
Brain Magnetic Resonance Imaging (MRI) in Spinal Muscular Atrophy: A Scoping Review
Abstract
Background:
5q Spinal Muscular Atrophy (SMA) is a prototypical lower motor neuron disorder. However, the characteristic early motor impairment raises the question on the scope of brain involvement with implications for further investigations on the brain as a potential therapeutic target.
Objective:
To review changes across the SMA clinical spectrum reported on brain magnetic resonance imaging (MRI).
Methods:
We conducted a scoping review of existing literature on PubMed and EMBASE. Two reviewers searched and retrieved relevant articles on magnetic resonance brain imaging in individuals with SMA censoring to April 2022. Full-text articles published in peer-reviewed journals or abstracts accepted to conferences in English and French were included.
Results:
Twelve articles were identified describing a total of 39 patients [age range: 11 days to 41 years old, type 0 (n = 5), type 1 (n = 4), type 2 (n = 2), type 3 (n = 22), type 4 (n = 6)]. All reported structural changes and did not explore other MRI modalities. In individuals with infantile onset SMA, cortical and subcortical brain abnormalities in white matter, basal ganglia, thalamus, hippocampus, and high intensity areas around lateral ventricles and thalami were reported over time. In individuals with later-onset SMA, reduced cerebellar and lobular volume were observed as well as increased grey matter density in motor areas.
Conclusions:
Limited data on brain imaging in SMA highlights both cortical and subcortical involvement in SMA, supporting the hypothesis that changes are not restricted to lower motor neuron pathways. Further studies are needed to determine the extent and prevalence of structural and functional brain changes across SMA types.
INTRODUCTION
Spinal Muscular Atrophy (SMA) describes a spectrum of genetic neuromuscular diseases characterized by the loss of alpha motor neurons in the brainstem and spinal cord. The most common form, 5q SMA, is caused by homozygous mutations or deletions in the survival motor neuron (SMN) 1 gene [1]. This leads to low production of full-length functional SMN protein from the SMN1 gene, however, this is partially compensated for by the paralogous SMN2 gene. The SMN protein is ubiquitously expressed and plays a critical role in various cell homeostatic pathways [2]. The requirement for SMN protein by the central nervous system is highest during embryonic and early postnatal development coinciding with neuromuscular development [3]. Given that significant levels of SMN protein have previously been identified in the brain [4], it is plausible that reduced expression affects motor regions of the nervous system. Indeed, impaired motor development early in life can potentially impact a critical window of brain maturation.
However, the impact of SMA on the brain remains poorly characterized. Early pathology reports from autopsy evaluations described macroscopic abnormalities in the brain, spinal cord, and muscle [5]. Moreover, few animal and human SMA studies point to brain involvement with evidence of regional and selective changes in brain morphology [6] and prominent involvement of thalamic and sensory neurons [7]. Brain changes have also been detected by structural magnetic resonance imaging (MRI). MRI is widely available and a non-invasive technique appropriate for characterizing structural brain changes. Currently, only one review of brain, cognition and language development in SMA type 1 is available, where MRI findings were also reviewed [8].
The aim of this review is to explore and summarize the current knowledge of brain imaging findings across SMA types and to identify existing gaps. The generated knowledge can guide future neuroimaging studies that aim to explore structural and functional brain changes across the SMA clinical spectrum, potentially paving the way for new pathways of intervention.
METHODS
The Joanna Briggs Institute (JBI) methodology for scoping reviews was used to conduct the review [9]. Results are presented following the Preferred Reporting Items for Systematic Reviews and Meta-Analyses extension for Scoping Reviews [10]. No a priori protocol was registered. Further information on the process can be obtained from the corresponding author on request.
Inclusion criteria
The inclusion criteria used to select the articles for the review were based on the population and concept elements reported below.
Population
We included articles addressed to 5q SMA. Articles published prior to 1991 were not included as they predated the identification of the SMN gene. Though few studies on SMA were published before 1991, it is unclear if the affected children and adults were genetically confirmed to have SMA. Hence, this selection of articles reflects a population with a genetically confirmed 5q SMA diagnosis or a confirmed diagnosis by muscle biopsy.
Concept
We selected articles that used brain MRI in humans. Other imaging methods such as computed tomography and ultrasound were excluded. MRI scans were the preferred neuroimaging tool as they are readily available in clinical settings and produce more detailed structural images compared to other imaging techniques.
Literature search
Published literature on brain imaging in 5q SMA was identified through database searches up to April 2022. Selected keywords were combined to create search strategies that were adjusted for each database. Articles were searched in PubMed/MEDLINE and Ovid EMBASE + EMBASE Classic from 1991–2022. To identify relevant articles, Medical Subject Headings (MeSH terms indexed on PubMed) and key terms were used such as ‘spinal muscular atrophy’, ‘Werdnig Hoffmann’, ‘Kugelberg-Welander’, ‘Dubowitz disease’, which were combined with acronyms for neuroimaging modalities such as ‘MRI’, ‘fRMI’, ‘DTI’, and ‘DWI’. Language was restricted to English and French, however it is possible that some articles in French were missed due to the use of English search terms. Retrieved literature was screened and all relevant articles were used for this review. The reference section of relevant articles was manually searched to identify additional articles of relevance.
Selection of studies
Articles were initially screened by two reviewers (NM and MO) based on titles and abstracts according to the population and concept elements previously described (data on brain magnetic resonance imaging) in SMA types 1 to 4. Full-text peer-reviewed articles and conference abstracts in English or French were selected. The articles were examined by two authors (NM and MO) and eligibility for inclusion was performed independently.
Data extraction and presentation
All relevant data to inform the scoping review objectives and questions were extracted and are summarized in Table 1. Study characteristics related to publication (reference), population (SMA type, sample size, sex, age), and study design were first extracted. The main findings of each article relevant to brain MRI were extracted as well as other clinical findings. Risk of bias assessment was performed by one certified author (MO) using the American Academy of Neurology framework [11].
Table 1
Neuroimaging reports in patients with spinal muscular atrophy (SMA)
| Reference | Study design MRI sequence | Sample (size, SMA type, sex, age range at time of MRI) | Brain MRI main findings | Other clinical findings |
| Vincent and McSweeney [12] | Case report Brain MRI | N = 1; SMA type 0; Female; 11 days | Normal brain | •Symptomatic at birth |
| •SMN2 copy number not specified | ||||
| •On permanent ventilation | ||||
| •MRI spine: normal | ||||
| Mendonca et al. [13] | Case series Longitudinal brain MRI T2-weighted image | N = 3; SMA type 0; 2 Males, 1 Female; 1–3years | •MRI (before 2 mo): supratentorial atrophy of subcortical predominance, tapered corpus callosum, widening sulcus and ventricles; normal signal intensity and volume of thalamus, basal ganglia, cerebellum, white matter. | •Confirmed SMA 0 with fetal onset of reduced movements and symptoms present at birth. |
| •MRI (follow up at 11mo, 1y, 3y respective): progressive supratentorial and hippocampal atrophy (pt 1 and 2), severe white matter reduction. Cerebellum was spared. | •All three had gastrostomy tube feeding exclusively | |||
| •Pt 2 and 3: symmetric hyperintense signals on T2 and FLAIR in putamen and thalamus. | •Whole exome sequencing and clinical exome performed- negative | |||
| •Pt 1: marked ventricular dilatation | •All three had tracheostomy and permanent ventilation. | |||
| •Pt 2: atrophy of caudate | •No clinical seizures, patient 1 and 3 had EEG showing sporadic epileptic discharges | |||
| Maeda et al. [14] | Case report Longitudinal brain MRI | N = 1; SMA type 0; Female; neonatal, 7 months, 2 years) | •Severe and diffuse CNS atrophy of supraten-torial and infratentorial brain and spinal cord | •Confirmed SMA 0 symptomatic at birth, with respiratory failure at birth requiring intubation |
| •MRI (neonatal): no findings | •1 copy of SMN2 | |||
| •MRI (follow up at 7mo, 2y): progressive atrophy of cerebral cortex, subcortical white matter, thalamus, and basal ganglia; cervical cord, brainstem and cerebellar volume reduced at 2 years. | •Apgars 1 and 6 at 1 and 5 minutes | |||
| •No evidence of HIE on MRI in neonatal period | ||||
| •Homozygous deletion in SMN1 and NAIP | ||||
| •Distal necrosis of digits at 4 months | ||||
| •Seizures by 2 years of age, EEG initially burst suppression pattern | ||||
| •MRI spine: cervical cord volume reduced at 2 years | ||||
| Oka et al. [15] | Case report brain MRI T1-weighted image | N = 1; SMA type 1B; Male; 7 years | •Diffuse cerebral atrophy; dilation of 3rd and lateral ventricles; white matter atrophy with hypoplastic corpus callosum | •Symptom onset 1st month of life •SMN2 copy number not specified •On mechanical ventilation |
| •High intensity areas around posterior horns of lateral ventricles on T2 | ||||
| •Normal cerebellum and brainstem | ||||
| •EMG: neurogenic | ||||
| •Muscle biopsy: large groups of small fibers intermingled with small groups of large fiber of type 1 | ||||
| •Auditory evoked potentials | ||||
| •EEG: normal at 1 month | ||||
| •Invasive permanent ventilation at 3 months of age | ||||
| •Developed focal seizures with impaired awareness at 6 years of age, EEG spikes and polyspikes in left centroparietal area | ||||
| •At 7 years: oculomotor palsy | ||||
| •SPECT at 7 years: multiple areas of moderately decreased perfusion including bilateral cerebellar cortex and vermis, right putamen, anterior portion of left frontal lobe, and posterior portion of right frontal lobe. | ||||
| Ito et al. [16] | Case report Brain MRI T2-weighted FLAIR image | N = 1; SMA type 1; Female; 6 years | •High signal intensity in anterolateral portions of bilateral thalami and around posterior horns of lateral ventricles on T2 and FLAIR. | •Severe bulbar palsy |
| •Symptom onset at 3 months of age | ||||
| •SMN2 copy number not specified | ||||
| •EMG neurogenic (at 3 months) | ||||
| •Permanent ventilation from 7 months of age | ||||
| •Normal perinatal history | ||||
| •MRI spine: normal | ||||
| •EEG: diffuse fast activity, no epileptiform abnormality | ||||
| •Normal BAER and VER | ||||
| Cneude et al. [17] | Case report | N = 1; SMA type 1; Male; 10 months | •Lesions in CNS (thalamus, cerebellum); severe cortical dysplasia | •Normal perinatal history with 1 minute Apgar of 10 |
| brain MRI | •Agyria of posterior cortex and anterior cortical pachygyria | •Symptom onset at 3 months of age | ||
| •SMN2 copy number not specified | ||||
| •EEG at 3 months showed hyposarrhythmia | ||||
| •FISH for Miller Dieker syndrome negative (17p13.3) | ||||
| •Karyotype: balanced translocation (maternal) of chromosome 13 &14 | ||||
| •On hydrocortisone, depakene and Sabril for seizures | ||||
| •On mechanical ventilation by 8 months of age | ||||
| •EMG at 8 months neurogenic | ||||
| •Died of RSV infection by 10 months, no autopsy | ||||
| Reference | Study design MRI sequence | Sample (size, SMA type, sex, age range at time of MRI) | Brain MRI main findings | Other clinical findings |
| Harding et al. [18] | Case repor Brain MRI | N = 1; SMA type 1; sex not specified; 18 days | •Normal brain MRI in neonatal period | •On mechanical ventilation from birth to death |
| •Post-mortem: neurodegeneration in motor cortex, nuclei pontis cerebellar nuclei, substantia nigra, locus ceruleus, and meynert’s nucleus. | •1 copy of SMN2 | |||
| •Joint contractures and extensive weakness at birth | ||||
| •EMG: diffuse fasciculations, no conduction responses | ||||
| Losito et al. [19] | Case report brain MRI T1-weighted image | N = 1; SMA type 2; Female; 12 months | •At 12 months: Delayed myelination; high intensity areas around posterior horns of lateral ventricles; corpus callosum dysplasia with a lipoma in the splenium, normal cerebral hemisphere, cerebellum, and brainstem | •Symptom onset 7 months |
| •Normal perinatal history | ||||
| •3 copies of SMN2 | ||||
| •MRI spine: normal | ||||
| •SPECT at 12 months: slight increase in choline peak | ||||
| •EEG normal at 12 months | ||||
| •HFMSE 11/66 | ||||
| Kang et al. [20] | Case series brain MRI | N = 4; SMA type 3; Female; 1–5 years | •Normal brain (patient 3 and 4) | •EMG: neurogenic in pt 2, 3, 4. |
| •SMN2 copy number not specified | ||||
| •Fasciculations noted in pt 1. | ||||
| •Asymmetric weakness and upper motor neuron signs on presentation | ||||
| •Pt 1 had muscle biopsy: neurogenic | ||||
| Olivieri et al. [21] | Case report brain MRI | N = 1; SMA type 3; Female; 2years | •Normal brain | •Ambulatory, with age of onset 10 months to 4 years |
| •3 copies of SMN2 | ||||
| •MRI spine: normal | ||||
| •EMG: neurogenic | ||||
| Querin et al. [22] | Prospective case-control Baseline brain MRI 3D T1-weighted image | N = 25 SMA and N = 25 age and sex matched controls; SMA type 3 (n = 19) and SMA type 4 (n = 6); 18–66 years (mean 41 years) | •Increased grey matter density in motor cortex and extra-motor regions; normal white matter | •No bulbar involvement |
| •Ambulant (n = 18); non-ambulant (n = 7) | ||||
| •4 copies of SMN2 (n = 10) | ||||
| •3 copies of SMN2 (n = 2) | ||||
| •SMN2 copy number not specified (n = 13) | ||||
| •MRI spine: significant grey matter atrophy in C2–C6 levels. | ||||
| •MFM (mean±SD): 60.51±33.56 % | ||||
| •Disease duration (mean±SD): 30.38±15.40 years | ||||
| De Borba et al. [23] | Case-control brain MRI 3D T1-weighted image | N = 25 SMA and N = 25 age and sex matched controls; SMA type 3 (n = 19) and SMA type 4 (n = 6); 18–66 years (mean 41 years) | •Significant smaller cerebellum volume; lobule grey matter significantly reduced | •Bulbar symptoms (1 patient) |
| •Cortical thickness of cerebellum not significant | •4 copies of SMN2 (n = 13) | |||
| •3 copies of SMN2 (n = 7) | ||||
| •SMN2 copy number not specified (n = 5) | ||||
| •SMAFRS (mean±SD): 37.4±8.4 | ||||
| •MFM (mean±SD): 60.51±33.56 |
Legend: EEG: Electroencephalography. HIE: Hypoxic ischemic encephalopathy. MRI: magnetic resonance imaging. SPECT: single-photon emission computerized tomography. SMAFRS: Spinal Muscular Atrophy Functional Rating Scale. MFM: Motor Function Measure. HFMSE: Hammersmith Functional Motor Scale Expanded. BAER : Brainstem auditory evoked reponse. VER: Visual evoked response.
RESULTS
Search results
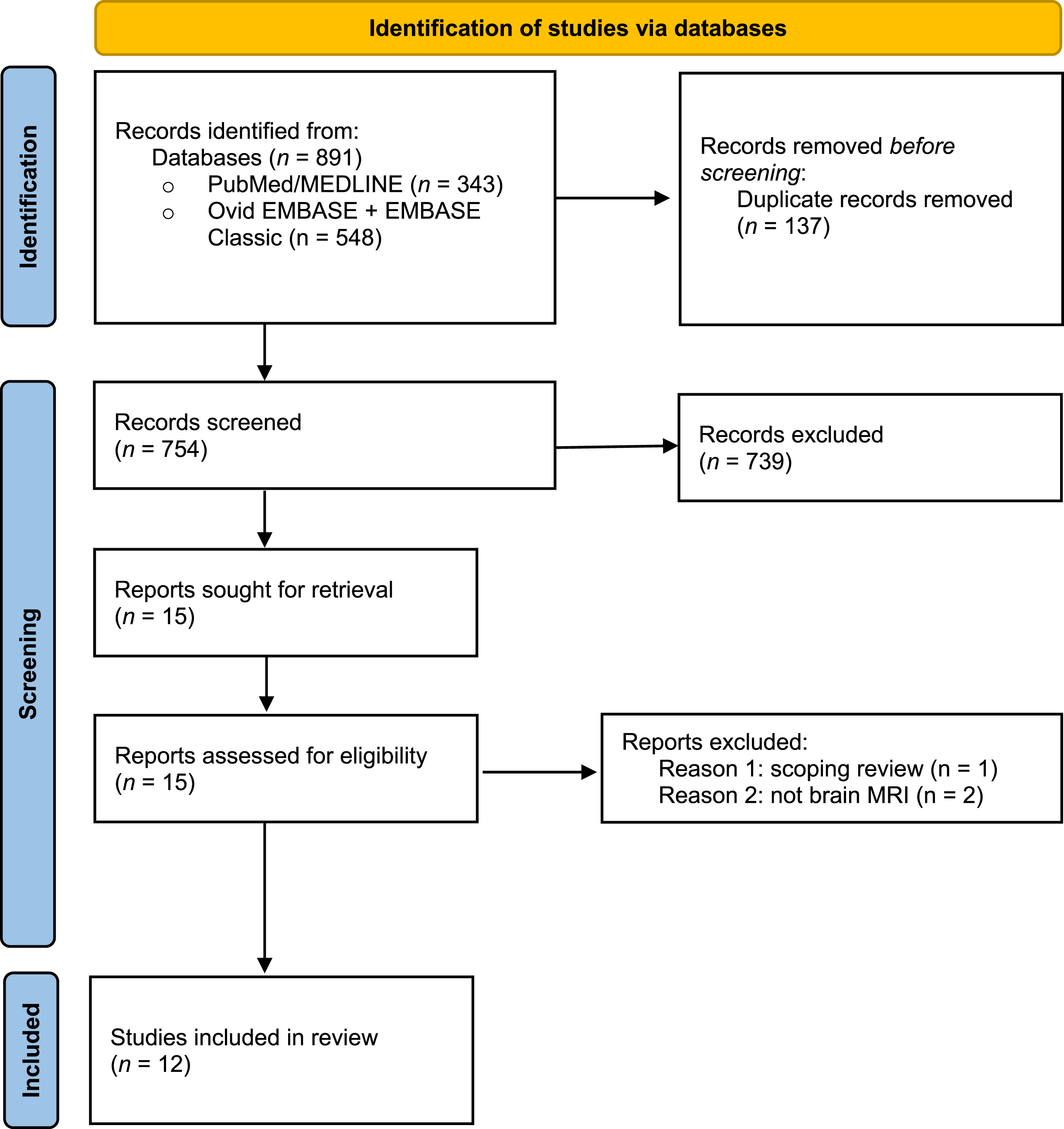
The initial search conducted in July 2021 identified 891 articles across the two databases that described neuroimaging findings in SMA. A total of 137 duplications were removed and after screening by title and abstract a total of 15 articles were included in the full-text screening. Twelve articles were included in the final review (Fig. 1). All articles included patients either genetically diagnosed and confirmed to have 5q SMA or diagnosed by muscle biopsy. An updated literature search was performed in April 2022, identifying no additional articles of interest.
Fig. 1
Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) Flow Diagram of the study.
SMA type 0
Three class IV studies reported on five patients with SMA type 0, showing progressive changes with prolonged survival. All infants had molecular diagnosis confirmation and fetal onset of symptoms. In one case report, no abnormalities were detected on brain MRI of a newborn despite a clinical diagnosis of moderate hypoxic ischemic encephalopathy (HIE) and undergoing therapeutic hypothermia. This infant was extubated and died at 11 days of age [12]. Two studies described a pattern of progressive atrophy and loss of white matter observed by MRI with prolonged survival in infants who remained on permanent ventilation [13, 14]. In a case series of three children under 3 years old, all with one copy of SMN2 and no documented hypoxic ischemic encephalopathy (HIE) at birth, Mendonça and colleagues initially described at 2 months supratentorial atrophy, a widening sulcus and ventricles, and a tapered corpus callosum. Imaging follow-up at 11 months (patient 3), 1 year (patient 1), and 3 years (patient 2) documented a marked progression of supratentorial and hippocampal atrophy and severe reduction in white matter [13]. Moreover, in a longitudinal MRI assessment as a neonate, at 7 months, and at 2 years of age without documented neonatal HIE, Maeda et. al observed progressive central nervous system (CNS) atrophy of the cerebral cortex, subcortical white matter, thalamus, and basal ganglia [14]. In contrast, Mendonça et. al reported no abnormalities in the thalamus and basal ganglia [13].
SMA type 1
Four class IV studies reported on four patients with SMA type 1. A case report of a 7-year-old patient described diffuse cerebral atrophy, dilation of the 3rd and lateral ventricles, and significant white matter atrophy as well as a hypoplastic corpus callosum [15]. T2-weighted images revealed high intensity areas around the posterior horns of the lateral ventricles. Similarly, Ito et al. reported high intensity areas around the posterior horns of the lateral ventricles based on T2-weighted and FLAIR (fluid attenuated inversion recovery) images and found high intensity areas in anterolateral portions of the bilateral thalami [16]. Moreover, lesions in the thalamus and cerebellum were observed in one patient [17]; however, the cerebellum and brainstem were normal in another patient [15]. No brain abnormalities were reported in another case of a newborn with type 1 SMA; yet postmortem examination revealed neurodegeneration in cortical and subcortical brain structures [18].
SMA type 2
Only one class IV case report of brain MRI in an infant with type 2 SMA was identified [19]. Delayed myelination with high intensity areas around the posterior horns of the lateral were found, however the cerebral hemisphere, cerebellum, and brainstem were normal.
SMA type 3 and 4
One class IV and two class III studies reported on 29 patients with SMA type 3 & 4. In a case series of four young children with type 3 SMA patients and one case report on two-year old, no structural brain abnormalities were observed on MRI [20, 21]. A larger prospective case control study of 25 type 3 and type 4 adult SMA patients described a greater cortical volume in those with type 4 SMA compared to type 3 SMA in the left motor cortex. Moreover, they found an increase in grey matter density in the bilateral motor cortex and extra-motor regions in both SMA types compared to control [22]. No white matter abnormalities were observed in either type 3 or 4. Using a similar case-control group, a significantly smaller total volume of cerebellar lobes was observed in SMA patients compared to controls. In particular, grey matter in one lobule was significantly smaller in volume compared to controls [23].
DISCUSSION
Brain neuroimaging findings are scarcely documented in patients with SMA. This scoping review serves to summarize the current state of evidence of brain involvement in children and adults with SMA as identified by MRI and to highlight important gaps to address with further research.
SMA type 0
Common neuropathological features marked in subcortical brain structures including the thalami, basal ganglia and cerebellum showed to be variably affected. The involvement of the thalami, substantia nigra and cuneate nuclei have been well documented in autopsy examinations [7, 24, 25], and the most severe cases of type 0 SMA additionally showed severe and diffuse CNS atrophy [13, 14], distinct from changes secondary to HIE. Imaging follow up in four type 0 patients (less than 3 years of age) highlighted widespread and progressive abnormalities over time: supratentorial brain atrophy with white matter reduction, severe hippocampal atrophy, and a thinned corpus callosum. Given that the SMN protein is highly expressed during fetal development up until early infancy in various brain structures of the nervous system [26], it could be hypothesized these significant abnormalities may be related to the reduction of SMN protein observed during this critical time period. In contrast, Vincent & McSweeney found no brain pathology in an infant of SMA type 0 in the neonatal period [12]. Most of the reported structural changes were observed over time, and they may be secondary to non SMN pathways in a chronically ill child.
SMA type 1 and 2
High signal intensity areas were observed around the lateral ventricles [15, 16, 19] and the thalami [16]. These could reflect unmyelinated areas or dysmyelination. A previous study found significant post-mortem cell death in the developing telencephalon at both the dorsal and ventral side of lateral ventricles [27]. It is not clear the type of cells described by the authors, but it is known that the ventricles are surrounded by ventricular and subventricular zones in which reside proliferating cells that contribute to white matter repopulation [28]. Based on the process of positive reinforcement in exuberance selection during development, the observed change in white matter is likely a consequence of a lack of myelin maturation during development. Lower SMN levels lead to motor neuron degeneration, which can lead to an absence of positive reinforcement in those connections and subsequent lack of myelin maturation. It would be less plausible for these observed changes to be caused by an active demyelination process. However, more research is needed. Abnormal development of the corpus callosum was also observed in two individuals [15, 19]. The corpus callosum is critical for inter-hemispheric communication and shows consistent reduced integrity in another motor neuron disorder, amyotrophic lateral sclerosis [29]. It is also important to note that all children were genetically confirmed for 5q SMA except for one child with SMA type 1 who underwent a muscle biopsy for diagnosis [15]. The authors performed additional diagnostic procedures and made longitudinal assessments of the child to further support this diagnosis. Nonetheless, without genetic confirmation of SMA, findings from this report should be interpreted with caution of potential misclassification bias.
In contrast to type 1 findings of significant abnormalities in cortical and subcortical structures, one case report of an infant with type 2 SMA revealed a normal brainstem, cerebral hemisphere and cerebellum besides white matter abnormalities [19]. The nature of type 1 SMA being the most severe may help explain these contrasting findings, however autopsy examinations in two other individuals with type 2 SMA revealed a reduced number of neurons in the primary motor cortex and fewer myelinated fibers in the spinal-pyramidal tract [30]. These differences could also be attributed to timing of the imaging, where some chronic changes could be missed in younger children.
SMA type 3 and 4
In later onset SMA (types 3 and 4) evidence of cerebellar pathology and changes in grey matter density in motor areas were identified in the largest adult cohorts of SMA patients [22, 23]. Increased cortical volume in the motor cortex of SMA type 4 compared to type 3 potentially suggests a reorganization of the grey matter in the motor cortex and an adaptive reorganization mechanism with progressing lower motor neuron degeneration [22]. Moreover, cerebellar function has historically been related to movement and balance and more recently in non-motor tasks such as sensorimotor control [31]. Cerebellar grey matter volume was found to be significantly reduced [23]. The three cerebellar lobules affected share the final target of proprioceptive output which is closely tied to the control of movement. In a recent cerebellar study in mice, researchers found significantly lower network activity in the cerebellum suggesting neuronal dysfunction that may follow volume loss [32].
These anomalies prompt inquiries about the potential impact of neurodegenerative and neurodevelopmental processes on the brain. SMN protein plays a crucial role in establishing the nervous system during embryonic and early postnatal stages, as well as maintaining motor neurons. Thus, it is plausible that the observed neurodegeneration may result from impaired neurodevelopmental processes. In studies of severely affected children with early-onset SMA, progressive degeneration across brain structures has been observed. In adults with late-onset SMA, cross-sectional examinations show an increase in gray matter and a reduction in cerebellar volume, indicating compensatory and degenerative processes. Since the SMN protein is reduced across the entire spectrum of the disease, both neurodevelopmental and neurodegenerative effects possibly overlap and contribute to the observed changes in the brain.
The clinical consequences of the brain abnormalities remain uncertain, with limited and variable data available from reported cases. The more widespread CNS atrophy seen in the most severe end of the clinical spectrum suggests a potential correlative relationship between disease severity and brain involvement. Interestingly, key motor regions such as thalamus, basal ganglia, cerebellum are often affected. In individuals with later-onset SMA, we observed different patterns of brain involvement, specifically increases in grey matter volumes in the motor cortex in patients with type 4 SMA compared to type 3 [22] as well as decreases in cerebellar volume [23]. The increase in grey matter volume suggests a compensatory process, and it was found to be positively correlated with longer disease duration and slower progression rates seen in type 4 SMA individuals. With cerebellar imaging findings, no significant correlations were found between clinical features or scales.
We provide a comprehensive overview of the available evidence of structural brain involvement detected by MRI across the SMA spectrum. A previous review published in 2021 had reported MRI findings only in SMA type 0/1 individuals [8] censoring their search to 2019. They identified three studies of brain MRI, all included in our review, while we identified an additional four studies in this population. They also concluded that several structures, including the thalami, basal ganglia, temporal and frontal cortices, hippocampi, and cerebellum were variably affected, with changes observed over time in more severely affected children.
Limitations
There are several limitations inherent to the literature reviewed in this scoping review. Case reports are particularly vulnerable to publication bias. Though we had reports of normal brain MRI findings in this scoping review, the overall limited number of studies identified may have been affected by publication bias.
Another limitation is the methodological quality of the papers. Only three studies out of twelve disclosed the type of MRI sequence (T1-weighted, T2-FLAIR, T2-weighted) used to capture the brain abnormalities. The type of sequence used is particularly important as different sequences are better suited to detect different structural abnormalities. It is also important to note the variability that comes with reading and analyzing MRI images. While many reports appear to have been obtained clinically by the site’s radiologist, no studies included in this review described the level of expertise of the radiologist analyzing the images. As such, it is possible that some abnormalities were missed or misinterpreted by some and not others. A standardized protocol for image acquisition and reporting should be used to ensure consistency and reliability. The small sample sizes, limited number of studies, and heterogeneity of neuropathological features present in some patients and not others, hampers the interpretation and generalizability of the findings.
CONCLUSION
SMA is increasingly recognized for its multisystemic features, however brain involvement remains under-investigated. This review provides a comprehensive description of the current findings on structural brain abnormalities in children and adults with SMA as identified by MRI. The current body of evidence is limited to case reports with high risk of bias, highlighting the gaps in knowledge. Future studies applying both quantitative and qualitative imaging techniques would be necessary to advance our understanding of the scope of brain involvement in SMA. This would pave the way for new pathways of interventions targeting the brain as adjunct to currently available disease-modifying therapies and potentially broaden the pool of disease-specific biomarkers for SMA.
ACKNOWLEDGMENTS
NM and AO are supported by the Research Institute of McGill University Health Centre Desjardins Child Health Research Fellowship.
6CONFLICT OF INTEREST
None.
REFERENCES
[1] | Lefebvre S ,et al. Correlation between severity and SMN protein level in spinal muscular atrophy, Nat Genet (1997) ; 16: (3): 265–69. doi: 10.1038/ng0797-265. |
[2] | Chaytow H , Huang Y-T , Gillingwater TH , Faller KME The role of survival motor neuron protein (SMN) in protein homeostasis, Cell Mol Life Sci (2018) ; 75: (21): 3877–94.. doi: 10.1007/s00018-018-2849-1. |
[3] | Bowerman M , et al. Therapeutic strategies for spinal muscular atrophy: SMN and beyond, Dis Model Mech (2017) ; 10: (8): 943–54. doi: 10.1242/dmm.030148. |
[4] | Briese M , Richter D-U , Sattelle DB , Ulfig N SMN, the product of the spinal muscular atrophy-determining gene, is expressed widely but selectively in the developing human forebrain, J Comp Neurol (2006) ; 497: (5): 808–16. doi: 10.1002/cne.21010. |
[5] | Harding BN , et al. Spectrum of neuropathophysiology in spinal muscular atrophy type I, J Neuropathol Exp Neurol (2015) ; 74: (1): 15–24. doi: 10.1097/NEN.0000000000000144. |
[6] | Wishart TM ,et al. SMN deficiency disrupts brain development in a mouse model of severe spinal muscular atrophy, Hum Mol Genet (2010) ; 19: (21): 4216–28. doi: 10.1093/hmg/ddq340. |
[7] | Towfighi J , Young RS , Ward RM Is Werdnig-Hoffmann disease a pure lower motor neuron disorder? Acta Neuropathol (Berl, Acta Neuropathol (Berl.) (1985) ; 65: (3–4): 270–80.. doi: 10.1007/BF00687008. |
[8] | Masson R , Brusa C , Scoto M , Baranello G cognition, and language development in spinal muscular atrophy type 1: A scoping review. Dev Med Child Neurol. https://doi.org/10.1111/dmcn.14798. |
[9] | Chapter 11: Scoping reviews - JBI Manual for Evidence Synthesis - JBI Global Wiki. https://jbi-global-wiki.refined.site/space/MANUAL/3283910770/Chapter+11%3A+Scoping+reviews (accessed Dec. 10, 2021). |
[10] | Tricco AC ,et al. PRISMA extension for scoping reviews (PRISMA-ScR): Checklist and explanation, Ann Intern Med (2018) ; 169: (7): 467–73. doi: 10.7326/M18-0850. |
[11] | Gronseth GS , Cox J , Gloss D , et al. Gronseth GS, Cox J, Gloss D, et al. On behalf of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology. Clinical Practice Guideline Process Manual, 2017 ed. Minneapolis, MN: The American Academy of Neurology, 2017. |
[12] | Vincent D , McSweeney N Spinal muscular atrophy type 0 with a congenital fracture: A case report. Dev Med Child Neurol. 2017. doi: 10.1111/dmcn.13347. |
[13] | Mendonça RH ,et al. Severe brain involvement in 5q spinal muscular atrophy type 0, Ann Neurol (2019) ; 86: (3): 458–62. https://doi.org/10.1002/ana.25549. |
[14] | Maeda K ,et al. Global central nervous system atrophy in spinal muscular atrophy type 0, Ann Neurol (2019) ; 86: (5): 801–2. doi: 10.1002/ana.25596. |
[15] | Oka A , Matsushita Y , Sakakihara Y , Momose T , Yanaginasawa M Spinal muscular atrophy with oculomotor palsy, epilepsy, and cerebellar hypoperfusion, Pediatr Neurol (1995) ; 12: (4): 365–9. doi: 10.1016/0887-8994(95)00058-N. |
[16] | Ito Y , et al. Thalamic lesions in a long-surviving child with spinal muscular atrophy type I: MRI and EEG findings, Brain Dev (2004) ; 26: (1): 53–6. doi: 10.1016/s0387-7604(03)00075-5. |
[17] | Cneude F , Sukno S , Boidein F , Dehouck MB , Bourlet A , Vittu G Cerebral agyria-pachygyria in a child with Werdnig-Hoffmann disease [French], Rev Neurol (Paris) (1999) ; 155: (8), 589–91. |
[18] | Harding B , Yum S , Chung W , Finkel R Spinal muscular atrophy, not only a motor neuron disorder: Phenotype genotype correlation. J Neuropathol Exp Neurol. 2011. doi: 10.1097/NEN.0b013e3182227917. |
[19] | Losito L , Gennaro L , Lucarelli E , Trabacca A Brain MRI abnormalities in a child with spinal muscular atrophy type II. Acta Neurol Belg. 2020. doi: 10.1007/s13760-020-01524-x. |
[20] | Kang PB , Krishnamoorthy KS , Jones RM , Shapiro FD , Darras BT Atypical presentations of spinal muscular atrophy type III (Kugelberg–Welander disease), Neuromuscul Disord (2006) ; 16: (8): 492–4. doi: 10.1016/j.nmd.2006.05.004. |
[21] | Olivieri AN , Iammarrone CS , Di Meglio M , Mauro A , Mellos A A peculiar case of lameness due to spinal muscular atrophy. Clin Exp Rheumatol. 2011. |
[22] | Querin G , et al. The spinal and cerebral profile of adult spinal-muscular atrophy: A multimodal imaging study, NeuroImage Clin (2019) ; 21: 101618.. doi: 10.1016/j.nicl.2018.101618. |
[23] | de Borba FC , Querin G , França MC , Pradat P-F Cerebellar degeneration in adult spinal muscular atrophy patients, J Neurol (2020) ; 267: (9): 2625–31. doi: 10.1007/s00415-020-09875-4. |
[24] | Devriendt K , et al. Clinical and molecular genetic features of congenital spinal muscular atrophy, Ann Neurol (1996) ; 40: (5): 731–8. doi: 10.1002/ana.410400509. |
[25] | Kuru S , Sakai M , Konagaya M , Yoshida M , Hashizume Y , Saito K An autopsy case of spinal muscular atrophy type III (Kugelberg-Welander disease), Neuropathol Off J Jpn Soc Neuropathol (2009) ; 29: (1): 63–7. doi: 10.1111/j.1440-1789.2008.00910.x. |
[26] | Giavazzi A , Setola V , Simonati A , Battaglia G Neuronal-specific roles of the survival motor neuron protein: Evidence from survival motor neuron expression pat-terns in the developing human central nervous system. J Neuropathol Exp Neurol (2006) ; 65: (3): 267–77. doi: 10.1097/01.jnen.0000205144.54457.a3. |
[27] | Liu H , Shafey D , Moores JN , Kothary R Neurodevelopmental consequences of Smn depletion in a mouse model of spinal muscular atrophy, J Neurosci Res (2010) ; 88: (1): 111–22. doi: 10.1002/jnr.22189. |
[28] | Hohsfield LA , et al. Subventricular zone/white matter microglia reconstitute the empty adult microglial niche in a dynamic wave, eLife.e8 10: : e66738. doi: 10.7554/eLife.66738. |
[29] | Filippini N , Douaud G , Mackay CE , Knight S , Talbot K , Turner MR Corpus callosum involvement is a consistent feature of amyotrophic lateral sclerosis, Neurology (2010) ; 75: (18): 1645–52. doi: 10.1212/WNL.0b013e3181fb84d1. |
[30] | Araki S , et al. Neuropathological analysis in spinal muscular atrophy type II, Acta Neuropathol (Berl) (2003) ; 106: (5): 441–8. doi: 10.1007/s00401-003-0743-9. |
[31] | Manto M , et al. Consensus paper: Roles of the cerebellum in motor control—the diversity of ideas on cerebellar involvement in movement, Cerebellum Lond Engl (2012) ; 11: (2): 457–87. doi: 10.1007/s12311-011-0331-9. |
[32] | Tharaneetharan A , et al. Functional abnormalities of cerebellum and motor cortex in spinal muscular atrophy mice, Neuroscience (2021) ; 452: 78–97. doi: 10.1016/j.neuroscience.2020.10.038. |


