
Intrafamilial Phenotypic Variability of Collagen VI-Related Myopathy Due to a New Mutation in the COL6A1 Gene
Abstract
A family of five male siblings (three survivors at 48, 53 and 58 years old; two deceased at 8 months old and 2.5 years old) demonstrating significant phenotypic variability ranging from intermediate to the myosclerotic like Bethlem myopathy is presented. Whole-exome sequencing (WES) identified a new homozygous missense mutation chr21:47402679 T > C in the canonical splice donor site of the second intron (c.227 + 2T>C) in the COL6A1 gene. mRNA analysis confirmed skipping of exon 2 encoding 925 amino-acids in 94–95% of resulting transcripts. Three sibs presented with intermediate phenotype of collagen VI-related dystrophies (48, 53 and 2.5 years old) while the fourth sibling (58 years old) was classified as Bethlem myopathy with spine rigidity. The two older siblings with the moderate progressive phenotype (48 and 53 years old) lost their ability to maintain a vertical posture caused by pronounced contractures of large joints, but continued to ambulate throughout life on fully bent legs without auxiliary means of support. Immunofluorescence analysis of dermal fibroblasts demonstrated that no type VI collagen was secreted in any of the siblings’ cells, regardless of clinical manifestations severity while fibroblast proliferation and colony formation ability was decreased. The detailed genetic and long term clinical data contribute to broadening the genotypic and phenotypic spectrum of COL6A1 related disease.
INTRODUCTION
Collagen VI-related myopathies are among the most frequently identified forms of congenital muscular dystrophies, including Bethlem myopathy (BTHLM1, Bethlem myopathy 1, OMIM # 158810), Ullrich myopathy (UCMD 1, Ullrich congenital muscular dystrophy 1, OMIM # 254090) and a number of intermediate clinical phenotypes [1, 2]. Collagen VI-related muscular dystrophies are the second most common form of congenital muscular dystrophy in Europe, Japan and Australia. For instance, the prevalence rate of UCMD and Bethlem myopathy in northern England is 0.13 and 0.77 per 100,000 individuals, respectively [3, 4]. Collagen VI-related myopathies form a single continuum, the most severe type of which is UCMD [5–7]. UCMD is characterized by an early onset, often from birth, manifesting as muscle weakness and hypotonia, proximal joint contractures, distal joint hypermobility, as well as a number of bone deformities such as kyphoscoliosis and torticollis. The mildest form of collagen VI-related myopathies is classical BTHLM characterized by distal as well as proximal contractures, proximal muscle weakness and remaining ambulation into adulthood [8, 9]. In addition, two opposing variants for BTHLM have been described: limb-girdle phenotype is characterized by late or no contractures [6] and myosclerotic myopathy is characterized by early, diffuse, progressive contractures resulting in severe limitation of movement of axial, proximal and distal joints [7, 10]. The CK activity level is either unchanged, or 1.5 –2-fold increased [11].
Collagen VI-related myopathies are caused by dominant as well as recessive mutations in COL6A1, COL6A2 (21q22.3) and COL6A3 (2q37) genes, en-coding for the corresponding subunits of type VI collagen: α1 (VI), α2 (VI), α3 (VI) [9, 12, 13].
In skeletal muscles type VI collagen provides interaction between muscle basement membranes and the extracellular matrix and is known to bind biglycan, types I and IV collagen and decorin [14]. In addition, disrupted induction of autophagy and the development of mitochondrial-mediated apoptosis with a d in myofibers has been reported in a mouse model of deficiency of α1(VI) [15, 16].
UCMD may be caused by recessive inheritance due to homozygous or compound heterozygous muta-tions; however, dominantly acting mutations in COL6A1 COL6A2 and COL6A3 have also been described and may be more common, in particular de novo mutations [17, 18]. Although the typical genetic mechanism of inheritance for Bethlem myopathy is dominant, recessive mutations have also been seen in patients [19–21]. An autosomal recessive inheritance mode has also been described specifically for the rare myosclerotic variant of BTHLM [7].
Inter-, intrafamilial, intergenerational phenotypic variability and disease progression in collagen VI-related myopathies are common. One of the reasons described for phenotypic variability is sporadic and parental mosaicism of dominantly acting collagen VI heterozygous mutations [22–24]. However, no significant phenotypic variability among siblings with homozygous mutations in the COL6A1 gene has been reported previously. We present the results of a familial observation of five siblings (aged 8 months, 2.5, 43, 53, 58 years) with distinct phenotypic variability (intermediate phenotype and BTHLM with spine rigidity) due to a new homozygous mutation in COL6A1. We undertook an in-depth comparison of phenotypic differences in three sibs (43, 53, 58 years old). We also describe remarkable adaptive changes at the late stage of the disease which allowed two siblings (43 and 53 years old) to continue ambulation throughout life on fully bent legs.
PATIENTS AND METHODS
Examinations
Examinations and tests were performed after in-formed consent was obtained. There was documented consanguinity of the parents in generation II in this Avarian family in Dagestan. Three out of five affected male siblings in generation III (Patient III:1, 48 years old, Patient III:2, 53 years old, and Patient III:3, 58 years old) with clinical manifestations of collagen VI-related myopathy were examined between 2016 and 2018. The two other affected siblings were not personally examined (Patient III:11, presented with floppy baby syndrome and died of respiratory failure at 8 months, Patient III:12 presented with early-onset severe proximal joint contractures and distal joint hypermobility and died as a result of a head injury at 2.5 years) (Fig. 1). We present here the results of the most current examination of the three surviving siblings along with data from medical records.
Fig. 1
Pedigree of the siblings’ family. Symbols in black represent sick family members; exclamation mark - family members personally examined by the author; mt –mutant allele; wt –wild type allele. The position of siblings when walking (a –Patient III:1, 48 years old, b –Patient III:2, 53 years old). The phenotype of siblings (Patient III:1, 48 years old –c, d, e, f, g, h, i; Patient III:2, 53 years old –J, k, l, m, n, o, p; Patient III:3, 58 years old –q, r, s, t, u, v, w). Written informed consents of patients are on file.

Laboratory tests and investigations
Serum CK levels were determined in all patients, X-rays of knee and ankle joints, spirography, ECG, echocardiography, EMG, MRI of pelvic and lower extremity muscles (T1-, T2-WI, T2-FatSat) were performed in all patients.
Genetic testing
Whole-exome sequencing (WES) was performed on DNA samples obtained from Patients III:1, III:2, III:3 using a pair-end method (2×90 base pairs) on the Illumina HiSeq 2000 platform using the SureSelectV4 target sequences enrichment system (51M). The average reading depth was 85× in all samples. Potentially pathogenic variants were identified in comparison with reference human genome databases and the changes detected were confirmed by standard methods (Sanger sequencing and PCR). Segregation analysis of the identified mutations was performed in the mother (II:2) of the siblings.
Primary dermal fibroblast culture
Biopsy samples (4 mm3) were taken from the skin behind-the-ear of patients III:1, III:2, and III:3 under local anesthesia (2% lidocaine solution). The biopsy samples were treated with 0.05% type II collagenase (Sigma, USA) at 37° C for 12 hours, then centrifuged at 200 g for 10 min. The cell pellet was resuspended and cultured in DMEM (Sigma, USA) supplemented with 10% fetal bovine serum (FBS) (HyClone, USA), and 20μg/ml gentamicin (Sigma, USA) at 37°C 5% CO2. Skin fibroblasts of a 26-year-old healthy individual were used as the control.
Fibroblast colony-formation efficacy (CFE-F)
CFE-F was evaluated at passage 1 under the standard procedure. The cell suspension was split into three 100 mm Petri dishes to obtain a clonal inoculum density of 3–4 cells/cm2 in DMEM (Sigma, USA) supplemented with 10% FBS (HyClone, USA), and 20μg/ml gentamicin (Sigma, USA). The Petri dishes were incubated in a CO2-incubator under saturated humidity conditions at 37°C in the 5% CO2 atmosphere for 14 days. Thereafter, the culture dishes with the pre-formed colonies were washed three times with PBS (pH 7.4) and fixed with 70% alcohol for 15 minutes at room temperature. Then alcohol residuals were removed by triple washing with distilled water, and the colonies were stained with a KaryoMAX® Giemsa Stain Stock Solution (Gibco, USA) at 37°C for 20 minutes. The dishes with stained colonies were thoroughly washed from excessive stain and dried at room temperature for 5–7 hours. Formed colonies were counted, the number and shape of their constituent cells determined. A total number of explanted cells in counted colonies only was more than 20. CFE-F was calculated according to the Fridenshtein’s equation for stromal progenitor cells: a pre-formed colonies to explanted cells ratio was multiplied by 100%. CFE-F values reflect individual fibroblast progenitor cell counts. When the colony formation efficacy ranges from 45 to 49% in men and from 36 to 45% in women, the regenerative potential is considered normal [25–26].
Fibroblast proliferative potential
All fibroblasts colonies were classified as dense, diffuse and mixed ones. The fibroblast proliferative potential (a proliferative index) was calculated by a colony distribution (dense, diffuse, mixed) using the formula: PI = [1 (DC)+2 (MC)+3 (DC1)]/100% [26], where PI is the proliferative index, DC –diffuse colonies, (%), MC –mixed colonies, (%), DC1 –dense colonies, (%).The proliferative potential depends on the number of mitotically active cells in the culture, which determines the rate of regenerative processes in the dermis. The proliferative potential of dermal fibroblasts is higher in denser colonies and less in diffuse ones. When the proliferative index ranges from 2.0 to 2.4 in men and from 1.8 to 2.0 in women, the proliferative potential is considered normal.
Immunofluorescent staining of skin-derived fibroblasts
Collagen VI was stained on confluent cells cultured with 0.25 mM ascorbic acid as previously reported by Hicks et al. (2008), and triton X-100 to permeabilize the cells in order to detect intracellular retention of COLVI [27]. Fibroblasts were incubated with collagen VI polyclonal antibodies (abcam, Ab6588, dilution –1:1000). Fluorescence was detected under an Axioplan 2 microscope (Zeiss, Germany) equipped with an HBO 100 mercury lamp (Zeiss). Single channel images and overlays were captured using the Metavue software (Molecular Devices).
Transcript analysis
Total RNA was extracted from fibroblast cultures using the ReliaPrep™ RNA Cell Miniprep System kit (Promega, France) and reverse transcribed into cDNA with the Superscrit RTIII kit (LifeTechnologies SAS, France) following the manufacturer’s recommendations. cDNA samples were subjected to PCR amplification using primers located in exons 1 and 3 of the COL6A1 gene (5’-ACCGTTAGTATGCGAGTTTCTGGCTGGGAGCAGGA-3’ and 5’-TCGGATAGTCAGTCGTTTATAGCGCAGTCGGTGTA-3’, respectively). RPLP0 was used as reference gene (5’-ATGTGGGCTTTGTGTTCACCPCR-3’ and 5’-TCCAGTCTTGATCAGCTGCA-3’). PCR products were analysed on 2% agarose gel and semi-quantification was performed using the ImageJ software.
Ethics statement
All procedures were performed after the patients signed an informed consent form as required by the Declaration of Helsinki (2013) and the study protocol (#AC-2348-082018) approved by the local Ethics Committee of the Dagestan State Medical Academy (Russia). All patients signed an informed consent form for publication of their medical data and photographs.
Description of clinical cases
All patients were born following normal pregnancies. The parents were second cousins and clinically unaffected. The proband’s father (II:1) died of stomach malignancy at the age of 75 years. The family history is otherwise negative for neuromuscular diseases (Table 1).
Table 1
Clinical characteristics and pulmonary function parameters of the sibs (Patient III:1, III:2, III:3)
| Characteristic (age of onset, years) | Patients | ||
| III:1 | III:2 | III:3 | |
| Beginning of independent walking | 3–4 | 3 | 2.5 |
| Running and jumping | Impossible | Impossible | Delay |
| Muscular weakness | 3 (m. quadr. | 3 (m. quadr. | 6–7 (m. quadr. |
| femor. 3/5 MRC) | femor. 3–4/5 MRC) | femor. 4/5 MRC) | |
| Difficulty in stair climbing | 3–4 | 3–4 | 33 |
| Difficulty in standing up from a sitting position | 6 | 7 | 40 |
| Means of support | 7–13 (crutches) | 10–13 (crutches) | Did not use |
| Muscle atrophy | Shoulder girdle, | Diffuse, shoulder | Shoulder girdle and |
| upper limbs | girdle mainly | upper limbs | |
| Knee joint contractures | 3–4 | 3–4 | 7–8 |
| Hip joints contractures | 3–4 | 3–4 | 7–8 |
| Elbow joint contractures | 13–14 | 13 | 18–19 |
| Shoulder joint contractures | 16–18 | 15–16 | 23–25 |
| Wrist joint contractures | 23–25 | 18 | 30–32 |
| Achilles tendon contractures | 13 | 13 | 16–17 |
| Joint hypermobility | Does not persist | Persists | Persists |
| Dyspnea, years | No | 40 | 40 |
| Pulmonary function parameters (at the last visit) | |||
| Age at last visit, y.o. | 48 | 53 | 58 |
| Tidal volume, ml (Δ% of N) | 200 (50) | 100 (25) | 150 (37.5) |
| VC, ml (Δ%, of proper VC) | 1400 (39.2) | 1100 (33.8) | 1800 (52.3) |
| Proper VC, ml. | 3572 | 3252 | 3444 |
| Inspiratory reserve volume, ml (Δ% of N) | 800 (57) | 700 (50) | 1000 (71) |
| Expiratory reserve volume, ml (Δ% of N) | 400 (40) | 300 (30) | 650 (65) |
Note: 1. VC is the vital capacity of the lungs; 2. Proper VC of the lungs = height (cm)×0.052 –age (years)×0.028 –3.2; 3. N expiratory reserve volume (ERV) –1000–1400 ml; 4. N inspiratory reserve volume (IRV) –1400 ml (IRV = VC –(RV + ERV)).
Review of history and medical records data for siblings III:1 and III:2 shows evidence of a similar disease onset at 3–4 years old and progression corresponding to the intermediate phenotype of collagen VI-related dystrophies while maintaining the ability to walk with bent knees and thigh joints from the age of 10–13 years (Fig. 1). The eldest sibling (Patient III:3) had a significantly lower rate of progression, the minimal severity of contractures, with a Trendelenburg gait retained and classified as BTHLM.
Clinical neurological examination (at the last visit)
Patient III:2 (53 years old) was extremely thin with a BMI of only 14.9 (less than 15.0: severely underweight). He had the most severe contractures, diffuse muscle atrophy with a tight feel to the muscles (the mid-upper arm circumference (MUAC) –16/15 cm; normal value for adult men greater than 23 cm [28]), that was a manifestation of intermediate phenotype of collagen VI-related dystrophies with a prominent diffuse contractural component. The youngest sibling (Patient III:1, 48 years old) had a normal trophic status with a BMI 23.31, but with a similar degree of prominent contractures, he had an earlier and more pronounced proximal muscle weakness and corresponded to intermediate phenotype of collagen VI-related myopathy with the predominance of a myopathic syndrome (MUAC –22.5/22.0 cm). The eldest sibling (Patient III:3, 58 years old) was very thin with a BMI being 17.01 (16.0–18.49), but with minimal limb contractures except for spinal (axial) rigidity and milder muscle weakness despite atrophic muscles (MUAC –19/18 cm), overall consistent with a Bethlem phenotype with spinal rigidity.
Distribution of contractures
All siblings examined had torticollis, cervico-thoracic scoliosis, cervical and lumbar spine rigidity due to asymmetric contractures in mm. scalenii, and m. sternocleidomastoideus, which were most pronounced in Patient III:2 (Fig. 1).
All siblings examined had contractures of large joints predominantly in the lower extremities which were most pronounced in Patient III:2. Flexion contractures in shoulder joints from 40 to 130° were caused by sclerotic shortening of mm. pectorales, and m. latissimus dorsi. Significant elbow flexion contractures up to 40–70° were due to m. brachioradialis and m. biceps brachii shortening. Wrist joints were characterized by flexion contractures due to sclerotic shortening of mm. flexor digitorum profundus et superficialis, m. and flexor carpi ulnaris, limiting the extension of the flexion position from 45° (Patient III:2), to 45° of the extensor position (Patient III:3), and hand supination to 15–60° and the wrist joint ulnar deviation to 10–45°. There were hip flexion contractures from 10 to 70° of the flexion position. Knee flexion contractures were most pronounced in siblings III:1 and III:2 (up to 60–110° of the flexion position), while being 10–15° in sibling III:3. All patients had equinovarus foot deformity (from 15 to 30°), most evident in sibling III:3. Patients III:2 and III:3 had a specific calcaneal prominence (Fig. 1).
Hypermobility of small joints of the upper limbs persisted throughout the life. Patients III:2 and III:3 had hyperextension up to 45° in distal interphalangeal joints of II–V fingers and up to 80–90° in interphalangeal joints of the thumbs (Fig. 1). Thus, hypermobility was minimal in Patient III:1 and most pronounced in Patient III:2.
Distribution of muscle strength and atrophy
All examined siblings had muscle weakness predominantly in the flexors of the neck, levators of the scapula, elbow extensors, digit flexors, thigh adductor muscles, lower leg extensors, and foot adductor muscles (3/5 MRC). Muscle weakness predominated in distal parts of the upper limbs (3–4/5 MRC), and in proximal ones of the lower extremities (3–4/5 MRC). It was most pronounced in Patient III:1, and least pronounced in Patient III:3 (Fig. 1).
Muscle atrophy and contractural changes were most intense in Patient III:2 and Patient III:3.
Upper extremity muscle atrophies were diffuse involving deltoid muscles to a least degree, where sclerotic changes resulted in the formation of 3–4 separate bundles (Fig. 3). Muscle atrophy was mainly observed in distal parts of the lower limbs.
Fig. 3
Collagen VI immunofluorescence in UCMD dermal fibroblasts (permeabilised (A) and non-permeabilised (B) conditions according to Hicks et al. (2008); CTRL –normal control, which has an abundance of well-organized collagen VI microfibrils showing a linear and unidirectional trend; III:1; III:2, III:3 –significant collagen VI rarefication with stained single extracellular microfibrils and intracellular protein retention were noticed in most cells. Dermal fibroblasts immunostained for matrix-deposited collagen VI (red) and with the nuclear stain DAPI (blue). Scale bar 50μm.
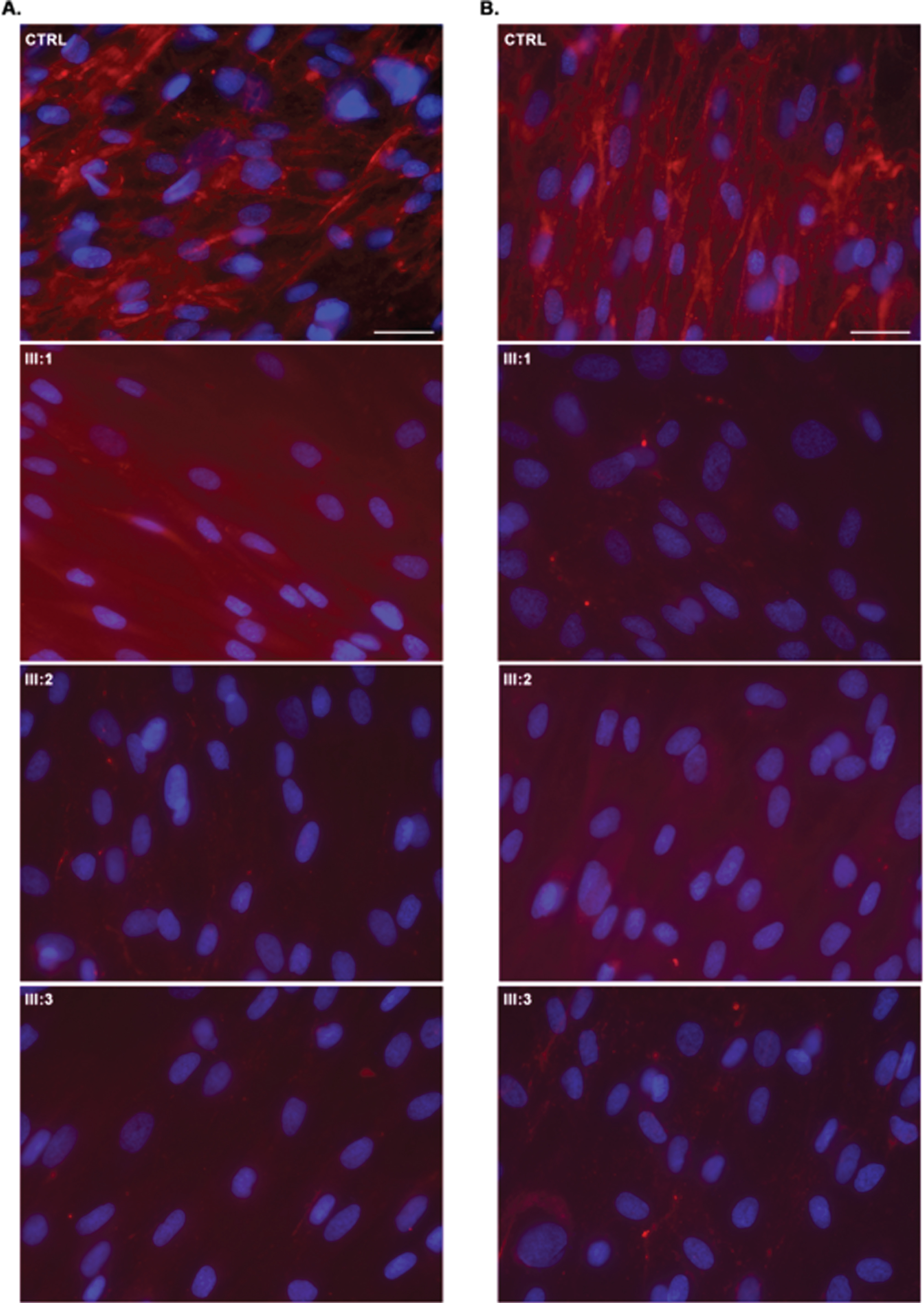
Fig. 4
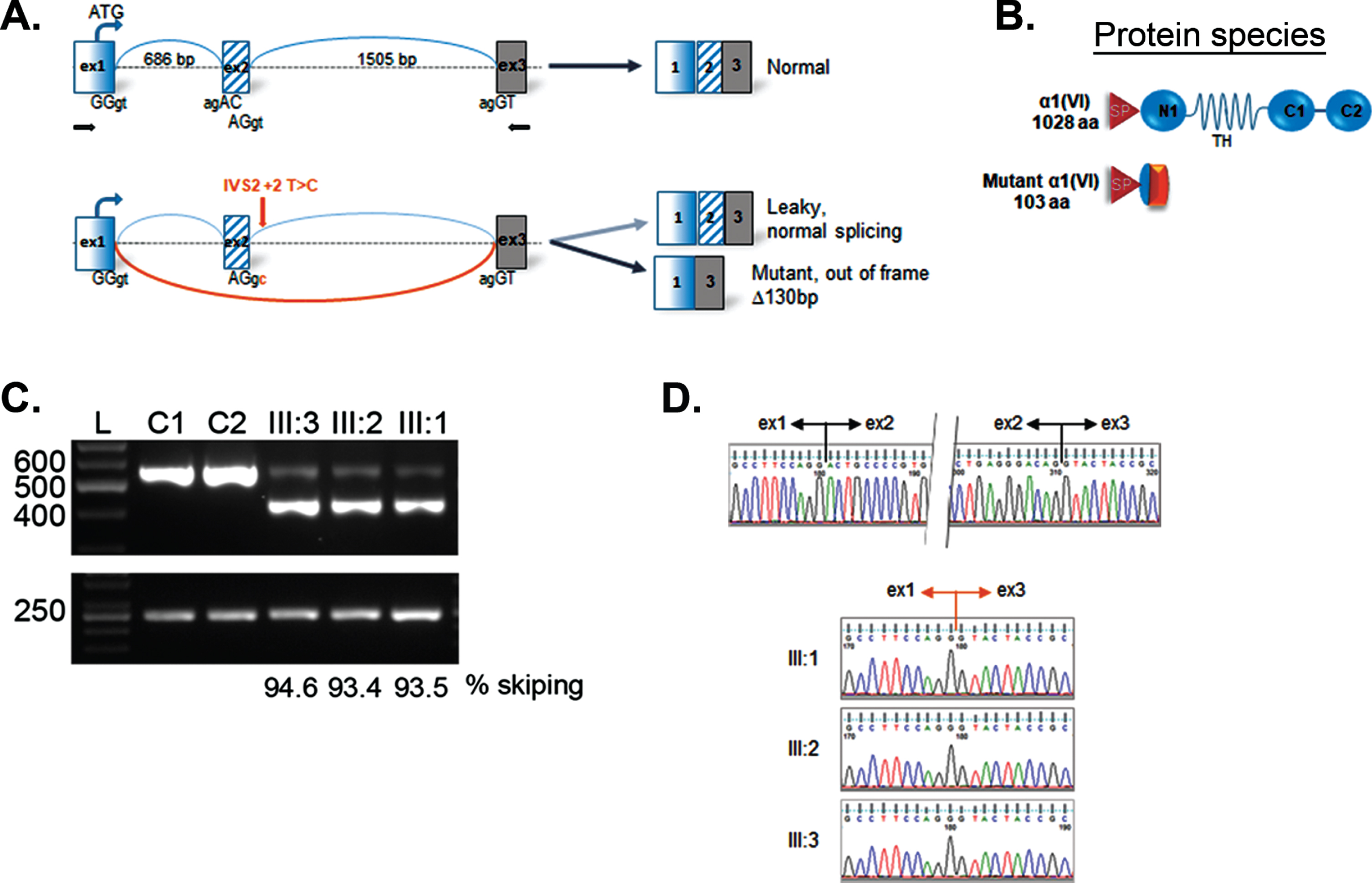
Skipping of exon 2 in COL6A1. Schematic representation of mutation effect on “leaky” splicing, which allows generating some normal transcripts and significant amount of mutant transcript with deletion of 2 exon having a size of 130 bp in patient (A); scheme of normal full-length (1028 aa) and mutant protein α1 (VI) 103 aa; SP –signal peptide; TH –triple helical domain; C1–C2 -terminal domains (B); RT-PCR gel showing an additional faster migrating band in the patient lanes (III:1; III:2; III:3), but not in the normal controls (C1, C2) (C); sequencing chromatogram of cDNA representing the upper normal band (top) and lower band in patient III:1, III:2, III:3 revealing the loss of sequence corresponding to exon 2. RPLP0 was used as reference gene (bottom) (D). Sequencing profiles showing the boundaries of exons 1/2 and 2/3 in a control individual upper panel), while the deletion of exon 2 is evidenced in the 3 patients (lower panels).
Tendon reflexes could not be elicited.
Skin findings
Type I follicular hyperkeratosis was most pronounced in Patient III:1, to a lesser extent in Patient III:2, while none observed in Patient III:3. In addition, all siblings had signs of seborrheic dermatitis and did not have abnormal kelloid formation (Fig. 3).
Blood biochemistry
Serum CK activity was slightly increased (235–280 U/l) in the patients.
MRI of the lower extremities
A MRI pattern showed typical signs of perifascial pronounced fatty infiltration, resulting in increased signal on T1 weighted images along the muscle periphery and around the central fascia (in m. rectus femoris), as well as more diffuse involvement of other muscles [29–31].
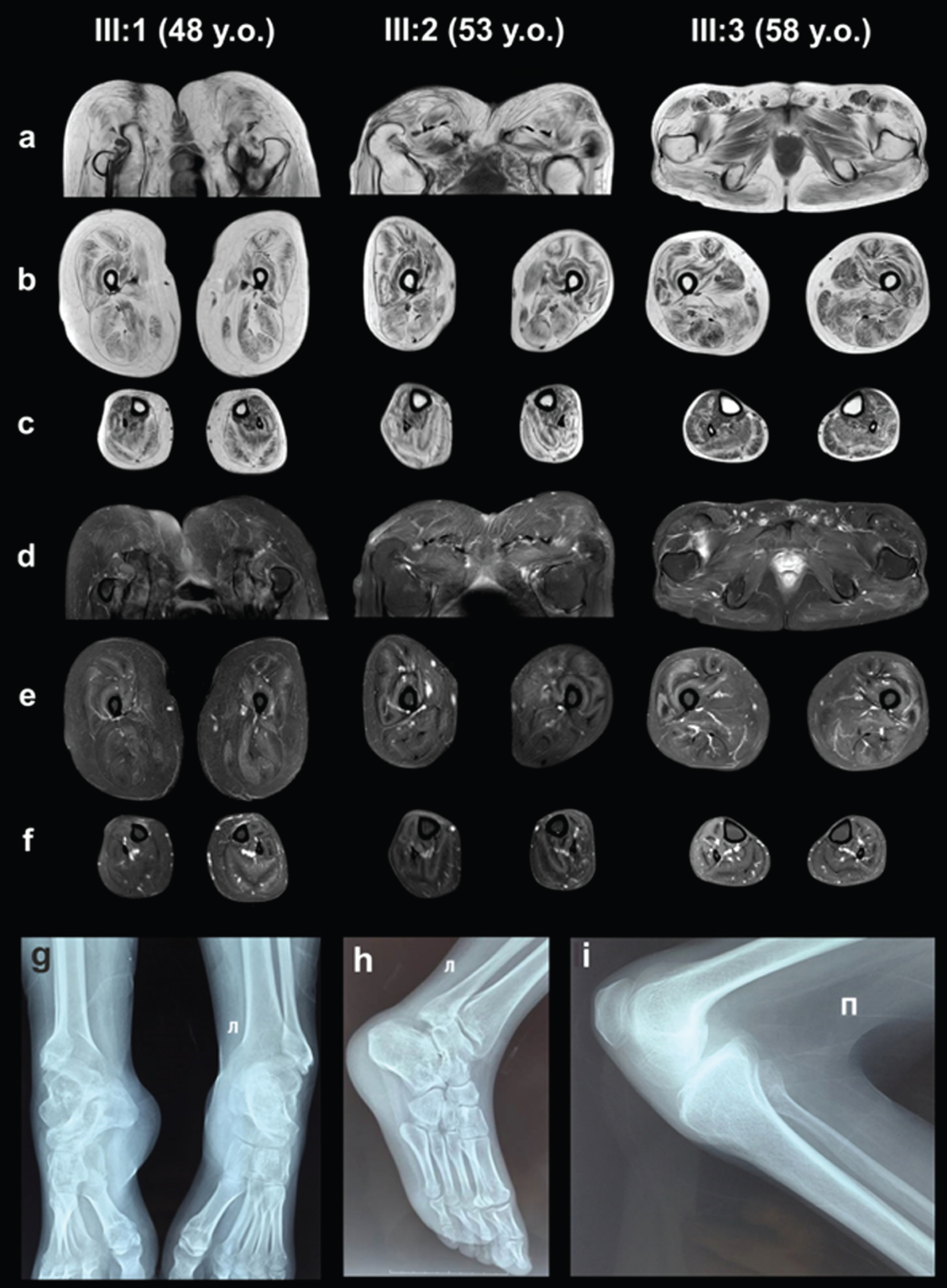
Fatty replacement was more pronounced in thigh and pelvic girdle muscles than in the calf ones predominantly involving the gluteus and thigh posterior muscles (stage 2B–3, according to E. Mercuri, 2002) [32]. Patients III:1 and III:2 had the most pronounced fatty infiltration. Yet, Patients III:1 and III:2 had a relatively hypertrophic appearance of m. rectus femoris, m. vastus lateralis, m. sartorius, m. semimembranosus, m. semitendinosus and m. biceps femoris caput longus, which thus appeared to be less involved, potentially related or contributing to ambulation with bent hip and knee joints. Patient III:3 had moderate atrophy of m. rectus femoris and m. vastus lateralis (Fig. 2).
Fig. 2
MRI of the pelvic girdle and lower extremities muscles (a, b, c –T1-WI, d, e, f –T2-WI FatSat). X-ray examination of the ankles (g, h –Patient III:1, 48 years old) and knee joint (i –Patient III:2, 53 years old).
Spirometry
Spirometry demonstrated a decrease in all basic pulmonary function parameters. The tidal volume was decreased by 50% (200 ml), 75% (100 ml) and 62% (150 ml) in patients III:1, III:2 and III:3, respectively (Table 1). Patients III:2 and III:3 had complained of dyspnea independent of body position from the age of 40. However, because of limited access to medical technology none of the patients had initiated non-invasive ventilatory support. Despite the untreated hypoventilation and living at high altitude (about 4100 m above sea level) they were able to maintain physical activity.
X-ray examination of knee and ankle joints
The results of X-ray scans showed chronic, knee-joint overloads and elevated patellae (an Insall-Salvati ratio of 1.4) in patients III:1 and III:2. The scans also uncovered signs of osteoarthritis in the ankle joints of patients III:1 and III:2. Lateral subluxations of the talus bones were combined with wedge-shaped deformations at the tibial-talus articular surfaces of patients III:1 and III:2 (Fig. 2).
Echocardiography
Patient III:2 had Stage 1 aortic valve insufficiency. All patients (III:1 –65%, III:2 –66%, III:3 –69%) had normal left ventricular ejection fraction as determined by Teichholz and normal blood pressure.
Electromyography
Needle electromyography demonstrated a myopathic pattern.
Genetic testing
Sequencing of Patient III:1, III:2 and III:3 DNA samples established a new homozygous mutation chr21:47402679T>C, leading to a change in the second intron canonical splice site (c.227 + 2T>C) in the COL6A1 gene. Sanger sequencing showed that the proband’s mother (II:2) is a carrier of heterozygous (c.227 + 2T>C) in the COL6A1 gene.
Dermal fibroblast culture
All patients had an abnormal collagen VI secretion by dermal fibroblasts, with severe to completely absent assembled matrix, associated with stained single extracellular microfibrils and intracellular retention of collagen VI precursors (Fig. 3).
There was a reduced CFE-F in all patients –by 30% in Patient III:1, by 35% in Patient III:2 and by 32% in Patient III:3 when compared to the control (45–49%) [26]. Reduced CFE-F indicates decreased fibroblast precursor cell counts in the patient’s fibroblasts and decreased regenerative potential of the dermal fibroblast population.
Patients III:2 and III:3 had a normal proliferation index, while it was slightly below the normal in Patient III:1. A decreased proliferative index indicates a low mitotically active cell count in the culture, and hence a decreased regeneration rate in Patient III:1.
mRNA analysis
mRNA analysis in all patients revealed exon 2 skipping containing 130 bp, coding for the beginning of the N1-domain. The skip results in an out-of frame transcript. We also detected full length transcripts at low percentage (III:1 –5.4%; III:2 –6.6%; III:3 –6.5%). Thus, the homozygous mutation in the COL6A1 gene leads to a laquoleakyraquo out-of-frame skip of mRNA with synthesis allowing for the generation of a small percentage of full length transcript expected to generate wild type protein.
DISCUSSION
Two out of three siblings (48 and 53 years old) had severe contractures of the proximal joints and might most closely correspond to an intermediate phenotype of collagen VI-related dystrophies (or the milder end of the “moderate progressive” phenotype of Brinas et al. [11]), because both patients were able to walk independently up to 7–8 years, followed by the ineffective use of crutches and the beginning of walking on bent legs due to progressive contractures, but relatively better preserved muscle strength [33]. The eldest one had BTHLM with spinal rigidity, in some aspects reminiscent of the myosclerosis phenotype described previously as caused by a homozygous nonsense COL6A2 mutation (p.Q819X) [7]. The coexistence of divergent phenotypes in siblings of the same family has been previously recognized in the COL6-related dystrophies [8]. Genetic analysis was not possible for patient III:12 (2.5 y.o.) due to his early death; however, he probably also had collagen VI-associated dystrophy, as he had an early-onset pronounced proximal joint contracture and distal hypermobility. Patient III:11 (8 m.o.) cannot be classified due to his early death from a respiratory failure, but hypotonia and distal hypermobility suggest the same disease.
It is noteworthy to highlight that patients aged 48 (III:1) and 53 (III:2) years could walk with bent knee and hip joints for 35–40 years, indicating that upright ambulation was predominantly impaired by contractures rather than diffuse weakness and attesting to considerable adaptive capabilities of these patients and their musculoskeletal system.
This family also highlights the considerable variability in the degree of joint contractures even with the same underlying collagen VI mutation. The mechanism of joint contractures in the collagen VI-related dystrophies is still not fully explored and might reside within the muscle connective tissue, but also in tendon and joint capsules. Clearly there must be additional genetic modifiers determining the extent of such contractures.
Despite considerable muscle atrophies in the upper limbs of siblings moving with the bent knee and hip joints (Patients III:1, III:2), there was hypertrophy of the posterior thigh muscles and some anterior thigh muscles (m. vastus lateralis, m. rectus femoris) in the lower extremities. Findings on muscle MRI were consistent with a collagen VI related dystrophy (perifascial involvement) and there were diffuse lesions in muscles that appeared hypertrophied [29, 31]. Partial sparing of the contractile properties of myofibrils within an otherwise affected muscle might help account for these adaptive capabilities of skeletal muscles. In context of an otherwise severely contractural phenotype [34], this may be allowing for the functional compensation seen in the siblings.
It is of interest that in this family the pulmonary function was relatively better preserved, compared to the functional severity of the skeletal muscle involvement. If the patients were within the UCMD phenotype one would have predicted earlier need for non-invasive ventilation at the latest at beginning of the second decade of life as previously reported [33, 35]. This supports our functional classification of the family in the intermediate to Bethlem range with less pulmonary involvement but with a considerable contractural phenotype. Therefore, restrictive changes of the chest may be in part independent on the severity of proximal joint contractures [36]. Three patients described did not obviously require non-invasive ventilation as he has adapted to physical activity without a wheelchair in high altitudes. However, a nocturnal polysomnography will be needed to fully assess his sleep oxygenation and possibility for CO2 retention.
The newly described homozygous chr21:47402679T>C mutation in the COL6A1 gene results in a change in the canonical splice donor site of the second intron (c.227 + 2T>C), which is likely to lead to the destruction of the splice site and the formation of an aberrant transcript with an out-of-frame skip of exon 2. In all patients described semi-quantitative PCR of fibroblast mRNA found a significant predominance of this aberrant transcript (93.4, 93.5 and 94.6%) which did not undergo a nonsense-mediated decay, but also confirmed the presence of full length wild type transcripts, presumably allowing for the production of some normal collagen VI. However, the predominant synthesis of severely truncated α1(VI) results in severe rarefication of extracellular microfibers and diffuse intracellular accumulation of type VI collagen precursors, detected in an immunofluorescence assay of permeabilised dermal fibroblasts. Thus, the mutation is “leaky” and therefore consistent with the overall milder presentation in this family compared to a full “null” situation, which would be predicated to result in a severe UCMD phenotype. However, we found no significant differences in the percentage of normal transcript formation among patients, to account for the milder phenotype in Patient III:3. In addition, all siblings had equally reduced fibroblast colony formation efficacy, which might suggest a genetically determined decrease of fibroblast progenitor cell counts, irrespective of the phenotype. While our findings appears to be in contrast to a previously reported correlation between a disease severity, the mutation type and the degree of decrease in collagen synthesis by fibroblasts [18, 37] it is possible that there are variations in the levels of normal collagen VI matrix formation in vivo in muscle and tendons that are not reflected in our in vitro assays.
CONCLUSION
This family observation demonstrates intrafamilial phenotypic and functional variability of collagen VI-related dystrophy presenting on the intermediate to Bethlem spectrum in patients with particularly pronounced differences in the contractural manifestations of the disease. It is caused by a newly described homozygous COL6A1 out-of-frame exon 2 skipping splice mutation. We demonstrate its leakiness allowing for the generation of some normal transcript. While the analysis on mRNA, collagen VI matrix secretion and proliferative ability did not reveal obvious differences that would explain the cause of intrafamilial phenotypic variability, more subtle differences in the degree of normal matrix deposition in vivo as well as the presence of yet to be identified genetic modifiers may be responsible for the variability observed in this family.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
The authors are grateful to E.A. Pomerantseva, the head of laquoGeneticOraquo laboratory (Moscow), and her employees (genetic testing) and editors for their help in working on the text of the article.
REFERENCES
[1] | Allamand V , Brinas L , Richard P , Stojkovic T , Quijano-Roy S , Bonne G . ColVI myopathies: Where do we stand, where do we go? Skelet Muscle. (2011) ;1: :30. doi:10.1186/2044-5040-1-30 |
[2] | Bonnemann CG . The collagen VI-related myopathies: muscle meets its matrix. Nat Rev Neurol. (2011) ;7: :379–90. doi:10.1038/nrneurol.2011.81 |
[3] | Norwood FL , Harling C , Chinnery PF , Eagle M , Bushby K , Straub V . Prevalence of genetic muscle disease in Northern England: In-depth analysis of a muscle clinic population. Brain. (2009) ;132: :3175–86. doi:10.1093/brain/awp236 |
[4] | Okada M , Kawahara G , Noguchi S , Sugie K , Murayama K , Nonaka I , Hayashi YK , Nishino I . Primary collagen VI deficiency is the second most common congenital muscular dystrophy in Japan. Neurology. (2007) ;69: :1035–42. doi:10.1212/01.wnl.0000271387.10404.4e |
[5] | Ullrich O . Kongenitale, atonisch-sklerotische Muskeldystrophie, ein weiterer Typus der heredodegenerativen Erkrankungen des neuromuskulären Systems. Zeitschrift für die gesamte Neurologie und Psychiatrie. (1930) ;126: :171–201. doi:10.1007/bf02864097 |
[6] | Scacheri PC , Gillanders EM , Subramony SH , Vedanarayanan V , Crowe CA , Thakore N , Bingler M , Hoffman EP . Novel mutations in collagen VI genes: Expansion of the Bethlem myopathy phenotype. Neurology. (2002) ;58: :593–602. doi:10.1212/wnl.58.4.593 |
[7] | Merlini L , Martoni E , Grumati P , Sabatelli P , Squarzoni S , Urciuolo A , Ferlini A , Gualandi F , Bonaldo P . Autosomal recessive myosclerosis myopathy is a collagen VI disorder. Neurology. (2008) ;71: :1245–53. doi:10.1212/01.wnl.0000327611.01687.5e |
[8] | Bonnemann CG . The collagen VI-related myopathies Ullrich congenital muscular dystrophy and Bethlem myopathy. Handb Clin Neurol. (2011) ;101: :81–96. doi:10.1016/b978-0-08-045031-5.00005-0 |
[9] | Lampe AK , Bushby KM . Collagen VI related muscle disorders. J Med Genet. (2005) ;42: :673–85. doi:10.1136/jmg.2002.002311 |
[10] | Bradley WG , Hudgson P , Gardner-Medwin D , Walton JN . The syndrome of myosclerosis. J Neurol Neurosurg Psychiatry. (1973) ;36: :651–60. doi:10.1136/jnnp.36.4.651 |
[11] | Brinas L , Richard P , Quijano-Roy S , Gartioux C , Ledeuil C , Lacene E , Makri S , Ferreiro A , Maugenre S , Topaloglu H , Haliloglu G , Penisson-Besnier I , Jeannet PY , Merlini L , Navarro C , Toutain A , Chaigne D , Desguerre I , de Die-Smulders C , Dunand M , Echenne B , Eymard B , Kuntzer T , Maincent K , Mayer M , Plessis G , Rivier F , Roelens F , Stojkovic T , Taratuto AL , Lubieniecki F , Monges S , Tranchant C , Viollet L , Romero NB , Estournet B , Guicheney P , Allamand V . Early onset collagen VI myopathies: Genetic and clinical correlations. Ann Neurol. (2010) ;68: :511–20. doi:10.1002/ana.22087 |
[12] | Foley AR , Hu Y , Zou Y , Yang M , Medne L , Leach M , Conlin LK , Spinner N , Shaikh TH , Falk M , Neumeyer AM , Bliss L , Tseng BS , Winder TL , Bonnemann CG . Large genomic deletions: A novel cause of Ullrich congenital muscular dystrophy. Ann Neurol. (2011) ;69: :206–11. doi:10.1002/ana.22283 |
[13] | Weil D , Mattei MG , Passage E , N’Guyen VC , Pribula-Conway D , Mann K , Deutzmann R , Timpl R , Chu ML . Cloning and chromosomal localization of human genes encoding the three chains of type VI collagen. Am J Hum Genet. (1988) ;42: :435–45. PMCID: PMC1715162 |
[14] | Kuo HJ , Maslen CL , Keene DR , Glanville RW . Type VI collagen anchors endothelial basement membranes by interacting with type IV collagen. J Biol Chem. (1997) ;272: :26522–9. doi:10.1074/jbc.272.42.26522 |
[15] | Irwin WA , Bergamin N , Sabatelli P , Reggiani C , Megighian A , Merlini L , Braghetta P , Columbaro M , Volpin D , Bressan GM , Bernardi P , Bonaldo P . Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat Genet. (2003) ;35: :367–71. doi:10.1038/ng1270 |
[16] | Bernardi P , Bonaldo P . Mitochondrial dysfunction and defective autophagy in the pathogenesis of collagen VI muscular dystrophies. Cold Spring Harb Perspect Biol. (2013) ;5: :a011387. doi:10.1101/cshperspect.a011387 |
[17] | Baker NL , Morgelin M , Peat R , Goemans N , North KN , Bateman JF , Lamande SR . Dominant collagen VI mutations are a common cause of Ullrich congenital muscular dystrophy. Hum Mol Genet. (2005) ;14: :279–93. doi:10.1093/hmg/ddi025 |
[18] | Bozorgmehr B , Kariminejad A , Nafissi S , Jebelli B , Andoni U , Gartioux C , Ledeuil C , Allamand V , Richard P , Kariminejad MH . Ullrich Congenital Muscular Dystrophy (UCMD): Clinical and Genetic Correlations. Iran J Child Neurol. (2013) ;7: :15–22. PMCID: PMC3943075 |
[19] | Foley AR , Hu Y , Zou Y , Columbus A , Shoffner J , Dunn DM , Weiss RB , Bonnemann CG . Autosomal recessive inheritance of classic Bethlem myopathy. Neuromuscul Disord. (2009) ;19: :813–17. doi:10.1016/j.nmd.2009.09.010 |
[20] | Gualandi F , Urciuolo A , Martoni E , Sabatelli P , Squarzoni S , Bovolenta M , Messina S , Mercuri E , Franchella A , Ferlini A , Bonaldo P , Merlini L . Autosomal recessive Bethlem myopathy. Neurology. (2009) ;73: :1883–91. doi:10.1212/WNL.0b013e3181c3fd2a |
[21] | Caria F , Cescon M , Gualandi F , Pichiecchio A , Rossi R , Rimessi P , Cotti Piccinelli S , Gallo Cassarino S , Gregorio I , Galvagni A , Ferlini A , Padovani A , Bonaldo P , Filosto M . Autosomal recessive Bethlem myopathy: A clinical, genetic and functional study. Neuromuscul Disord. (2019) ;29: :657–63. doi:10.1016/j.nmd.2019.07.007 |
[22] | Armaroli A , Trabanelli C , Scotton C , Venturoli A , Selvatici R , Brisca G , Merlini L , Bruno C , Ferlini A , Gualandi F . Paternal germline mosaicism in collagen VI related myopathies. Eur J Paediatr Neurol. (2015) ;19: :533–6. doi:10.1016/j.ejpn.2015.04.002 |
[23] | D’Amico A , Fattori F , Tasca G , Petrini S , Gualandi F , Bruselles A , D’Oria V , Verardo M , Carrozzo R , Niceta M , Udd B , Ferlini A , Tartaglia M , Bertini E . Somatic mosaicism represents an underestimated event underlying collagen 6-related disorders. Eur J Paediatr Neurol. (2017) ;21: :873–83. doi:10.1016/j.ejpn.2017.07.009 |
[24] | Donkervoort S , Hu Y , Stojkovic T , Voermans NC , Foley AR , Leach ME , Dastgir J , Bolduc V , Cullup T , de Becdelievre A , Yang L , Su H , Meilleur K , Schindler AB , Kamsteeg EJ , Richard P , Butterfield RJ , Winder TL , Crawford TO , Weiss RB , Muntoni F , Allamand V , Bonnemann CG . Mosaicism for dominant collagen 6 mutations as a cause for intrafamilial phenotypic variability. Hum Mutat. (2015) ;36: :48–56. doi:10.1002/humu.22691 |
[25] | Latsinik NV , Grosheva AG , Narovlianskiı AN , Pavlenko RG , Fridenshteın AI . Klonal’naia priroda koloniı˘ fibroblastov, obrazuemykh stromal’nymi kostnomozgovymi kletkami v kul’turakh. Biulleten’ eksperimental’noi biologii i meditsiny. (1987) ;103: :356–8. PMID: 3828519 |
[26] | Zorin V , Zorina A , Smetanina N , Kopnin P , Ozerov IV , Leonov S , Isaev A , Klokov D , Osipov AN . Diffuse colonies of human skin fibroblasts in relation to cellular senescence and proliferation. Aging (Albany NY). (2017) ;9: :1404–13. doi:10.18632/aging.101240 |
[27] | Hicks AKLD , Barresi R , et al. A refined diagnostic algorithm for Bethlem myopathy. Neurology. (2008) ;1192–9. doi:10.1212/01.wnl.0000307749.66438.6d |
[28] | James WP , Mascie-Taylor GC , Norgan NG , Bistrian BR , Shetty PS , Ferro-Luzzi A . The value of arm circumference measurements in assessing chronic energy deficiency in Third World adults. Eur J Clin Nutr. (1994) ;48: :883–94. PMID: 7889897 |
[29] | Mercuri E , Lampe A , Allsop J , Knight R , Pane M , Kinali M , Bonnemann C , Flanigan K , Lapini I , Bushby K , Pepe G , Muntoni F . Muscle MRI in Ullrich congenital muscular dystrophy and Bethlem myopathy. Neuromuscul Disord. (2005) ;15: :303–10. doi:10.1016/j.nmd.2005.01.004 |
[30] | Deconinck N , Richard P , Allamand V , Behin A , Laforet P , Ferreiro A , de Becdelievre A , Ledeuil C , Gartioux C , Nelson I , Carlier RY , Carlier P , Wahbi K , Romero N , Zabot MT , Bouhour F , Tiffreau V , Lacour A , Eymard B , Stojkovic T . Bethlem myopathy: Long-term follow-up identifies COL6 mutations predicting severe clinical evolution. J Neurol Neurosurg Psychiatry. (2005) ;86: :1337–46. doi:10.1136/jnnp-2013-307245 |
[31] | Fu J , Zheng YM , Jin SQ , Yi JF , Liu XJ , Lyn H , Wang ZX , Zhang W , Xiao JX , Yuan Y . “Target” and “Sandwich” Signs in Thigh Muscles have High Diagnostic Values for Collagen VI-related Myopathies. Chin Med J (Engl). (2016) ;129: :1811–6. doi:10.4103/0366-6999.186638 |
[32] | Mercuri E , Cini C , Counsell S , Allsop J , Zolkipli Z , Jungbluth H , Sewry C , Brown SC , Pepe G , Muntoni F . Muscle MRI findings in a three-generation family affected by Bethlem myopathy. Eur J Paediatr Neurol. (2002) ;6: :309–14. doi:10.1016/s1090-3798(02)90618-5 |
[33] | Foley AR , Quijano-Roy S , Collins J , Straub V , McCallum M , Deconinck N , Mercuri E , Pane M , D’Amico A , Bertini E , North K , Ryan MM , Richard P , Allamand V , Hicks D , Lamande S , Hu Y , Gualandi F , Auh S , Muntoni F , Bonnemann CG . Natural history of pulmonary function in collagen VI-related myopathies. Brain. (2013) ;136: :3625–33. doi:10.1093/brain/awt284 |
[34] | Blaauw B , Agatea L , Toniolo L , Canato M , Quarta M , Dyar KA , Danieli-Betto D , Betto R , Schiaffino S , Reggiani C . Eccentric contractions lead to myofibrillar dysfunction in muscular dystrophy. J Appl Physiol. (1985) ;108: (2010):105–11. doi: 10.1152/japplphysiol.00803.2009 |
[35] | Quijano-Roy S , Khirani S , Colella M , Ramirez A , Aloui S , Wehbi S , de Becdelievre A , Carlier RY , Allamand V , Richard P , Azzi V , Estournet B , Fauroux B . Diaphragmatic dysfunction in Collagen VI myopathies. Neuromuscul Disord. (2014) ;24: :125–33. doi:10.1016/j.nmd.2013.11.002 |
[36] | Nadeau A , Kinali M , Main M , Jimenez-Mallebrera C , Aloysius A , Clement E , North B , Manzur AY , Robb SA , Mercuri E , Muntoni F . Natural history of Ullrich congenital muscular dystrophy. Neurology. (2009) ;73: :25–31. doi:10.1212/WNL.0b013e3181aae851 |
[37] | Hicks D , Lampe AK , Barresi R , Charlton R , Fiorillo C , Bonnemann CG , Hudson J , Sutton R , Lochmuller H , Straub V , Bushby K . A refined diagnostic algorithm for Bethlem myopathy. Neurology. (2008) ;70: :1192–9. doi:10.1212/01.wnl.0000307749.66438.6d |


