
Safety and Treatment Effects of Nusinersen in Longstanding Adult 5q-SMA Type 3 – A Prospective Observational Study
Abstract
Objective:
Spinal muscular atrophy (SMA) is a progressive autosomal recessive motor neuron disease caused by loss of the SMN1 gene. Based on randomized clinical trials in children with SMA type 1 and 2, Nusinersen has been approved as the first treatment for all types of SMA, including adults with SMA type 3.
Methods:
We evaluated the safety and treatment effects of Nusinersen in longstanding adult 5q-SMA type 3. Patients were treated with intrathecal loading doses at day 1, 14, 28 and 63, followed by maintenance dose every four months up to 300 days. We monitored the patients within SMArtCARE, a prospective open-label outcome study for disease progression, side effects and treatment efficacy, encompassing clinical examination including MRC sum score, vital capacity in sitting position (VC, VC % pred.), ALS Functional Rating Scale (ALS-FRS), 6-Minute-Walk-Test (6MWT), Revised Upper Limb Module (RULM), and Hammersmith Functional Rating Scale (HFMSE). We also measured biomarkers in the spinal fluid (phosphorylated neurofilament heavy chain pNFH, neuron-specific enolase NSE, proteins, ß-Amyloid 1–40, ß-Amyloid 1–42, tau and phospho-tau) and creatine kinase (CK). Assessments were performed at baseline, day 63 (V4), day 180 (V5) and day 300 (V6). For statistical analysis, we compared baseline to V4, V5 and V6, using the paired sample t-test. When there were significant differences, we added cohen's d and effect size r for evaluation of clinical meaningfulness.
Results:
19 patients were included, 17 of them have completed the observation period of 10 months (day 300, V6). Patients were aged 18 to 59 years with disease duration ranging from 6 to 53 years. Except for the 6MWT, the RULM and the peak cough flow, there were no relevant significant changes in all functional outcome assessments at V4, V5 or V6, compared to baseline. For the 6MWT, there was a statistically significant improvement at visit 5 and at visit 6. RULM-score increased significantly at V6, and peak cough flow at visit 5. In biomarker studies, there was a significant decline in NSE and pTAU as well as a slight increase in proteins. In safety analysis, overall, Nusinersen applications were well tolerated. Eleven patients reported adverse events that were related to the study procedures, comprising back pain in seven patients and post-lumbar-puncture headache following intrathecal administration in four patients. Post-lumbar-puncture headache was reported in three females and one male, in total eleven times of 108 punctures (10%). No serious adverse events occurred.
Conclusions:
This prospective observational study indicates a mild treatment effect in adults with long-standing SMA3 after 10 months of treatment with Nusinersen, which had never occurred in the natural history of the disease. In our cohort, the most significant outcome measures were the 6MWT with statistically significant changes after day 180 and day 300, RULM after day 300 and peak cough flow after day 180.
INTRODUCTION
Spinal muscular atrophy is an autosomal recessive motor neuron disease caused by loss of the survival motor neuron 1 gene (SMN1) resulting in severe and progressive muscular atrophy and weakness. SMA has been categorized into five subtypes (0–4) based on the age of onset, the severity of motor decline and life expectancy. SMA3 comprises the milder end of the spectrum with an age of onset between 18 months and 3 years (type 3a), >age 3 years (type 3b), and >12 years of age (type 3c); patients typically achieve the motor milestone of walking (“walkers”) at least for some time in life [1]. Natural history studies have shown a slowly progressive decline of motor function across all subtypes [2]. Therapeutic development in SMA aims at increasing the full-length SMN protein by enhancing the SMN2 gene expression, by the inclusion of exon 7 in SMN2 transcripts, by stabilizing the SMN protein or by replacing the defect SMN1 gene [3]. In June 2017, FDA and EMA have approved the antisense oligonucleotide Nusinersen as the first treatment for all SMA subtypes without age restriction. Nusinersen alters the splicing of SMN2 pre-mRNA to increase the production of full-length SMN protein [4, 5]. Although most prominent treatment effects have been observed in the earlier stages of the disease and in patients up to 15 years of age [6], mouse models of adult spinal muscular atrophy demonstrated positive treatment effects in the later course of the disease [7]. However, so far, there is no data from clinical trials or real-life data in adult SMA3 patients; therefore, health care systems cover only the cost for SMA type 1 and 2 patients up to 18 years of age in numerous countries. Until now, it remains unclear which outcome measures are most suitable in adult patients for assessment of treatment effects. We report on treatment effects and safety during routine treatment of intrathecal administered Nusinersen in SMA3 patients ≥18 years of age with long-standing disease.
METHODS
Setting
In this prospective observational monocenter study, we evaluated the safety and treatment effects of Nusinersen in longstanding adult 5q-SMA type 3 patients. All Patients had a genetically confirmed diagnosis of 5q-SMA3 and were treated between October 2017 and May 2019. A current genetic report including SMN2 copy number was mandatory for treatment initiation. Data were collected during routine patient visits and items for data collection are aligned with the international consensus for SMA registries. All patients gave written informed consent.
Treatment schedule
All patients were treated with intrathecal loading doses of 12 mg Nusinersen at day 1 (Baseline), day 14 (visit 1), day 28 (visit 2) and day 63 (visit 3), followed by maintenance doses every four months (day 180, visit 5 and day 300, visit 6). Intrathecal injections were performed with standard access and without imaging control.
Outcome variables
Motor function
Patients were monitored within the SMArtCARE (www.smartcare.de) outcome study for disease progression, side effects and treatment efficacy by clinical examination and monitoring of respiratory function by assessment of vital capacity (VC). All data were collected during routine patient visits at the time points baseline, visit 4, visit 5 and visit 6. Items for data collection are aligned with the international consensus for SMA registries. Clinical assessment of motor function included Expanded Hammersmith Functional Rating Scale Expanded (HFMSE [8]), 6-Minute-Walk-Test (6MWT [9]), Revised Upper Limb Module (RULM [10]) and ALS Functional Rating Scale (ALSFRS [11]). For RULM, HFMSE and 6MWT, a physical therapist was trained within the SMArtCARE initiative. Additionally, Medical Research Council (MRC) scales (0–5) were used to assess muscle function [12].
Pulmonary function
Spirometry and measurement of peak cough flow (PCF) were performed in accordance with the standards and recommendations of the statement of the American Thoracic Society and the European Respiratory Society [13, 14]. For spirometry, three attempts for forced expiratory vital capacity (VC) were recorded (in litres and % predicted of normal) and the best test result in % predicted of normal was used for analysis. For PCF, three measurements were performed and the best test result in litres/min was used for analysis.
Laboratory testing
Biomarkers were tested at baseline, visit 4, visit 5 and visit 6. The cerebral fluid was analyzed for protein content, phosphorylated heavy chain neurofilament concentration (pNfH), neuron-specific enolase (NSE), β-amyloid 1–40 and 1–42 as well as tau peptides and phosphorylated tau peptides – all resembling specific biomarkers of neuroaxonal damage in neurodegenerative diseases [15, 16]. As there are limited or even no reports on other neurochemical markers in CSF in adult SMA patients other than pNfH, we have added NSE, ß-amyloid 1–40 and 1–42 as well as tau peptides and p-tau peptides. Overall, the selection of these biomarkers is based on the results of previous studies with neurodegenerative diseases. pNfH has been shown to be a reliable marker in motor neuron diseases, such as amyotrophic lateral sclerosis and SMA type 1 and 2 [15], but not in adult SMA3 [17]. The elevation of pNfH indicates an axonal loss and a correlation between neurofilament level and disease progression is suspected [15, 18]. NSE was previously described as a biomarker of axonal damage and as a prognostic factor for hypoxic encephalopathy and Creutzfeld-Jakob disease [19, 20]. Typical biomarkers for subcortical axonal degeneration are amyloid ß 1–40 and 1–42, tau-peptides and p-tau-peptides, and are known to be associated with dementia. For tau protein, it was recently shown in a mouse model and in vitro model of human spinal cord samples, that tau phosphorylation is significantly increased in SMA motor neurons, indicating that hyperphosphorylation of non-aggregating tau may contribute to motor neuron degeneration [21]. We also assessed creatine kinase (CK) levels at every time point.
Statistical analysis
Demographic and baseline data for medical history and SMA3 disease history were summarized. We calculated descriptive and exploratory analysis, where applicable. For comparing baseline to V4, V5 and V6, we used a paired sample t-test. When there were significant differences, we added cohen's d and effect size r for evaluation of meaningfulness. Correlation analysis was performed for the statistical relationship between SMN2 copies and age of onset and wheelchair dependency, using independent samples t-test or chi-square test, where applicable. Significance was set at a level of α≤0.05. For statistical analysis, we used SPSS Statistics 25 and Microsoft Excel 2016. Data analysis is carried out independently of commercial partners [22].
SAFETY
Safety evaluation was based on individual data for AEs, clinical laboratory findings, and vital sign parameters. Clinical laboratory tests included liver enzymes (AST, ALT, γGT), creatinine and creatine kinase (CK). Vital sign parameters included blood pressure, heart rate and peripheral oxygen saturation. Adverse events were collected from baseline until day 300 (visit 6) or end of participation plus one week. They were categorized by severity, seriousness and relationship to the administration of Nusinersen. In addition, all patients underwent one native cerebral magnetic resonance imaging (cMRI) scan during clinical routine after an official warning of Nusinersen-related communicating hydrocephalus in September 2018.
DATA AVAILABILITY
SMArtCARE is a joint initiative of neurologists, child neurologists, and patients’ organizations. The aim of SMArtCARE is to collect longitudinal “real-world data” on all available SMA patients independent of their actual treatment regime as disease-specific SMA registry. For this purpose, an online platform for SMA patients for health-care providers in German-speaking countries was provided [22].
RESULTS
We included 19 adult patients with genetically confirmed SMA3 into this prospective observational study. Two patients withdrew their consent after the fourth injection due to solely subjective safety concerns regarding hydrocephalus development and planned pregnancy and were therefore lost to follow up at visit 4 (Table 1).
Table 1
Patient’s demographics
| Demographics mean±SD (median; range) | smn2 3 copies (N = 4) | smn2 4 copies (N = 15) | total (N = 19) |
| Gender [female:male] | 1:3 | 6:9 | 7:12 |
| BMI [kg/m2] | 26.75±4.79 (25.50; 23–33) | 23.47±5.48 (23.00; 14–34) | 24.16±5.39 (23.00; 14–34) |
| age of onset [y] | 8.00±7.12 (7.00; 2–16) | 11.53±9.69 (12.00; 1–40) | 10.79±9.15 (12.00; 1–40) |
| age at diagnosis [y] | 13.25±6.90 (15.00; 4–19) | 22.53±13.12 (22.00; 5–50) | 20.58±12.53 (18.00; 4–50) |
| age at start of therapy [y] | 27.75±4.27 (29.00; 22–31) | 37.07±12.38 (39.00; 18–59) | 35.11±11.72 (34.00; 18–59) |
| Duration of disease [y] | 19.75±10.05 (25.5; 6–29) | 25.53±12.61 (27.00; 8–53) | 24.32±12.10 (25.0; 6–53) |
m = male; f = female; SMN2 = survival motor neuron 2; y = years; BMI = body mass index; SD = standard deviation; n = number.
Baseline demographics and characteristics
Twelve (63%) patients were male, seven (37%) female. At baseline, the median (SD) age was 34 years (11.72) ranging from 18 to 59 years at the beginning of therapy (mean 35 years). The median disease duration was 25.0 years (12.1) ranging from 6 to 53 years (mean 24.3 years). The median age at disease onset was 12 years (9.2) with a range from 1 to 40 years (mean 10.8 years) (Table 1, Fig. 1). Three patients suffered from scoliosis. None of our patients had previous spinal surgery with rod implantation; however, one patient had incomplete paraplegia of the legs, following a car accident in 2011 with T12 and L1 fractures. Seven patients (37%) were wheelchair-dependent, 12 (63%) ambulant (including one patient with the ability to walk independently only a few single steps). The genetic analysis detected three copies of SMN2 in four patients and four copies in 15 of our patients. Except for vital capacity % predicted of normal (p = 0.044), the SMN2 copy number did not correlate with any other assessment of baseline characteristics like age of onset (p = 0.860), BMI (0.852), the status of ambulation (p = 0.372), motor function tests or peak cough flow. In addition, serum biomarkers showed no significant difference between patients with 3 or 4 SMN2-copies (tables not provided). Baseline demographics, characteristics, motor function tests, biomarker assessments and statistical analyses are provided in Tables 1–3 and Fig. 1.
Fig.1
Age of symptom onset, age at diagnosis and duration of disease for patients with 3 and 4 SMN2 copies. Boxplots of symptom onset, age at diagnosis and duration of disease for patients with 3 and 4 SMN2 copies. Black circles indicate outliers with patient numbers. There were no significant differences between the groups with 3 or 4 SNM2 copies in the age of onset, age at diagnosis and disease duration until the start of first treatment with Nusinersen (α> 0.05).
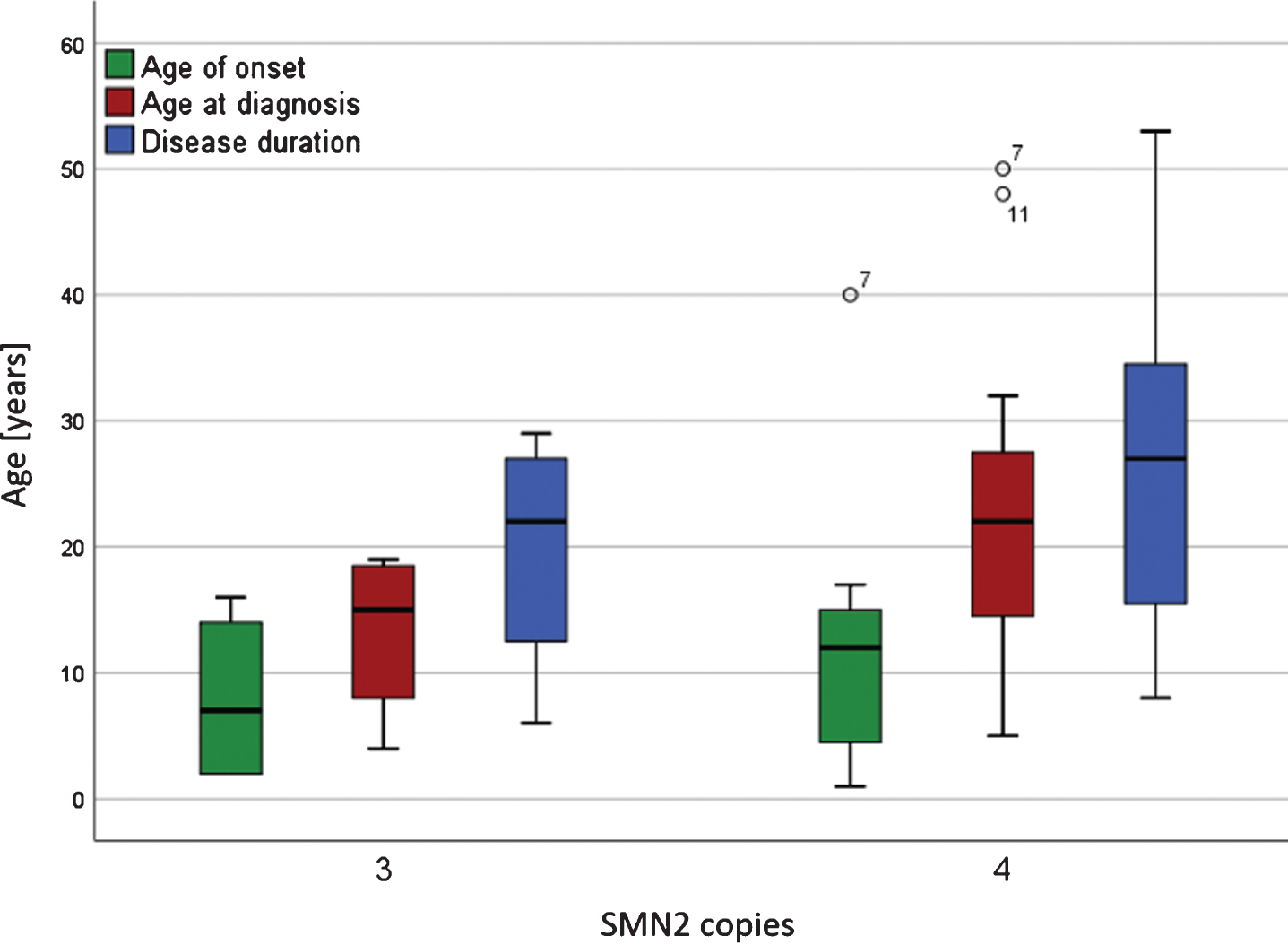
Table 2
Functional outcome measures at baseline, visit 4 (day 63), visit 5 (day 180) and visit 6 (day 300)
| Outcome measures Mean±SD (median; range; N) | baseline | V4 | V5 | V6 | p-value baseline-V4 | p-value baseline-V5 | p-value baseline-V6 |
| RULM [score] | 32.32±7.39 | 32.58±7.31 | 32.76±7.31 | 33.06±7.33 | 0.096 | 0.163 | 0.048 |
| (37.00; 11–37; 19) | (37.00; 12–37; 19) | (37.00; 12–37; 17) | (37.00; 12–37; 16) | d = 0.1; r = 0.05 | |||
| HFMSE [score] | 35.16±21.14 | 36.84±20.65 | 38.59±20.13 | 39.50±20.58 | 0.112 | 0.153 | 0.201 |
| (42.00; 0–64; 19) | (44.00; 0–64; 19) | (46.00; 0–64; 17) | (46.50; 100–613; 12) | ||||
| 6MWT[m] | 369.50±126.62 | 384.73±131.80 | 378.83±147.17 | 377.75±156.60 | 0.058 | 0.002 | 0.010 |
| (389.00; 181–550; 10) | (405.00; 175–608; 11) | (410.00; 135–610; 12) | (429.50; 400–613; 12) | d = 0.1; r = 0.03 | d = 0.1; r = 0.03 | ||
| ALSFRS [score] | 32.17±4.94 | 32.65±4.68 | 32.57±5.58 | 33.07±5.56 | 0.875 | 0.255 | 0.729 |
| (34.00; 23–38; 18) | (34.00; 23–83; 17) | (34.00; 20–39; 14) | (34.00; 20–39; 15) | ||||
| FVC [% ] | 94.54±15.45 | 96.31±16.50 | 98.52±14.48 | 99.54±12.42 | |||
| (91.8; 59–122; 17) | (92.80; 58–126; 19) | (94.70; 79–128; 17) | (96.25; 83–129; 16) | 0.317 | 0.329 | 0.335 | |
| Peak cough flow [l/min] | 376.11±183.12 | 430.00±160.48 | 461.54±147.24 | 435.63±125.23 | 0.311 | 0.044 | 0.285 |
| (345.00; 86–800; 14) | (355.00; 230–800; 14) | (450.00; 300–800; 13) | (450.00; 260–575; 8) | d = 0.5; r = 0.25 | |||
| MRC [sum score] | 113.95±22.91(115.00; | 115.37±22.67(115.00; | 117.65±24.85(119.00; | 118.50±24.63(119.50; | |||
| 46–154; 19) | 46–154; 19) | 46–154; 17) | 58–158; 16) | 0.490 | 0.209 | 0.214 |
m = meter; l = liter, n.d. = not done; RULM = Revised Upper Limb Module; HFMSE = Hammersmith Functional Motor Scale Expanded; 6MWT = 6-Minute Walk Test; ALSFRS = Amyotrophic Lateral Sclerosis Functional Rating Scale; FVC = Forced Vital Capacity.
Table 3
Biomarkers analyzed at baseline, visit 4 (day 63), visit 5 (day 180) and visit 6 (day 300)
| Outcome measures mean±SD (median; range; N) | Baseline | V4 | V5 | V6 | p-value baseline-V4 | p-value baseline-V5 | p-value baseline-V6 |
| βAmyloid 1–42 [pg/ml] | 795.06±308.55 | 825.26±262.74 | 811.35±304.24 | 838.87±280.35 | 0.705 | 0.721 | 0.688 |
| (786.73; 292.71–1470.00; 18) | (851.13; 360.28–1240.00; 19) | (784.87; 149.09–1570.00; 17) | (736.15; 459.68–1616.00; 16) | ||||
| βAmyloid 1–40 [pg/ml] | 10762.33±4296.74 | 11064.84±3080.05 | 10916.12±3583.42 | 11109.44±3612.89 | 0.863 | 0.672 | 0.771 |
| (11391.00; 1939.00–19171.00; 18) | (10560.00; 5940.00–16732.00; 19) | (10535.00; 3870.00–18324.00; 17) | (10836.50; 6214.00–21513.00; 16) | ||||
| TAU-protein [pg/ml] | 155.56±64.50 | 154.74±62.01 | 171.99±85.43 | 183.87±77.30 | 0.979 | 0.645 | 0.320 |
| (135.91; 100.00–364.20; 17) | (141.29; 100.00–366.66; 19) | (149.57; 100.00–450.69; 15) | (151.01; 119.08–421.50; 15) | ||||
| pTAU-protein [pg/ml] | 56.29±32.75 | 43.33±11.19 | 44.23±15.87 | 40.49±12.13 | 0.035 | 0.016 | |
| (46.88; 18.96–129.94; 17) | (43.36; 23.45–70.24; 18) | (44.31; 23.05–90.06; 16) | (36.28; 28.67–76.15; 15) | d = 0.5; r = 0.26 | 0.103 | d = 0.6; r = 0.31 | |
| NSE [ng/ml] | 10.96±3.20 | 10.36±4.00 | 9.88±4.16 | 9.43±2.76 | 0.013 | 0.007 | 0.005 |
| (10.40; 7.80–17.10; 11) | (8.00; 6.60–19.40; 17) | (7.60; 6.10–1940; 16) | (8.20; 6.30–14.40; 15) | d = 0.2; r = 0.08 | d = 0.3; r = 0.14 | d = 0.5; r = 0.25 | |
| pNfH [pg/ml] | <62,5 in all patients | <62,5 in all patients | <62,5 in all patients | 306.25±381.86 | |||
| (98.0; 63–966; 17) | n/a | n/a | n/a | ||||
| Protenis [mg/dl] | 37.39±12.07 | 38.05±11.62 | 40.41±15.51 | 41.07±13.26 | 0.141 | 0.053 | 0.31 |
| (34.00; 19.00–60.00; 18) | (34.00; 20.00–60.00; 19) | (36.00; 22.00–82.00; 17) | (43.00; 22.00–65.00; 15) | d = 0.2; r = 0.11 | d = 0.3; r = 0.14 | ||
| CK [U/l] | 448.68±375.74 | 419.00±307.30 | 486.12±354.40 | 490.25±358.13 | |||
| (350.00; 68.00–1419.00; 19) | (357.00; 70.00–1203.00; 19) | (413.00; 99.00–1321.00; 17) | (424.50; 98.00–1418.00; 16) | 0.445 | 0.861 | 0.644 |
pg = picograms, ml = millilitres, ng = nanogram, U = Units, l = liter, d = Cohen’s d, r = effect size r, pTAU-protein = phosphorylated Tau-protein, NSE = neuron-specific enolase, pNfH = phosphorylated neurofilaments heavy chain, CK = creatinekinase.
Baseline functional assessments
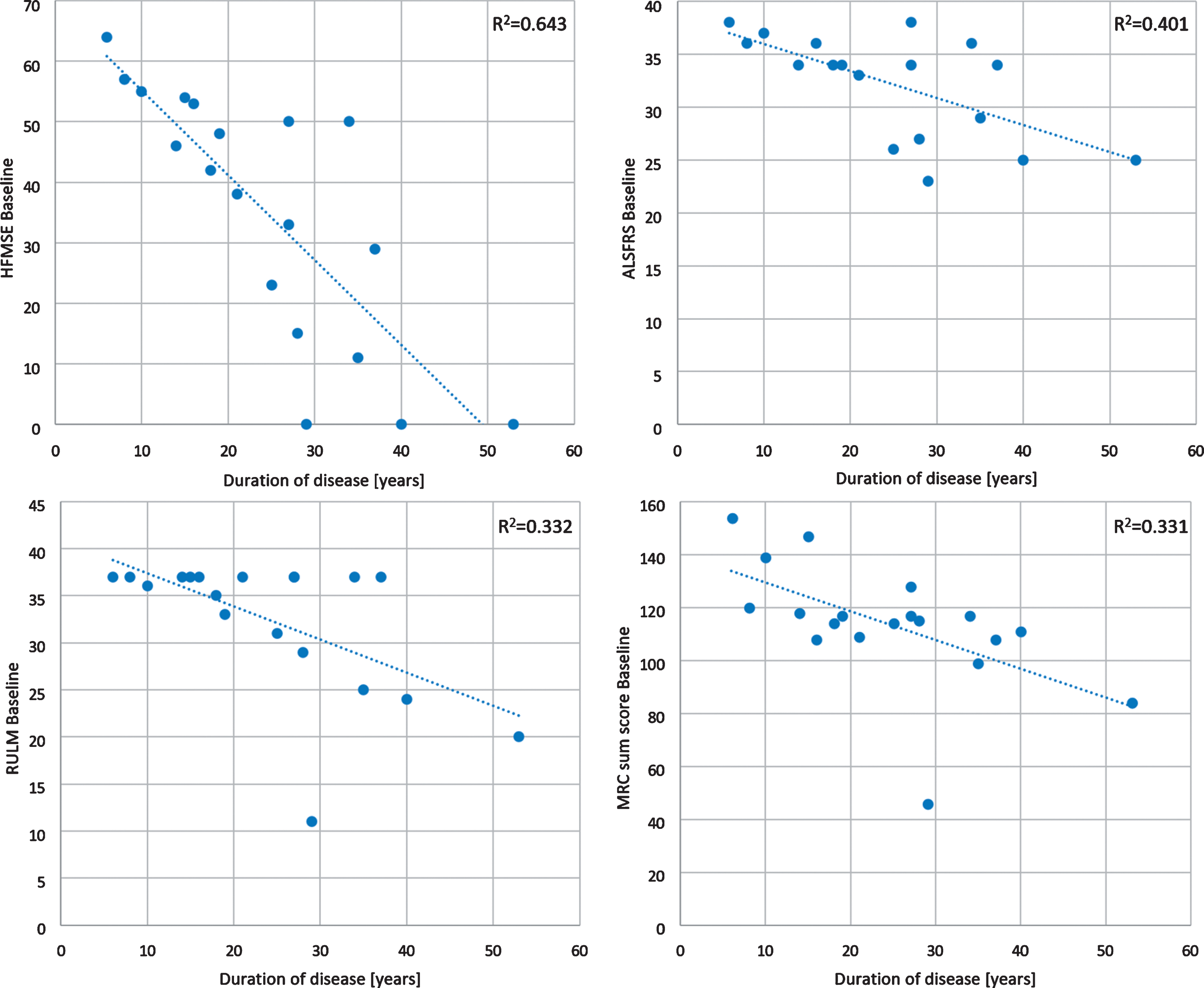
The HFMSE ranged from 11 to 64 points (median 47, SD 15.68, mean 42) and the RULM ranged from 11 to 37 points (median 37, SD 7.46, mean 33). The 6MWT was performed in 11 ambulant patients. Distances ranged from 181 to 550 m (median distance 389 m, SD 126.6, mean 370). Percent predicted of vital capacity ranged from 59% to 122% (median 92, SD 15.45, mean 95) (Table 2). A longer duration of the disease had a negative impact on motor function tests without significant difference between both groups with 3 or 4 SMN2 copies (p = 0.411). This was most evident in HFMSE (R2 = 0.643), lesser in ALSFRS (R2 = 0.401) and lowest in RULM and MRC sum score (R2 = 0.332 and R2 = 0.331) (Fig. 2).
Fig.2
Scatterplots of baseline motor function tests. RULM = Revised Upper Limb Module; HFMSE = Hammersmith Functional Motor Scale Expanded; ALSFRS = Amyotrophic Lateral Sclerosis Functional Rating Scale; MRC = Medical Research Council score for assessing muscle weakness. The MRC sum score was calculated by summing 14 muscles/regions, which ranges from 0 (complete paralysis) to 160 (normal strength): neck flexors and extensors, deltoid, biceps brachii, triceps brachii, hand extensors and hand flexors, finger extensors and finger flexors, iliopsoas, quadriceps, hamstring muscles, as well as foot extensors and flexors.
Safety
Intrathecal administration of Nusinersen was generally well tolerated; Post lumbar-puncture headache was reported in three females and one male, in total eleven times of 108 punctures (10%). 1 patient reported fatigue one time after one intrathecal application of Nusinersen. Seven patients suffered from lower back pain within 48 hours following intrathecal injection. None of the patients revealed clinical or radiological signs and symptoms of hydrocephalus communicans. Safety laboratory tests were unremarkable, especially without signs for nephrotoxicity.
Treatment effects
We could observe statistically significant improvements in the 6MWT when comparing baseline data with visit 5 (p = 0.002) and visit 6 (p = 0.010), but both with negligible effect sizes (d = 0.1 and r = 0.03, for both visits). The improvement in distance ranged from 24 m to 83 m in seven patients. The walking distance was stable in one patient (patient 17, 285 m), and two patients (patients 3 and 15) showed a non-significant decline at visit 6 (loss of 7 m and 14 m). The mean of the 6MWT was 403 meters (SD 136.23), ranging from 172 to 613 m. The mean improvement of 6MWT in our 11 ambulant patients was 8,25 m (median 40,5 m) (Fig. 3).
Fig.3
Boxplot 6MWT [meters]. Boxplots of the 6MWT at baseline, visit 4, visit 5 and visit 6. There were statistically significant differences between baseline compared to visit 5 and visit 6.
![Boxplot 6MWT [meters]. Boxplots of the 6MWT at baseline, visit 4, visit 5 and visit 6. There were statistically significant differences between baseline compared to visit 5 and visit 6.](https://ip.ios.semcs.net:443/media/jnd/2019/6-4/jnd-6-4-jnd190416/jnd-6-jnd190416-g003.jpg)
For the assessment of the upper limb module RULM, comparison between baseline and V4 and V5 did not change significantly, but V6 showed a significant improvement in this outcome measure, but also with a negligible effect size (p = 0.048; d = 0.1, r = 0.05). In detail, we could observe an improvement in 6/17 patients (3/11 ambulant with 1–2 score points, 3/6 non-ambulant with 1–2 score points), while nine patients (8 ambulant, 1 non-ambulant) remained stable. In two non-ambulant patients, RULM decreased by –1 to –2 points. In respiratory assessments, only PCF changed statistically significant when comparing baseline to visit 5 (p = 0.044) with a moderate effect size (d = 0.5; r = 0.25).
In summary, except for the 6MWT, the RULM in functional assessments and the PCF in respiratory assessments, there were no relevant significant changes in all other functional outcome measurements at V4, V5 or V6, compared to baseline (Table 2). Although there was no significant change in our data for HFMSE, in sub-analysis we could observe an improvement at visit 6 in 7 out of 17 patients with a maximum increase of 15 points. Four patients remained stable and five patients declined from –1 to –6 points. The median score increased from 47 to 49 (SD 14.63) with a range from 10 to 62 points. In addition, no relevant changes were seen in ALSFRS (Table 2) and MRC grading. Vital capacity ranged from 83%–129% of normal at baseline (median 96, SD 12.3) and did not change significantly during the observation period.
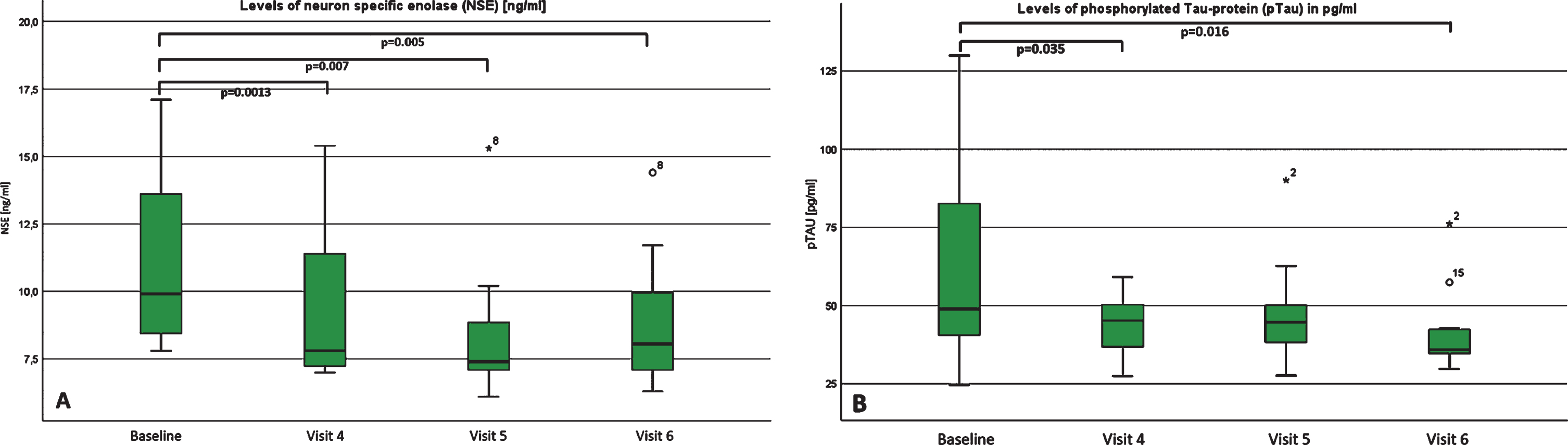
Most notably in biomarker assessments of cerebral fluid, levels of neuron-specific enolase (NSE) decreased continuously and showed a significant reduction at visit 4, 5 and 6, with a moderate effect size (for visit 6: p = 0.005; d = 0.5; r = 0.25)(Fig. 4a). While levels of tau-protein increased steadily but not significant, levels of phosphorylated Tau-protein decreased and were significantly changed at visit 4 and visit 6 with a moderate to high effect size (p = 0.016; d = 0.6; r = 0.3)(Fig. 4b). The levels of phosphorylated neurofilaments heavy chain (pNfH) were below the detection limit in all patients at baseline, visit 4 and visit 5. Interestingly, in four of our patients, pNfH levels became detectable at visit 6. In two of them, a negligible elevation of 66 pg/ml and 63 pg/ml was found. In one, we detected a mild elevation of 130 pg/ml and in another patient, a very high elevation of 966 pg/ml was found – which was clearly above the diagnostic cut-off level of 560 pg/mL for ALS. At this point, we cannot explain this high elevation with absolute certainty. Because of only 2 relevant elevations at visit 6 in all of our patients, we did not include statistical analysis. Also interestingly, the protein level increase was statistically significant, but the effect size was negligible (p = 0.031; d = 0.3; r = 0.14). ß-amyloid 1–40 and 1–42 and tau-protein remained almost unchanged. In addition, CK-levels in whole blood also did not show a statistically significant change over time.
Fig.4
A and B: Levels of (A) pTau and (B) NSE at baseline, visit 4, visit 5 and visit 6. Boxplots of the levels of (A) neuron specific enolase (NSE) and (B) phosphorylated TAU-Protein (pTau) at baseline, visit 4, visit 5 and visit 6, both measured in cerebral fluid. For pTAU, there was a significant decrease between baseline and visit 4 as well as visit 6. For NSE, we could detect a significant decrease in all visits compared to baseline.
Notably, tendon reflexes of the upper extremities became re-obtainable in two patients (patient 2 and 9) at visit 6, along with the ability to climb 12 steps without assistance in patient 2, and with improved stair-climbing performance in patient 9. Because this was only a clinical observation, we did not include any additional clinical tests or statistical analysis.
DISCUSSION
To our knowledge, this is the first report on a larger cohort on the safety and efficacy of Nusinersen treatment in adult 5q-SMA3 patients so far. In our cohort, there were no significant differences between the patient groups with three or four SMN2 copies as regards age of symptom onset, age of diagnosis and disease duration, but the phenotypic presentation at baseline differed considerably between the patients, irrespective of the copy number (seven were wheelchair-dependent and twelve were ambulant), and patient numbers were low. However, other disease-modifying factors may contribute to the progression of the disease. For evaluation of treatment effects, we performed well-established SMA validated outcome measures (RULM, HFMSE, 6MWT, MRC) over a treatment period of 300 days as well as assessments of pulmonary function and biomarkers in the cerebral fluid. As expected, a longer duration of the disease had a negative impact on motor function tests with the highest impact in HFMSE. This supports the general assumption, that an early start of treatment may be beneficial by decelerating the progression of muscle weakness. In our cohort, functional assessments revealed statistically significant improvements in the 6MWT at visits 5 and 6, the RULM at visit 6 and PCF at visit 5 - all indicating a trend to a mild improvement and/or stabilization of motor functions. Truly, since the effect size of the significant changes in motor function tests were all negligible, the improvements have to be interpreted with care. However, considering the chronic progressive deterioration in the natural history of the disease, stabilization or even improvement and respiratory function tests over such a short period of time is significant. Most importantly, 7 out of 11 ambulant patients achieved an improvement in the 6MWT between 31 and 83 m. Compared to other trials in chronic progressive muscle weakness, such as in Duchenne muscular dystrophy during eteplirsen treatment, or in children with spinal muscular atrophy following treatment with nusinersen, an improvement of 30 m had been defined as clinically meaningful [23, 24]. In a natural history cohort of 38 SMA3 patients, results of 6MWT ranged between 75 and 510 m (mean 294.91) at baseline and between 50 and 611 m (mean 293.41 m) at 12 months, similar to our cohort. The mean change in distance between the baseline and 12 months was –1.46, ranging from –183 to 131.8 meters. These changes were not correlated with age or baseline values even though younger patients reaching puberty had a relatively higher risk of showing the deterioration of more than 30 m compared to older patients [25]. In a larger cohort of 73 SMA3 patients, the overall mean change in 6MWT was –7.8 m/y, with a more prominent loss of –20.8 m/y in the age group of 11–19 years, and a milder decline of –9.7 m/y in the age group of >20 years [26]. However, improvement of 6MWT in SMA natural history cohorts has never been described so far, and no training effect has ever been described in natural history.
Although not statistically significant, there was an improvement of HFMSE score in 41% of patients and stability in 24% of patients. Natural history in mild SMA 2/3 phenotypes suggests a stable course of the disease over 12 months with a slow functional decline measured by HFMSE beyond one year of observation [27]. Cross-sectional and retrospective studies in SMA 2/3 have shown a mean loss of 0.5 HFSME points per year [2, 28], while a ≥3 point change in the HFMSE is usually seen as clinically meaningful [29]. Noteworthy, although statistically not significant, nine patients in our cohort showed improvement in HFSME, five of them with >3 points. In the majority of our patients, RULM showed stabilization.
Interestingly, age and disease duration did not have a negative impact on the treatment response; however, these findings may be due to a selection bias, since the milder end of the SMA3 spectrum was treated in our cohort. Even though this observation needs to be interpreted with caution with regard to the small sample size, the open design and the lack of statistical significance Nusinersen treatment in patients with a preserved good motor function seems to be worthwhile even in long-standing disease. This assumption is not only true with regard to stabilization of the disease but also aiming at an additional gain of motor function. These findings urgently need to be further evaluated within the SMArtCARE initiative within the next years in a larger patient cohort. A longitudinal observation of SMA3b patients revealed a slow deterioration by one MRC grade within five years [30]. However, MRC grades did not show relevant changes since the start of treatment in our cohort.
Since the therapy with Nusinersen has been approved for SMA across all types and age groups, randomized, placebo-controlled clinical trials may no longer be feasible and ethical and gain of scientific knowledge will largely depend on real-life data. Outcome measures must be sensitive enough to assess changes in the course of the disease in treated and untreated adult SMA3 patients; HFMSE, 6MWT proved valid across a broad range of SMA types, whereas RULM, MRC grading and ALS-FRS score seem to be too rough to detect changes in the same time intervals.
Post-lumbar-puncture headache was reported eleven times of a total of 108 punctures (10%) in a total of 4/19 patients (3 of them female), which is equal to previously described numbers in the literature (5–10% after an atraumatic puncture) [31, 32]. In our cohort, side effects decreased with ongoing injections. None of our patients developed a hydrocephalus communicans. Drop-outs in our study were solely for personal reasons, and not due to any treatment side effects.
We added biomarker assessments of cerebral fluid to assess effects of Nusinersen on axonal damage in adult SMA patients. Neurofilaments as a marker of neuroaxonal damage were below the measurable range in all our patients at baseline prior to therapy. This is in contrast to paediatric SMA patients, where strongly elevated pNfH levels were observed prior to therapy, which decreased with progression and age with and without therapy. However, the decrease was more rapid under Nusinersen therapy than in sham-injected controls, which led to the proposal of pNfH as a potential biomarker [33]. Interestingly, pNfH became detectable in four of our patients following treatment. One of them showed levels compatible with degenerative motor neuron disease in adults at 996 pg/ml at visit 6. This patient also presented newly obtainable reflex levels and improvement of 53 meters in 6MWT under treatment (patient 2). This observation raises doubt if the level of neurofilaments could serve as a prognostic or confirmative marker in terms of treatment response in adult patients with SMA. In addition, levels of NSE and pTAU-protein decreased continuously and showed a significant reduction at visit 6, also indicating an alleviation of axonal injury over time. Although the effect size was insignificant, a slight continuous increase was also seen for protein levels, which can be interpreted as an effect caused by recurrent trauma by the lumbar puncture. Overall, our biomarker studies provide evidence that neuroaxonal damage decreased under therapy. Consistent with previous studies, pNfH levels were not detectable at baseline in all of our patients. Notably, a relevant increase was found in two patients (130 pg/ml and 966 pg/ml) at visit 6. Since this only applied to two patients, it is hard to draw clear and substantial conclusions. Very recently, an increase of pNfH in a subgroup of treated SMA patients along with a slight increase of proteins and neurofilament light chain and without correlation to clinical assessments was observed by another study. The authors speculated, that this elevation may be due to the change in the CSF flow, which might be attributed to the Nusinersen injections [17]. As there were no significant changes in motor function tests in our two patients, deterioration of axonal loss seems to be unlikely. However, additional studies are needed to assess the significance and usefulness of neurochemical markers for treatment efficacy evaluation.
Our observation has several limitations; treatment duration is still too short to assess long-term treatment efficacy and sustainability of improvement, while our small cohort with heterogeneous phenotypes (ambulant and non-ambulant SMA3 patients with a wide range of disease duration) may impede the interpretation of our data. Nevertheless, even in SMA3 patients with a long-standing disease course up to 53 years and a median disease duration of 24 years, improvement of motor function exceeded our expectations of mere disease stabilization, justifying treatment beyond childhood and early adolescence.
The optimal time for a meaningful intervention is not yet fully elucidated, particularly at which point irreversibility of decline precludes any meaningful therapeutic response. Importantly, treatment response should not be based on the improvement of motor function alone, but focus on preventing all aspects of disease progression. This may include additional objective measurements of disease progression, like quantitative high-resolution muscle ultrasound or MRI of muscle and peripheral nerve to indicate early nerve tissue changes and to detect and measure the progression of muscular atrophy and nerve tissue changes in response to the treatment [34–36]. In addition, suitable questionnaires for patient-related outcomes have to be developed to assess the efficacy of future treatments. The clinical benefit in patients with the long-standing disease will have to be further evaluated regarding efficacy and side effects in the years to come; therefore, real-world data in adult SMA3 will be crucial to justify ongoing treatment.
FINANCIAL DISCLOSURES
Simone Thiele, Julia Stauber, Miriam Hiebeler, Eva Greckl, Kristina Stahl report no disclosures.
Astrid Pechmann received funding from Biogen for educational activities.
Hanns Lochmüller: Consultancy and financial support for research projects and clinical trials from AMO Pharma, Biogen, Desitin, GW Pharma, Pfizer, PTC Therapeutics, Roche, Santhera, Sarepta, Satellos and Ultragenyx. Editor-in-chief for the Journal of Neuromuscular Disease (IOS Press)
Benedikt Schoser has served on advisory boards for Amicus Therapeutics, Audentes, Nexien, Lupin, Valerion, Vertex, and Sanofi Genzyme; he has undertaken contracted unrestricted research for Greenovation Biopharm, Sanofi Genzyme, Nexien; and has received honoraria from Octapharm and Kedrion, and travel expenses from Amicus and Sanofi Genzyme.
Janbernd Kirschner received funding from Avexis, Biogen, and Roche for advisory activities and clinical research activities related to spinal muscular atrophy.
Maggie C. Walter has served on advisory boards for Avexis, Biogen, Novartis, Roche, Santhera, Sarepta, PTC Therapeutics, Ultragenyx, Wave Sciences, received funding for Travel or Speaker Honoraria from Novartis, Biogen, Ultragenyx, Santhera, PTC Therapeutics, and worked as an ad-hoc consultant for AskBio, Audentes Therapeutics, Biogen Pharma GmbH, Fulcrum Therapeutics, GLG Consult, Guidepoint Global, Gruenenthal Pharma, Novartis, PTC Therapeutics.
Stephan Wenninger has served on advisory boards for Alexion Pharma, CSL Behring and Sanofi Genzyme GmbH. He received funding for Travel or Speaker Honoraria from Sanofi-Aventis Germany GmbH; SH Glykogenose Gesellschaft, Germany; AbbVie Germany GmbH; Recordati Pharma GmbH, Germany; CSL-Behring GmbH, Germany; Alexion Pharma Germany GmbH; Desitin Germany.
STATISTICAL ANALYSIS
Stephan Wenninger, Kristina Stahl.
STUDY FUNDED
n.a.
Appendices
Appendix 1.
Authors
| Name | Location | Role | Contribution |
| Maggie C. Walter | Friedrich-Baur-Institute, Ludwig-Maximilians-University of Munich | Corresponding Author | Analysis and interpretation of data, manuscript draft, critical revision of the manuscript for intellectual content, final manuscript draft |
| Stephan Wenninger | Friedrich-Baur-Institute, Ludwig-Maximilians-University of Munich | Author | Statistical analysis and interpretation of data, discussion of results, critical revision of the manuscript for intellectual content, final manuscript draft |
| Simone Thiele | Friedrich-Baur-Institute, Ludwig-Maximilians-University of Munich | Author | Patient acquisition and examination, acquisition of data, analysis and interpretation of data, final manuscript draft |
| Julia Stauber | Friedrich-Baur-Institute, Ludwig-Maximilians-University of Munich | Author | Patient acquisition and examination, acquisition of data, analysis and interpretation of data, first manuscript draft |
| Miriam Hiebeler | Friedrich-Baur-Institute, Ludwig-Maximilians-University of Munich | Author | Patient acquisition and examination, acquisition of data, critical revision of the manuscript for intellectual content |
| Eva Greckl | Friedrich-Baur-Institute, Ludwig-Maximilians-University of Munich | Author | Patient scoring, acquisition of data, critical revision of the manuscript for intellectual content |
| Kristina Stahl | Friedrich-Baur-Institute, Ludwig-Maximilians-University of Munich | Author | Statistical analysis and interpretation of data, discussion of results |
| Astrid Pechmann | Dept, of Neuropediatrics and Muscle Disorders, Medical Center-University of Freiburg, Faculty of Medicine, University of Freiburg | Author | Analysis and interpretation of data, critical revision of the manuscript for intellectual content |
| Hanns Lochmüller | Children’s Hospital of Eastern Ontario Research Institute; Division of Neurology, Department of Medicine, The Ottawa Hospital; and Brain and Mind Research Institute, University of Ottawa, Ottawa, Canada | Author | Analysis and interpretation of data, discussion of results, critical revision of the manuscript for intellectual content |
| Janbernd Kirschner | Dept, of Neuropediatrics and Muscle Disorders, Medical Center-University of Freiburg, Faculty of Medicine, University of Freiburg | Author | Analysis and interpretation of data, discussion of results, critical revision of the manuscript for intellectual content |
| Benedikt Schoser | Friedrich-Baur-Institute, Ludwig-Maximilians-University of Munich | Author | Analysis and interpretation of data, discussion of results, critical revision of the manuscript for intellectual content |
ACKNOWLEDGMENT
We thank our patients and their families for their cooperation and contribution. We thank H. Saodiy, I. Hirschmann and F. Walter for scientific assistance and database research.
REFERENCES
[1] | Farrar MA , et al. Emerging therapies and challenges in spinal muscular atrophy. Ann Neurol. (2017) ;81: (3):355–68. |
[2] | Wadman RI , et al. Muscle strength and motor function throughout life in a cross-sectional cohort of 180 patients with spinal muscular atrophy types 1c-4. Eur J Neurol. (2018) ;25: (3):512–8. |
[3] | Bowerman M , et al. Therapeutic strategies for spinal muscular atrophy: SMN and beyond. Dis Model Mech. (2017) ;10: (8):943–54. |
[4] | Finkel RS , et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. (2017) ;377: (18):1723–32. |
[5] | Mercuri E , et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N Engl J Med. (2018) ;378: (7):625–35. |
[6] | Day J , Swoboda K , Darras B . Nusinersen experience in teenagers and and young adults with spinal muscular atrophy. in Muscular Dystrophy Association Clinical Conference, (2018) . |
[7] | Feng Z , et al. Pharmacologically induced mouse model of adult spinal muscular atrophy to evaluate effectiveness of therapeutics after disease onset. Human Molecular Genetics. (2016) ;25: (5):964–75. |
[8] | O’Hagen JM , et al. An expanded version of the hammersmith functional motor scale for SMA II and III patients. Neuromuscul Disord. (2007) ;17: (9-10):693–7. |
[9] | Dunaway S , et al. Physical therapy services received by individuals with spinal muscular atrophy (SMA). J Pediatr Rehabil Med. (2016) ;9: (1):35–44. |
[10] | Mazzone ES , et al. Revised upper limb module for spinal muscular atrophy: Development of a new module. Muscle Nerve. (2017) ;55: (6):869–74. |
[11] | Bakker LA , et al. Assessment of the factorial validity and reliability of the ALSFRS-R: A revision of its measurement model. J Neurol. (2017) ;264: (7):1413–20. |
[12] | Compston A . Aids to the investigation of peripheral nerve injuries. Medical Research Council: Nerve Injuries Research Committee. His Majesty’s Stationery Office: 1942, p. 48 (iii) and 74 figures and 7 diagrams; with aids to the examination of the peripheral nervous system. By Michael O’Brien for the Guarantors of Brain. Saunders Elsevier: 2010, p. [8] 64 and 94 Figures. Brain. (2010) ;133: (10):2838–44. |
[13] | Aggarwal AN , Agarwal R . The new ATS/ERS guidelines for assessing the spirometric severity of restrictive lung disease differ from previous standards. Respirology. (2007) ;12: (5):759–62. |
[14] | Criée C , et al. Leitlinie zur spirometrie leitlinie der deutschen atemwegsliga, der deutschen gesellschaft für pneumologie und beatmungsmedizin und der deutschen gesellschaft für arbeitsmedizin und umweltmedizin zur spirometrie. Pneumologie. (2015) ;69: (3):147–64. |
[15] | Lehnert S , et al. Multicentre quality control evaluation of different biomarker candidates for amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. (2014) ;15: (5-6):344–50. |
[16] | Steinacker P , et al. Neurofilaments in the diagnosis of motoneuron diseases: A prospective study on 455 patients. J Neurol Neurosurg Psychiatry. (2016) ;87: (1):12–20. |
[17] | Wurster CD , et al. Neurochemical markers in CSF of adolescent and adult SMA patients undergoing nusinersen treatment. Ther Adv Neurol Disord. (2019) ;12: , 1756286419846058. |
[18] | Khalil M , et al. Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol. (2018) ;14: (10):577–89. |
[19] | Aksamit AJ Jr , Preissner CM , Homburger HA . Quantitation of 14-3-3 and neuron-specific enolase proteins in CSF in Creutzfeldt-Jakob disease. Neurology. (2001) ;57: (4):728–30. |
[20] | Storm C . Biomarkers after resuscitation: Relevance in daily clinical practice for prognosis estimation and definition of therapeutic goals. Med Klin Intensivmed Notfmed. (2019) ;114: (4):313–8. |
[21] | Miller N , et al. Non-aggregating tau phosphorylation by cyclin-dependent kinase 5 contributes to motor neuron degeneration in spinal muscular atrophy. J Neurosci. (2015) ;35: (15):6038–50. |
[22] | Pechmann A , et al. SMArtCARE - A platform to collect real-life outcome data of patients with spinal muscular atrophy. Orphanet J Rare Dis. (2019) ;14: (1):18. |
[23] | Mendell JR , et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol. (2013) ;74: (5):637–47. |
[24] | Montes J , et al. Nusinersen improves walking distance and reduces fatigue in later-onset spinal muscular atrophy. Muscle Nerve. (2019) . |
[25] | Mazzone E , et al. Six minute walk test in type III spinal muscular atrophy: A 12month longitudinal study. Neuromuscul Disord. (2013) ;23: (8):624–8. |
[26] | Montes J , et al. Ambulatory function in spinal muscular atrophy: Age-related patterns of progression. PLoS One. (2018) ;13: (6):e0199657. |
[27] | Kaufmann P , et al. Prospective cohort study of spinal muscular atrophy types 2 and 3. Neurology. (2012) ;79: (18):1889–97. |
[28] | Mercuri E , et al. Patterns of disease progression in type 2 and 3 SMA: Implications for clinical trials. Neuromuscul Disord. (2016) ;26: (2):126–31. |
[29] | Swoboda KJ , et al. SMA CARNI-VAL trial part I: Double-blind, randomized, placebo-controlled trial of L-carnitine and valproic acid in spinal muscular atrophy. PLoS One. (2010) ;5: (8):e12140. |
[30] | Deymeer F , et al. Natural history of SMA IIIb: Muscle strength decreases in a predictable sequence and magnitude. Neurology. (2008) ;71: (9):644–9. |
[31] | Armon C , et al. Addendum to assessment: Prevention of post-lumbar puncture headaches: Report of the Therapeutics and technology assessment subcommittee of the american academy of neurology. Neurology. (2005) ;65: (4):510–2. |
[32] | Lavi R , et al. Standard vs atraumatic Whitacre needle for diagnostic lumbar puncture: A randomized trial. Neurology. (2006) ;67: (8):1492–4. |
[33] | Winter B , et al. Neurofilaments and tau in CSF in an infant with SMA type 1 treated with nusinersen. J Neurol Neurosurg Psychiatry. (2019) . |
[34] | Kollmer J , et al. Quantitative MR neurography biomarkers in 5q-linked spinal muscular atrophy. Neurology. (2019) ;93: (7):e653–e664. |
[35] | Durmus H , et al. Muscle magnetic resonance imaging in spinal muscular atrophy type Selective and progressive involvement. Muscle Nerve. (2017) ;55: (5):651–6. |
[36] | Wu JS , Darras BT , Rutkove SB . Assessing spinal muscular atrophy with quantitative ultrasound. Neurology. (2010) ;75: (6):526–31. |


