | Type 1 fibre predominance | Absent | Present | Cores>CNM | KBTBD13 | |
Abbreviations: CNM – centronuclear myopathy; CFTD- Congenital myopathy with fibre type disproportion; MGT – Modified Gomori Trichome; NA – Not Available; NADH/SDH – Reduced nicotinamide adenine dinucleotide/Succinate dehydrogenase.
Mutational analysis:
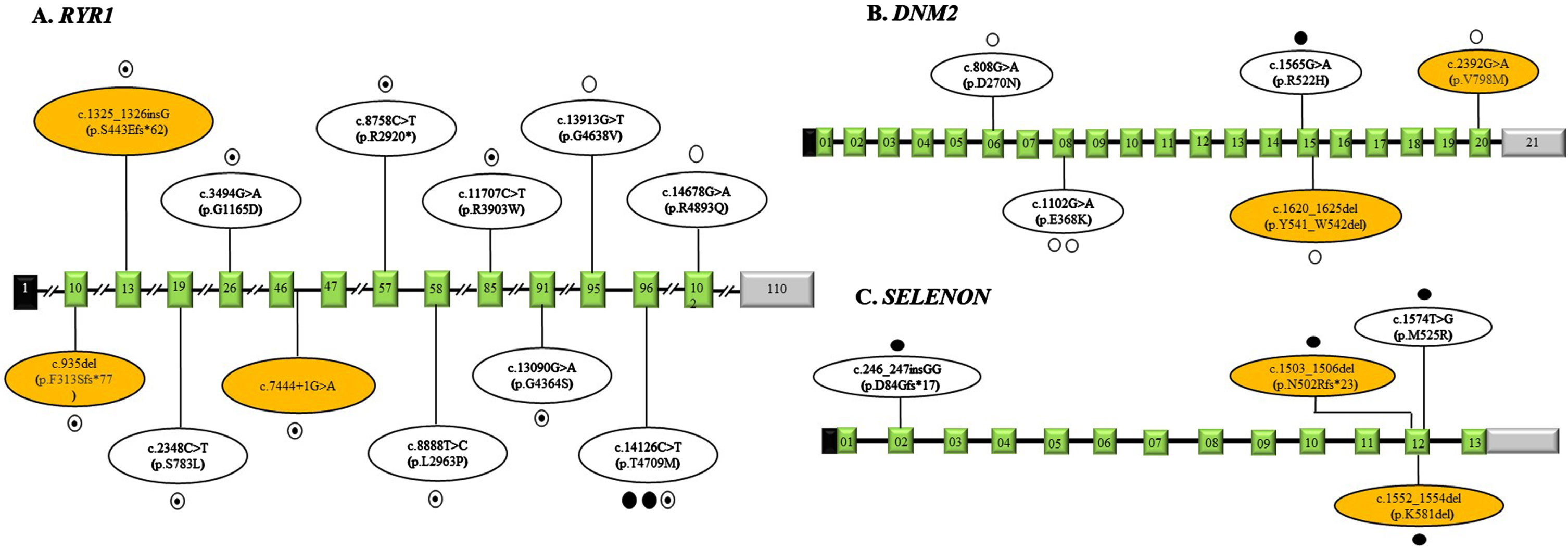
Among 31 patients, 41.9% of them had homozygous mutations, 19.4% had compound heterozygous, and the rest, 38.7%, had heterozygous mutations. A total of 33 different variants were observed in the CM-related genes. The majority of them among these 33 variants were missense variants (48.5%, 16/33), followed by frameshift (18.2%, 6/33), splice site (15.2%, 5/33), nonsense (9.1%, 3/33) and in-frame deletion (9.1%, 3/33). Among these 33 variants, 23 (69.7%) were previously reported and 10 (30.3%) were novel variants. Overall, 90.1% of variants (30/33) were classified as either pathogenic/ likely pathogenic, and the rest, 9.9% (3/23), were variants of uncertain significance (VUS). Among children with VUS, (i) P-17 with DNM2 mutation had muscle biopsy features consistent with CM supporting the diagnosis, (ii) P-31 had two variants in ACTA1 (Likely pathogenic) and MYH2 (VUS). Though the clinical phenotype was matching, due to lack of functional studies, pathogenicity among these two could not be ascertained, and (iii) P-12 had variant in DNM2 gene with matching clinical phenotype, as this was a novel variant, considered to be causative. A summary of the genetic variations observed in this study is shown in Table 3. The highest number of mutation variants were detected in RYR1 (36.4%), followed by DNM2 (15.2%), SELENON (12.1%), NEB (9.1%), KBTBD13 (6.1%), and MYPN (6.1%). Others were MYH2 , TPM3, ACTA1, MTM1, and TPM2, one (3%) each. The distribution of mutations identified in this study in the genes RYR1, DNM2, and SELENON are shown in Fig. 3. One variant in RYR1 (c.14126 C>T; p.Thr4709Met) was observed in three patients, homozygous state in two (P-7 and P-8) and heterozygous in one (P-2). In the subject P-31 heterozygous variants were detected in both MYH2 (c.5579 C>T; p.Thr1860Met; pathogenic) and ACTA1 (c.275_277del; p.Phe92del; likely pathogenic).
Table 3
Summary of the genetic variants observed in our congenital myopathy cohort
| Cases | Gene | Pattern of | #OMIM | Variant in HGVS | Location | Variant | Zygosity | Variant classification | Frequency in | HGMD ID | ClinVar | ||
| Inheritance | format | Consequence | as per ACMG / AMP Criteria (2015) | Population Databases | |||||||||
| Interpretation | Variation ID | Phenotype | |||||||||||
| P- 1 | RYR1 | AR | 255320 | NM_000540.3:c.936delCNP_000531.2:p.F313Sfs*77 | Exon 10 of 106 | Frameshift | Het | PathogenicPM2 PVS1 PP4 | gnomAD exomes-NovelgnomAD genomes-NovelInhouse database- Nove | NR | NR | NR | NR |
| NM_000540.3: c.11707 C>TNP_000531.2: p.R3903W | Exon 85 of 106 | Missense | Het | Likely PathogenicPM2 PM1 PP2 PM5 PP4 | gnomAD exomes-3individuals only in het stategnomAD genomes-NovelInhouse database- Novel | NR | NR | NR | NR | ||||
| P- 2 | RYR1 | AR | 255320 | NM_000540.3: c.1326dupGNP_000531.2: p.S443Efs*62 | Exon 13 of 106 | Frameshift | Het | PathogenicPM2 PVS1 PP4 | gnomAD exomes-3individuals only in het stategnomAD genomes-NovelInhouse database- Novel | NR | NR | NR | NR |
| NM_000540.3: c.14126 C>TNP_000531.2: p.T4709M | Exon 96 of 106 | Missense | Het | Likely Pathogenic PP2 PP3 PM5 PP4 BS1 BS2 | gnomAD exomes-36individuals only in het state gnomAD genomes-7 individuals only in het stateInhouse database-1 individuals only in homo state | CM073322 | Pathogenic/Likely Pathogenic | RCV000555087/RCV000763429 | RYR1-RD/CCO/CFTD/MMD | ||||
| P- 3 | RYR1 | AR | 255320 | NM_000540.3: c.2348 C>TNP_000531.2: p.S783L | Exon 19 of 106 | Missense | Het | Likely PathogenicPM2 PP2 PP3 PP4_Moderate | gnomAD exomes-3individuals only in het stategnomAD genomes-NovelInhouse database-Novel | NR | VUS | RCV000553747 | RYR1-RD |
| NM_000540.3: c.8888T>CNP_000531.2: p.L2963P | Exon 58 of 106 | Missense | Het | Likley PathogenicPM2 PP2 PP3 PS1 PP4 | gnomAD exomes-4individuals only in het state gnomAD genomes-NovelInhouse database-1 individuals only in het state | CM136222 | Pathogenic | RCV000796219 | RYR1-RD | ||||
| P- 4 | RYR1 | AR | 255320 | NM_000540.3: c.3494 G>ANP_000531.2: p.G1165D | Exon 26 of 106 | Missense | Het | Likely PathogenicPM1 PP2 PP3 PS1 PP4 BS2 | gnomAD exomes-10individuals only in het stategnomAD genomes-2 individuals only in het stateInhouse database-7 individuals only in het state | CM117526 | VUS | RCV001237337 | RYR1-RD |
| NM_000540.3: c.7444 + 1 G>A | Intron 46 of 105 | Splice Donor | Het | PathogenicPM2 PVS1 PP4 | gnomAD exomes-NovelgnomAD genomes-NovelInhouse database-1 individuals only in het state | NR | NR | NR | NR | ||||
| P- 5 | RYR1 | AR | 255320 | NM_000540.3: c.8758 C>TNP_000531.2: p.R2920* | Exon 57 of 106 | Stop Gained | Het | PathogenicPM2 PVS1 PP4 | gnomAD exomes-Novel gnomAD genomes-Novel Inhouse database- Novel | CM144265 | NR | NR | NR |
| NM_000540.3: c.13090 G>ANP_000531.2: p.G4364S | Exon 91 of 106 | Missense | Het | Likely PathogenicPM2 PM1 PP2 PP3 PM5 PP4_Moderate | gnomAD exomes-NovelgnomAD genomes-NovelInhouse database- Novel | NR | VUS | RCV000731208 | NP | ||||
| P- 6 | RYR1 | AD | 117000 | NM_000540.3: c.13913 G>TNP_000531.2: p.G4638V | Exon 95 of 106 | Missense | Het | PathogenicPM2 PM1 PP2 PP3 PM5 PP4_Strong | gnomAD exomes-Novel gnomAD genomes-Novel Inhouse database- Novel | NR | Likely pathogenic/Pathogenic | RCV001198930/ RCV000056188 | CFTD/ CCO |
| P- 7 P- 8 | RYR1 | AR | 255320 | NM_000540.3: c.14126 C>TNP_000531.2: p.T4709M | Exon 96 of 106 | Missense | Homo | Likely Pathogenic PP2 PP3 PM5 PP4_Strong BS1 BS2 | gnomAD exomes-36individuals only in het stategnomAD genomes-7 individuals only in het stateInhouse database-Novel | CM073322 | Pathogenic/Likely Pathogenic | RCV000555087/RCV000763429 | RYR1-RD/CCO/CFTD/MMD |
| P- 9 | RYR1 | AD | 117000 | NM_000540.3: c.14678 G>ANP_000531.2: p.R4893Q | Exon 102 of 106 | Missense | Het | Pathogenic PM2 PM1 PP2 PP3 PM5 PP4_Strong | gnomAD exomes-NovelgnomAD genomes-Novel Inhouse database-Novel | CM030713 | Pathogenic/Pathogenic | RCV001218792/RCV000056235 | RYR1-RD/ CCD |
| P- 10 | NEB | AR | 256030 | NM_001271208.2: c.1569 + 1 G>A | Intron 17 of 182 | Splice Donor | Homo | PathogenicPM2 PVS1 PP4 | gnomAD exomes-14 individuals only in het stategnomAD genomes-Novel Inhouse database- Novel | CS1413956 | Pathogenic | RCV000667222 | NEM2 |
| P- 11 | NEB | AR | 256030 | NM_001271208.2: c.10612 C>TNP_001258137.2:p.R3538* | Exon 73 of 183 | Stop Gained | Het | PathogenicPM2 PVS1 PP5 PP4 | gnomAD exomes-7 individuals only in het state gnomAD genomes-Novel Inhouse database- Novel | NR | Likely Pathogenic | RCV000986840 | NEM2 |
| NM_001271208.2:c.24407_24410dupTGTTNP_001258137.2:p.L8137Ffs*18 | Exon 173 of 183 | Frameshift | Het | PathogenicPM2 PVS1 PP5 PP4 | gnomAD exomes-9 individuals only in het stategnomAD genomes-NovelInhouse database-Novel | NR | Pathogenic/Likely Pathogenic | RCV000790962/RCV000781649 | NEM2/ NEM | ||||
| P- 12 | DNM2 | AD | 160150 | NM_001005360.3:c.808 G>ANP_001005360.1:p.D270N | Exon 6 of 21 | Missense | Het | VUSPM2 PP2 PP3 PP4 | gnomAD exomes-28 individuals only in het stategnomAD genomes- 2 individuals only in het stateInhouse database-Novel | NR | VUS | RCV001772330/RCV001122233 | NP/ ADCM |
| P- 13 P- 14 | DNM2 | AD | 160150 | NM_001005360.3:c.1102 G>ANP_001005360.1:p.E368K | Exon 8 of 21 | Missense | Het | PathogenicPM2 PM1 PP2 PP3 PS1 PP4 | gnomAD exomes-NovelgnomAD genomes-Novel Inhouse database- Novel | CM053834 | Pathogenic/Pathogenic | RCV000554046/RCV000145898 | CNM/ CMTDIB |
| P- 15 | DNM2 | AR | 160150 | NM_001005360.3:c.1565 G>ANP_001005360.1:p.R522H | Exon 15 of 21 | Missense | Het | Pathogenic PM2 PM1 PP2 PP3 PS1 PM5 PP4 | gnomAD exomes-NovelgnomAD genomes-Novel Inhouse database-Novel | CM102028 | Pathogenic/Pathogenic | RCV000552861/RCV000679888 | CMTDIB/ CNM1 |
| P- 16 | DNM2 | AD | 160150 | NM_001005360.3:c.1622_1627delACTGGTNP_001005360.1:p.Y541_W542del | Exon 15 of 21 | In-frame Deletion | Het | Likely pathogenic PM2 PM4 PP3 PP4 | gnomAD exomes-Novel gnomAD genomes-Novel Inhouse database- novel | NR | NR | NR | NR |
| P- 17 | DNM2 | AD | 160150 | NM_001005360.3:c.2392 G>ANP_001005360.1:p.V798M | Exon 20 of 21 | Missense | Het | VUS PP2 PP4_Strong | gnomAD exomes- 7 individuals only in het stategnomAD genomes-Novel Inhouse database- Novel | NR | NR | NR | NR |
| P- 18 | SELENON | AR | 602771 | NM_020451.3:c.249_250dupGGNP_065184.2:p.D84Gfs*17 | Exon 2 of 13 | Frameshift | Homo | Pathogenic PM2 PVS1 PP5 PP4 | gnomAD exomes- 13 individuals only in het stategnomAD genomes- 1 individual in het state Inhouse database- 1 individual in het state | NR | Pathogenic/Pathogenic | RCV000799500/RCV000627410 | RSMD1/ NP |
| P- 19 | SELENON | AR | 602771 | NM_020451.3: c.1505_1508delACCA NP_065184.2: p.N502Rfs*23 | Exon 12 of 13 | Frameshift | Homo | Likley Pathogenic PM2 PVS1 PP4 | gnomAD exomes-Novel gnomAD genomes-Novel Inhouse database- Novel | NR | NR | NR | NR |
| P- 20 | SELENON | AR | 602771 | NM_020451.3: c.1552_1554delAAG NP_065184.2: p.K518del | Exon 12 of 13 | In-frame Deletion | Homo | Likely Pathogenic PM2 PM4 PP3 PP4 | gnomAD exomes-Novel gnomAD genomes-Novel Inhouse database- Novel | NR | NR | NR | NR |
| P- 21 | SELENON | AR | 602771 | NM_020451.3:c.1574T>GNP_065184.2:p.M525R | Exon 12 of 13 | Missense | Homo | Likely PathogenicPP3 PM5 PP4_Strong | gnomAD exomes-14 individuals only in het state gnomAD genomes-1 individuals only in het state Inhouse database- Novel | NR | VUS | RCV001326195 | RSMD1 |
| P- 22 | MYPN | AR | 617336 | NM_032578.4:c.1973 + 1 G>C | Intron 10 of 19 | Splice donor | Homo | Pathogenic PM2 PVS1 PP4 | gnomAD exomes-NovelgnomAD genomes-Novel Inhouse database- 2 individuals only in het state | NR | NR | NR | NR |
| P- 23 | MYPN | AR | 617336 | NM_032578.4:c.1974-2A>C | Intron 10 of 19 | Splice Acceptor | Homo | Pathogenic PM2 PVS1_Strong PP4_Strong | gnomAD exomes-NovelgnomAD genomes-Novel Inhouse database- 4 individuals only in het state | NR | NR | NR | NR |
| P- 24 P- 25 | TPM3 | AR | 609284 | NM_152263.4:c.856T>ANP_689476.2:p.*286Kext*57 | Exon 10 of 10 | Stop Loss | Homo | Likely Pathogenic PM2 PM4 PP4_Strong | gnomAD exomes-NovelgnomAD genomes-NovelInhouse database- Novel | NR | NR | NR | NR |
| P- 26 | TPM2 | AD | 609285 | NM_003289.4:c.415_417delGAGNP_003280.2:p.E139del | Exon 4 of 9 | In-frame Deletion | Het | Likely PathogenicPM2 PM4 PP3 PP5 PM5 PP4_Moderate | gnomAD exomes-NovelgnomAD genomes-Novel Inhouse database- Novel | NR | Pathogenic | RCV000013281/RCV000500415 | CAPM2/NEM4 |
| P- 27 | MTM1 | AR | 310400 | NM_000252.3:c.688T>CNP_000243.1:p.W230R | Exon 9 of 15 | Missense | Homo | PathogenicPM2 PM1 PP2 PP3 PS1 PP4 | gnomAD exomes-NovelgnomAD genomes-Novel Inhouse database- Novel | CM050296 | Pathogenic/Likely pathogenic | RCV000146479 | CNMX |
| P- 28 | KBTBD13 | AD | 609273 | NM_001101362.3:c.477delCNP_001094832.1:p.V160* | Exon 1 of 1 | Frameshift | Het | Likely PathogenicPM2 PP4_Strong | gnomAD exomes- 1 individuals only in het state gnomAD genomes- 2 individuals only in het state Inhouse database- Novel | NR | VUS | RCV000532534 | NEM6 |
| P- 29 P- 30 | KBTBD13 | AD | 609273 | NM_001101362.3:c.677A>GNP_001094832.1:p.E226G | Exon 1 of 1 | Missense | Het | Likely PathogenicPM2 PP3 PP4_Strong | gnomAD exomes- 4 individuals only in het state gnomAD genomes- novel Inhouse database- 2 individuals only in het state | CM184977 | VUS | RCV001891752 | NEM6 |
| P- 31 | ACTA1 | AD | 161800 | NM_001100.4:c.275_277delTCTNP_001091.1:p.F92del | Exon 3 of 7 | In-frame Deletion | Het | Likely PathogenicPM2 PM1 PM4 PP3 PP5 PM5 PP4_Moderate | gnomAD exomes-NovelgnomAD genomes-NovelInhouse database- Novel | NR | Pathogenic | RCV000691025 | NEM3 |
| MYH2 | 605637 | NM_017534.6:c.5579 C>TNP_060004.3:p.T1860M | Exon 39 of 40 | Missense | Het | VUSPP2 PP3 PP4 | gnomAD exomes- 79 individuals only in het stategnomAD genomes-10 individuals only in het state Inhouse database- 1 individuals only in het state | NR | VUS | RCV000538870 | MYPOP | ||
Abbreviations: AR- Autosomal Recessive, AD- Autosomal Dominant,ACMG- American College of Medical Genetics; Ex-Exon; Hom- Homozygous; VUS- Variant of uncertain significance; RYR1-RD: RYR1-Related Disorders; NP- Not provided; NR-Not reported; Het- Heterozygous; CCO-Central core myopathy; CFTD-Congenital myopathy with fiber type disproportion; MMD – Multiminicore disease; In- Intron; SS – Splice site; NS- Nonsense; CCD – Central core myopathy; NEM – Nemaline myopathy; ADCM-Autosomal dominant centronuclear myopathy; CNM – Centronuclear myopathy; CMTDIB-Charcot-Marie-Tooth disease, dominant intermediate B;IFD-In-frame deletion; RSMD1 – Eichsfeld type congenital muscular dystrophy; CAPM2-Cap myopathy 2; CNMX- Severe X-linked myotubular myopathy; MYPOP – Myopathy, proximal, and ophthalmoplegia; OMIM – Online Mendelian Inheritance in Man.
Fig. 3
Schematic representation of the variations identified in our study in RYR1 gene (Panel A), DNM2 gene (Panel B) and SELENON gene (Panel C) with corresponding exons. The exons are represented as boxes with respective exonic numbers with non-coding regions shaded in black and grey at the ends. The novel variants are shaded in orange color. The filled dots represent homozygous variations, unfilled dots represent heterozygous variations, semi filled dot represent compound heterozygous variations and each dot represents number of variations.
Clinical follow-up details:
A total of 77.4% of children were available for follow-up. The median duration of follow-up was 4.5 (Range 0.5–11) years, and the median age at the last follow-up was 13 (Range 3–35) years. Among those on follow-up, 20.8% could walk independently without any support, 50% were walking with minimal assistance, and 20.8% required one-person support to get up from the floor and to climb stairs. Nonetheless, all were independent for activities of daily living. Two (8.3%) children expired (P-17, DNM2; P-18, SELENON) due to respiratory failure at the age of 20 years and 12 years respectively.
Phenotype – genotype correlation of selective CM
Overall, 61.3% of children had biallelic, and 38.7% had monoallelic patterns of gene mutation. The phenotypic details of some of the common genetic CMs observed in our cohort are described in Table 4.
Table 4
Table showing comparison of clinical features of common sub-types of congenital myopathy observed in our cohort
| Variables | RYR1 (n = 9) | DNM2 (n = 6) | SELENON (n = 4) | KBTBD13 (n = 3) |
| Age at onset (Years) (Median;IQR) | 1(IQR:1-2) | 11.0(IQR:8.8-13.3) | 3.5(IQR:1.8-5.8) | 1.5(IQR:1.3-4.3) |
| Duration (Years) (Median;IQR) | 4.0(IQR:3-14) | 2.0(IQR:2-11) | 7.5(IQR:7-9.8) | 8.5(IQR:7.3-9.3) |
| Age at presentation (Years) (Median;IQR) | 9.0(IQR:6-16) | 14.5(IQR:13-17.5) | 11.5(IQR:8-16.8) | 14.0(IQR:12-14.5) |
| M: F | 4 : 5 | 3 : 3 | 1 : 3 | 2 : 1 |
| Pattern of weakness | ||||
| Proximal and distal | 44.4% | 50% | 50% | 33.3% |
| Proximal | 55.6% | 50% | 50% | 0 |
| Distal | 0 | 0 | 0 | 66.4% |
| Axial weakness | 11.1% | 16.6% | 0 | 0 |
| Facial weakness | 44.4% | 50% | 50% | 100% |
| Ptosis | 33.3% | 66.6% | 50% | 33.3% |
| Myopathic facies | 66.6% | 16.6% | 75% | 0 |
| External Ophthalmoplegia | 44.4% | 16.6% | 0 | 33.3% |
| Contractures | 11.1% | 16.6% | 50% | 33.3% |
| Consanguinity | 44.4% | 16.6% | 25% | 33.3% |
| CK (U/L) (Mean±SD) | 189.6±277.9 | 1950.3±3581.2 | 461.7±705.3 | 97±83.5 |
| Histopathology | 33.3% (All CNM) | 66.6% (All CNM) | 0 | 33.3% (Core myopathy) |
Abbreviations: CNM- centronuclear myopathy; CK: Creatine phosphokinase; IQR- Inter quartile range; M- Male; n- number of patients; F- Female; U/L: SD- Standard Deviation; U/L-units per liter.
RYR1; Congenital myopathy 1A/congenital myopathy 1B [OMIM # 117000; 255320] (n = 9).
Among the children with RYR1 mutation, 89% had onset in the first two years of life, with a median age at presentation being 9 (IQR:6-16) years. About 44.4% of children had both proximal and distal weakness, and the remaining 55.6% had only proximal limb weakness in limb-girdle pattern. Nearly 66.6% of children had myopathic facies and external ophthalmoplegia was observed in 44.4%. Malignant hyperthermia and respiratory infections were not observed in our cohort. The mean CK was 189.6±277.9 U/L. Histopathology showed features of centronuclear myopathy in those who underwent muscle biopsy (33.3%). Nearly 77.7% of children had biallelic and 22.2% had monoallelic gene mutation. All the children were followed for a median duration of 4 (Range 1-7) years, with the median age at the last follow-up being 12 (Range 6-22) years. These children were ambulant, requiring minimal assistance to walk, and none of them were wheelchair-bound.
DNM2; Centronuclear myopathy 1 [OMIM# 160150] (n = 6).
Patients with Centronuclear myopathy 1 had a later age of presentation with a median age of onset of 14.5 (IQR:13–17.5) years. All except one (83.6%) had normal motor developmental milestones. Clinical phenotype was myopathy in all (100%) and the pattern of weakness was both proximal and distal in 50% of children and isolated limb girdle weakness in the remaining 50%. Ophthalmoparesis was seen in only 16.6% of children. Muscle biopsy was done in 66% and findings were suggestive of centronuclear myopathy. The mean CK was 1950.3±3581.2 U/L. All except one (83.6%) had autosomal dominant (AD) pattern of inheritance. One child died at the age of 20 years due to respiratory infection after seven years of follow-up, and the remaining were ambulant during the last follow-up. The median follow-up duration was 7 (Range 0.5–7) years, and the median age at the last follow-up was 23.0 (range 13–35) years.
SELENON; Congenital myopathy 3 with rigid spine [OMIM #602771] (n = 4).
The median age at onset of symptoms in children with SELENON mutation was 3.5 (IQR:1.8–5.8) years. The median age at presentation was 11.5 (IQR:8–16.8) years, with features of proximal and distal limb weakness in 50%, and the remaining 50% had only limb-girdle pattern of weakness, of which one (25%) had recurrent respiratory infections. The majority (75%) had myopathic facies. Scoliosis was present in two (50%) children, of which one had the feature of a rigid spine. The mean CK was 461.8±705.3 U/L. All children had AR pattern of inheritance. One child died at the age of 12 years due to recurrent chest infections and respiratory failure.
KBTBD13; Nemaline myopathy 6 [OMIM # 609273) (n = 3).
Three children with KBTBD13 mutation presented with motor predominant delay. AD inheritance pattern was observed in all. They had difficulty in getting up from the floor with the median age at onset of this symptom being 1.5 (IQR:1.3–4.3) years. The median age at presentation to us was 14 (IQR:12–14.25) years. On examination, features of proximal and distal limb weakness were present in all (100%) and with sluggish tendon reflexes. The mean CK was 97±83.5 U/L. At one year follow-up, the clinical status has remained the same.
Other Congenital Myopathies: (n = 9).
Two children with NEB(P-10,11) and TPM3(P-24,25) gene mutation each had presented with motor predominant delay in the first two years of life. Children with TPM3 mutation had myopathic faces with bifacial weakness and generalized hypotonia. Children with MYPN mutation (P-22,23) presented with features of limb-girdle weakness. A child with ACTA1 mutation (P-31) had manifested with bulbar weakness at onset associated with developmental delay. Antenatal history of decreased foetal movements was present in a child with TPM2 mutation (P-26). This child had recurrent respiratory infections and later on, developed limb-girdle weakness and had myopathic facial features (elongated facies, low set ears, high arch palate, and bifacial weakness) with exaggerated lumbar lordosis. An adolescent girl with MTM1 mutation (P-27), had presented with difficulty in running at 20 years of age, who at the time of birth had a history of hypophonia and bulbar weakness and had features of non-fatigable asymmetric ptosis and elbow contracture with proximal limb weakness. Most of these patients gradually gained motor milestones and were able to walk with or without assistance.
DISCUSSION
In the present study, we attempted to describe the clinical profile, histopathology, mutational analysis and follow up of a large pediatric cohort with genetically confirmed CM from a single neurology centre, at south India. As this is the first such study from Asian region, we have compared our results with European population. Largely, patients were from southern part followed by eastern part of the country. The predominant clinical manifestations observed were both proximal and distal muscle weakness, in the majority associated with motor-predominant delay followed by facial weakness, ophthalmoparesis, and skeletal abnormalities. In our cohort, centronuclear myopathy was the commonest histopathological subtype, and RYR1 gene-associated CM was the commonest genetic subtype, which was in coherence with the previously published literature [9, 16]. Most of the patients in our study had a slow progression with majority (around 90%) being ambulant with or without assistance at the last median follow-up of 4.5 years. It is interesting to note that only one-fifth of the children referred to us had an initial referral diagnosis of CM, highlighting the lack of awareness among clinicians regarding CM.
The median age at onset of symptoms was the second year of life in our cohort, unlike previous studies where onset in the neonatal/infantile period is common. Antenatal or neonatal onset ranged between 56% to 76% in prior studies, with nearly one-third requiring respiratory support [11, 16]. The later age at the onset of symptoms in our cohort could probably be due to institutional bias with the lack of neonatal care at our institute. The other plausible explanation could be the lesser proportion of patients with mutations in ACTA1 and MTM1 genes, which tend to have an earlier onset, and the majority die by the first year of life, as observed in a previous study [16]. Most of the children had delayed developmental milestones similar to the Denmark study, except in centronuclear myopathy 1, where the majority had normal milestones with later age of onset [10]. Proximal or proximo-distal weakness observed in most were similar to the observations reported in European studies [9, 11]. It is also worthwhile to note that two of three patients with KBTBD13 mutation had distal predominant weakness. Children with respiratory distress requiring ventilatory support were less commonly observed in our study, in contrast to the study from London, where nearly 25–30% of the children required assisted ventilation [9, 16]. This could be due to the difference in patient population, genetic spectrum, institutional bias, and non-availability of follow-up in some patients. Axial weakness was observed in children with mutations in RYR1, DNM2, and TPM2 genes. While facial weakness and contractures were non-specific and observed in all the genes, ptosis was pronounced in those with RYR1, DNM2 and SELENON gene mutations. In comparison to the other non-RYR1 subtypes of myopathies, consanguinity and external ophthalmoplegia were commonly observed in RYR1 myopathy. Similarly, features of bulbar palsy and siblings being affected are more often observed in non-RYR1 subtypes of CM. However, there were no significant differences in the clinical profile among RYR1 and non-RYR1 subtypes of congenital myopathies. The mean CK value was mildly elevated, up to threefold, with the highest in those with DNM2 mutations.
As we included only those with genetically confirmed CMs, histopathology was available in less than half of the cohort, with the commonest subtype observed being centronuclear myopathy. Nonetheless, core myopathies have been reported frequently in studies from UK and Spain [9, 11]. The genetic heterogeneity described in CMs was evident in our cohort, with mutations in both RYR1 and DNM2 resulting in the same histopathology, i.e., centronuclear myopathy. Nemaline myopathy was seen in those with TPM3 mutations. Core myopathy was noticed in patients with mutations in MTM1 and KBTBD13 genes. Interestingly, two of our patients also had dual pathology with the presence of both rods and cores, which were delineated clearly with genetic testing as nemaline myopathy 2 and congenital myopathy 4A/congenital myopathy 4B. Previously, such dual pathology has been reported in RYR1 myopathy [17]. At times, even genetically established myopathy can have non-specific histopathological observations, as evidenced by prior studies where these changes ranged from 3.8% to 22% [9, 11]. These variations can also be due to dynamic changes based on the site of the biopsy, and the findings can be absent if a biopsy is done very early in the course of the illness, highlighting the development of new histopathological changes with aging [18]. Owing to these variations, it is essential to have a more comprehensive approach for making the diagnosis of CM. The yield of genetic testing in histology-confirmed CMs ranged from 56% to 79% across studies from the UK, Spain, and Denmark [9–11]. The yield was reportedly higher in those with specific histopathology findings (presence of cores, nemaline rods, central nuclei, or fibre type disproportion).
RYR1 mutation was the most common subtype of CM in our cohort. Similar observations were noted in all previously reported large global cohorts both in children [10, 19] and adults [20]. The higher prevalence of Congenital myopathy 1A/congenital myopathy 1B (RYR1) in most of the studies could be due to complete sequencing of this large gene, unlike nebulin, which is often difficult to identify in conventional studies [9]. Congenital myopathy 1A/congenital myopathy 1B (RYR1) has been described as having both AD and AR patterns of inheritance and sometimes with sporadic mutations [21, 22]. Centronuclear myopathy 1 (DNM2) was the second most common subtype, similar to the observations of the Denmark study [10], and unlike the study from the United Kingdom, where none had DNM2 mutation [9]. Also, it is important to note that all centronuclear myopathy 1 (DNM2) children were sporadic, which strengthens the hypothesis of de novo mutations in these subgroups [23]. SELENON myopathy was the third most common subtype of CM, unlike the Spanish population where TTN myopathy was seen in higher proportion, while SELENON mutations were absent. Variation in the proportion of various genetic subtypes in the study population of our groups, including those from the United Kingdom and Spain, suggests geographic variability among different subtypes of CM, though RYR1 mutation remains the most prevalent globally.
The majority of our children had an AR pattern of inheritance. In Congenital myopathy 1A/congenital myopathy 1B (RYR1), both recessive and dominant patterns were observed, and there was not much difference between these two subtypes. However, the sample was too small to make any inference. However, severely affected phenotype was observed in recessive forms in previous studies [10]. AD pattern was observed mainly in DNM2 and KBTBD13 gene-associated myopathy. An Italian study also showed de-novo dominant pattern of inheritance in DNM2 myopathy [23]. Similar observations have been made in Nemaline myopathy 6 (KBTBD13). The type of gene mutations was varied and mainly had missense and frameshift mutations, and no single mutation hot spot was observed, similar to the results observed in the study from Denmark, United Kingdom [9, 10]. However, studies with larger samples of each genetic subtype are needed for any inference. Table 5 summarizes the previously published literature and our findings on genetically confirmed cases of congenital myopathy.
Table 5
A comparison table describing the summary of previously published literature on genetically confirmed cases of congenital myopathy and our study
| Parameters | Maggi et al., 2013 [9] | Colombo et al., 2015 [16] | Witting et al., 2017 [10] | Benito et al., 2021 [11] | Present study |
| Place | United Kingdom | London | Denmark | Spain | India |
| No of subjects | 66 | 125 | 82 | 104 | 31 |
| Male: Female | 37 : 29 | 61 : 64 | 41 : 41 | 57 : 47 | 14 : 17 |
| Age at onset (years) | 0.8 | Neonatal onset 76% | NA* | Neonatal onset-56% | 2 |
| Age at presentation (years) | 4.7 | Neonatal presentation-60% Infantile presentation 16% | 27.8 | 12.5 | 13 |
| Pattern of weakness | |||||
| Proximal limb weakness | 71.2% | NA | NA | 55% | 35.4% |
| Proximal and distal limb weakness | 6.1% | 40% | 54.8% | ||
| Distal predominant weakness | – | – | 10% | ||
| Axial predominant weakness | 19.7% | – | 10% | ||
| Ophthalmoplegia | 10.6% | 20.7% | 12.5% | 54.8% | |
| Facial involvement | NA | 64.6% | 66% | 64.5% | |
| Bulbar palsy | 36.4% | 46.4% | NA | 14.9% | 3.2% |
| Skeletal abnormalities | 13.6% | 40% | 25.6% | 42% | 19.3% |
| Histopathology | |||||
| Number of subjects | 54/66 | 104/125 | 46/82 | 95/104 | 12/31 |
| Core myopathy | 54% | 37.5% | 17% | 42% | 16.6% |
| Nemaline rod myopathy | 17% | 31.8% | 15% | 16% | 16.6% |
| Myotubular/centronuclear myopathy | 13% | 17.3% | 18% | 14% | 66.6% |
| Congenital fibre type disproportion | 4% | 4.8% | 33% | 3% | No |
| Isolated type 1 predominance | 11% | 4.8% | No | No | No |
| Mixed core–rod myopathy | 2% | No | No | No | No |
| Nonspecific myopathic changes | 11.1% | 3.8% | 17% | 22% | No |
| Gene mutations | |||||
| Number of subjects | 44/66 | 99/125 | 46/82 | 65/104 | 31/31 |
| RYR1 | 59% | 44.4% | 22.0% | 23.1% | 29% |
| ACTA1 | 16% | 17.2% | 4.8% | 1.9% | 3.2% |
| SELENON | 16% | 16.2% | 3.6% | – | 12.9% |
| MTM1 | 5% | 8.1% | 3.6% | 6.7% | 3.2% |
| NEB | 2% | 8.1% | 7.3% | 3.8% | 6.5% |
| TPM3 | 2% | 2.1% | 4.8% | 1.9% | 6.5% |
| DNM2 | – | 1% | 7.3% | 2.9% | 19.4% |
| TTN | – | – | 1.2% | 7.7% | – |
| TPM2 | – | 1% | Included in TPM3 | 1.9% | 3.2% |
| KBTBD13 | – | – | – | – | 9.7% |
| SELENON | – | – | – | 6.7% | – |
| MYH3 | – | – | – | 1% | – |
| MYPN | – | – | – | – | 6.5% |
| Follow up | |||||
| Number of subjects | 66/66 | 125/125 | 80/82 | NA | 24/31 |
| Median duration of follow up (years) | 5 | 10 | NA | 4.5 (IQR:3-6.5) | |
| Stable | 86% | – | 73.8% | 94% | |
| Deteriorated | 6% | – | 15% | ||
| Expired | 8% | 12% | – | 5.9% |
*NA- Not available.
Among children, who were available for follow-up, all continued to be independent for their daily activities, highlighting the slowly progressive nature of the CMs irrespective of subtype. Similar observations were noted in studies from Denmark and the United Kingdom [9, 10]. In contrast to this, a higher mortality rate was noted in a study from London, especially those with neonatal onset and specific subtypes like Congenital myopathy-2A/2B/2 C (ACTA1) and X-linked centronuclear myopathy (MTM1), probably due to severity of illness and associated respiratory dysfunction and infections. This suggests a hypothesis that those CMs with later onset tend to have slow progression.
The strengths of this study are (i) This is the first Indian study on phenotypic and genotypic characterization of genetically confirmed CM patients while also describing the follow-up and natural history, (ii) one of the large cohorts of various sub-types of genetically confirmed CM, reported from Asian region, and (iii) This study also emphasizes the utility of genetic testing in children with suspected congenital myopathy.
The study limitations include (i) inherent flaws of a retrospective study like lack of uniformity in clinical assessments, data collection and short follow up, (ii) detailed muscle charting and functional testing could not be done as majority were young, not co-operative, (iii) assessment of co-morbidities like cardiac involvement, respiratory function and cognitive levels in all the children were not available, (iv) current pediatric cohort cannot be considered as a reflection of the prevalence of CM in the country as majority of patients were from southern parts of the country which could be due to institutional bias,(v) long term follow up with inclusion of adults would have been ideal for better understanding of disease course, (vi) segregation analysis and functional validation of the variants were not done and (vii) lack of histopathological confirmation in few cases.
FUTURE DIRECTIONS
The current study provides in-depth insights into the genetic spectrum of CMs from the Indian subcontinent. The advances in genetic therapeutics seen particularly in the field of neuromuscular diseases provide ample signs that patients with CM may also be future candidates. Knowledge about the genetic spectrum and maintenance of CM registries from India will ensure trial readiness as and when novel gene therapy options become available. With a unique gene pool that is ethnically and genetically diverse, multicentric registries from across the country would be the way forward.
CONCLUSIONS
Our study gives an insight into the clinical presentations and genetic mutational patterns of congenital myopathy from Indian sub-continent. Majority had symptom onset by 2 years of age with centronuclear myopathy being the most common histological classification. Mutations in RYR1 followed by DNM2 genes were the leading pathogenic variants identified. Majority were independent for their activities of daily living during the last follow-up, highlighting the fact that the disease has slow progression irrespective of the genotype.
ACKNOWLEDGMENTS – FUNDING
Nil.
CONFLICT OF INTEREST
Nil.
REFERENCES
[1] | Bornemann A , Goebel HH . Congenital myopathies. Brain Pathol. (2001) ;11: :206–17. doi: 10.1111/j.1750-3639.2001.tb00393.x. |
[2] | North KN , Wang CH , Clarke N , Jungbluth H , Vainzof M , Dowling JJ , et al. International Standard of Care Committee for Congenital Myopathies. Approach to the diagnosis of congenital myopathies. NeuromusculDisord. (2014) ;24: (2):97–116. doi: 10.1016/j.nmd.2013.11.003. |
[3] | Huang K , Bi FF , Yang H . A Systematic Review and Meta-Analysis of the Prevalence of Congenital Myopathy. Front Neurol. (2021) ;12: :761636. doi: 10.3389/fneur.2021.761636. |
[4] | Tubridy N , Fontaine B , Eymard B . Congenital myopathies and congenital muscular dystrophies. CurrOpin Neurol. (2001) ;14: (5):575–82. doi: 10.1097/00019052-200110000-00005. |
[5] | Claeys KG . Congenital myopathies: an update. Dev Med Child Neurol. (2020) ;62: (3):297–302. doi: 10.1111/dmcn.14365. |
[6] | Magee KR , Shy GM . A new congenital non-progressive myopathy. Brain. (1956) ;79: :610–21. doi: 10.1093/brain/79.4.610. |
[7] | Cassandrini D , Trovato R , Rubegni A , Lenzi S , Fiorillo C , Baldacci J , et al. Italian Network on Congenital Myopathies. Congenital myopathies: clinical phenotypes and new diagnostic tools. Ital J Pediatr. (2017) ;43: (1):101. doi: 10.1186/s13052-017-0419-z. |
[8] | Bonne G , Rivier F , Hamroun D . The 2018 version of the gene table of monogenic neuromuscular disorders (nuclear genome). NeuromusculDisord. (2017) ;27: (12):1152–1183. doi: 10.1016/j.nmd.2017.10.005. |
[9] | Maggi L , Scoto M , Cirak S , Robb SA , Klein A , Lillis S , et al. Congenital myopathies–clinical features and frequency of individual subtypes diagnosed over a 5-year period in the United Kingdom. NeuromusculDisord. (2013) ;23: (3):195–205. doi: 10.1016/j.nmd.2013.01.004. |
[10] | Witting N , Werlauff U , Duno M , Vissing J . Phenotypes, genotypes, and prevalence of congenital myopathies older than 5 years in Denmark. Neurol Genet. (2017) ;3: (2):e140. doi: 10.1212/NXG.0000000000000140. |
[11] | Natera-de Benito D , Ortez C , Jou C , Jimenez-Mallebrera C , Codina A , Carrera-García L , et al. The Phenotype and Genotype of Congenital Myopathies Based on a Large Pediatric Cohort. Pediatr Neurol. (2021) ;115: :50–65. doi: 10.1016/j.pediatrneurol.2020.11.002. |
[12] | Li H , Durbin R . Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. (2010) ;26: (5):589–95. doi: 10.1093/bioinformatics/btp698. |
[13] | McKenna A , Hanna M , Banks E , Sivachenko A , Cibulskis K , Kernytsky A , et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. (2010) ;20: (9):1297–303. doi: 10.1101/gr.107524.110. |
[14] | McLaren W , Pritchard B , Rios D , Chen Y , Flicek P , Cunningham F . Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. (2010) ;26: (16):2069–70. doi: 10.1093/bioinformatics/btq330. |
[15] | Richards S , Aziz N , Bale S , Bick D , Das S , Gastier-Foster J , et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) ;17: (5):405–23.27. doi: 10.1038/gim.2015.30. |
[16] | Colombo I , Scoto M , Manzur AY , Robb SA , Maggi L , Gowda V , et al. Congenital myopathies: Natural history of a large pediatric cohort. Neurology. (2015) ;84: (1):28–35. doi: 10.1212/WNL.0000000000001110. |
[17] | Scacheri PC , Hoffffman EP , Fratkin JD , Semino-Mora C , Senchak A , Davis MR , et al. A novel ryanodine receptor gene mutation causing both cores and rods in congenital myopathy. Neurology. (2000) ;55: :1689–96. doi: 10.1212/wnl.55.11.1689. |
[18] | Rocha J , Taipa R , Melo Pires M , Oliveira J , Santos R , Santos M . Ryanodine myopathies without central cores–clinical, histopathologic, and genetic description of three cases. Pediatr Neurol. (2014) ;51: (2):275–8. doi: 10.1016/j.pediatrneurol.2014.04.024. |
[19] | Amburgey K , McNamara N , Bennett LR , McCormick ME , Acsadi G , Dowling JJ . Prevalence of congenital myopathies in a representative pediatric united states population. Ann Neurol. (2011) ;70: :662–665. doi: 10.1002/ana.22510. |
[20] | Nicolau S , Liewluck T , Tracy JA , Laughlin RS , Milone M . Congenital myopathies in the adult neuromuscular clinic: Diagnostic challenges and pitfalls. Neurol Genet. (2019) ;5: (4):e341. doi: 10.1212/NXG.0000000000000341. |
[21] | Amburgey K , Bailey A , Hwang JH , Tarnopolsky MA , Bonnemann CG , Medne L , et al. Genotype-phenotype correlations in recessive RYR1-related myopathies. Orphanet J Rare Dis.117. (2013) ;8: :117. doi: 10.1186/1750-1172-8-117. |
[22] | Todd JJ , Razaqyar MS , Witherspoon JW , Lawal TA , Mankodi A , Chrismer IC , et al. Novel variants in individuals with RYR1-related congenital myopathies: genetic, laboratory, and clinical findings. Front Neurol. (2018) ;9: :118. doi: 10.3389/fneur.2018.00118. |
[23] | Catteruccia M , Fattori F , Codemo V , Ruggiero L , Maggi L , Tasca G , et al. Centronuclear myopathy related to dynamin 2 mutations: clinical, morphological, muscle imaging and genetic features of an Italian cohort. NeuromusculDisord. (2013) ;23: (3):229–38. doi: 10.1016/j.nmd.2012.12.009. |
[24] | Sambuughin N , Yau KS , Olivé M , Duff RM , Bayarsaikhan M , Lu S , et al. Dominant mutations in KBTBD13, a member of the BTB/Kelch family, cause nemaline myopathy with cores. American Journal of Human Genetics. (2010) ;87: (6):842–847. doi: 10.1016/j.ajhg.2010.10.020. |


