
A Mutation in the Mitochondrial Aspartate/Glutamate Carrier Leads to a More Oxidizing Intramitochondrial Environment and an Inflammatory Myopathy in Dutch Shepherd Dogs
Abstract
Background:
Inflammatory myopathies are characterized by infiltration of inflammatory cells into muscle. Typically, immune-mediated disorders such as polymyositis, dermatomyositis and inclusion body myositis are diagnosed.
Objective:
A small family of dogs with early onset muscle weakness and inflammatory muscle biopsies were investigated for an underlying genetic cause.
Methods:
Following the histopathological diagnosis of inflammatory myopathy, mutational analysis including whole genome sequencing, functional transport studies of the mutated and wild-type proteins, and metabolomic analysis were performed.
Results:
Whole genome resequencing identified a pathological variant in the SLC25A12 gene, resulting in a leucine to proline substitution at amino acid 349 in the mitochondrial aspartate-glutamate transporter known as the neuron and muscle specific aspartate glutamate carrier 1 (AGC1). Functionally reconstituting recombinant wild-type and mutant AGC1 into liposomes demonstrated a dramatic decrease in AGC1 transport activity and inability to transfer reducing equivalents from the cytosol into mitochondria. Targeted, broad-spectrum metabolomic analysis from affected and control muscles demonstrated a proinflammatory milieu and strong support for oxidative stress.
Conclusions:
This study provides the first description of a metabolic mechanism in which ablated mitochondrial glutamate transport markedly reduced the import of reducing equivalents into mitochondria and produced a highly oxidizing and proinflammatory muscle environment and an inflammatory myopathy.
INTRODUCTION
Similar to human inflammatory myopathies (IMs), canine IMs are a large group of disorders characterized by infiltration of inflammatory cells into muscle [1]. The most common IMs in humans are immune-mediated and include polymyositis (PM), dermatomyositis and inclusion body myositis. In dogs, the immune-mediated IMs masticatory myositis and PM, and myositis caused by infectious diseases, usually protozoal infection, are most common. Many breeds of dogs can be affected with IMs and in one large study of 200 cases, with the exception of a higher incidence in Boxer and Newfoundland breeds, the affected breed distribution approximated the American Kennel Club statistics [2]. Recently, a breed associated IM has been described in Vizsla dogs [3]. Clinical signs of generalized IMs include muscle weakness, a stiff short-strided gait, and muscle atrophy. This clinical presentation, however, is not specific for IM and myopathies including dystrophies and other congenital myopathies need to be considered in the differential diagnosis. The serum creatine kinase (CK) activity, a marker of muscle damage, is persistently but variably elevated in inflammatory, dystrophic and necrotizing myopathies. Histopathology for confirmation of cellular infiltrates and immunostaining of muscle biopsy specimens to phenotype inflammatory cells and localize major histocompatibility antigens is required for confirmation of an IM.
A small family of young Dutch Shepherd dogs, a breed not previously identified with IM, was presented to local veterinarians for evaluation of progressive clinical signs of generalized muscle weakness and muscle atrophy beginning at 3 to 9 months of age. Clinical evaluations including general physical and neurological examinations supported a neuromuscular disease. Histopathology of muscle biopsies confirmed an inflammatory myopathy and immune-mediated, infectious and dystrophic disorders were considered. Given the early onset of muscle weakness and presence of related affected dogs, an underlying genetic cause was suspected. A genome-wide association (GWA) strategy to map a chromosomal locus, followed by whole genome sequencing (WGS), identified a probable SLC25A12 gene mutation. Transport studies including functional reconstitution of the recombinant AGC1 in liposomes, coupled with metabolomic investigations of muscles from affected Dutch Shepherd and control dogs, confirmed a dramatic decrease of AGC1 transport activity and the presence of a proinflammatory milieu in muscles from affected dogs. This report is the first description of a SLC25A12 variant resulting in an inflammatory myopathy in any species and reveals a complex pattern of metabolomic changes associated with this variant.
METHODS
Animals
Five related Dutch Shepherd dogs, 4 males and 1 female, were presented to the referring veterinarians for clinical evaluation of generalized progressive weakness and stiff stilted gait beginning at 3 to 9 months of age. Serum CK activities were persistently elevated ranging from 800 to 2500 IU/L (reference <200 IU/L). All affected dogs were euthanized by 2 years of age for clinical progression of weakness. Diagnostic muscle biopsy specimens were collected by the referring veterinarians on the five affected dogs and evaluated at the Comparative Neuromuscular Laboratory (CNL, University of California San Diego (UCSD) following informed consent from all owners. The samples used for the initial genetic studies comprised a pedigree of Dutch Shepherd dogs including the 5 affected dogs, 2 parents and 3 full-sibs. In addition, 44 other close relatives, 130 other “unrelated” Dutch Shepherds, and 56 dogs of other breeds (Belgian Malinois, Belgian Shepherd, Belgian Tervuren, German Shepherd, Border Collie, Australian Shepherd, and Havanese) were included in the follow-up genetic studies. Metabolomics studies included extracts obtained from archived frozen muscle biopsies from the 5 Dutch Shepherd cases, as well as 20 cryopreserved normal muscle biopsies from a specimen archive maintained at UCSD. The Institutional Animal Care and Use Committee (IACUC) of the University of Minnesota, in accordance with national animal welfare guidelines, also approved the study.
Histopathology, Histochemistry and Immunohistochemistry
Biopsies from the biceps femoris, vastus lateralis and triceps muscles were collected under general inhalational anesthesia by an open biopsy procedure. The biopsy specimens were either unfixed and shipped under refrigeration to the CNL by an express service, then snap frozen in isopentane pre-cooled in liquid nitrogen, or immersion fixed in 10% buffered formalin. Cryosections (8μm) were subjected to a standard panel of histological and histochemical stains and reactions including succinic dehydrogenase (SDH) and cytochrome C oxidase (COX) [4]. Immunofluorescent staining to phenotype cellular infiltrates, for localization of MHC I and II antigens, and for localization of dysferlin, a protein associated with limb-girdle muscular dystrophy type 2B, was performed using defined monoclonal antibodies as previously described [5].
SNP array genotyping
Genomic DNA was isolated from whole blood or muscle using the Gentra PureGene blood kit (Qiagen). The DNA sample set for genotyping on the Illumina CanineHD BeadChip consisted of the 5 cases (histopathology-confirmed) and 5 phenotypically normal controls (2 parents, 3 full-sibs). This array assays 173,662 SNP markers in which 97,134 SNPs remained after pruning of genotyping data as described previously [6]. We assumed an autosomal recessive model and utilized a homozygosity mapping approach with the GENO test in PLINK [7], in which SNPs that were homozygous in the cases and heterozygous in parents, and homozygous for the alternative allele or heterozygous in unaffected sibs, were identified. Haplotypes around the significantly associated locus were constructed using fast PHASE [8]. All genome positions refer to the CanFam3.1 reference sequence assembly.
Whole genome sequencing and variant identification
WGS of one case was performed with a PCR-free library on an Illumina HiSeq 2500 (Illumina, San Diego, USA). Briefly, a fragment library with average insert size of ∼680 bp was prepared from which ∼125 million 2×125 bp paired-end reads were generated, corresponding to roughly 17x coverage. The reads were mapped against the dog reference genome assembly (CanFam3.1) as described [6]. Variants identified in the critical interval on chromosome 36 in the case were compared to those of control genomes from 639 dogs of 126 diverse breeds and 8 wolves found in the Dog Biomedical Variant Database Consortium (DBVDC) database. Variant effects were predicted with MutPred2 [9], SNPs&GO [10], and Provean [11]. These reads have been made publicly available at NCBI’s Short Read Archive at http://www.ncbi.nlm.nih.gov/bioproject/563058.
Genotyping
Genotyping for the SLC25A12 mutation was performed via PCR-RFLP using PCR primers 5’-TGCTGCTGGAGAGATAAAATCA-3’ and 5’-TCCACTGCAGTTGTTCTCTGA and restriction enzyme BglII.
Comparative modeling
Multiple sequence alignment of mitochondrial aspartate/glutamate carrier 1 (AGC1) orthologs from various species and other mitochondrial carriers of C. lupus was obtained using ClustalW.13. Modeller was used to obtain a 3D structural model of C. lupus AGC1 using the structure of the bovine BtAAC1 (protein data bank accession code: 1okc) as template for the comparative modeling.
Construction of the expression plasmids and bacterial expression
The coding sequence for SLC25A12 variant X2 (XM_535962.5), mutated at position c.1046T>C, was amplified by PCR from the muscle cDNA of the same proband that underwent WGS. The oligonucleotide primers were synthesized corresponding to the extremities of the coding sequence with additional BamHI and HindIII sites. The amplified product was cloned into the pMW7 expression vector and transformed into E. coli TOP 10 cells. Transformants were selected on ampicillin (100μg/ml) and screened by direct colony PCR and by restriction digestion of purified plasmids. Finally, the sequence of the insert was verified. The WT coding sequence of canine SLC25A12 was obtained from the mutated sequence by site-directed mutagenesis using the QuikChange® Site-Directed Mutagenesis Kit from Stratagene.
AGC1 WT and its mutated form (p.L349P) were overexpressed at 30°C in E. coli C0214(DE3) [12]. Inclusion bodies were purified on a sucrose density gradient [13], washed at 4°C with TE buffer (10 mM Tris— HCl, 1 mM EDTA pH 6.5), then twice with a buffer containing Triton X-114 (3%, w/v), 1 mM EDTA and 10 mM PIPES— KOH pH 6.5, and once again with TE buffer.
Reconstitution into liposomes and transport measurements
WT and p.L349P mutant AGC1 were solubilized in 1.9% sarkosyl (w/v), and a small residue was removed by centrifugation (258 000 g, 1 h). Solubilized proteins were diluted 9-fold with buffer (10 mM PIPES— KOH pH 6.5, 1 mM EDTA) and then reconstituted into liposomes in the presence of glutamate 20 mM [14]. The amount of protein incorporated into liposomes was measured as described [15]. In all cases, it was approximately 20% of the protein added to the reconstitution mixture. Transport at 25°C was started by adding [14C]aspartate or [14C]glutamate to the proteoliposomes and terminated by addition of 20 mM pyridoxal 5′-phosphate and bathophenanthroline [14]. In controls, inhibitors were added with the labeled substrate. Entrapped radioactivity was counted [14].
Sample processing for metabolomics
Previously frozen diagnostic muscle biopsy specimens (the 5 clinically affected Dutch Shepherd dogs and 20 archived histologically normal muscles from various breeds) were stored at – 80°C until further processed. Samples were scraped into Petri dishes then transferred to liquid nitrogen and ground to powder in a pre-chilled mortar. The powered tissue (15–50 mg) was weighed and mixed with stable isotope internal standards and incubated at room temperature for 10 min to permit metabolite interaction with binding proteins. Pre-chilled extraction buffer containing methanol:acetonitrile:water (43:43:16, v/v/v) was then added at a 9:1 ratio (μl/mg) to the powdered samples to produce a final solvent ratio of 40:40:20 (v/v/v). The samples were incubated on crushed ice for 10 min and centrifuged at 16,000 g for 10 min at 4°C. The supernatant was transferred to a new tube and kept at – 80°C for further analysis.
Metabolomics analysis
Targeted, broad-spectrum metabolomic analysis was performed as described previously [16]. Briefly, the samples were analyzed on a Shimadzu LC-20AD UHPLC system coupled with a SCIEX QTRAP 5500 triple quadrupole mass spectrometer (LC-MS/MS). Ten microliters of extract were injected and the metabolites were separated on a Shodex polymer based NH2 HPLC column (250 mm×2.0 mm, 4μm, Showa Denko, USA) held at 25°C. The MS was performed by scheduled multiple reaction monitoring (sMRM) in both negative and positive mode with rapid polarity switching (50 ms). A total of 630 endogenous metabolites covering 63 biochemical pathways were targeted. 383 metabolites were measurable in all samples. The compound-dependent parameters were optimized using the purified standards. The sphingomyelins were also reconfirmed by analyzing them on a Kinetex PFP reverse phase column (150 mm×2.1 mm, 1.7μm, Phenomenex). Measurements of 27 stable isotope-labeled standards and 50 endogenous metabolites were monitored daily for process control. Reproducibility was quantified by calculating the intra-day and inter-day correlations and median of the relative standard deviations (RSD). The chromatographic peaks were manually inspected and integrated semi-automatically using MultiQuant 3.0 (SCIEX, USA).
Statistical analysis
Metabolomic peak area under the curve (AUC) data were log2-transformed, scaled by control standard deviations, and the resulting Z-scores (
RESULTS
Clinical signs, histopathology, histochemistry and immunofluorescence staining confirmed a diagnosis of an immune-mediated polymyositis
Five related Dutch Shepherd dogs with progressive muscle weakness and generalized muscle atrophy beginning at 3 to 9 months of age were euthanized by 2 years of age (Fig. 1A). Clinical signs included muscle tremors, pelvic limb stiffness, weakness progressing to inability to walk, and severe muscle atrophy. Serum CK activity was moderately elevated (ranging from 800–2,500 IU/L (reference values <200 IU/L) in all affected dogs, indicating myofiber damage. As far as could be determined, affected dogs did not have behavioral abnormalities or seizure activity, interacted with littermates and other dogs, and were visual with no ocular abnormalities noted. As myopathic changes predominated, extensive evaluations by a veterinary neurologist for central nervous system disease or by an ophthalmologist for ocular disease were not performed.
Fig. 1
A. A Dutch shepherd dog affected with the p.L349P mutation showed a myopathic stance including a hunched back and muscle wasting B. Representative H&E stained cryosection of a biopsy from the biceps femoris muscle showed excessive variability in myofiber size, numerous atrophic fibers having a round shape, multifocal areas of mixed mononuclear cell infiltrations having an endomysial and perimysial distribution, scattered necrotic fibers undergoing phagocytosis and moderate endomysial fibrosis. C. Cryosections were reacted and stained for localization of the mitochondrial specific oxidative enzymes succinic dehydrogenase (SDH), cytochrome C oxidase (COX) and for combined SDH and COX. No specific abnormalities were identified that would support an abnormal number or distribution of mitochondria, or fibers devoid of oxidative activity. D. Infiltrating cells identified by immunofluorescent microscopy were predominantly CD3+ T cells, with variable numbers of CD4+ and CD8+ T cells. Scattered CD11c+ macrophage/dendritic cells and rare CD21+ B lymphocytes were also noted. Several regenerating myofibers were localized with the antibody against developmental myosin heavy chain (dMHC). Both MHC class I and MHC class II antigens were expressed on the sarcolemma of morphologically normal and abnormal myofibers independent of foci of cellular infiltrates. Staining for dysferlin, which can be associated with inflammatory changes in limb girdle muscular dystrophy type 2B, was similar to control muscle.
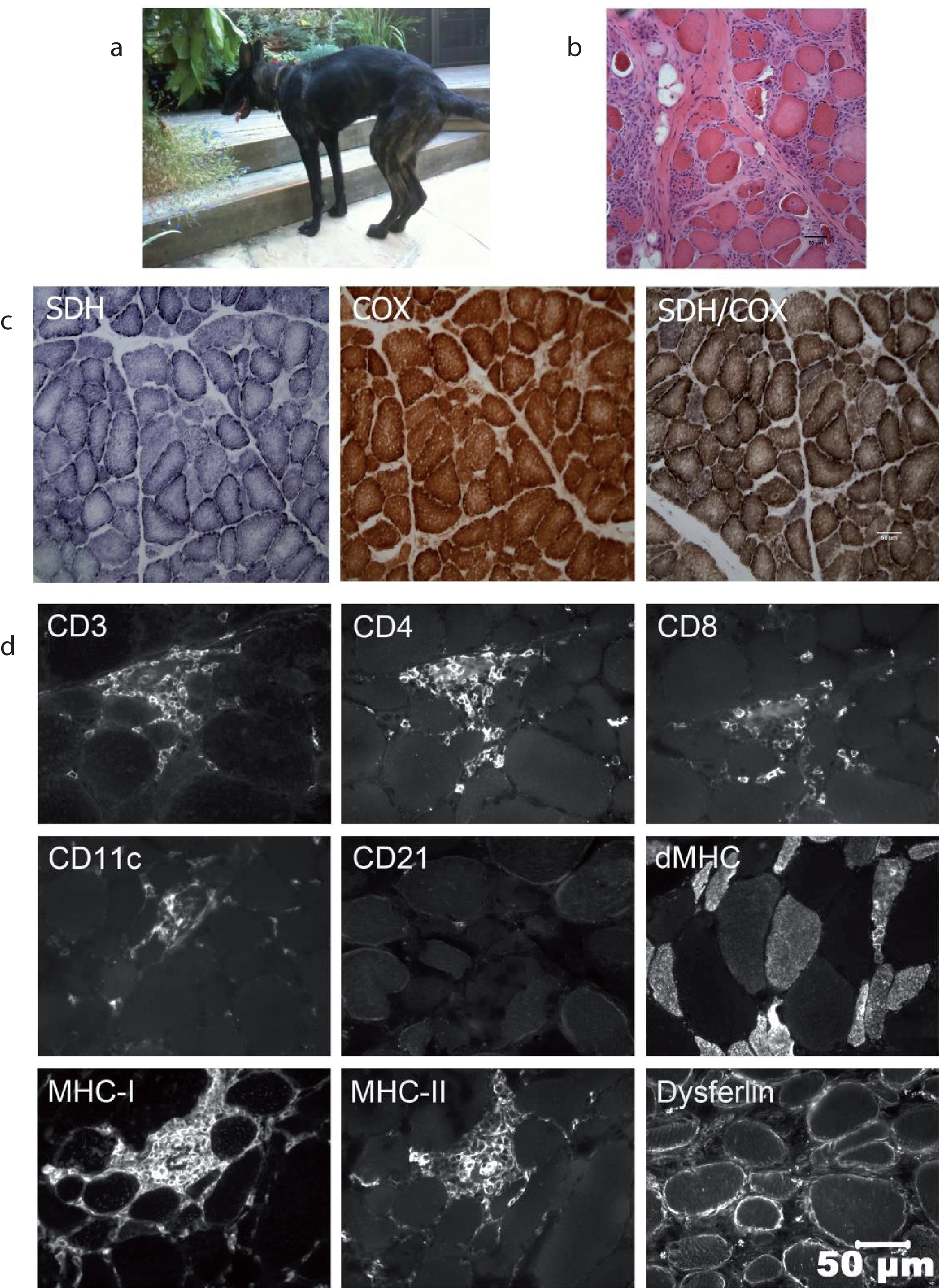
Histopathologic examination of multiple thoracic and pelvic limb muscles from each affected dog confirmed an inflammatory myopathy (myositis) with excessive variability in myofiber size, numerous atrophic fibers having a round shape, and multifocal areas of mixed mononuclear cell infiltrations having an endomysial and perimysial distribution, scattered necrotic fibers undergoing phagocytosis, and moderate endomysial fibrosis (Fig. 1B). Histochemical staining for the mitochondrial specific enzymes SDH and COX did not reveal any specific mitochondrial abnormalities (Fig. 1C).
Infiltrating cells identified by immunofluorescent microscopy were predominantly CD3+ T cells, with variable numbers of CD4+ and CD8+ T cells (Fig. 1D). Scattered CD11c+ macrophage/dendritic cells and rare CD21+ B lymphocytes were also noted. Several regenerating myofibers were localized with the antibody against developmental myosin heavy chain. Both MHC class I and MHC class II antigens were expressed on the sarcolemma of both morphologically normal and abnormal myofibers independent of foci of cellular infiltrates, but myofibers themselves were negative for both MHC class I and II antigens. Inflammatory cells and endothelial cells also expressed both MHC class I and MHC class II antigens. Staining for dysferlin, which can be associated with inflammatory changes in limb girdle muscular dystrophy type 2B, was similar to control muscle.
Genome locus mapping and whole genome resequencing identified a leucine to proline substitution in codon 349 of the aspartate-glutamate carrier 1 (AGC1)
The 5 cases were derived from 2 Dutch Shepherd litters produced by the same sire with two different females. All 3 parents shared a common ancestor within 4 generations that, coupled with the severity and early onset of clinical signs, indicated a recessive mode of inheritance was most likely (Fig. 2). GENO model testing with PLINK identified chromosomal segments located on CFA 26 (4 SNPs) and CFA 36 (105 SNPs) that met significance criteria for a recessive trait. However, haplotype analysis identified CFA36 as the single chromosome of interest, with all 5 cases sharing a single homozygous haplotype, and parents being heterozygous for that same haplotype. Three subregions were defined (chr36:3,992,378–6,163,588 Mb, ch36:7,277,053–7, 393,969 Mb, and chr36:11,576,547–18,215,489 Mb) (Fig. 3). These critical intervals included more than 140 protein-coding genes.
Fig. 2
Pedigree depiction of 5 cases from 2 litters resulting from the same sire and two different females. Genotypes for the SLC25A12 mutation identified in this study are provided and a putative female founder is indicated with red shading.
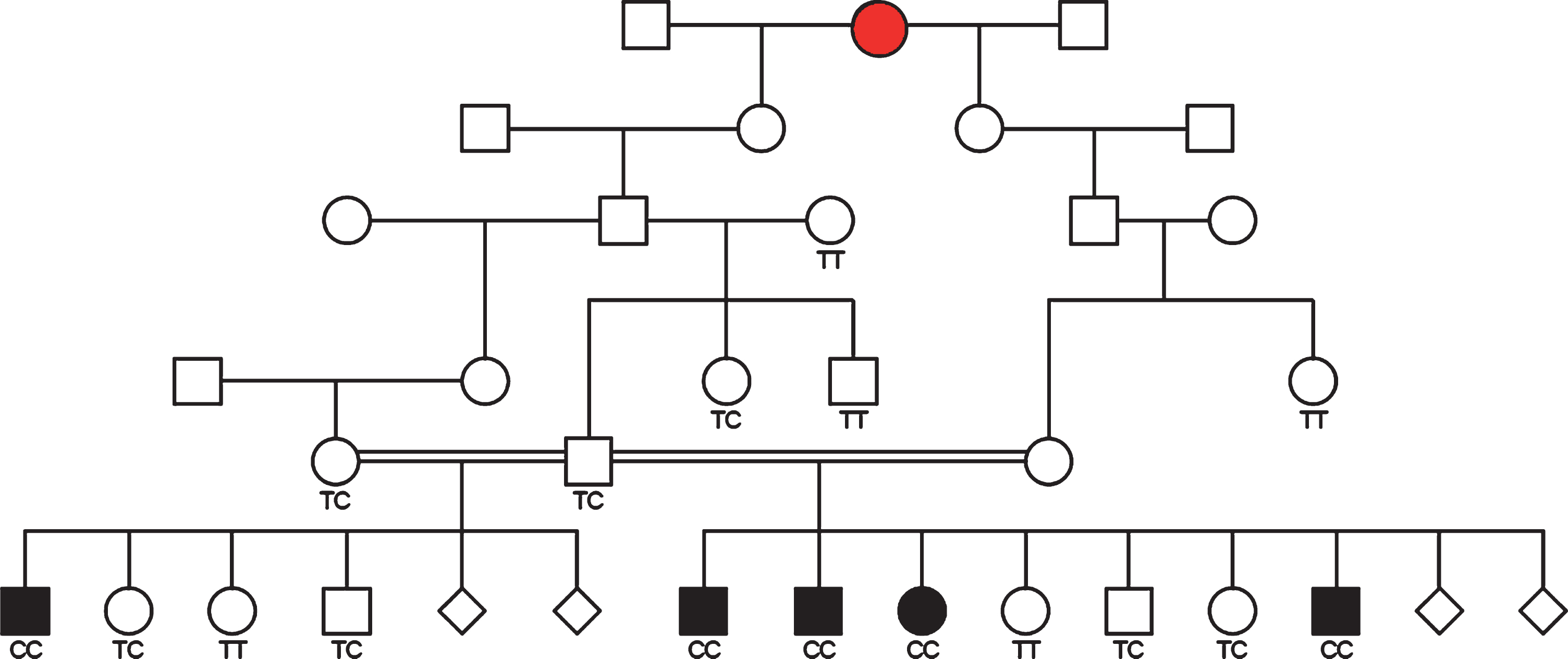
Fig. 3
Haplotypes from the region on CFA36 identified by homozygosity mapping. Included are the 5 cases with histopathology-confirmed myopathy (red) and 5 healthy controls >2 years old. The controls consisted of 2 of parents and 3 full-sibs (blue). Both haplotypes from each individual are provided, with 3 areas of homozygosity in the cases observed as clear areas underneath the nucleotide positions.
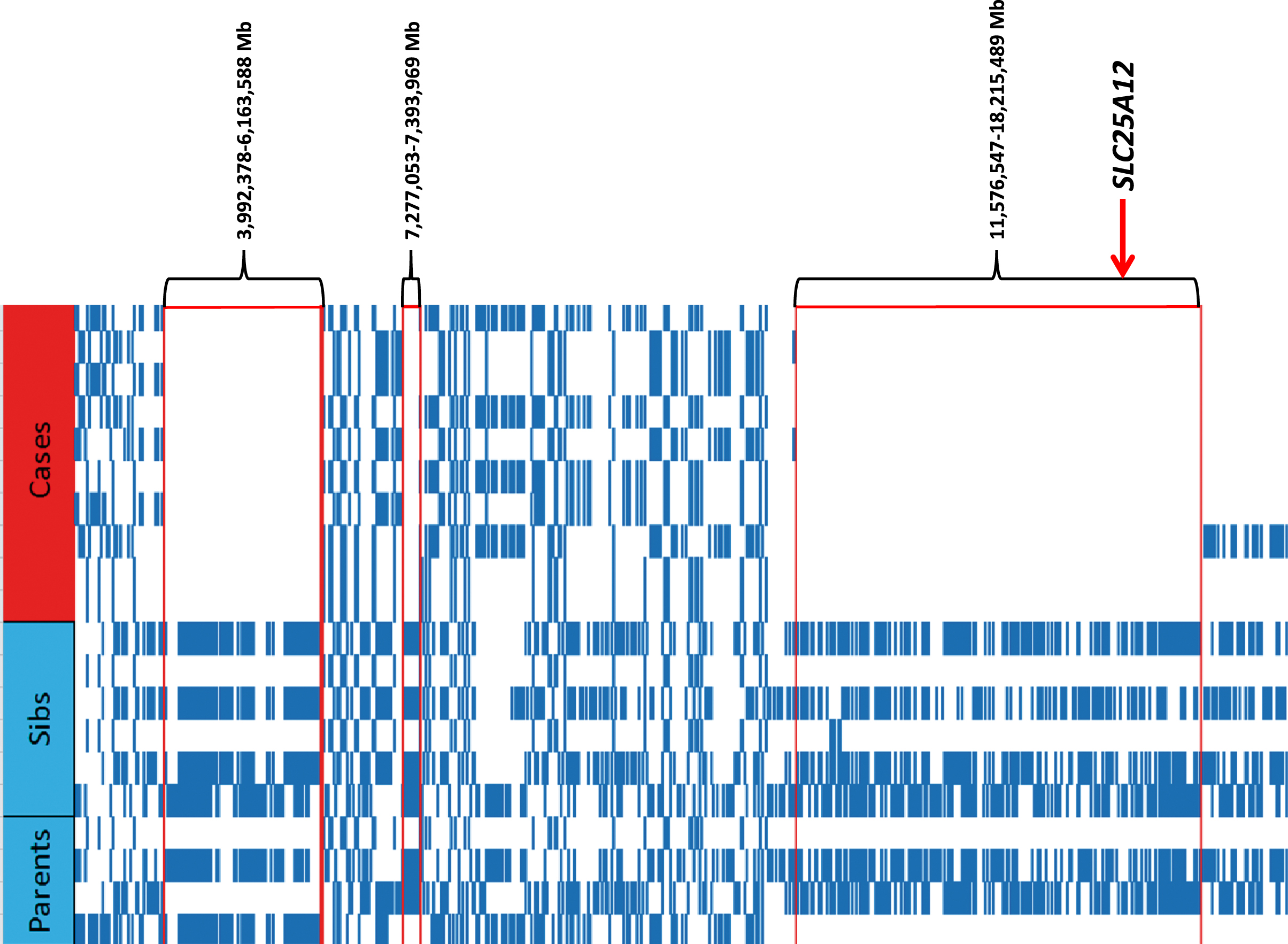
Using WGS from a case, and assuming autosomal recessive inheritance (keeping only homozygous variants), the number of potential causative variants in the region of interest on CFA36 was reduced to 17,522. From there, limiting the search to only those variants that resulted in a change to the protein coding sequence within genes in these regions left 31 variants. After filtering these protein-coding homozygous variants against a dog genetic variant database (see Methods) only one was unique to the sequenced case; a leucine to proline mutation (chr36:g.16,219,219A>G; c.1046T>C; p.L349P) within the SLC25A12 gene.
The p.L349P residue belongs to the first transmembrane α-helix of the carrier protein (Fig. 4A) and corresponds to the fourth residue of the signature motif PX[D/E]XX[K/R]X[R/K], which is characteristic of the mitochondrial carrier family, and is part of the matrix-gate area. Leucine at position 349 is strongly conserved in the gene homologues (Fig. 4B upper panel). Considering the amino acid frequency at the equivalent position in the 51 paralogues of Canis lupus familiaris, it is one of the most frequent amino acids along with Thr, Arg and Val (Fig. 4B lower panel). Proline is an alpha helix breaker and the leucine to proline variant would disrupt the alpha helix formation in this highly conserved area. Accordingly, the p.L349P mutation was predicted to be pathogenic by MutPred2 (score of 0.885, scale of 0.00 – 1.00), disease causing by SNPs&GO (score of 0.96, scale 0.00 – 1.00), and deleterious by Provean (score – 6.58, less than – 2.5 is considered deleterious).
Follow-up genotyping revealed that all 5 cases were homozygous for the mutant P349 allele, both available parents of the cases were heterozygous, 4 sibs to the cases were heterozygous, 2 sibs were homozygous for the L349 allele, and 4 first degree relatives were either heterozygous or homozygous for the reference allele (Fig. 3). 37 other siblings and close relatives were heterozygous or homozygous for the reference allele (not shown). The mutant allele was not found in 130 “unrelated” Dutch Shepherds from the USA and Europe, nor in 56 dogs from Dutch Shepherd related breeds (Belgian Malinois, Belgian Shepherd, Belgian Tervuren, German Shepherd) or breeds with a similar haplotype over the SLC25A12 locus (Border Collie, Australian Shepherd, Havanese).
The SLC25A12 variant resulted in marked impairment of transport activity and strong evidence for the pathogenicity of the mutation
To test the functional relevance of the identified mutation, the transport activity of the p.L349P AGC1 mutant was measured upon reconstitution of purified recombinant protein into liposomes. As shown in Fig. 4C, the substitution of L349 with proline causes a strong impairment of transport activity compared to WT AGC, as the mutant form of AGC1 was unable to transport aspartate or glutamate in exchange with internal glutamate. The differences in measured transport activity were not due to differences in protein content in liposomes between WT AGC1 and mutant p.L349P (data not shown). These results strongly argue in favor of the L to P substitution being pathogenic.
Fig. 4
Biochemical and bioinformatic analysis of the SLC25A12 variant. A. Structural homology model of the C. lupus C-terminal domain of the mitochondrial aspartate/glutamate carrier 1 (AGC1). The amino acid position of Leu349 that is mutated in Pro is shown in the box. B. Sequence alignment of AGC1 from different organisms (upper panel) and other members of the mitochondrial carrier family in Canis lupus familiaris (lower panel). C. Functional characterization of wild-type (WT) and mutated AGC1 form. The uptake rate of (14C) glutamate or (14C) aspartate was measured by adding 1 mM of radiolabeled glutamate or aspartate to liposomes reconstituted with purified WT AGC or with the mutant p.L349P and containing 20 mM of glutamate. The transport reaction was terminated after 1 min by adding 20mM pyridoxal 5′-phosphate and bathophenanthroline. The means and SDs from three independent experiments are shown.

Metabolomic analysis of skeletal muscle identified a proinflammatory milieu and strong support for oxidative stress
We further assessed the functional consequences of the AGC1/SLC25A12 mutation by performing a metabolomic analysis on muscle biopsy specimens from affected and control dogs. 383 of the 630 targeted metabolites were measurable in all muscle samples. Of the 383 measured metabolites, 52 were significantly different between affected and control samples. These 52 metabolites resulted from disturbances in 22 biochemical pathways that were then ranked by multivariate significance measured by VIP scores (Table 1). About 88% of the metabolic abnormalities resulted in an increase (46 of 52 metabolites), while about 12% of the abnormalities were from metabolites that were decreased (Table 1). The overall pattern of metabolic changes was visualized in Cytoscape (Fig. S1).
Table 1
Rank order of discriminating biochemical pathways
| No. | Pathway Name | Measured Metabolites in the Pathway (N) | Expected Pathway Proportion (P = N/383) | Expected Hits in Sample of 52 (P * 52) | Observed Hits in the Top 52 Metabolites | Fold Enrichment (Obs/Exp) | Impact (Sum VIP Scores) | Fraction of Impact Explained (% of 96.8) | Increased | Decreased |
| 1 | Phospholipid Metabolism | 75 | 0.196 | 10.2 | 13 | 1.3 | 25.63 | 26% | 12 | 1 |
| 2 | Glycosphingolipid Metabolism | 10 | 0.026 | 1.4 | 4 | 2.9 | 7.44 | 8% | 2 | 2 |
| 3 | Purine Metabolism | 24 | 0.063 | 3.3 | 4 | 1.2 | 6.48 | 7% | 4 | 0 |
| 4 | SAM, SAH, Methionine, Cysteine, Glutathione | 13 | 0.034 | 1.8 | 3 | 1.7 | 6.01 | 6% | 3 | 0 |
| 5 | Sphingomyelin Metabolism | 36 | 0.094 | 4.9 | 3 | 0.6 | 5.78 | 6% | 3 | 0 |
| 6 | GABA, Glutamate, Arginine, Ornithine, Proline | 5 | 0.013 | 0.7 | 3 | 4.4 | 5.07 | 5% | 3 | 0 |
| 7 | Tryptophan, Kynurenine, Serotonin, Melatonin | 5 | 0.013 | 0.7 | 2 | 2.9 | 4.50 | 5% | 2 | 0 |
| 8 | Krebs Cycle | 13 | 0.034 | 1.8 | 2 | 1.1 | 4.05 | 4% | 2 | 0 |
| 9 | Histidine, Histamine, Carnosine Metabolism | 4 | 0.010 | 0.5 | 2 | 3.7 | 3.56 | 4% | 2 | 0 |
| 10 | Lysine Metabolism | 3 | 0.008 | 0.4 | 2 | 4.9 | 3.48 | 4% | 2 | 0 |
| 11 | Nitric Oxide, Superoxide, Peroxide Metabolism | 2 | 0.005 | 0.3 | 2 | 7.4 | 3.29 | 3% | 1 | 1 |
| 12 | 1-Carbon, Folate Metabolism | 6 | 0.016 | 0.8 | 1 | 1.2 | 2.59 | 3% | 1 | 0 |
| 13 | Amino-Sugar, Galactose, &Non-Glucose | 6 | 0.016 | 0.8 | 1 | 1.2 | 2.25 | 2% | 1 | 0 |
| 14 | Plasmalogen Metabolism | 7 | 0.018 | 1.0 | 1 | 1.1 | 1.83 | 2% | 0 | 1 |
| 15 | Pyrimidine Metabolism | 14 | 0.037 | 1.9 | 1 | 0.5 | 1.81 | 2% | 1 | 0 |
| 16 | Vitamin B1 (Thiamine) Metabolism | 2 | 0.005 | 0.3 | 1 | 3.7 | 1.70 | 2% | 1 | 0 |
| 17 | Cardiolipin Metabolism | 8 | 0.021 | 1.1 | 1 | 0.9 | 1.67 | 2% | 1 | 0 |
| 18 | Vitamin C (Ascorbate) Metabolism | 2 | 0.005 | 0.3 | 1 | 3.7 | 1.65 | 2% | 1 | 0 |
| 19 | Fatty Acid Oxidation and Synthesis | 39 | 0.102 | 5.3 | 1 | 0.2 | 1.64 | 2% | 0 | 1 |
| 20 | Bioamines and Neurotransmitter Metabolism | 7 | 0.018 | 1.0 | 1 | 1.1 | 1.62 | 2% | 1 | 0 |
| 21 | Glycolysis and Gluconeogenesis Metabolism | 11 | 0.029 | 1.5 | 1 | 0.7 | 1.61 | 2% | 1 | 0 |
| 22 | Microbiome Metabolism | 12 | 0.031 | 1.6 | 1 | 0.6 | 1.60 | 2% | 1 | 0 |
| 23 | Urea Cycle | 2 | 0.005 | 0.3 | 1 | 3.7 | 1.54 | 2% | 1 | 0 |
| Subtotal | 46 | 6 | ||||||||
| Total | 52 |
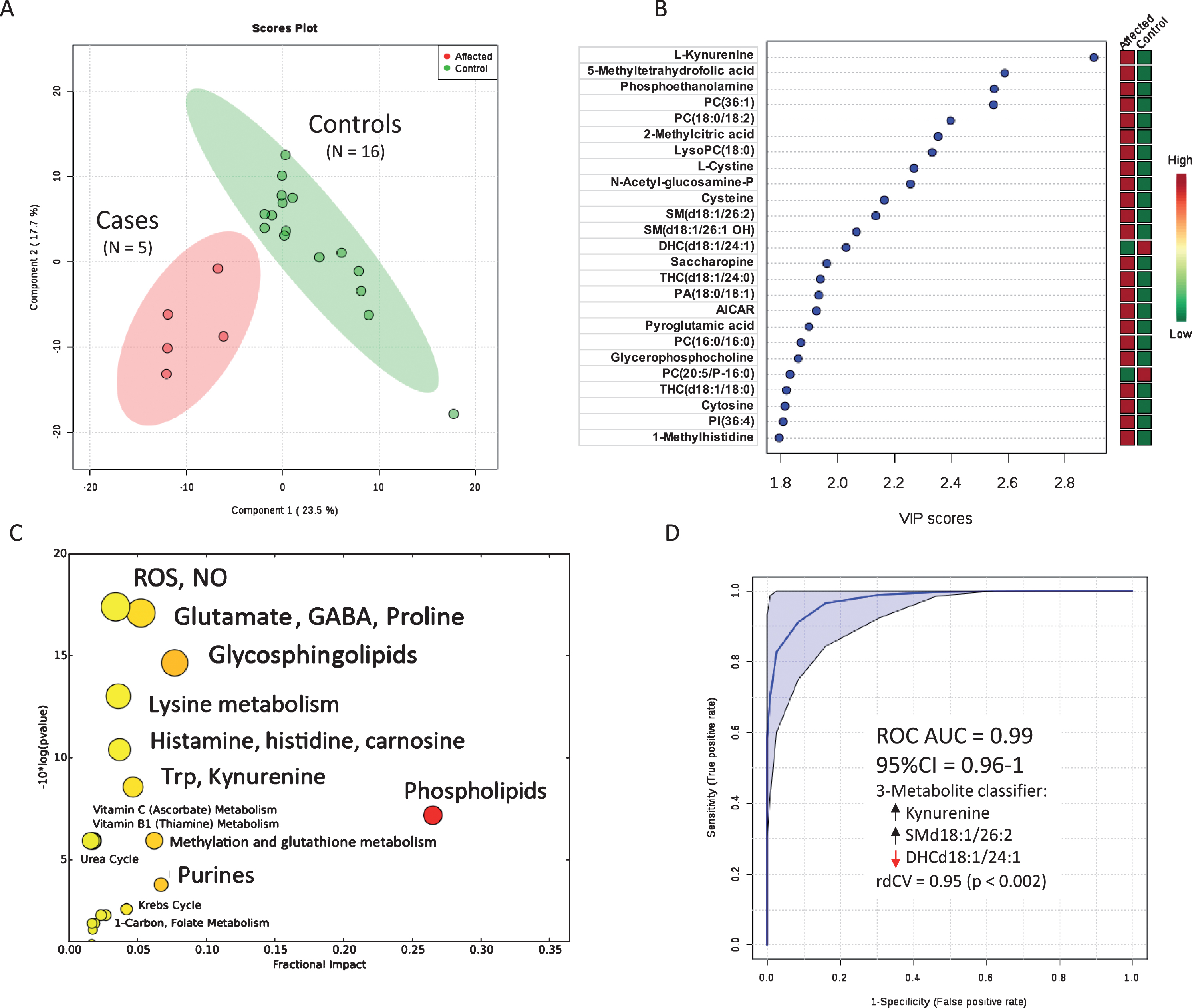
Multivariate analysis of the metabolomic results showed a clear separation of affected and control muscle metabotypes (Fig. 5A). The top 25 metabolite differences were then ranked by VIP scores (Fig. 5B). The observed changes in metabolomic profile can be divided into primary and secondary effects. Summaries of the specific primary and secondary metabolic differences, according to pathways, are as follows.
Fig. 5
Metabolic features of skeletal muscle samples of Dutch shepherds with the p.L349P AGC1 mutation. A. Metabolic discrimination of cases and controls by multivariate analysis. B. Rank order of metabolite differences. C. Bubble plot of significance and fraction impact of specific metabolic pathways. D. Receiver operator characteristic (ROC) curve analysis of the accuracy of 3 metabolites as a diagnostic classifier to distinguish cases and controls. 3- metabolites (increased kynurenine, increased sphingomyelin SM(d18:1/26:2), and decreased dihexosylceramide DHC(d18:1/24:1) showed over 90% accuracy in this sample (N = 5 cases and 16 controls). Repeated double cross validation (rdCV) with bootstrapping 100 times showed an accuracy of 0.95 (p < 0.002).
Glutamate, GABA, aspartate and lysine
A primary effect of the AGC1 mutation was an increase in muscle glutamate and glutamine (Table 1, Fig. 5C and Fig. 6 A,B). These results can be correlated with the inhibition of glutamate transport and reducing potential in the form of NADH from the cytosol into mitochondria via the malate aspartate shuttle. GABA was also increased (Fig. 6C). GABA is synthesized from glutamate by glutamate decarboxylase. No change in aspartate was found (Fig. 6D). Two products of intramitochondrial lysine metabolism, saccharopine and aminoadipic acid, were increased (Fig. 6L). The oxidation of lysine in mitochondria can be used as an alternative source of NADH and FADH2 via the enzymes saccharopine dehydrogenase [24], and glutaryl CoA dehydrogenase, respectively.
Markers of oxidative stress
A major direct effect of the AGC1 mutation, which decreases the net import of reducing equivalents (NADH) into mitochondria, was an increase in mitochondrial redox potential. The pathway disturbance with the greatest statistical support was reactive oxygen, nitrogen, and sulfur species (Fig. 5C). Both cysteine and its oxidized disulfide cystine were increased (Fig. 6E,F). Oxidized glutathione, the disulfide of the tripeptide of glutamate, cysteine, and glycine, was elevated (Fig. 6G), as were each of the three amino acids required for its synthesis. Pyroglutamate, also known as 5-oxoproline (Fig. 6H), formed in the glutathione cycle via gamma-glutamyl cyclotransferase in regenerating and rapidly growing cells, was also found to increase. Asymmetric dimethylarginine (ADMA) is a natural inhibitor of nitric oxide synthase and NO production, and is typically increased under conditions of chronic NO synthesis. Dimethylarginine was increased in affected muscle samples (Figs. 5, 6I). Kynurenine was the single most increased metabolite measured by multivariate analysis (Figs. 5B, 6J). Kynurenine is produced via the inflammatory activation of the indoleamine 2,3-dioxygenase (IDO) pathway of tryptophan metabolism. Allantoin, the oxidized product of uric acid, was also increased (Figs. 5, 6K).
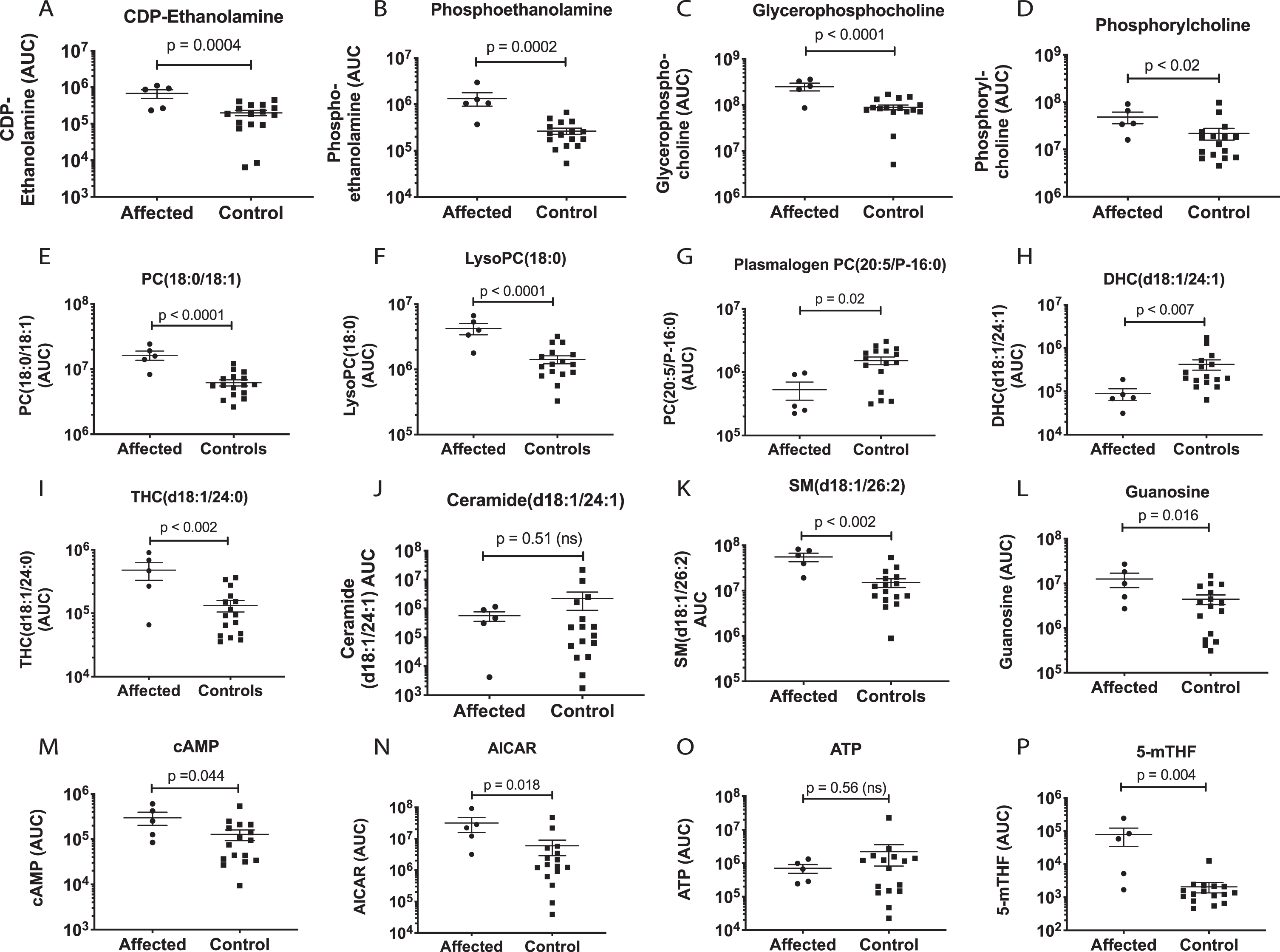
Purines and pyrimidines
Purines were heavily impacted by the AGC1 mutation (Table 1, Fig. 6). The release of purine and pyrimidine nucleotides like ATP and UTP from stressed cells is an early step in activating the cell danger response [25]. Once in the extracellular space, these nucleotides and their metabolites can activate up to 19 purinergic receptors that play an important role in neuroprotection, inflammation, and neurodegeneration [26]. Muscle guanosine (Fig. 7L), cyclic AMP (cAMP, Fig. 7M), and AICAR (Fig. 7N) were increased, while ATP was unchanged (Fig. 7O).
Fig. 6
Metabolic effects on glutamate, redox, and inflammation pathways. P values were calculated by univariate, non-parametric Mann-Whitney U test.
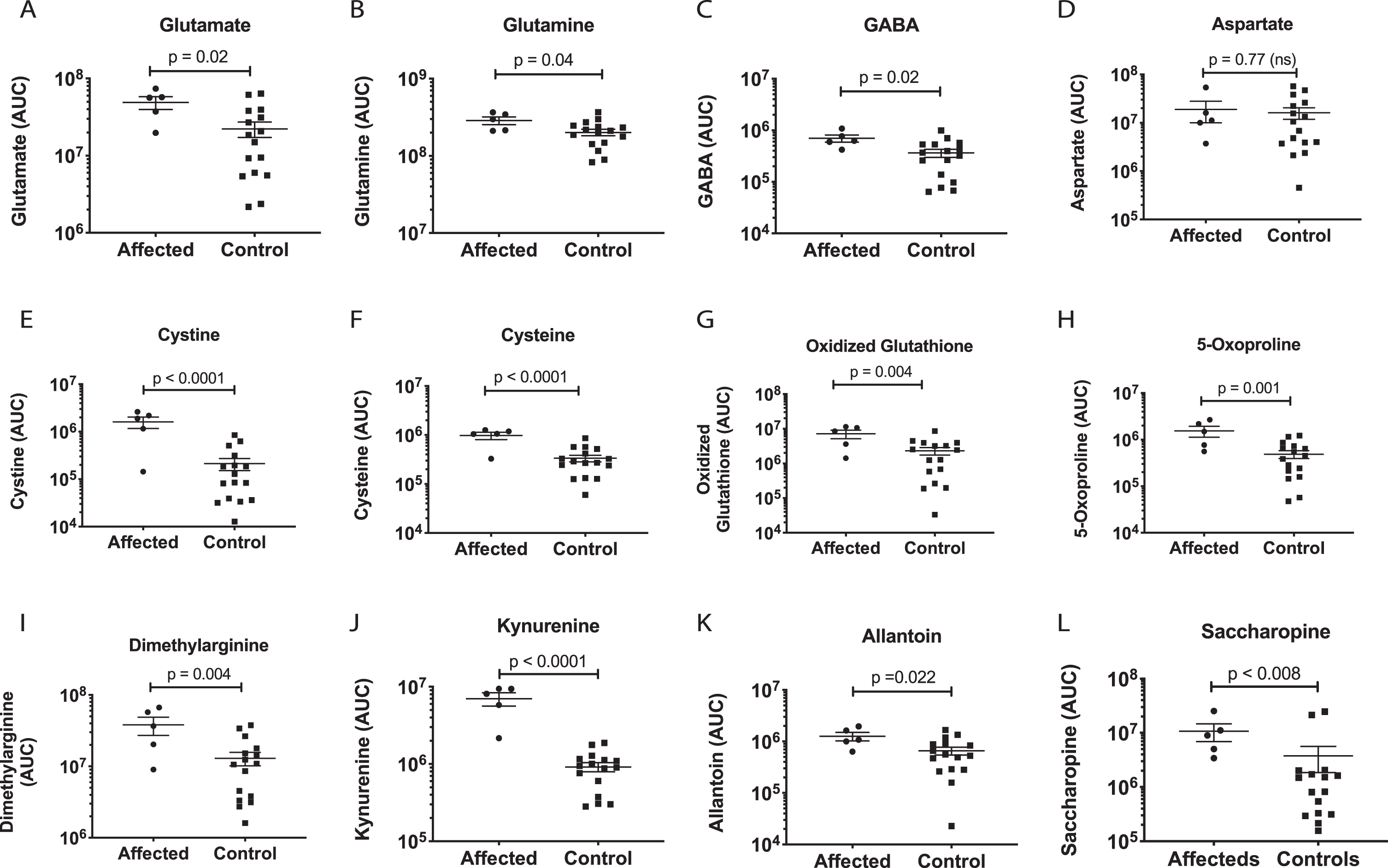
Fig. 7
Metabolic effects on phospholipid. sphingolipid, and purine pathways. P values were calculated by univariate, non-parametric Mann-Whitney U test.
Metabolomic Signatures of AGC1-Associated Myositis
We tested the utility of metabolomic analysis in characterizing the muscle biochemistry in AGC1 mutated vs control animals using receiver operator characteristic (ROC) curve analysis (Fig. 5D). In this small sample, we found that a classifier of just three metabolites including kynurenine (Fig. 6J), sphingomyelin d18:1/26:2 (SMd18:1/26:2; Fig. 7K) and dihexosylceramide d18:1/24:1 (DHCd18:1/24:1; Fig. 7H) could discriminate between cases and controls with an accuracy over 90% (Fig. 5D). Larger sample sizes would be needed to confirm these findings and to test for other possible predictive metabolomic combinations.
DISCUSSION
The SLC25 genes encode a superfamily of mitochondrial membrane-embedded transporters known as the mitochondrial carrier family or the SLC25 protein family [27, 28]. To date, 21 human inherited diseases associated with mitochondrial transporters of the SLC25 family resulting in metabolic diseases and defective mitochondrial energy production, have been well characterized biochemically and genetically [29–37]. The disorders are autosomal recessive except for two; a dominant progressive external ophthalmoplegia [29] and Gorlin-Chaudhry syndrome [33, 34].
The SLC25A12 gene encodes isoform 1 of the AGC that in humans is highly expressed in neurons, heart and skeletal muscle. The aspartate-glutamate carrier catalyzes the export of mitochondrial aspartate in exchange with cytosolic glutamate plus a proton across the inner mitochondrial membrane. Stimulated by cytosolic calcium [38], and together with the oxoglutarate carrier (OGC), AGC1 plays a key role in the malate-aspartate shuttle (MAS) which is the main biochemical pathway to transfer NADH reducing equivalents from the cytosol to mitochondria [39]. The MAS is essential for the glycolytic pathway by preserving the cytosolic redox state (NADH/NAD+ ratio) and thus contributes to oxidative metabolism and efficient ATP production.
It is clear from our functional transport and metabolomic studies that the p.L349P mutation in the SLC25A12 gene results in impaired function of the AGC1 and the MAS. As a result of this impaired transport, mitochondrial redox becomes more oxidizing with fewer reducing equivalents imported into the mitochondria via NADH. MAS defects lead to progressive intramitochondrial oxidation as NADH/NAD+ and NADPH/NADP+ ratios fall. The fall in intramitochondrial NADH/NAD+ ratio results in a highly oxidizing and proinflammatory muscle milieu. This is in contrast to the primary disorders of oxidative phosphorylation like complex I and complex IV deficiency leading to Leigh Syndrome [40], in which the mitochondria cannot consume the NADH that comes in and the intramitochondrial NADH/NAD+ ratio rises.
The extra electrons in the extramitochondrial cytosol creates a problem for the cell. Since mitochondria cannot import as much glutamate through AGC1, extramitochondrial glutamate levels increase. High extramitochondrial glutamate levels make the cells more prone to excitotoxic calcium release through glutaminergic pathways. Intracellular oxidative changes also promote the conversion of glutamate to its oxidized cyclic form, pyroglutamate (aka 5-oxoproline). Oxidation also increases oxidized glutathione (a marker for oxidative stress) and oxidized cystine. Further consequences of the failure to import adequate glutamate into mitochondria is the disposal of excessive glutamate and electrons via cytosolic pathways affecting phospholipid synthesis (ER) and glycosphingolipid synthesis (ER and Golgi).
Human patients carrying loss-of-function and hypomorphic SLC25A12 mutations present with central nervous system manifestations including developmental delay, epilepsy, hypotonia, and hypomyelination varying from a severe manifestation [41] to less severe symptoms [42]. The mutated residues [41, 42] are both highly conserved in aspartate glutamate carriers from different organisms, as is the mutated residue described here. All mutations cause an impairment of transport activity resulting from involvement in substrate binding [41] or in the closing and opening of the carrier at the matrix side [42 and this manuscript].
SLC25A12 knock-out mice show a milder phenotype with failure to thrive, progressive weakness, tremor, cerebral hypomyelination [43] and vision defects [44]. Physical and neurological examinations in the affected dogs were consistent with a primary neuromuscular disease without a history of seizures, behavioral changes or blindness. Brain imaging and thorough ophthalmologic examinations were not performed in the family of dogs described here so subtle CNS changes may have been missed. Interestingly, a congenital myasthenic syndrome associated with impaired neuromuscular transmission has been associated with another member of the SLC25 family, the mitochondrial citrate carrier. In a zebrafish model of this congenital myasthenic syndrome, malfunction of the neuromuscular junction was described along with variable involvement of the eye, brain and heart [45].
The variable clinical phenotypes may be explained by the different metabolic contexts rather than by the effects on carrier activity alone, which is virtually abolished in all cases. The primary localization of clinical signs to muscle in this family of affected dogs could be due to the highly oxidative nature of canine muscle metabolism and the relative paucity of classical type 2B (fast twitch glycolytic) muscle fibers in canine limb and trunk muscles [46, 47]. Fast type 2A fibers (fast twitch oxidative glycolytic) are generally associated with aerobic oxidative metabolism, and dog muscles are known to rely heavily on mitochondrial ATP production, a metabolic feature that contributes to the high resistance to fatigue in long-lasting exercise [46, 50]. In contrast, type 1 (slow twitch oxidative, fatigue resistant) and to a lesser extent type 2A fibers (both oxidative and glycolytic) rely on fast glucose utilization which requires an efficient MAS to shuttle the reducing equivalents formed in the cytosol into mitochondria. The impaired function of the AGC1 and the MAS resulting from the SLC25A12 mutation in this highly oxidative environment was shown in this study to result in marked oxidative stress and a proinflammatory muscle milieu. The effect on type 2B (glycolytic) fibers by the AGC1 mutation and impaired MAS should be minimal.
The findings in this study also have diagnostic and treatment implications. Based on results of histopathology and immunostaining, and before the p.L349P variant had been identified, a diagnosis of immune-mediated polymyositis had been made and immunosuppressive treatments initiated with no clinical improvement. The presence of CD3+, CD4+ and CD8+ T lymphocytes, and upregulation of major histocompatibility antigens MHCI and MHCII would be consistent with current clinical definitions of polymyositis [1]. The trigger(s) for immune-mediated diseases are currently not known, but unmasking of previously unexposed antigens, post-translational modifications, bystander effects or cross-reacting immune responses with infectious agents have been postulated. This study suggests that an underlying metabolic defect could result in a proinflammatory milieu and strong oxidative stress. This study also expands the spectrum of etiologies of inflammatory myopathies and pathologists should be aware of the possibility of an underlying mitochondrial disorder associated with myositis.
While AGC1 is a mitochondrial protein, its effects on the electron transport chain and oxidative phosphorylation are indirect and mediated by resulting metabolic changes. Transient myositis with mononuclear cell infiltration has been described in certain primary mitochondrial diseases [48]. However, chronic inflammatory myopathy is not a feature of any of the over 350 genetic forms of oxphos diseases [49]. In contrast to the classical mitochondrial disorders that result from mutations that directly affect the electron transport chain, several secondary mitochondrial disorders do present as a chronic inflammatory myopathy. Two of these are polymyositis with mitochondrial pathology, and sporadic inclusion body myositis (sIBM) [50]. Interestingly, a recent metabolomic analysis of sIBM identified many of the same metabolites that were found to be abnormal in this study. These included increased kynurenine, adenosine, cytosine, citrulline, dimethylarginine, and glutamate [51].
With regard to treatment, it is unlikely that any treatment would completely neutralize the metabolic effects of this mitochondrial myopathy, and in cases of autoimmune or inflammatory myopathy with mitochondrial abnormalities, it has been found that the response to immunosuppressive therapy is generally poor [52]. However, metabolomic analysis opens the possibility of a different approach to treatment. Given the strong impact on cellular redox, glutathione, and 1-carbon metabolism, the possibility exists that a treatment approach that includes CoQ10, N-acetylcysteine (NAC), carnitine, acetyl-L-carnitine (ALCAR), and alpha-lipoic acid (ALA) with or without folinic acid (leucovorin calcium), might mitigate or slow the clinical progression. The increased production of purines like adenosine, guanosine, and inosine that was found might contribute to the inflammatory milieu after release from cells through purinergic signaling. For example, if extracellular ATP is increased, P2X7 and P2Y receptors can be activated, leading to inflammation, T-cell recruitment, and Th17 dysregulation [53, 54]. If purinergic signaling is found to be an important driver of inflammation in the AGC1 mutant animals, antipurinergic therapy might represent an emerging opportunity for therapy [55–57].
Limitations
The small sample size of just 5 dogs carrying the homozygous p.L349P mutation in AGC1 limited the statistical power of the metabolomic analysis. Even so, all 52 reported metabolic differences had VIP scores above 1.5, and a mean false discovery rate of 24% (range = 0.001–0.37; Table S1). Another limitation was that the specific skeletal muscle biopsied varied among and within both affected and control animals. The frozen storage time at –80°C varied between a mean of 1.6 (±2.2) years for controls and 4.4 (±2.1) years (p < 0.02) for the affected samples.
CONCLUSIONS
The homozygous p.L349P substitution in the mitochondrial aspartate-glutamate carrier 1 (AGC1) occurred at a site that was evolutionarily conserved throughout eukaryotes (Fig. 4B), and was found to markedly decrease the functional transport activity of the expressed protein (Fig. 4C). In contrast to primary mitochondrial disorders that have a highly reducing intramitochondrial redox potential manifested as an increase in the NADH/NAD+ ratio, metabolomic analysis of AGC1 mutant muscle samples showed a strongly oxidizing intramitochondrial redox potential, with a decrease in the NADH/NAD+ ratio. Although classical histochemical staining for mitochondrial respiratory chain enzymes like cytochrome c oxidase and succinate dehydrogenase were normal, secondary mitochondrial abnormalities led to the development of a strongly oxidizing and proinflammatory muscle milieu.
SUPPORTING INFORMATION
Table S1. Discriminating metabolites. (XLS)
Table S2. Raw metabolomic data. (CSV)
Figure S1. Cytoscape map of disturbed metabolic pathways.
Supplemental Results and References. Covers metabolites in Figures 6 and 7 not covered in the main manuscript.
CONFLICTS OF INTEREST
All co-authors have nothing to declare. No conflicts or competing interests.
ETHICS APPROVALS
Not applicable.
ACKNOWLEDGMENTS
Support for this study was provided by grant GGP11139 from Comitato Telethon Fondazione Onlus and the MFAxMCD InterOmics Flag Project of the Consiglio Nazionale delle Ricerche. Support for the metabolomic analysis was provided in part by the UCSD Christini Fund. The authors thank Karen Wroblewski DVM, Allen Animal Hospital, Livonia, MI for bringing this family of dogs to our attention.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JND-190421.
REFERENCES
[1] | Shelton GD . From dog to man: The broad spectrum of inflammatory myopathies. Neuromuscul Disord. (2007) ;17: :663–70. |
[2] | Evans J , Levesque D , Shelton GD . Canine inflammatory myopathies: A clinicopathologic review of 200 cases. J Vet Intern Med. (2004) ;18: :679–91. |
[3] | Massey J , Rothwell S , Rusbridge C , et al. Association of an MHC Class II haplotype with increased risk of polymyositis in Hungarian VizslaDogs. PLoS One. (2013) ;8: :e56490. doi 10.1371/journpone.0056490 |
[4] | Dubowitz V , Sewry CA , Oldfors A . Histological and histochemical stains and reactions. In Dubowitz V , Sewry CA , Oldfors A eds. Muscle Biopsy: A practical approach. 4th ed. Oxford: Saunders Elsevier, (2013) , pp. 16–27. |
[5] | Guo LT , Moore SA , Forcales S , Engvall E , Shelton GD . Evaluation of commercial dysferlin antibodies on canine, mouse and human skeletal muscle. Neuromuscul Disord. (2010) ;20: :820–5. doi: 10.1016/j.nmd.2010.07.278 |
[6] | Becker D , Minor KM , Letko A . et al. A GJA9 frameshift variant is associated with polyneuropathy in Leonberger dogs. BMC Genomics. (2017) ;18: :662. doi: 10.1186/s12864-017-4081-z |
[7] | Purcell S , Neale B , Todd-Brown K , et al. PLINK, a tool set for whole-genome association and population-based linkage analysis. Am J Hum Genet. (2007) ;81: :559–75. |
[8] | Scheet P , Stephens M . A fast and flexible statistical model for large-scale population genotype data: Applications to inferring missing genotypes and haplotypic phase. Am J Hum Genet. (2006) ;78: :629–44. |
[9] | Pejaver V , Urresti J , Lugo-Martinez J . MutPred2: Inferring the molecular and phenotypic impact of amino acid variants. bioRxiv. (2017) ;134981. Doi: htpps://doi.org/10.1101/134981. |
[10] | Calabrese R , Capriotti E , Fariselli P , Martelli PL , Casadio R . Functional annotations improve the predictive score of human disease- related mutations in proteins. Hum Mutat. (2009) ;30: :1237–44. doi: 10.1002/humu.21047 |
[11] | Choi Y , Chan AP . PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. (2015) ;31: :2745–7. |
[12] | Fiermonte G , Dolce V , Palmieri L , Ventura M , Runswick MJ , Palmieri F , Walker JE . Identification of the human mitochondrial oxodicarboxylate carrier: Bacterial expression, reconstitution, functional characterization, tissue distribution and chromosomal location. J Biol Chem. (2001) ;276: :8225–30. |
[13] | Fiermonte G , Walker JE , Palmieri F . Abundant bacterial expression and reconstitution of an intrinsic membrane-transport protein from bovine mitochondria. Biochem J. (1993) ;294: :293–9. |
[14] | Palmieri F , Indiveri C , Bisaccia C , Iacobazzi V . Mitochondrial metabolite carrier proteins: Purification, reconstitution, and transport studies. Methods Enzymol. (1995) ;260: :349–69. |
[15] | Porcelli V , Fiermonte G , Longo A , Palmieri F . The human gene SLC25A29, of solute carrier family 25, encodes a mitochondrial transporter of basic amino acids. J Biol Chem. (2014) ;289: :13374–84. doi: 10.1074/jbc.M114.547448. Epub 2014 Mar 20. |
[16] | Li K , Naviaux JC , Bright AT , Wang L , Naviaux RK . A robust, single-injection method for targeted, broad-spectrum plasma metabolomics. Metabolomics. (2017) ;13: :122. doi 10.1007/s11306-017-1264-1. |
[17] | Xia J , Sinelnikov IV , Han B , Wishart DS . MetaboAnalyst 3.0-making metabolomics more meaningful. Nucleic Acids Res. (2015) ;43: :W251–7. doi: 10.1093/nar/gkv380. Epub 2015 Apr 20. |
[18] | Chong J , Soufan O , Li C , et al. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. (2018) ;46: (W1):W486–94. doi 10.1093/nar/gky310 |
[19] | Benjamini Y , Hochberg Y . Controlling the false discovery rate— a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B-Methodological. (1995) ;57: :289–300. |
[20] | Storey JD . The positive false discovery rate: A Bayesian interpretation and the q-value. The Annals of Statistics. (2003) ;31: :2013–35. |
[21] | Breiman L . Random forests. Machine Learning. (2001) ;45: :5–32. |
[22] | Filzmoser P , Liebmann B , Varmuza K . Repeated double cross validation. J Chemometrics. (2009) ;23: :160–71. |
[23] | Szymanska E , Saccenti E , Smilde AK , Westerhuis JA . Double-check: Validation of diagnostic statistics for PLS-DA models in metabolomics studies. Metabolomics. (2012) ;8: :3–16. |
[24] | Pink DB , Gatrell SK , Elango R , et al. Lysine alpha-ketoglutarate reductase, but not saccharopine dehydrogenase, is subject to substrate inhibition in pig liver. Nutr Res. (2011) ;31: :544–54. doi: 10.1016/j.nutres.2011.06.001 |
[25] | Naviaux RK . Metabolic features of the cell danger response. Mitochondrion. (2014) ;16: :7–17. |
[26] | Burnstock G . An introduction to the roles of purinergic signalling in neurodegeneration, neuroprotection and neuroregeneration. Neuropharmacology. (2016) ;104: :4–17. doi: 10.1016/j.neuropharm.2015.05.031. Epub 2015 Jun 6. |
[27] | Palmieri F . The mitochondrial transporter family (SLC25): Physiological and pathological implications. Pflugers Arch. (2004) ;447: :689–709. |
[28] | Palmieri F . The mitochondrial transporter family SLC Identification, properties and physiopathology. Mol Aspects Med. (2013) ;34: :465–84. doi: 10.1016/j.neuropharm.2015.05.031. Epub 2015 Jun 6. |
[29] | Palmieri F . Mitochondrial transporters of the SLC25 family and associated diseases: A review. J Inherit Metab Dis. (2014) ;37: :565–75. doi: 10.1007/s10545-014-9708-5. Epub 2014 May 6. |
[30] | Kishita Y , Pajak A , Bolar AJ , et al. Intra-mitochondrial methylation deficiency due to mutations in SLC25A26. Am J Hum Genet. (2015) ;97: :761–8. doi: 10.1016/j.ajhg.2015.09.013. Epub 2015 Oct 29. |
[31] | Shamseldin HE , Smith LL , Kentab A , et al. Mutation of the mitochondrial carrier SLC25A42 causes a novel form of mitochondrial myopathy in humans. Hum Genet. (2016) ;135: :21–30. doi: 10.1007/s00439-015-1608-8. Epub 2015 Nov 5. |
[32] | Schiff M , Veauville-Merllie A , Su CH , et al. SLC25A32 Mutations and riboflavin-responsive exercise intolerance. N Engl J Med. (2016) ;374: :795–7. doi: 10.1056/NEJMc1513610 |
[33] | Ehmke N , Graul-Neumann L , Smorag L , et al. De Novo mutations in SLC25A24 cause a craniosynostosis syndrome with hypertrichosis, progeroid appearance, and mitochondrial dysfunction. Am J Hum Genet. (2017) ;101: :833–43. doi: 10.1016/j.ajhg.2017.09.016 |
[34] | Writzl K , Maver A , Kovacic L , et al. De Novo Mutations in SLC25A24 cause a disorder characterized by early aging, bone dysplasia, characteristic face, and early demise. Am J Hum Genet. (2017) ;101: :844–55. doi: 10.1016/j.ajhg.2017.09.017 |
[35] | Boczonadi V , King MS , Smith AC , et al. Mitochondrial oxodicarboxylate carrier deficiency is associated with mitochondrial DNA depletion and spinal muscular atrophy-like disease. Genet Med. (2018) ;20: :1224–35. doi: 10.1038/s41436-019-0506-1 |
[36] | Khan S , Ansar M , Khan AK , et al. A homozygous missense mutation in SLC25A16 associated with autosomal recessive isolated fingernail dysplasia in a Pakistani family. Br J Dermatol. (2018) ;178: :556–6. doi: 10.1111/bjd.15661. Epub 2017 Dec 1. |
[37] | Punzi G , Porcelli V , Ruggiu M , et al. SLC25A10 biallelic mutations in intractable epileptic encephalopathy with complex I deficiency. Hum Mol Genet. (2018) ;27: :499–504. doi: 10.1093/hmg/ddx419 |
[38] | Palmieri L , Pardo B , Lasorsa FM , et al. Citrin and aralar1 are Ca(2+)-stimulated aspartate/glutamate transporters in mitochondria. EMBO J. (2001) ;20: :5060–9. |
[39] | Napolioni V , Persico AM , Porcelli V , Palmieri L . The mitochondrial aspartate/glutamate carrier AGC1 and calcium homeostasis: Physiological links and abnormaliaties in autism. Mol Neurobiol. (2011) ;44: :83–92. doi: 10.1007/s12035-011-8192-2. Epub 2011 Jun 21. |
[40] | Robinson BL , De Meirleir L , Glerum M , Sherwood G , Becker L . Clinical presentation of mitochondrial respiratory chain defects in NADH-coenzyme Q reductase and cytochrome oxidase: Clues to pathogenesis of Leigh disease. J Pediatr. (1987) ;10: :216–22. |
[41] | Wibom R , Lasorsa FM , Tohonen V , et al. AGC1 deficiency associated with global cerebral hypomyelination. N Engl J Med. (2009) ;361: :489–95. doi: 10.1056/NEJMoa0900591 |
[42] | Falk MJ , Li D , Gai X , et al. AGC1 deficiency causes infantile epilepsy, abnormal myelination, and reduced N-acetylaspartate. JIMD Rep. (2014) ;14: :77–85. doi: 10.1007/8904_2014_314 |
[43] | Jalil MA , Begum L , Contreras L . et al. Reduced N-acetylaspartate levels in mice lacking aralar, a brain- and muscle-type mitochondrial aspartate-glutamate carrier. J Biol Chem. (2005) ;280: :31333–9. |
[44] | Contreras L , Ramirez L , Du J , Hurley JB , Satrustegui J , de la Villa P . Deficient glucose and glutamine metabolism in Aralar/AGC1/Slc25a12 knockout mice contributes to altered visual function. Mol Vis. (2016) ;22: :1198–212. |
[45] | Chaouch A , Porcelli V , Cox D , et al. Mutations in the mitochondrial citrate carrier SLC25A1 are associated with impaired neuromuscular transmission. J Neuromuscul Dis. (2014) ;1: :75–90. |
[46] | Snow DH , Billeter R , Mascarello F , Carpene E , Rowlerson A , Jenny E . No classical type IIB fibres in dog skeletal muscle. Histochemistry. (1982) ;75: :53–65. |
[47] | Toniolo L , Maccatrozzo L , Patruno M , et al. Fiber types in canine muscles: Myosin isoform expression and functional characterization. Am J Physiol Cell Physiol. (2007) ;292: :1915–26. |
[48] | Mancuso M , Orsucci D , Lenco EC , Ricci G , Ali G , Servadio A , et al. An “inflammatory” mitochondrial myopathy. A case report. Neurouscul Disord. (2013) ;23: (11):907–10. doi: 10.1016/j.nmd.2013.07.011 |
[49] | McCormick EM , Muraresku CC , Falk MJ . Mitochondrial genomics: A complex field now coming of age. Curr Genet Med Rep. (2018) ;6: (2):52–61. doi: 10.1007/s40142-018-0137-x |
[50] | Papadimas GK , Kokkinis C , Xirou S , Chrysanthou M , Kararizou E , Papadopoulos C . Polymyositis with mitochondrial pathology or atypical form of sporadic inclusion body myositis: Case series and review of the literature. Rheumatol Int. (2019) ;39: (8):1459–66. doi: 10.1007/s40142-018-0137-x |
[51] | Buzkova J , Nikkanen J , Ahola S , Hakonen AH , Sevastianova K , Hovinen T . et al. Metabolomes of mitochondrial diseases and inclusion body myositis patients: Treatment targets and biomarkers. EMBO Mol Med. (2018) ;10: (12):pii:e9091. doi: 10.15252/emmm.201809091 |
[52] | Varadhachary AS , Weihl CC , Pestronk A . Mitochondrial pathology in immune and inflammatory myopathies. Curr Opin Rheumatol. (2010) ;22: (6):651–7. doi: 10.1097/BOR.0b013e32833f108a |
[53] | Ledderose C , Bao Y , Ledderose S , Woehrle T , Heinisch M , Yip L , et al. Mitochondrial dysfunction, depleted purinergic signaling, and defective T cell vigilance and immune defense. J Infect Dis. (2016) ;213: (3):456–64. doi: 10.1093/infdis/jiv373 |
[54] | Fernandez D , Flores-Santibanez F , Neifa J , Osorio-Barrios F , Tejon G , Nunez S , et al. Purinergic signaling as a regulator of Th17 cell plasticity. PloS One. (2016) ;11: (6):e0157889. doi: 10.1371/journal.pone.0157889 |
[55] | Naviaux RK . Metabolic features and regulation of the healing cycle-A new model for chronic disease pathogenesis and treatment. Mitochondrion. (2019) ;46: :278–97. doi: 10.1016/j.mito.2018.08.001 |
[56] | Allard B , Longhi MS , Robson SC , Stagg J . The ectonucleotidases CD39 and CD Novel checkpoint inhibitor targets. Immunol Rev. (2017) ;276: (1):121–44. doi: 10.1111/imr.12528 |
[57] | Eltzschig HK , Sitkovsky MV , Robson SC . Purinergic signaling during inflammation. New Eng J Med. (2012) ;367: (24):322–33. doi: 10.1056/NEJMra1205750 |


