
Molecular and Clinical Characteristics of a National Cohort of Paediatric Duchenne Muscular Dystrophy Patients in Norway
Abstract
Background:
As new gene-related treatment options for Duchenne muscular dystrophy (DMD) are being developed, precise information about the patients’ genetic diagnosis and knowledge about the diversities of natural history in DMD is vital.
Objective:
To obtain detailed insight into the genetic and clinical characteristics of paediatric DMD in Norway.
Methods:
94 boys with DMD, aged 0–18 years, were identified over a period of 3.5 years, yielding a national prevalence of 13.5×10–5 boys. 73 boys (78%) were recruited to full genetic and clinical or limited (genetic only) evaluation.
Results:
Molecular analysis disclosed 64% deletions, 18% duplications and 18% point mutations. The mean age of diagnosis was 3.9±2.0 years. 78% were treated with glucocorticoids from age 5.8±1.5 years. 23 boys (35%) had lost ambulation at an age of 10.7±2.0 years. 17% were treated for left ventricular dysfunction from age 12.1±3.0 years and 12% had received night-time non-invasive positive pressure ventilation from age 13.0±2.5 years.
Conclusions:
The distribution of mutation types and sites was similar to previous studies but with more duplications and fewer point mutations. Any genotype-phenotype correlations were not uncovered. The boys were diagnosed early but there is still diagnostic delay among boys presenting with late motor development. Glucocorticoid treatment was widespread, especially among the younger boys. The clinical results of this comprehensive nationwide study highlight the large variability of disease progression in DMD.
INTRODUCTION
Duchenne muscular dystrophy (DMD) is a severe, hereditary and progressive neuromuscular disorder, which almost exclusively affects boys. Although it is a rare disease, DMD is the most common muscular dystrophy in children, affecting 1:3500–1:6000 live male births [1, 2]. The disease presents in childhood and is caused by a mutation in the DMD gene, located on chromosome Xp21 [3]. To date, more than 5400 unique public DNA variants have been recorded in the Leiden Open Variation Database [4, 5]. The mutation leads to a deficient production of dystrophin, a structural protein which contributes to the stabilisation of the sarcolemma during muscle contraction or stretch [6]. As a general rule, any mutation that disrupts the reading frame for the translation of mRNA into dystrophin or introduces a premature stop codon, appears to cause a near total absence of functional dystrophin and the severe DMD phenotype, while a mutation that preserves the reading frame will typically give rise to the milder phenotype Becker muscular dystrophy (BMD) [4].
A definite link between the type, site or size of a mutation on the very large DMD gene and muscle function phenotype has been difficult to establish [7, 8], although a favourable clinical profile of single deletion of exon 45 compared to other single exon deletions is established [9–11]. A possible link has been found between the effect of the mutation on the production of central nervous system dystrophin isoforms and cognitive and neuropsychiatric difficulties [12, 13].
Comprehensive national and global genetic databases and patient registries have been established over the past 15 years [4, 14, 15]. Knowledge about the specific mutations of the DMD population is vital for the identification of patients eligible for new treatment options. Observational studies of phenotypic variations in disease progression are however also necessary in order to evaluate both standards of care and the results of clinical trials.
In Norway, the multidisciplinary care of children with chronic disease is based on services from 17 local hospitals throughout the country and 4 regional centres linked to the University Hospitals in Oslo, Bergen, Trondheim and Tromso. Because of the systematic and comprehensive manner of healthcare delivery in Norway, with a population of approximately 5 million, it is possible to conduct good quality and nationwide epidemiological research.
However, a coherent and complete overview of the DMD population in Norway has been lacking.
The aim of the present article is to describe the incidence, molecular diagnosis, treatment and ambulatory and cardiopulmonary function of the Norwegian paediatric DMD population, and to evaluate possible genotype/phenotype correlations.
MATERIALS AND METHODS
Recruitment
All local and regional treatment centres in Norway were requested to identify the number and age of their DMD patients under the age of 18. Based on consent from a legal guardian, or from the patient himself if older than 16 years and consent competent, obtained via their local paediatric neurologist; candidates received an information pack including consent form from the study group. Inclusion required the return of written consent.
The study was approved by the Regional Ethics Committee and the Privacy ombudsman of Oslo University Hospital, and registered with ClinicalTrials.gov, number NCT01963897.
Enrolment opened on 01.07.2013 and closed on 31.12.2016. During this period, 94 cases of DMD under the age of 18 years were identified. Prior to analyses, DMD cases were defined by both 1) mutation of the DMD gene and/or dystrophin deficiency consistent with DMD found on muscle biopsy, and 2) DMD phenotype; i.e. non-ambulant after 13 years in steroid-naïve cases or after 16 years in steroid treated cases.
Where the phenotypic severity of younger participants could not be conclusively determined, boys were included if all of the following criteria were met: a) their mutation was out-of-frame; and/or there was evidence of dystrophin deficiency consistent with DMD on muscle biopsy; and b) suspicion of DMD was raised before the age of 3 years based on delayed motor development, abnormal gait, elevated transaminases, speech delay or a family history of DMD; or they were diagnosed before the age of 5 years; and c) Creatine kinase (CK) levels were above 9 000 U/L.
65 boys (69%) agreed to participate in the study. 63 (95%) of these boys and their parents or guardians met with the investigator on one, two or three occasions over a period of three years.
In addition, with the patients or, when needed, the caregivers consent, information about DMD mutations, year of birth and county of residence was collected from an additional 8 cases (8.5%), leaving us with genetic and basic demographic information on 73 boys, or 78% of the total paediatric DMD population in Norway.
Data collection and analysis
Participants were subjected to physical examination and extensive laboratory and selected radiologic evaluations as listed in the appendix. The examinations took place at Oslo University Hospital, or at the boys’ local rehabilitation unit in conjunction with routine appointments, in cases where this was more convenient for the boys and their caregivers.
Cardiopulmonary assessments were performed by specialist paediatricians at the participants’ local or regional centres as part of their routine follow-up. Assessments included electrocardiograms (ECG), echocardiograms, spirometry tests and overnight sleep polygraphy.
Medical records were collected from all participants’ local or regional care centres. In two cases, information was based on their medical records alone.
Laboratory samples were, most often, collected and analysed at Oslo University Hospital. In cases where the boys were examined at their local rehabilitation unit, blood samples were collected locally and sent to Oslo University Hospital for analyses.
Molecular diagnoses were established by multiplex ligation-dependent probe amplification (MLPA) (n = 46), multiplex PCR (n = 4), Chamberlain and Begg test (n = 1) or sequencing (n = 22). We used the Humgen DMD exonic deletions/duplications reading-frame checker 1.9 [16] to analyse the predicted effect on the reading-frame of deletions and duplications. All sequence variants found in our study population were submitted to the ClinVar database [14], and exonic deletions and duplications to Leiden Muscular dystrophy pages [4].
Data collection and clinical examinations were performed by the first author (EA). A research assistant exchanged the participants’ personal data for consecutive case numbers. Data were analysed using the IBM SPSS Version 23 software. Demographic data and clinical characteristics were subjected to descriptive and between groups analyses of variance (ANOVA). When our data met the required assumptions of normal distribution and sufficient sample size, Pearson correlation or a one-way between groups ANOVA and post-hoc comparisons by Turkey HSD tests were performed. Otherwise, non-parametric methods were used; Spearman’s Rank Order Correlation, Mann-Whitney U test, Kruskal-Wallis test or Fisher’s exact test for statistical significance.
RESULTS
Prevalence & incidence
At the start of the study, there were 77 known cases of DMD under the age of 18 years in Norway. At the end of our study, 17 new cases had been identified, and 15 boys had become adults.
Based on population data from Statistics Norway [17], the prevalence of DMD in Norway at the start and finish of the study was 0.013% and 0.014% respectively, or 13.5×10–5 (95% CI: 10.6–16.4), or one in 7418 boys under the age of 18 years. The mean annual incidence rate among boys born in Norway in the period 2000–2009 was 19.7×10–5 (95% CI: 15.1–24.9), or one in 4967 live male births, or 6 new cases per year.
Non-participants
We have limited information about the 21 boys (22% of the DMD population) who declined participation in our study. Information about their phenotype, clinical status or steroid use is lacking.
There was a statistically significant difference in age at invitation between full participants (n = 65), limited participants (n = 8) and non-participants (n = 21). The age at invitation for non-participants (12.3±5.1 years) was significantly higher than the age of both full participants (9.3±3.9 years) and limited participants (7.4±4.4 years).
Demographics
DMD boys were identified in 18 of 19 Norwegian counties. Participants represented all ages between 2 and 18 years, with a mean age of 11.0±4.0 years.
Five pairs of siblings and two first cousins were enrolled in our study. In addition, two of the 65 participants had a non-participating brother with DMD, while three boys knew about other non-recruited cases of DMD in their biological family.
Molecular characteristics
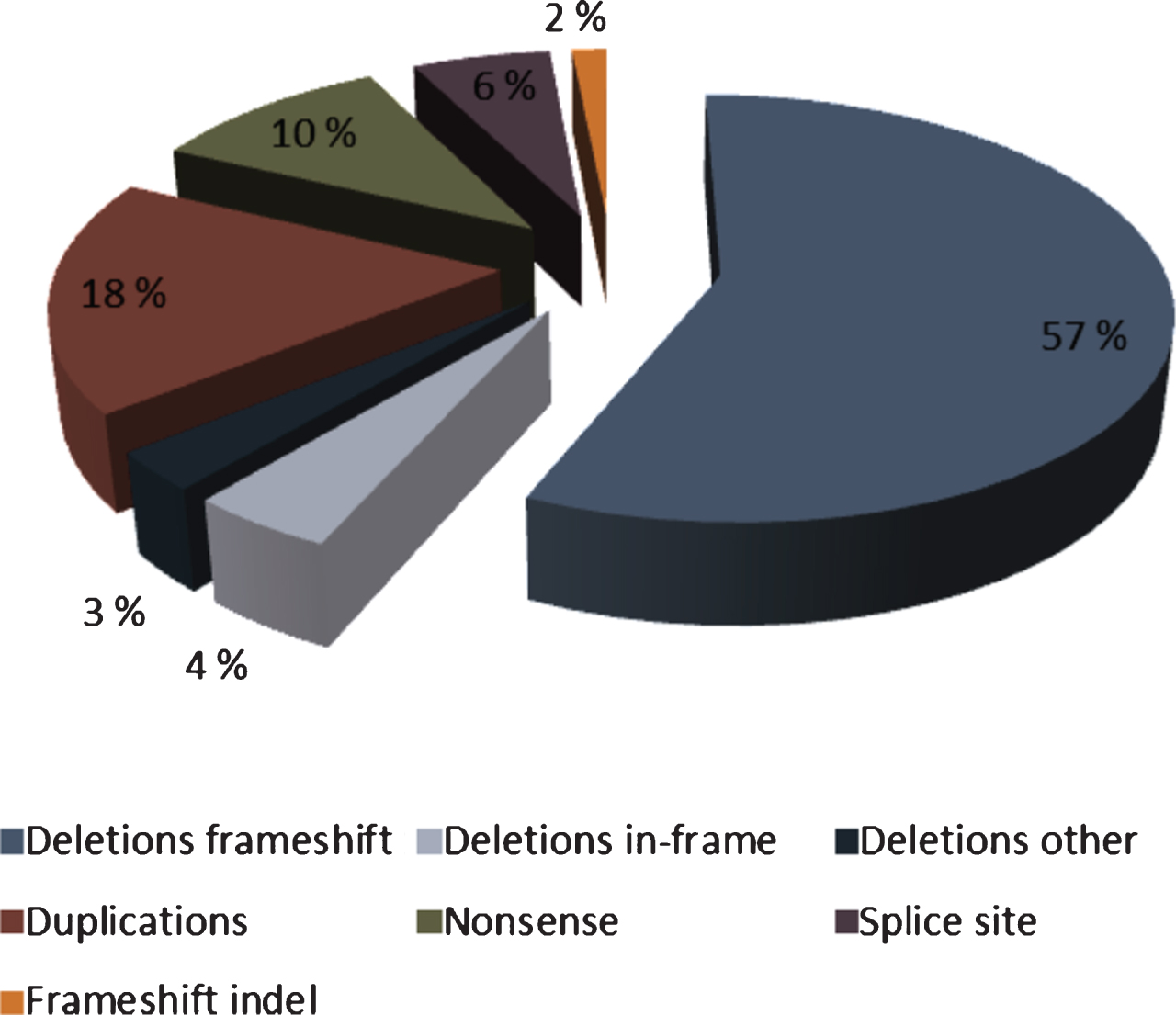
The genetic foundation for the diagnosis was established for all participants; 73 boys from 67 individual families. In order to avoid a skewed presentation of genotype variations, only one proband from each family was included in genotype analyses. However, all 73 cases were included in genotype-phenotype analyses, as there were signs of phenotypical variability even among brothers with identical mutations. Types, sites and sizes of mutations are illustrated in Fig. 1. The relative percentages of mutation types are illustrated in Fig. 2.
Fig.1
Graphic overview of the type and site of mutations in 67 Norwegian paediatric DMD families. Mutations affecting exons 1 through 79 of the DMD gene. Schematic locations of the actin-binding, central rod, cysteine-rich and C-terminal domains are marked (top) [4]. Duplications (red), frameshift deletions (blue), in-frame deletions (light blue), other deletions (dark blue), nonsense mutations(green), splice site mutations (purple) and frameshift indels (orange) are marked for each family. Mutation hotspots are marked as yellow circles.
![Graphic overview of the type and site of mutations in 67 Norwegian paediatric DMD families. Mutations affecting exons 1 through 79 of the DMD gene. Schematic locations of the actin-binding, central rod, cysteine-rich and C-terminal domains are marked (top) [4]. Duplications (red), frameshift deletions (blue), in-frame deletions (light blue), other deletions (dark blue), nonsense mutations(green), splice site mutations (purple) and frameshift indels (orange) are marked for each family. Mutation hotspots are marked as yellow circles.](https://ip.ios.semcs.net:443/media/jnd/2019/6-3/jnd-6-3-jnd190402/jnd-6-jnd190402-g001.jpg)
Fig.2
Relative percentage of mutation types in the Norwegian paediatric DMD population. Colours of mutation types correspond to Fig. 1. Total percentage of frameshift, in-frame and other deletions is 64%.
Exonic deletions
Deletion was the most frequent mutation in our population. We found 32 different deletions in 43 families (64%), 27 of which (84%) affected the reading frame. Each of seven deletions, all affecting exon 46–55 in various combinations, recurred in two families. Single deletion of exon 45 and deletion of exon 45–50 were both found in three individual families.
Three of 32 deletions (9%) were in-frame, affecting exon 3–13, 3–20 and 10–13 respectively. All three boys had a CK above 16 000 U/L at diagnosis and motor developmental delay noted before 3 years of age. Two lost ambulation at 8.0 and 9.5 years respectively, while one boy, with inconclusive biopsy findings, was treated with Prednisolone and was ambulant at 10 years.
In two cases (6%), the effect on the reading-frame could not be predicted, affecting Dp427c – exon 9, including the Dp427 m muscle promoter, and exon 60–79 respectively, due to deletion of the natural start- and stopcodon and regulatory elements. Both boys follow the typical clinical course of DMD.
Deletions were found throughout the DMD gene, but with a non-random distribution clustered around exons 3–18 (14%) and 44–55 (44%). Deletions in the proximal cluster tended to be larger and more dispersed than deletions in the distal cluster. The overall size of deletions ranged from one to 32 exons.
Exonic duplications
Eleven unique duplications were found in 12 families (18%). The Humgen DMD exonic deletions/duplications reading-frame checker [16] predicted that 10 (91%) were frameshift mutations, whereas the effect on the reading frame could not predicted in one mutation affecting Dp427c – exon 2. The two boys in this family displayed typical DMD phenotypes, and lost ambulation at 12 and 14 years respectively, despite glucocorticoid treatment. As a rule, it may be difficult to predict the effect of duplications on the reading frame, as the exact position of the insert is not known and the consequence on the reading frame is thus not certain [8].
Duplications ranged from one to 32 exons, and were found in exons 1–62. The distribution of duplications along the gene was more dispersed than the deletions, although slightly more commonly affecting exon 2–17.
Point mutations
We found 6 different nonsense mutations, introducing premature stopcodons, in 7 families (10%), 4 splice site mutations (6%) and one frameshift indel (2%). Point mutations were dispersed throughout exon 3–67, but not in the C-terminal domain. A nonsense mutation of exon 36 was found in two individual families. There were no other recurrent point mutations.
The diagnostic process
Age at diagnosis
The mean age of diagnosis was 3.9±2.0 years; while 72% had their diagnosis established by the time they reached 5 years. There was no correlation between birth year and age of diagnosis.
Eighteen boys (28%) were diagnosed after the age of 5 years. Sixteen of them presented with delayed motor development or gait disorder and two with elevated transaminases. Genetic analysis of these 18 cases revealed 10 (56%) frameshift deletions, 2 (11%) frameshift duplications, 5 (28%) nonsense mutations and 1 (5%) in-frame deletion.
The age at diagnosis varied across mutation types, where boys with nonsense mutations were diagnosed late, at a mean age of 5.1±2.9 years while duplications and deletions were found at 3.5±2.1 and 3.9±1.8 years, respectively. However, the numbers are small and the differences in age at diagnosis for mutations type were not significant.
Delayed motor development was the most common reason for diagnostic referral. The reasons for diagnostic referral and the mean age of diagnosis for each reason are summarized in Table 1.
Table 1
Reasons for diagnostic referral leading to the diagnosis of DMD in the Norwegian paediatric DMD population
| Reasons for diagnostic referral | N (%) | Mean age at diagnosis (years (SD)) | Min | Max |
| Delayed motor development | 35 (53.8) | 4.3 (1.9) | 0.6 | 8.6 |
| Abnormal gait | 11 (16.9) | 4.5 (1.6) | 2.6 | 7.0 |
| Family history | 8 (12.3) | 2.8 (3.2) | 0.0 | 10.0 |
| Elevated transaminases | 6 (9.2) | 3.8 (1.7) | 2.1 | 6.2 |
| High CK | 4 (6.2) | 2.8 (1.3) | 1.5 | 4.4 |
| Speech delay | 1 (1.5) | 3.1 | ||
| Total | 65 (100) |
Age presented in years.
Muscle biopsy
A muscle biopsy was performed as part of the diagnostic process in 32 of 65 cases (49%) in the period from year 2000 to 2016. The procedure was more commonly performed in cases of point mutations; 75% of nonsense and splice site mutations. In patients with exonic deletion or duplication, a muscle biopsy had been performed in 46% and 38% of cases respectively.
Between year 2000 and 2010, a muscle biopsy was performed in 26 of 39 cases (67%). During this earlier period, a muscle biopsy was deemed not possible or not expedient for other reasons in six cases (15%), while the procedure was considered unnecessary due to a positive family history in seven cases (18%). In the period from 2011 to 2016, following the publication of international consensus guidelines [18], the number of muscle biopsies was reduced to 6 of 26 cases (23%).
Glucocorticoid (GC) treatment
Treatment with glucocorticoids (GCs) became gradually more common in DMD in Norway from year 2000, as convincing data on prolonged ambulation and reduction in the incidence of scoliosis with GCs emerged [19, 20]. 51/65 boys (78%) in our study population were, or had previously been, treated with GCs. The mean age of initiating treatment was 5.8 years (SD±1.5).
The Norwegian DMD patients are, as a general rule, on daily GC regimens. In accordance with consensus guidelines, recommended starting doses are 0.75 mg/kg/d for prednisolone and 0.9 mg/kg/d for deflazacort [21]. The mean actual prescribed doses in our study population were 0.49±0.13 mg/kg/d of prednisolone or 0.49±0.20 mg/kg/d of deflazacort. The younger boys received higher doses per kg bodyweight than the older participants. The majority of treated cases were first prescribed Prednisolone, the first drug of choice by the Norwegian medicinal regulatory authorities. At the time of our final inquiry, 19/65 boys (29%) were prescribed Prednisolone, while 26/65 (40%) had switched to deflazacort. Thus, the boys in the deflazacort group were older (mean age 12.0±2.9 years) than the Prednisolone group (mean age 9.8±3.1 years). Reports of a possible favourable side-effect profile of deflazacort regarding reduced weight gain [22], was the given reason for the switch in all cases. We do not have sufficient cases or individual data to report an effect of a switch from Prednisolone to deflazacort on weight in our population.
Six boys (9%), with a mean age of 15.9±2.9 years, had stopped GC treatment due to unacceptable side effects, namely excessive weight-gain, pubertal delay, cataracts and/or psychiatric difficulties (behavioural changes and psychotic episodes). All six boys were initially treated with Prednisolone. Four were prescribed deflazacort before treatment cessation.
Fourteen boys (22%) had never received GC treatment. Six of these boys were less than 4 years old and considered to be too young for treatment. One eight-year-old had only recently been diagnosed. The parents of seven boys, with a mean age of 13.3±3.9 years, had declined treatment due to concerns about long-term side effects.
Ambulatory development
Age at time of independent ambulation
The mean age at time of independent ambulation was 18±6 months. Eight boys (13%) did not walk independently until the age of two years or more. Frameshift deletions affecting one or more exons in the 46–79 range were found in seven of these boys, while one had a duplication of exon 10–11.
Loss of ambulation (LoA)
Twenty-three boys (35.4%) had lost ambulation by the time of their final study visit. The mean age at loss of ambulation (LoA) for these boys was 10.7±2.0 years. There was an apparent difference in groups, where the GC treated boys lost ambulation 1.7 years later than the non-GC group, and with much wider variance, as illustrated in Table 2. However, the difference was not significant.
Table 2
Glucocorticoid (GC) regimen at the time of Loss of ambulation (LoA)
| GC treatment | n | % of total | Mean age at LoA (years)±SD | Min | Max |
| No | 7 | 30.4 | 9.9±1.2 | 8.0 | 11.9 |
| Yes | 16 | 69.6 | 11.1±2.1 | 7.9 | 14.8 |
| Total | 23 | 100.0 |
SD: Standard deviation.
There was an apparent difference in age of loss of ambulation across mutation types, where boys with deletions were at a mean age of 10.1±1.8 years at LoA, vs 11.4±2.0 years in point mutations and 11.3±2.6 years in the duplication group. However, the difference did not reach statistical significance in our study population.
There was no significant correlation between the ages of loss of ambulation and independent ambulation or age at diagnosis.
Cardiopulmonary function
Pulmonary function
Twelve of 65 boys (19%) had access to a coughing device, but only seven (11%) used their device regularly. Commonly, the non-compliers had not felt the need for the device.
Night-time hypoventilation or obstructive sleep apnoea was diagnosed in eight boys (12%), the youngest of whom was 12.3 years at the time of study examination. All eight boys had been prescribed night-time non-invasive positive pressure ventilation (NIPPV) at a mean age of 13.0±2.5 years. Only 50% of the boys used their respiratory support on a regular basis. The non-compliers commonly explained their resistance with difficulties in mentally accepting the device or physical discomfort related to ill-fitting masks.
Due to insufficient power, any significant correlation between time of initiation of NIPPV and GC treatment or time of LoA could not be determined. We did not identify any specific site or type of mutation more prevalent in boys treated for night-time ventilatory insufficiency.
Cardiac function
Eleven (17%) boys were treated for left ventricle systolic dysfunction by their paediatric cardiologist. Ventricular dysfunction was defined by a reduced ejaculation fraction (EF) on echocardiogram. No cardiac rhythm abnormalities were noted in our population.
ACE-inhibitors were the first drug of choice in all but one of these 11 boys, initiated at a mean age of 12.1±3.0 years. β-blockers had been added in four cases and aldosterone antagonists in two.
Of the 11 boys receiving cardio protective treatment, nine were treated with GCs. Possible correlations between the ages of initiation of cardio protective treatment and initiation of steroid treatment or current steroid treatment, or between the age of initiation of cardio protective treatment and mutation type, could not be established due to insufficient power.
Additionally, four boys (6%) were treated for hypertension, defined as systemic blood pressure >90 perc for age and height [23]. All four were being treated with deflazacort, and all four were prescribed ACE-inhibitors for their hypertension at a median age of 11.1 years (IQR 10.6, 13.3). As the numbers were small, any increased incidence of hypertension in patients treated with deflazacort compared to prednisolone could not be examined.
DISCUSSION
The prevalence of DMD under the age of 18 in Norway at the time of our study is comparable to 16.8×10–5 (CI 11.4–23.8) boys under 16 years found in Western Sweden [24], 12.76×10–5 (CI 8.26–18.84) under 20 years found in Estonia [25] and the pooled totals of 12.6×10–5 (CI 9.04–17.5) found in a meta-analysis by Mah et al. [26]. Our estimate includes the 21 boys who declined participation, and there may be misdiagnosed cases among them. However, based on the high level of genetic confirmation combined with a DMD phenotype in the recruited sample, we believe that our estimate of correctly diagnosed cases is fairly accurate. There were no deaths recorded among the under 18 DMD population during the study period.
The incidence of DMD in Norway corresponds to 21.5×10–5 (CI 12.5–34.5) in Western Sweden in 1979–1986 [24] and 18.8×10–5 (CI 12.4–25.2) in Denmark in 1992–1996 [27]. We based our estimate on male births from year 2000 to 2009. The youngest boys in this birth cohort would be 8 years old in 2017, and considering the natural history and severity of the disease, combined with excellent cooperation from all the 21 local and regional multidisciplinary care centres in the country, we do not believe that there are undiagnosed or misdiagnosed cases within the birth cohort.
The relative frequencies of different DMD mutations in our cohort are similar to those found in previous studies [7, 8], but we noted slightly more duplications and fewer point mutations in the Norwegian cohort. In accordance with previous studies, deletions are clustered in two “hot-spots”, with 44% of all deletions located in the distal hot-spot and with single exon 45 or exon 45–50 deletion being the most frequent mutations [4, 7]. Also in accordance with previous studies, deletions in the proximal hot-spot are larger and more dispersed than in the distal cluster [7, 28]. In our study material, no mutation is larger than 32 exons. The number of in-frame deletions corresponds with frequencies found in international databases [15]. They are all located in the actin-binding and central rod domains. Frequent exceptions to the reading-frame rule in these domains are previously described [4].
We were unable to establish a significant correlation between mutation site, size or type and clinical milestones in our study. The anticipated relative protective effect of single exon 45 deletion [10, 11] compared to other single exon mutations could not be explored in our cohort, as all three boys with single exon 45 deletions in our cohort were younger than seven years old.
Interestingly, even brothers and first cousins, who carried the same mutation and received the same standards of care, varied in motor function and cardiopulmonary disease. This illustrates the probable role of genetic modifiers in DMD disease progression [29].
Theoretically, antisense-mediated exon skipping could be applicable to 83% of all DMD mutations, with skipping of exon 51 (13.0%), exon 45 (8.1%), exon 53 (7.7%) and exon 44 (6.2%) covering 35% of the DMD population [30]. In our study, only 6 of 67 individual mutations (8.9%) were potentially amendable to skipping of exon 51, while skipping of exon 45 or 52 would theoretically apply to 5 mutations (7.5%) each. Skipping of exon 44, 46 or 53 would potentially apply to 4 mutations (6.0%) each.
Read-through treatment of nonsense mutations (ataluren) is commercially available in Norway for ambulant DMD patients over the age of 2 years, although not regularly reimbursed. In our study population, 8/73 boys (11%) were found to have nonsense mutations. Only one was ambulant at the time of our study. Ataluren treatment had not yet been initiated at that time.
Previous retrospective studies have reported a mean age of diagnosis of 4.0–4.9 years [7, 31, 32]. The lower age at diagnosis found in our cohort may reflect recent advances in genetic analysis as well as raised clinical awareness. However, boys with incidentally discovered elevated transaminases, high CK, speech delay or a positive family history of DMD were diagnosed early, while the mean age of diagnosis following delayed motor development or gait disorder in our study population was 4.3 and 4.5 years respectively. Reviewing the medical records of the participants, there are indications of concern raised by parents, community health centres or kindergarten from an early age in many late-diagnosed cases. The mean age of independent ambulation was 18 months. Thus, many of the late-diagnosed cases could have been diagnosed earlier if CK was analysed routinely in boys with delayed motor development, i.e. not walking independently at 18 months, as suggested already 40 years ago [33].
Boys with nonsense mutations were diagnosed with DMD 1.4 years later than deletions and duplications, although in most cases, the tentative diagnosis of dystrophinopathy was established before the mutation was found, based on clinical findings and muscle biopsy. This reflects recent advances in the diagnostic process. Until 2015, Norwegian laboratories did not routinely perform DMD sequencing, and further analyses following negative MLPA or multiplex PCR had to be performed abroad. In most cases, a muscle biopsy was required before further genetic analyses could be performed, as reflected by the higher frequencies of muscle biopsies performed on boys with nonsense or splice site mutations in our study. With improved genetic diagnostic technology, and following the publication of international consensus guidelines [18], a muscle biopsy is reserved for atypical cases of unclear molecular diagnosis or phenotypic uncertainty, considerably reducing the use of this procedure as a part of the diagnostic process of DMD in Norway.
A significant delay of LoA with steroid treatment has been found by larger studies [10, 34]. In our cohort, the trend showed prolonged ambulation by 1.7 years in the GC treated group but the difference was not significant, possibly due to insufficient power. Similarly, a correlation between GC treatment and the development of night-time hypoventilation or left ventricular dysfunction have been reported elsewhere [22, 34, 35] but was not revealed in our study.
Data on the development of hypoventilation and left ventricular dysfunction in this study are based on retrospective data from planned annual patient visits at a number of different hospitals. The data are extracted from medical records in order to counter the effects of subjective retrospective memory in reports from the participants or caregivers. Retrospective records nevertheless carry the risk of containing erroneous data that may influence the results of statistical analyses. The timing of events is extracted indirectly based on time of initiation of treatment, rather than directly based on onset of signs of disease. The criteria for the diagnoses of hypoventilation or left ventricular dysfunction were not uniformly defined and may have varied between clinicians.
It is possible that the boys in the GC treated group, who tended to be younger, were diagnosed with and treated for cardiac and respiratory complications of DMD relatively earlier than the non-GC group, in accordance with international recommendations of a more proactive attitude towards these issues in recent years [36–39]. The non-participating group was significantly older than the participants in our study. Decline of participation among the older boys probably reflects the increasing disease burden with age in DMD. Higher participation of older boys would possibly have affected the power of the analyses, in particular regarding LoA and cardiopulmonary dysfunction.
Norway is a small country, and Duchenne muscular dystrophy is a rare disease. As a consequence, data were often too limited to meet the requirements of parametric statistical methods. Non-parametric statistics are less sensitive and our findings may have been hampered by insufficient sample sizes. These limitations of a small national dataset illustrate the value of international collaboration through federated database systems that would allow research based on collected national cohorts without compromising national data protection regulations.
This study provides the first analysis of a nationwide cohort of paediatric DMD in Norway. Participation is high and the participants represent all ages from 2–18 years and from all health regions in Norway. The genetic analyses provide important insight into the molecular basis of disease, in preparation for mutation specific research and treatment options. The clinical results highlight the phenotypic variability of DMD and constitute a valuable contribution to the existing international research that forms the foundation of the understanding of natural history of DMD.
CONFLICTS OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
The authors would like to thank the participating DMD boys and their families for their time and patience. The authors would also like to thank the Paediatric neurologists and physiotherapists at all Child rehabilitation clinics in Norway for their assistance in patient recruitment, and in particular dr. Jasmina Tajsic at University Hospital of North Norway, dr Espen Lien at Trondheim University Hospital, dr Kathrine T. Leversen and dr Nanette Mjellem at Bergen University Hospital, and dr Marit Mork at Stavanger University Hospital for their kind assistance in patient recruitment and data collection.
The project was funded by the National Research Council (NFR), with supplementary funding from Erik Allums legat. The funding sources were not involved in the study design, data collection, data interpretation or submission of this article.
Appendices
Appendix
Appendix. Study variables in the paediatric Duchenne muscular dystrophy Norway study 2013–2016
| Background information | |
| Demographics | County |
| Family history DMD | |
| Family history delayed puberty (biological father) | |
| Genotype | Mutation type, site and size, |
| Laboratory and mode of investigation | |
| Muscle biopsy | Dystrophin activity |
| Phenotype | Age at independent ambulation |
| Age at diagnosis | |
| Age at GC treatment initiation | |
| Age at initiation of night splints | |
| Age at initiation of physiotherapy | |
| Age at loss of ambulation | |
| Annual history | Annual investigations |
| Activities and participation | Physical examination |
| Physiotherapy (frequency) | Growth (height /weight /BMI, arm span if non-ambulant) |
| Pool therapy (frequency) | Pubertal development (Tanner) |
| Assistance in kindergarten / school | Testicular volume (orchidometer) |
| Assistance at home / care arrangements | Blood pressure |
| Leisure activities (sports and other) | Scoliosis (clinical evaluation) |
| Contractures | |
| North Star Ambulatory Assessment if ambulatory and >5 years of age | |
| Vignos &Brooke motor function assessment | |
| Physical adaptations and equipment | Laboratory investigations |
| School physical environment adjustments | Haemoglobin, white cell count, differential, thrombocytes, |
| Home physical environment adjustments | Sodium, Potassium, Magnesium, Phosphate, free Calcium, |
| Wheel chairs, other seating, standing device | CK, AST, ALT, ALP (total and bone specific) |
| Other equipment (for daily living and leisure) | HbA1c, creatinine, urea, glucose |
| Ventilatory support (NIPPV / cough device) | 1,25-(OH)2 - vitamin D, 25-(OH)-vitamin D, |
| Osteocalcin, leptin, PTH, CTX, PINP | |
| IGF-1, IGF-binding protein 3, growth hormone | |
| Testosterone, FSH, LH, SHBG | |
| Biobank 2 test vials | |
| Natural functions | Radiology |
| Sleep (insomnia, awakening, hypersomnia) | Left hand and wrist (Greylich &Pyle skeletal age) |
| Nutrition, eating, chewing, swallowing (gastrostomy) | Vertebra on clinical suspicion of fracture |
| Gastroesophageal reflux | Spinal on clinical suspicion of scoliosis |
| Oral hygiene / dentistry | Dual energy X-ray absorptiometry (DEXA scan) |
| Constipation or faecal incontinence | |
| Urinary incontinence (day / night) | |
| Cardiopulmonary function | Quality of life and mental health |
| Spirometry status | Parental occupation and economic support |
| Sleep polygraphy | Siblings |
| Cardiologic assessments Blood pressure assessments Ophthalmologic assessments if GC treated | PedsQL Generic Norwegian translation (all participants and caregivers if fluent in Norwegian language) SDQ-Nor (participants 11–17 years, caregiver 4–16 years if fluent in Norwegian language) |
| Medication | |
| Prednisolone/deflazacort doses | |
| Vitamin D / Calcium supplements | |
| Cardio protective treatment | |
| Other medication or supplements | |
| Bone health | |
| Radiologically confirmed neuromuscular scoliosis Fractures (long bone / vertebra) | |
| Orthopaedic surgery |
REFERENCES
[1] | Mendell JR , Shilling C , Leslie ND , Flanigan KM , al-Dahhak R , Gastier-Foster J , et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol. (2012) ;71: :304–13. |
[2] | Emery AEH Population frequencies of inherited neuromuscular diseases—A world survey. Neuromuscul Disord. (1991) ;1: :19–29. |
[3] | Koenig M , Hoffman E , Bertelson C , Monaco AP , Feener C , Kunkel LM Complete cloning of the duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. (1987) ;50: :509–17. |
[4] | Aartsma-Rus A , Van Deutekom JC , Fokkema IF , Van Ommen GJ , Den Dunnen JT Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve. (2006) ;34: :135–44. |
[5] | Leiden Open Variation Database. [Gene variant database] [accessed 10.06.2019]. Available from: https://www.LOVD.nl/DMD. |
[6] | Ervasti JM Costameres: The achilles’ heel of herculean muscle. J Biol Chem. (2003) ;278: :13591–4. |
[7] | Magri F , Govoni A , D’Angelo MG , Del Bo R , Ghezzi S , Sandra G , et al. Genotype and phenotype characterization in a large dystrophinopathic cohort with extended follow-up. J Neurol (2011) ;258: :1610–23. |
[8] | Juan-Mateu J , Gonzalez-Quereda L , Rodriguez MJ , Baena M , Verdura E , Nascimento A , et al. DMD mutations in 576 dystrophinopathy families: A step forward in genotype-phenotype correlations. PLoS One. (2015) ;10: :e0135189. |
[9] | van den Bergen JC , Ginjaar HB , Niks EH , Aartsma-Rus A , Verschuuren JJ Prolonged ambulation in duchenne patients with a mutation amenable to exon 44 skipping. J Neuromuscul Dis. (2014) ;1: :91–4. |
[10] | Koeks Z , Bladen CL , Salgado D , van Zwet E , Pogoryelova O , McMacken G , et al. Clinical outcomes in duchenne muscular dystrophy: A study of patients from the TREAT-NMD DMD global database. Neuromuscul Disord. (2017) ;4: :293–306. |
[11] | Wang RT , Barthelemy F , Martin AS , Douine ED , Eskin A , Lucas A , et al. DMD genotype correlations from the Duchenne Registry: Endogenous exon skipping is a factor in prolonged ambulation for individuals with a defined mutation subtype. Hum Mutat. (2018) ;39: :1193–202. |
[12] | Taylor PJ , Betts GT , Maroulis S , Gilissen C , Pedersen RL , Mowat DR , et al. Dystrophin gene mutation location and the risk of cognitive impairment in Duchenne muscular dystrophy. PLoS One. (2010) ;5: :e8803. |
[13] | Ricotti V , Mandy WP , Scoto M , Pane M , Deconinck N , Messina S , et al. Neurodevelopmental, emotional, and behavioural problems in Duchenne muscular dystrophy in relation to underlying dystrophin gene mutations. Dev Med Child Neurol. (2016) ;58: :77–84. |
[14] | National Center for Biotechnology Information. ClinVar. [Online database] 2018 [accessed 2018 Jan 15]. Available from: http://www.ncbi.nlm.nih.gov/clinvar/. |
[15] | Bladen CL , Salgado D , Monges S , Foncuberta ME , Kekou K , Kosma K , et al. The TREAT-NMD DMD global database: Analysis of more than 7,000 duchenne muscular dystrophy mutations. Hum Mutat. (2015) ;36: :395–402. |
[16] | Centre for Human and Clinical Genetics, Leiden University Medical Center. Humgen DMD exonic deletions/duplications reading-frame checker 1.9 [Online database] [accessed 2017 Nov 14]. Available from: http://www.humgen.nl/scripts/DMD_frame.php. |
[17] | Statistics Norway. 07459 Population, by sex and one-year age groups. 1 January (M) 1986 - 2017. [Internet] 2018 [accessed 2018 Jan 11]. Available from: http://www.ssb.no/en/statbank/table/07459?rxid=0141c90d-6fc8-458f-b5b3-ac4c4dc854b2. |
[18] | Bushby K , Finkel R , Birnkrant DJ , Case LE , Clemens PR , Cripe L , et al. Diagnosis and management of Duchenne muscular dystrophy, part Diagnosis, and pharmacological and psychosocial management. Lancet Neurol. (2010) ;9: :77–93. |
[19] | DeSilva S , Drachman DB , Mellits D , Kuncl RW Prednisone treatment in duchenne muscular dystrophy: Long-term benefit. Arch Neurol. (1987) ;44: :818–22. |
[20] | Dubowitz V Special centennial workshop– 101st ENMC international workshop: Therapeutic possibilities in duchenne muscular dystrophy, 30th November-2nd December Naarden, The Netherlands. Neuromuscul Disord. (2002) ;12: :421–31. |
[21] | Birnkrant DJ , Bushby K , Bann CM , Apkon SD , Blackwell A , Brumbaugh D , et al. Diagnosis and management of Duchenne muscular dystrophy, part Diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. (2018) ;17: :251–67. |
[22] | Matthews E , Brassington R , Kuntzer T , Jichi F , Manzur AY Corticosteroids for the treatment of Duchenne muscular dystrophy. Cochrane Database Syst Rev. (2016) : CD003725. |
[23] | Flynn JT , Kaelber DC , Baker-Smith CM , Blowey D , Carroll AE , Daniels SR , et al. Clinical practice guideline for screening and management of high blood pressure in children and adolescents. Pediatrics. (2017) ;140. |
[24] | Darin N , Tulinius M Neuromuscular disorders in childhood: A descriptive epidemiological study from western Sweden. Neuromuscul Disord. (2000) ;10: :1–9. |
[25] | Talkop Ü-A , Kahre T , Napa A , Talvik I , Sööt A , Piirsoo A , et al. A descriptive epidemiological study of Duchenne muscular dystrophy in childhood in Estonia. Eur J Paediatr Neurol. (2003) ;7: :221–6. |
[26] | Mah JK , Korngut L , Dykeman J , Day L , Pringsheim T , Jette N A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul Disord. (2014) ;24: :482–91. |
[27] | Jeppesen J , Green A , Steffensen BF , Rahbek J The Duchenne muscular dystrophy population in Denmark, 1977–2001: Prevalence, incidence and survival in relation to the introduction of ventilator use. Neuromuscul Disord. (2003) ;13: :804–12. |
[28] | Tuffery-Giraud S , Beroud C , Leturcq F , Yaou RB , Hamroun D , Michel-Calemard L , et al. Genotype-phenotype analysis in 2,405 patients with a dystrophinopathy using the UMD-DMD database: A model of nationwide knowledgebase. Hum Mutat. (2009) ;30: :934–45. |
[29] | Hightower RM , Alexander MS Genetic modifiers of Duchenne and facioscapulohumeral muscular dystrophies. Muscle Nerve. (2018) ;57: :6–15. |
[30] | Aartsma-Rus A , Fokkema I , Verschuuren J , Ginjaar I , Van Deutekom J , Van Ommen G , et al. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum Mutat. (2009) ;30: :293–9. |
[31] | Bushby KMD , Hill A , Steele JG Failure of early diagnosis in symptomatic Duchenne muscular dystrophy. Lancet. (1999) ;353: :557–8. |
[32] | Ciafaloni E , Fox DJ , Pandya S , Westfield CP , Puzhankara S , Romitti PA , et al. Delayed diagnosis in duchenne muscular dystrophy: Data from the Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet). J Pediatr. (2009) ;155: :380–5. |
[33] | Gardner-Medwin D , Bundey S , Green S Early diagnosis of Duchenne muscular dystrophy. Lancet. (1978) ;311: :1102. |
[34] | McDonald CM , Henricson EK , Abresch RT , Duong T , Joyce NC , Hu F , et al. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: A prospective cohort study. Lancet. (2018) ;391: :451–61. |
[35] | Barber BJ , Andrews JG , Lu Z , West NA , Meaney FJ , Price ET , et al. Oral corticosteroids and onset of cardiomyopathy in Duchenne muscular dystrophy. J Pediatr. (2013) ;163: :1080-4.e1. |
[36] | Bushby K , Finkel R , Birnkrant DJ , Case LE , Clemens PR , Cripe L , et al. Diagnosis and management of Duchenne muscular dystrophy, part Implementation of multidisciplinary care. Lancet Neurol. (2010) ;9: :177–89. |
[37] | Spurney C , Shimizu R , Hache LP , Kolski H , Gordish-Dressman H , Clemens PR , et al. CINRG Duchenne Natural History Study demonstrates insufficient diagnosis and treatment of cardiomyopathy in Duchenne muscular dystrophy. Muscle Nerve. (2014) ;50: :250–6. |
[38] | D’Amario D , Amodeo A , Adorisio R , Tiziano FD , Leone AM , Perri G , et al. A current approach to heart failure in Duchenne muscular dystrophy. Heart. (2017) ;103: :1770–9. |
[39] | Birnkrant DJ , Bushby K , Bann CM , Alman BA , Apkon SD , Blackwell A , et al. Diagnosis and management of Duchenne muscular dystrophy, part Respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. (2018) ;17: :347–61. |


