
Discovery of Novel Therapeutics for Muscular Dystrophies using Zebrafish Phenotypic Screens
Abstract
The recent availability and development of mutant and transgenic zebrafish strains that model human muscular dystrophies has created new research opportunities for therapeutic development. Not only do these models mimic many pathological aspects of human dystrophies, but their small size, large clutch sizes, rapid ex utero development, body transparency, and genetic tractability enable research approaches that would be inconceivable with mammalian model systems. Here we discuss the use of zebrafish models of muscular dystrophy to rapidly screen hundreds to thousands of bioactive compounds in order to identify novel therapeutic candidates that modulate pathologic phenotypes. We review the justification and rationale behind this unbiased approach, including how zebrafish screens have identified FDA-approved drugs that are candidates for treating Duchenne and limb girdle muscular dystrophies. Not only can these drugs be re-purposed for treating dystrophies in a fraction of the time and cost of new drug development, but their identification has revealed novel, unexpected directions for future therapy development. Phenotype-driven zebrafish drug screens are an important compliment to the more established mammalian, target-based approaches for rapidly developing and validating therapeutics for muscular dystrophies.
INTRODUCTION
Muscular dystrophies (MDs) are inherited primary diseases of skeletal muscle affecting approximately 17 in every 100,000 individuals worldwide [1]. The three most common MDs in adults, in decreasing prevalence, are myotonic dystrophy, facioscapulohumeral dystrophy (FSHD), and the limb girdle muscular dystrophies (LGMDs) [2, 3]. The most common MD in children is Duchenne muscular dystrophy (DMD), an X-linked disorder with an incidence of 20–30 cases per 100,000 live male births [1, 2, 4]. As a general rule, MDs are progressive in nature with affected muscles degenerating and becoming weaker over time. In the case of DMD, proximal muscle weakness becomes evident in early childhood, progressing to loss of ambulation by the early teens. Other muscle groups are eventually affected with patients succumbing to respiratory or cardiac failure.
Current treatments for DMD, including corticosteroids to reduce muscle loss, assistive ventilation to help preserve diaphragm contractility, and afterload reduction to enhance cardiac performance, have extended the period that patients remain ambulatory, and have enabled some patients to live into their mid-30’s and beyond [5]. Progress towards eventual cures is continuing as gene delivery, gene editing, and antisense technologies have advanced novel protein restoration or replacement therapies into clinical trials [6]. Major hurdles remain including poor efficacy, inadequate delivery, limited applicability to only some mutations and patients, and lack of rescue for muscles that have undergone years of extensive degeneration.
Many of the symptoms associated with MDs result from downstream, non-genetic events, such as muscle fiber atrophy, fibrosis, inflammation, oxidative stress, Ca2+ dysregulation, and blood flow impairment [7–9]. Existing or novel pharmacological compounds that target these events, such as promoting muscle hypertrophy, restoring Ca2+-homeostasis, or that function as vasodilators, anti-inflammatories, antioxidants, or antifibrotics are all under investigation as potential therapeutic treatments [8].
There is renewed emphasis on using phenotypic approaches to discover and develop new therapeutics for human diseases [10, 11]. Zebrafish represent one of the few vertebrate models currently amenable to a phenotype-based drug discovery strategy [12]. The expanding availability of zebrafish models for different MDs now makes this approach a realistic option for identifying novel downstream therapeutics for many of these diseases. We review the rationale and strategies behind this approach. Using DMD and LGMD type 2I (LGMD2I) as examples, we discuss how this approach has been used to identify FDA-approved drugs that may be suitable for repurposing as DMD or LGMD2I therapies. Finally, we review how these studies have uncovered novel pathways and mechanisms that may be attractive targets for future therapy development.
PHENOTYPE-DRIVEN DRUG DISCOVERY
Developing a new FDA-approved drug from initial concept through clinical trials can take 10–12 years at an estimated cost of one billion dollars [11]. Over the past 30 years, drug discovery has often focused on identifying compounds that target a specific molecule or a defined disease pathway. The initial stage of this target-based approach is to evaluate a compound’s ability to modulate in vitro systems, such as isolated cells or tissues. After a compound has been identified that impacts the molecules or pathways of interest, its toxicity, off-target effects, and therapeutic benefits are evaluated in a pre-clinical animal model.
Living organisms are more complex than cell and tissue systems and many lead compounds identified by target-based approaches fail when advanced to pre-clinical or clinical studies. This failure rate has been estimated as 90% or more [10]. Consequently, considerable time, capital, and human resources are invested in the developmental stages of a candidate compound that has a low probability of ever becoming an approved therapeutic.
Prior to the 1990’s, most new drugs were discovered using phenotypic criteria and there is currently a renewed interest in using this type of holistic approach for drug development [10, 11]. The initial focus of a phenotype-driven or chemogenomics strategy is identifying compounds that are safe and therapeutically effective under in vivo conditions [12, 13]. This usually involves treating model organisms with thousands of compounds in order to identify the few that modulate a disease phenotype or marker. After non-toxic compounds that reverse a pathological phenotype have been identified, the focus shifts to target deconvolution and identification of the mechanisms of action.
A phenotype-driven approach offers several advantages over target-based drug discovery [10, 11]. For instance, compounds that have poor bioavailability, exhibit unacceptable off-target effects, or which are toxic are eliminated early in the discovery process. Because there is no a priori bias as to what targets or pathways are therapeutically important, novel and unexpected disease mechanisms may be revealed for further exploration. Finally, phenotypic screens are an important approach for repurposing existing drugs for new therapeutic uses. This can result in a new therapeutic receiving FDA approval in as little as 2-3 years at a fraction of the cost associated with developing an entirely new drug [11].
ZEBRAFISH AS A MODEL FOR DRUG DISCOVERY
The large-scale nature of drug screens requires the use of a model organism with distinctive, easily assayed phenotypes that can be raised rapidly and maintained economically in large numbers. Phenotype-based drug discovery is therefore incompatible with mammalian disease models due to the prohibitive time, staffing, infrastructure, and costs required to bring these models to scale. This limits phenotypic screens to small, prolific, and rapidly developing invertebrates, such as flies (D. melanogaster) and worms (C. elegans), and vertebrates, like frogs (Xenopus laevis) and the zebrafish (Danio rerio). While frogs have the advantage of being tetrapods that are evolutionary closer to humans, there are a number of compelling reasons why zebrafish are currently the overwhelming choice for large-scale phenotypic approaches to human drug development.
Zebrafish have a rich history as a model of vertebrate biology [14]. The zebrafish was the first vertebrate to be cloned [15] and throughout the 1980’s and 90’s found widespread use as a model of vertebrate development [16–18]. The first large-scale forward-genetic examination of vertebrate genotype-phenotype relationships was carried out in the mid-1990’s using zebrafish [19]. A few years later, zebrafish were used in the first large-scale screen designed to identify small molecules that were capable of modifying a vertebrates phenotype [20]. That same year, morpholino antisense oligonucleotides injected into zebrafish embryos were used to develop “knock-down” models of human disease [21].
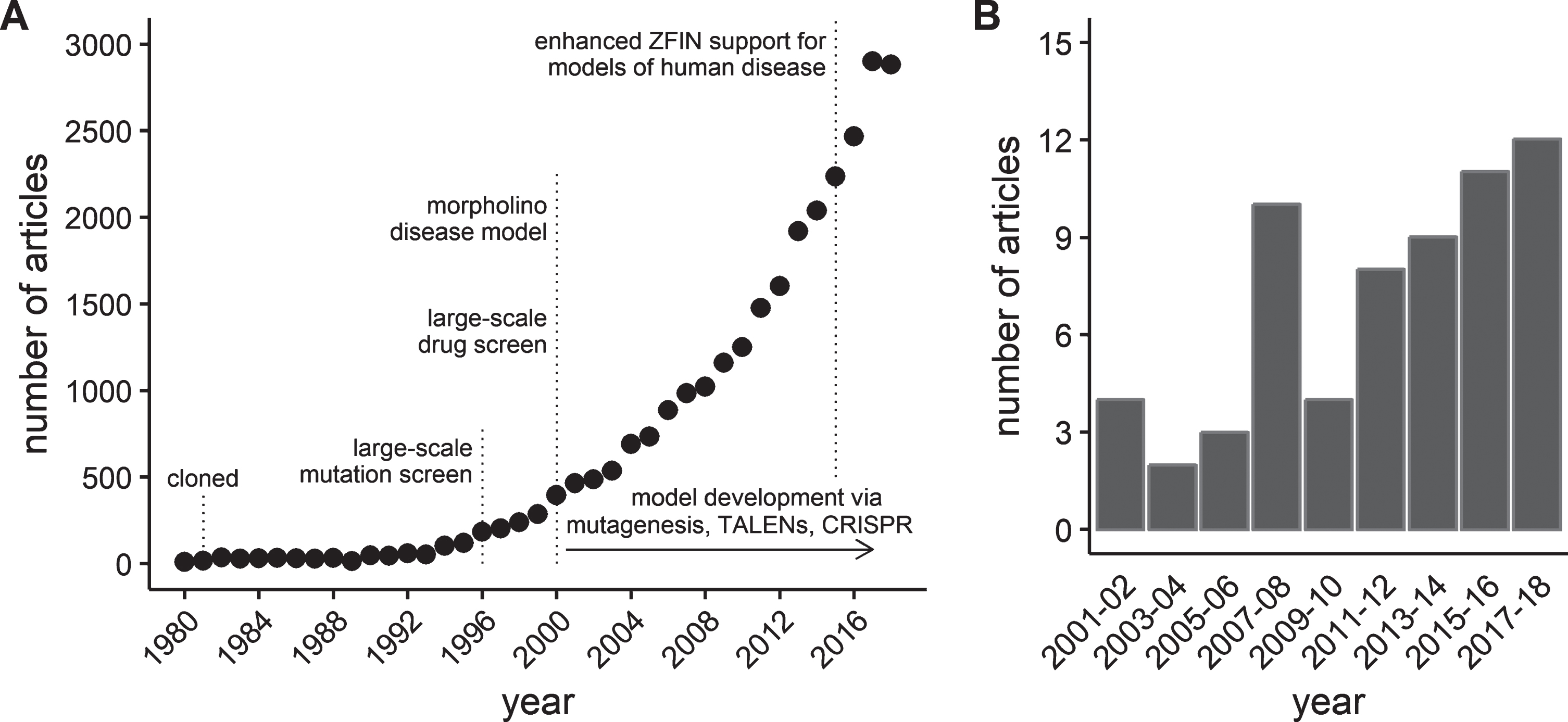
This work kindled a steady rise in the interest in the use of zebrafish as a model organism [22]. In the mid- to late 1990’s, an on-line database of zebrafish genetics and development, the Zebrafish Information Network (ZFIN, http://zfin.org) was established to provide a resource for the rapidly expanding zebrafish scientific community [23]. A measure of the growth in the zebrafish model is illustrated by the exponential increase in the number of zebrafish journal articles curated by ZFIN (Fig. 1-A). Similar growth has been reported by others using different databases or inclusion criteria [22, 24]. Using zebrafish to study skeletal muscle function and disease is a relatively new application of the model. However, zebrafish offer numerous advantages that make them an important compliment to pre-clinical mammalian models of muscle disease. These advantages are beginning to be recognized by the field, leading to a rise in interest in zebrafish models of dystrophies and other muscle diseases (Fig. 1-B).
Fig.1
Growth of journal articles pertaining to zebrafish and muscular dystrophy research. Data obtained by searching the publication database maintained by the Zebrafish Information Network (ZFIN). (A) The number of journal articles by publication year from 1980 through 2018. (B) The number of journal articles containing the term “muscular dystrophy” in either the title or as a keyword between 2001 and 2018. Results grouped into 2 year periods to reduce noise.
Genetic and drug target conservation
Over 80% of genes associated with human disease and morbidity have an orthologue in zebrafish [25]. Skeletal muscle is a highly conserved organ, so in terms of MDs, the homologies between zebrafish and humans is likely even greater. In fact, orthologues for almost all of the genes linked to human dystrophies have been identified in the zebrafish [26]. This suggests that almost any human MD could be modeled by genetic manipulation in zebrafish. Currently, there are dozens of mutant zebrafish lines available that serve as models of human MDs and non-dystrophic myopathies [27–29].
In addition to genetic conservation between species, drug targets must also be conserved if compounds effective in one species are expected to be effective in another. Drug targets appear to be well conserved between zebrafish and humans. For instance, many neuroactive drugs, compounds affecting cardiac repolarization, and cell cycle regulators have similar effects in zebrafish and humans [30–33].
Genetic tractability
The extensive use of zebrafish as a vertebrate model for the past three decades has stimulated the development of robust methodologies for manipulating the fish genome [34]. In forward screens, male fish are exposed to a potent mutagen, such as N-ethyl-nitrosourea (ENU), and the offspring evaluated for a selected phenotype. For example, in the first large-scale phenotype screen described in the last section, the offspring from ENU mutagenized males were evaluated for mobility defects [35]. Four mutants that displayed impaired swimming also showed a progressive deterioration of the axial muscle. Mutations in three of these mutants were subsequently mapped to genes associated with human MDs, providing some of the first fish models for studying muscle disease [36–38].
Reverse genetics is centered on the targeting of a specific gene and observing the subsequent effects on phenotype. Transient knock-down of a protein of interest can be achieved by using antisense oligonucleotides to temporally inhibit the translation of the targeted gene. This approach to protein knockdown is very straightforward in zebrafish as oligonucleotides can be injected directly into 1–4 cell embryos. The targeted protein is absent or reduced in very early larvae prior to oligonucleotide dilution and degradation. Antisense strategies can therefore be used to rapidly test a preliminary hypothesis or confirm a previous result. For example, antisense oligonucleotides were used to knock-down dystrophin expression in zebrafish embryos in order to evaluate whether changes in morphology and behavior were consistent with DMD prior to creation of a dystrophin-deficient mutant model [39].
Permanent knock-out of a protein can be accomplished using gene editing approaches. Zinc-finger nucleases (ZFNs), which bind to a specific 3-nucleotide long target sequence, and transcription activator-like nucleases (TALENs), which bind to a single nucleotide, induce double-stranded breaks and have been used to effectively knock down protein expression in zebrafish [40–43]. The simpler and more efficient CRISPR-Cas system is now supplanting ZFN’s and TALENs as an approach for introducing targeted mutations in the zebrafish genome [44]. These genomic editing tools allow for efficient disruption of a single or multiple genes and the generation of novel zebrafish models of muscular dystrophies.
Complications can arise when generating novel zebrafish models. Ancestral zebrafish underwent two whole genome duplication events before the divergence of fish and mammals and another episode after the teleost radiation [45, 46]. In addition to this, the average duplication rate in zebrafish is higher than in any other vertebrate studied due to their exceptional retention of duplicated genes [47]. Duplicate genes have been identified for several muscle proteins, including filamin C and titin, and likely exist for other proteins associated with human muscle disease [36].
ZEBRAFISH DRUG SCREENS
Zebrafish-based drug screens have identified novel compounds that modulate pathological phenotypes arising from a wide range of diseases, including cancer, bone marrow failure, cardiac and vascular abnormalities, metabolic disease, and behavioral disorders [12, 13]. Here we briefly review screening strategies and libraries. Greater detail can be obtained from recent reviews that focus on experimental design considerations, compound libraries, methods and protocols, and interpretation of findings [12, 13, 48, 49].
Strategies
Screening strategies can be very straightforward when zebrafish disease models have clear and specific morphological phenotypes that parallel a human disease. Because of the optical clarity of zebrafish eggs and larvae, compounds that affect anatomical and structural features, such as muscle tissue disorganization, can be identified using basic light microscopy. Zebrafish also display robust stereotypic behaviors early in development. For instance, spontaneous coiling, ability to hatch on time, or swimming behavior can be very useful as screening criteria as they represent the integrated output of several physiological systems.
In order to screen drugs that influence expression levels of specific target genes, transgenic fish can be created with promoter regions for target genes driving expression of enhanced green fluorescent protein (EGFP) cDNA. Detection of EGFP signals in the transgenic fish can then be used to monitor the expression level of the gene of interest. This approach can reveal pathways involving expression of the target gene and lead to new approaches to drug development.
Manual manipulation of larvae and scoring of phenotype may become prohibitive in large-scale screens. This step can be streamlined by automated devices that can pick up and orientate a single larvae for image acquisition and other procedures before dispensing the larvae into an individual well of a multi-well plate [50]. Even smaller-scale manual manipulation and scoring can benefit from using relatively simple microfludic devices or casts to facilitate rapid and reproducible positioning of larvae [51–53].
Chemical libraries
Many chemical companies offer compound libraries for drug screening. Academic facilities (e.g. the ICCB-Longwood Screening Facility at Harvard Medical School, https://iccb.med.harvard.edu) may offer commercial and custom drug libraries at reduced cost. Libraries are typically supplied in 96–384 well plate formats and can range from tens up to tens of thousands of compounds. Most zebrafish drug library screens evaluate between 1,000 to 5,000 compounds [13].
Libraries can be chosen based on the specific aims of the screen. Libraries comprised of compounds derived from natural substances provide a high degree of chemical diversity and may be appropriate for lead compound screening. In order to identify therapeutic compounds and their potential mechanisms of action, libraries comprised of well-characterized bioactive compounds that target a wide range of biochemical processes or mechanisms are available. For drug repurposing, investigators can choose libraries of FDA-approved compounds. Several of these drug libraries also include compounds that were shown to be safe but which failed to show efficacy in the latter stages of the approval process. Compounds identified using these libraries will have well-characterized pharmacokinetic and pharmacodynamic properties, which may expedite their translation into clinical use. Finally, custom libraries can be created that consist of proprietary compounds.
DISCOVERING SMALL MOLECULE THERAPEUTICS FOR DMD
Skeletal muscle fibers produce force that is directed longitudinally through the myotendineous junction and laterally into the extracellular matrix [54]. One mechanism by which these lateral forces are transmitted across the sarcolemma is via the dystrophin associated protein complex (DAPC). Several components of the DAPC bind sequentially to form a link between the force-bearing actin cytoskeleton and the basal lamina. A critical component in this complex is dystrophin, a large, flexible intracellular spectrin-like protein that binds strongly to γ-actin, linking it to the integral DAPC component β-dystroglycan [55].
Duchenne muscular dystrophy occurs when out-of-frame deletions, duplications, and insertions in the dystrophin gene result in the loss of functional dystrophin at the sarcolemma [56, 57]. In-frame mutations that result in transcription of a shortened, partially functional dystrophin protein are associated with the milder Becker muscular dystrophy [58].
The down-stream physiological consequences of dystrophin-deficiency are multifaceted, interrelated, and often self-propagating [9]. In the absence of dystrophin, the normal transmission of force across the sarcolemma is perturbed. This can lead to sarcomeric instability, loss of sarcolemma integrity, and muscle fiber damage [59–61]. Intracellular Ca2+ levels rise in damaged fibers either as a direct result of mechanical perturbations to the sarcolemma or due to dysregulation of mechanosensitive channels [62–64]. Elevations in intracellular Ca2+ can activate proteases and phospholipases, further exacerbating damage to the cell [65, 66].
The DAPC also serves as a mechano-signaling locus in muscle fibers. Dystrophin interacts with the microtubular network and the loss of this interaction leads to elevated production of reactive oxygen species (ROS) and intracellular Ca2+ dysregulation [67, 68]. ROS can activate stretch-activated channels leading to further influx of Ca2+ into the cell [69]. In the absence of dystrophin, mechanical stretch can activate the NF-κB pathway and raise levels of inflammatory cytokines [70].
Finally, the DAPC is linked to critical enzymatic properties of muscle cells. Neuronal nitric oxide synthase (nNOS) is bound to the central rod region of dystrophin via interactions with syntrophin [71]. Loss of dystrophin, and the subsequent displacement of nNOS from its subsarcolemma localization blunts NO-induced vasodilation during contractile activity, leading to inadequate tissue perfusion and muscle tissue damage [71–73].
Because these damaging mechanisms and pathways are chronically activated, the normal cell regenerative processes present in muscle fibers are eventually overwhelmed resulting in a loss of myofibers and the consequent replacement by fibrotic tissue [74]. At some point, the loss of functional contractile tissue reaches a critical threshold resulting in impaired locomotion, ventilation, and cardiac function.
These down-stream consequences of dystrophin deficiency provide a variety of targets for slowing progression of the disease or as compliments to gene- and protein-based therapies that may not completely restore all of the functions of full-length dystrophin. Targeting downstream events has the advantage of being applicable to all DMD patients as benefits would be independent of the underlying mutation. Large-scale screens using dystrophin-deficient zebrafish have proved to be a rapid strategy for identifying approved drugs that could be repositioned to target these mechanisms and to discover novel compounds to modulate these pathological events.
Zebrafish models of DMD
During the first large-scale vertebrate genetic screen, four zebrafish mutants were characterized by their abnormal swimming performance coupled with progressive axial muscle degeneration [35]. One of these mutants, sapje, was subsequently found to have a nonsense mutation in exon 4 of the zebrafish orthologue to the human dystrophin gene [37, 75]. A second dystrophin mutant, sapje-like, was identified during a large-scale zebrafish screen [76]. In sapje-like, the mutation is at the exon 62 donor splice junction.
The absence of dystrophin is lethal in the sapje and sapje-like lines. Mortality begins to deviate from wild-type larvae around 12 dpf, with only a small subset of dystrophin-deficient larvae surviving to 30 dpf, and none reaching reproductive age [77]. Consequently, lines are maintained by mating heterozygotes as the zebrafish dmd gene is autosomal. Because the gene is recessive, only 25% of a clutch will be dystrophin-null.
Fortunately, these dystrophin null mutants can be readily identified at 3-4 dpf using a non-lethal optical technique that takes advantage of the transparent nature of zebrafish larvae and relies on the birefringent properties of skeletal muscle [35, 78, 79]. By placing larvae between two polarizing filters arranged to cancel light transmission, the highly ordered structure of the myofilaments will refract or rotate light causing the axial muscle of the larvae to appear illuminated (Fig. 2). Wild type or unaffected larvae will show a bright, highly ordered birefringence pattern consistent with the regular geometry of the myofilament lattice. In contrast, the somites of a dystrophic or affected larvae will be randomly pocketed with regions of reduced birefringence.
Fig.2
Identification of dystrophic zebrafish larvae using a birefringence assay.

These regions of abnormal birefringence represent gaps and breaks in the myofilament lattice formed by the retraction of myofibers whose dystrophin-null ends have become detached from the myosepta and are characteristic of models of muscular dystrophy like sapje and sapje-like. In contrast, models of non-dystrophic congenital myopathies exhibit reduced overall levels of birefringence without gaps or breaks [81, 82]. In sapje, these gaps and breaks in the myofilament lattice have been linked to the precocious muscular activity of larvae [83], consistent with the idea that dystrophin-deficient muscles are susceptible to the mechanical strains generated during contractile activity [84]. Consequently, early sapje and sapje-like zebrafish larvae have easily identifiable morphological features that support the role of dystrophin as a sarcomeric stabilizer and which can serve as a rapid, non-lethal assay of dystrophin-deficiency.
Dystrophin deficient zebrafish muscles also show signs of inflammation, fibrosis, and degeneration∖regeneration consistent with mammalian models of DMD. By 28 dpf, muscle fibers of sapje zebrafish are infiltrated with neutrophils and macrophages and show interstitial deposits of collagen [80]. These larvae also show increased proliferation of muscle satellite cells although this is insufficient to restore myofibers to the areas of muscle damage [80].
Because the gaps and breaks in the axial muscles of dystrophin-deficient zebrafish larvae represent areas of hypercontracted or absent contractile material, these areas would no longer be expected to be functional. Consistent with this are findings that muscles of dystrophin-deficient larvae exhibit substantial weakness [85, 86]. Unlike the mildly affected mdx mouse model of DMD, where dystrophic limb muscles may hypertrophy resulting in normal absolute force and a modest reduction in force normalized to muscle cross-sectional area [87–89], the axial muscle of dystrophin-deficient zebrafish shows a reduction in both absolute and normalized force [85, 86]. Furthermore, these force deficits are roughly 2-fold greater than those observed for limb muscles of the mdx mouse and are more in line with the force deficits reported for skeletal muscles of dystrophin-deficient canines [90, 91] or the double knockout dystrophin-utrophin mouse [92].
In addition to lower force, axial muscles of affected sapje zebrafish also show a heightened sensitivity to acute mechanical strain [85]. This response is consistent with the role of dystrophin as a mechanical stabilizer and is qualitatively similar to that observed for muscles of mouse and canine models of DMD [60, 91, 93–97]. Thus, the two functional hallmarks of dystrophic mammalian muscle, a reduction in peak force and a heightened susceptibility to mechanical strain, are replicated in dystrophin-deficient zebrafish models.
Screens to discover DMD therapeutics
In order to identify putative therapeutic compounds for treating DMD, Kawahara and colleagues [77, 98] used sapje and sapje-like dystrophin-null zebrafish to screen the Prestwick 1 Collection, the NINDS 2 Compound Library, and the Known Bioactives 2012 Library (Table 1). The 2640 compounds making up these libraries cover almost 50% of all FDA-approved drugs.
Table 1
Chemical libraries that have been used to screen dystrophic zebrafish embryos or larvae
| Name | Company | no. chemicals | Notes |
| 1a. Prestwick 1 Collection | Prestwick Chemical | 1280 | 95% approved drugs |
| 1b. Prestwick 2 Collection | Prestwick Chemical | 1120 | >85% marketed drugs |
| 2. NINDS 2 Compound Library | ENZO Life Sciences | 1040 | 75% FDA-approved drugs |
| 3. ICCBL Known Bioactives 2012 | ENZO Life Sciences | 480 | targets biochemical pathways |
| 4. FDA Approved Drugs | ENZO Life Sciences | 640 | 100% FDA-approved drugs |
Note that the composition and number of chemicals in a library can change over time.
A schematic of the screening strategy is illustrated in Fig. 3. In an initial screen, pools containing eight chemicals were applied to 20 randomly selected 1 dpf embryos from heterozygous sapje parents. Recall that 25% of these embryos would be expected to be dystrophin-null and would show abnormal morphology at 4 dpf as revealed by the birefringence assay. It was reasoned that this percentage would be unchanged if all eight compounds of a pool had no effect on the dystrophic phenotype. However, if one or more compounds in the pool had a therapeutic effect, then the percentage of larvae showing abnormal birefringence would be reduced.
Fig.3
A summary of the work-flow used by Kawahara et al. [77] to screen for novel drug candidates. Similar work-flows have been used by others to identify potential therapeutics for DMD and LGMD2I [99, 120]. See text for details.
![A summary of the work-flow used by Kawahara et al. [77] to screen for novel drug candidates. Similar work-flows have been used by others to identify potential therapeutics for DMD and LGMD2I [99, 120]. See text for details.](https://ip.ios.semcs.net:443/media/jnd/2019/6-3/jnd-6-3-jnd190389/jnd-6-jnd190389-g003.jpg)
In the secondary screen, each of the individual compounds in a pool that reduced the percentage of morphologically affected fish was tested separately using the same screening strategy. When a positive hit was detected, the genotype of all 20 embryos in the well was evaluated to confirm that the fish rescued by the compound were dystrophin-null. Fifteen compounds that restored normal birefringence to a majority of dystrophin-null mutants were identified in this secondary screen (Table 2).
Table 2
Compounds that reduced the incidence of the dystrophic phenotype in zebrafish models of Duchenne muscular dystrophy
| Therapeutic group, mechanism | Compound (PubChem CID) | % affected | Survival | Reference |
| PDE inhibitors | aminophylline (9433) | 5–17.5 | extended | [77, 98, 99] |
| ibudilast (3671) | 15 | [77] | ||
| rolipram (5092) | 17 | [77] | ||
| dipyridamole (3108) | 17 | [77] | ||
| sildenafil citrate (5212) | 15 | extended | [98] | |
| dopamine agonists | ergotamine (8223) | 0 | [99] | |
| ropinirole (5095) | 0–16 | [99] | ||
| pergolide (47811) | 5 | [99] | ||
| serotonin, SSRI’s | serotonin (5202) | 0 | [99] | |
| fluoxetine (3386) | 0 | mixed | [99] | |
| paroxetine (43815) | 17 | [99] | ||
| chelating agents | pentetic acid (3053) | 7.5–10 | extended | [77] |
| flunarizine (941361) | 10 | [99] | ||
| steriods | androsterone acetate (6432116) | 7.5–18.4 | [98] | |
| equilin (223368) | 5–10 | no effect | [77] | |
| antihistamines | conessine (441082) | 10–12.5 | toxic | [77] |
| homochlorcyclizine dihydrochloride (656590) | 10–12.5 | toxic | [77] | |
| lipid synthesis inhibitor | cerulenin (5282054) | 2.5–10 | extended | [98] |
| immunosuppressent | crassin acetate (5355441) | 2.5–12.5 | extended | [98] |
| anti-inflammatory | epirizole (3242) | 10–15 | extended | [77] |
| antiprotozoal | nitromide (4511) | 7.5–12.5 | [98] | |
| antioxidant | pomiferin (4871) | 10–12.5 | [98] | |
| smooth muscle relaxant | propantheline bromide (9279) | 12.5–15 | [98] | |
| cardiotonic | proscillaridin A (5284613) | 10 | toxic | [77] |
| vasoconstrictor | 9a,11b-prostaglandin F2 (5280886) | 7.5–10 | no effect | [98] |
Embryos were treated starting at 1 dpf. Compounds that reduced the incidence of the affected dystrophic phenotype at 4 dpf by 30% or more, i.e. from the expected 25% of larvae to 17.5% or less, are compiled in the table. Some compounds were subsequently tested for their effect on the survival of 1 dpf [99] or 4 dpf [98, 99] affected larvae. Abbreviations: dpf, days post-fertilization; PDE, phosphodiesterase; SSRI, selective serotonin re-uptake inhibitor.
To address whether the therapeutic candidates identified in the short-term screen could reverse pathology once it had become established, a long-term screen was conducted in which 4-5 dpf sapje larvae displaying abnormal birefringence were treated with several candidate compounds and followed for several weeks [77, 98]. By 20 dpf, mortality of the untreated affected larvae was about twice that of their wild-type siblings. Six compounds extended survival of over half of the affected larvae (Table 2). These longer surviving dystrophic larvae also showed improved muscle morphology and more extensive vascularization of axial tail muscle [98]. Immunoblotting indicated that enhanced survival, more extensive vascularization, and normalization of muscle morphology all occurred in the absence of any detectable dystrophin expression.
PDE inhibitors
One class of compounds revealed by these screens were phosphodiesterase (PDE) inhibitors. Aminophylline, a nonspecific PDE inhibitor, and sildenafil citrate, a PDE5 inhibitor, were two of the six compounds that improved muscle morphology, increased vascularization, and extended survival of dystrophin-null larvae [98]. The benefits of aminophylline in rescuing dystrophin-deficient sapje larvae were subsequently confirmed by an independent group conducting an unbiased screen of the ENZO FDA Approved Drug library [99]. To further investigate this class of therapeutics, five additional PDE inhibitors have been evaluated in dystrophic zebrafish [77]. The PDE4 inhibitors ibudilast and rolipram, and the PDE5 inhibitor dipyridamole, reduced the dystrophic phenotype in 4 dpf sapje larvae (Table 2), although not as effectively as either aminophylline or sildenafil. In contrast, enoximone and milrinone, PDE3 inhibitors, had no therapeutic effect.
The identification of PDE inhibitors as candidate therapeutics was significant for several reasons. Targeted drug discovery approaches, focusing on the nitric oxide– cyclic guanosine monophosphate pathways, revealed that the PDE5 inhibitors sildenafil and tadalafil reduced skeletal and cardiac muscle dysfunction and damage in mouse and canine models of DMD [73, 100–103]. The identification of PDE inhibitors by independent laboratories using unbiased phenotypic screening approaches validates that large-scale dystrophic zebrafish screens take advantage of target and mechanistic conservation between the zebrafish and well-accepted pre-clinical mammalian DMD models.
Selective serotonin re-uptake inhibitors
Waugh et al. [99] screened the ENZO FDA Approved Drug library and reported that 1 dpf sapje larvae treated with the selective serotonin re-uptake inhibitor fluoxetine all lived to 17 dpf, a time when none of the untreated sapje larvae remained alive. The therapeutic effects of fluoxetine were rapidly reversible as larvae initially treated and asymptomatic rapidly developed pathology when treatment was interrupted. However, fluoxetine treatment had to be initiated early in development in order to be effective as delaying treatment until the dystrophic phenotype became apparent had no effect on survival (Table 2). The available evidence therefore suggests that fluoxetine can prevent the establishment of dystrophy but may not be able to reverse the pathology once it has become established. It is noteworthy that serotonin was discovered to reduce muscle degeneration in a C. elegans model of dystrophin-deficiency [104] but not when evaluated in the mdx mouse [105].
Ca2+ antagonists
Treatments to reduce excessive intracellular Ca2+ in muscles of mdx mice and DMD patients have had mixed results but therapeutically targeting Ca2+ homeostasis remains an area of active investigation [8]. Two putative Ca2+ antagonists were identified in screens of dystrophic zebrafish, pentetic acid from the Prestwick library [77] and flunarizine from the ENZO FDA Approved Drug library [99].
Pentetic acid (diethylenetriaminepentaacetic acid or DTPA) is a metal chelator, similar to EDTA. Pentetic acid is used clinically to treat heavy metal poisoning, contamination by radioactive material, and when complexed with gadolinium, as a contrasting agent during MRI. We are unaware of any pre-clinical investigations or clinical trials using pentetic acid as a DMD therapeutic.
Flunarizine is a selective channel blocker used for treating occulsive vascular disease and neurological disorders, such as migraine, vertigo, and epilepsy. A small, one-year clinical trial of flunarizine showed no effect on any outcome measure of 5–15 year old boys diagnosed with DMD [106].
DISCOVERING SMALL MOLECULE THERAPEUTICS FOR LGMD2I
Dystrophin links intracellular actin to β-dystroglycan, a DAPC component embedded in the sarcolemma. β-dystroglycan is in turn linked to laminin-2 of the extracellular matrix via the glycoprotein α-dystroglycan. These linkages allow the DAPC to span the sarcolemma from intracellular actin, through dystrophin, β-dystroglycan, α-dystroglycan, and laminin-2 to the basal laminae. α-Dystroglycan is a highly glycosylated protein. Hypoglycosylation leads to a reduction in the affinity of α-dystroglycan for laminin and the loss of muscle fiber integrity and ultrastructure [107–109].
Mutations to the human fukutin-related-protein (FKRP) gene, which encodes a putative glycosyltransferase modulating α-dystroglycan glycosylation, are associated with a wide spectrum of pathological phenotypes [110, 111]. For instance, the p.Leu276Ile mutation is linked to reductions in the expression of α-dystroglycan and laminin α2 [111]. Patients with this mutation experience weakness of the hip, shoulder, and neck muscles, have difficulty performing daily activities, such as walking, running, and climbing stairs, and may eventually develop respiratory problems, cardiomyopathy, or cognitive deficits [111–113].
Current treatment for LGMD2I patients includes the use of cortiocsteroids, alone or in combination with the osteoporosis drug Fosamax, to maintain muscle strength [114, 115]. There are significant side effects to these treatments such as weight gain and inflammation. Novel therapies, such as over-expression of FKRP via adeno-associated viral vectors or autologous muscle stem cell transplantation have shown some promise but there are still significant issues regarding body wide expression and correct dosing amounts [116–119]. Thus, there is a need for novel therapeutic treatments to complement gene and cell-based therapies for LGMD2I patients.
Zebrafish screens for LGMD2I therapeutics
Zebrafish co-injected with human FKRP p.Leu276Ile mutation mRNA and an anti-sense morpholino oligonucleotide targeting the endogenous zebrafish fkrp gene showed a mild dystrophic phenotype, consistent with the human disorder [109]. Based on these findings, a zebrafish model of LGMD2I was developed using heat-shock inducible expression of the human p.Leu276Ile mutation in an FKRP-deficient zebrafish background [120]. This fish was characterized by abnormal birefringence indicative of myofibrillar disorganization, impaired spontaneous and touch-evoked swimming performance, reduced axial muscle tetanic force, eye and brain malformations, pericardiac edema, and a reduced lifespan.
In order to identify potential new therapeutics, a two-stage screening approach, similar to the method previously used on sapje and sapje-like larvae [77, 121], was used to identify small molecules that reduced the pathological phenotypes of the LGMD2I zebrafish model [120]. The first stage of the screen identified 20 unique compounds from the Prestwick 2 collection that reduced the occurrence of abnormal birefringence, eye and brain abnormalities, pericardiac edema, and depressed vitality from an expected 50% of larvae to <10% (Table 3).
Table 3
Compounds that reduced the pathological phenotype of a zebrafish model of LGMD2I
| Therapeutic group/mechanism | Compound (PubChem CID) |
| antibiotics | ornidazole (28061) |
| sulfacetamide sodium hydrate (6419954) | |
| flumequine (3374) | |
| dihydrostreptomycin sulfate (6419912) | |
| metampicillin sodium salt (25195407) | |
| aztreonam (5742832) | |
| channel blockers/inhibitors | calycanthine (5392245) |
| ethosuximide (3291) | |
| kawain (5281565) | |
| dopamine antagonists | perphenazine (4748) |
| thioproperazine dimesylate (92178) | |
| α1 adrenergic receptor agonists | oxymetazoline hydrochloride (21114704) |
| metaraminol bitartrate (51371362) | |
| muscarinic antagonists | ipratropium bromide (11553369) |
| pirenzepine dihydrochloride (45114102) | |
| anti-neoplastics | betulin (72326) |
| paclitaxel (36314) | |
| smooth muscle relaxant | alverine citrate salt (21718) |
| skeletal muscle relaxant | chlorphensin carbamate (2724) |
| glucocorticoid | rimexolone (5311412) |
| triterpenoid nutraceutical | ursolic acid (64965) |
| metal chelator | pentetic acid (3053) |
| others † | citric acid (311) |
| triethanolamine (7618) |
Compounds identified by Serafini et al. [120]. Chemicals were screened at 2 dpf, 1 day after heat shock-inducible over-expression of a human FKRP transgene. By 5 dpf all compounds listed had reduced the percentage of larvae displaying the pathological phenotype (abnormal muscle birefringence, head and eye malformations, pericardiac edema) from approximately 50% to ≤10%. †Additional compounds screened because of their structural similarity to pentetic acid. Abbreviations: PDE, phosphodiesterase inhibitor.
Pentetic acid
One of the most effective compounds in reducing the pathological phenotype in the LGMD2I zebrafish model was the chelator pentetic acid. This compound has previously been identified as a potential therapeutic for dystrophin-deficient fish [77]. To further investigate the role of chelation in the LGMD2I model, 7 chemicals that were structurally similar to pentetic acid were evaluated in a follow-up screen. This screen identified two additional compounds that rescued LGMD2I larvae, triethanolamine and citric acid (Table 3), both of which have metal chelating properties. These findings suggest that therapies directed at manipulating intracellular Ca2+ levels may be therapeutic for LGMD2I.
Ursolic acid
Another novel compound identified in this screen was ursolic acid, a pentacyclic triterpenoid found in many plants. Pentacyclic triterpenoids are steroid-like compounds that are structurally similar to cardiac glycosides such as ouabain or digitoxin. Cardiac glycosides have a positive inotropic effect on cardiac contractility by increasing intracellular Ca2+ through an inhibition of the Na+, K+-ATPase. In fact, ursolic acid was recently reported to be a moderate inhibitor of porcine Na+, K+-ATPase [122].
Ursolic acid has also been reported to induce skeletal muscle hypertrophy, increase grip strength, improve hindlimb muscle contractility, and reduce muscle wasting in mice subjected to fasting or muscle denervation [123]. These effects were associated with the down-regulation of mRNA’s associated with atrophy, such as atrogin-1 and MuRF-1, and with an increase in Akt phosphorylation. Thus, a reasonable hypothesis is that ursolic acid targets mechanisms regulating both cardiac and skeletal muscle contractility in the zebrafish LGMD2I model.
FUTURE DIRECTIONS
With appropriate modifications, the screening methodology and principles reviewed could be adapted and applied to other dystrophies and myopathies. Future work will be needed to address some of the complications and limitations of present screening approaches.
Libraries
To date, screens have used libraries comprised of FDA approved drugs in order to identify compounds that could be re-purposed as therapeutics for DMD or LGMD2I. Other libraries are available for addressing different research goals. For instance, libraries have been developed specifically for discovering novel lead compounds or identifying compounds targeting relevant biological mechanisms or pathways. The use of these alternative libraries represents an untapped resource for discovering new therapeutics for muscle disease.
Outcome measures
Visually assessing birefringence has been the most common approach for determining larval muscle phenotype in large-scale screening. This qualitative measure involves a degree of subjectivity that makes it difficult to distinguish graded responses to potential therapeutics. The birefringence assay is also limited to a narrow window of larval development, from about 3-4 dpf until 7–10 dpf when the appearance of pigmentation and scales reduces larval transparency. Limitations of the birefringence assay could be addressed in several ways. For instance, image analysis could be used to quantify changes in birefringence [78, 79]. Alternatively, monitoring reporter genes, proteins, or metabolites [124], assessing behavioral outcomes like locomotion [78], or directly measuring muscle function [85, 125, 126] could be used as outcome measures. The critical barrier to the use of any alternative approach is whether it can be incorporated into a high throughput work flow.
CONCLUSIONS
Zebrafish occupy a unique niche in the spectrum of model organisms available for biomedical research [24]. As vertebrates, they have orthologues to most of the human disease associated genes and to almost all of the genes linked to muscular dystrophies, yet they possess many of the characteristics of invertebrate model systems, including small size, rapid development, and genetic tractability. These characteristics have enabled investigators to generate zebrafish strains that replicate the phenotype of many human diseases and to use these models in large-scale chemical screens to identify compounds that modify phenotype. The availability of zebrafish models of muscular dystrophies now enables this approach to be used to identify candidate therapeutics targeting the secondary, downstream or non-genetic effects of these diseases. These unbiased phenotypic screens provide an important compliment to existing approaches for developing novel therapies for muscular dystrophies.
ACKNOWLEDGMENTS
Work described in this review, and support for the authors, was funded by grants R01AR064300, R01AR044345, R01HD075802 and P50HD060848 from the National Institutes of Health, by Muscular Dystrophy Association of U.S.A. grants MDA381140 and MDA602235, and by the Anderson Family Foundation. Lou Kunkel is a consultant for Sarepta and Dyne Therapeutics.
REFERENCES
[1] | Emery AE . Population frequencies of inherited neuromuscular diseases–a world survey. Neuromuscul Disord. (1991) ;1: :19–29. |
[2] | Mah JK , Korngut L , Dykeman J , Day L , Pringsheim T , Jette N . A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul Disord. (2014) ;24: :482–91. doi: 10.1016/j.nmd.2014.03.008 |
[3] | Deenen JCW , Horlings CGC , Verschuuren JJGM , Verbeek ALM , van Engelen BGM . The epidemiology of neuromuscular disorders: A comprehensive overview of the literature. J Neuromuscul Dis. (2015) ;2: :73–85. |
[4] | Mendell JR , Shilling C , Leslie ND , Flanigan KM , al-Dahhak R , Gastier-Foster J , et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol. (2012) ;71: :304–13. doi: 10.1002/ana.23528 |
[5] | Koeks Z , Bladen CL , Salgado D , van Zwet E , Pogoryelova O , McMacken G , et al. Clinical outcomes in Duchenne muscular dystrophy: A study of patients from the TREAT-NMD DMD global database. J Neuromuscul Dis. (2017) ;4: :293–306. doi: 10.3233/JND-170280 |
[6] | Guiraud S , Aartsma-Rus A , Vieira NM , Davies KE , van Ommen G-JB , Kunkel LM . The pathogenesis and therapy of muscular dystrophies. Annu Rev Genomics Hum Genet. (2015) ;16: :281–308. doi: 10.1146/annurev-genom-090314-025003 |
[7] | Tidball JG , Wehling-Henricks M . Evolving therapeutic strategies for Duchenne muscular dystrophy: Targeting downstream events. Pediatr Res. (2004) ;56: 831–41. doi: 10.1203/01.PDR.0000145578.01985.D0 |
[8] | Spinazzola JM , Kunkel LM . Pharmacological therapeutics targeting the secondary defects and downstream pathology of Duchenne muscular dystrophy. Expert Opin Orphan Drugs.. (2016) ;4: :1179–94. 10.1080/21678707.2016.1240613 |
[9] | Allen DG , Whitehead NP , Froehner SC . Absence of dystrophin disrupts skeletal muscle signaling: Roles of Ca2+, reactive oxygen species, and nitric oxide in the development of muscular dystrophy. Physiol Rev. (2016) ;96: :253–305. 10.1152/physrev.00007.2015 |
[10] | Kell DB . Finding novel pharmaceuticals in the systems biology era using multiple effective drug targets, phenotypic screening and knowledge of transporters: Where drug discovery went wrong and how to fix it. FEBS J. (2013) ;280: :5957–80. 10.1111/febs.12268 |
[11] | Zheng W , Thorne N , McKew JC . Phenotypic screens as a renewed approach for drug discovery. Drug Discov Today. (2013) ;18: :1067–73. 10.1016/j.drudis.2013.07.001 |
[12] | Wiley DS , Redfield SE , Zon LI . Chemical screening in zebrafish for novel biological and therapeutic discovery. Methods Cell Biol. (2017) ;138: :651–79. 10.1016/bs.mcb.2016.10.004 |
[13] | Rennekamp AJ , Peterson RT . 15 years of zebrafish chemical screening. Curr Opin Chem Biol. (2015) ;24: :58–70. 10.1016/j.cbpa.2014.10.025 |
[14] | Grunwald DJ , Eisen JS . Headwaters of the zebrafish – emergence of a new model vertebrate. Nat Rev Genet. (2002) ;3: :717–24. 10.1038/nrg892 |
[15] | Streisinger G , Walker C , Dower N , Knauber D , Singer F . Production of clones of homozygous diploid zebra fish (Brachydanio Rerio). Nature. (1981) ;291: :293–6. |
[16] | Kimmel CB , Ballard WW , Kimmel SR , Ullmann B , Schilling TF . Stages of embryonic development of the zebrafish. Dev Dyn.. (1995) ;203: :253–310. doi: 10.1002/aja.1002030302 |
[17] | Parichy DM , Elizondo MR , Mills MG , Gordon TN , Engeszer RE . Normal table of postembryonic zebrafish development: Staging by externally visible anatomy of the living fish. Dev Dyn.. (2009) ;238: :2975–3015. doi: 10.1002/dvdy.22113 |
[18] | Stickney HL , Barresi MJ , Devoto SH . Somite development in zebrafish. Dev Dyn. (2000) ;219: :287–303. doi: 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1065>3.0.CO;2-A |
[19] | Nüsslein-Volhard C . The zebrafish issue of Development. Development. (2012) ;139: :4099–103. doi: 10.1242/dev.085217 |
[20] | Peterson RT , Link BA , Dowling JE , Schreiber SL . Small molecule developmental screens reveal the logic and timing of vertebrate development. Proc Natl Acad Sci USA. (2000) ;97: :12965–9. doi: 10.1073/pnas.97.24.12965 |
[21] | Nasevicius A , Ekker SC . Effective targeted gene ’knockdown’ in zebrafish. Nat Genet. (2000) ;26: :216–20. doi: 10.1038/79951 |
[22] | Kinth P , Mahesh G , Panwar Y . Mapping of zebrafish research: A global outlook. Zebrafish. (2013) ;10: :510–7. doi: 10.1089/zeb.2012.0854 |
[23] | Westerfield M , Doerry E , Douglas S . Zebrafish in the Net. Trends Genet. (1999) ;15: :248–9. |
[24] | Lieschke GJ , Currie PD . Animal models of human disease: Zebrafish swim into view. Nat Rev Genet. (2007) ;8: :353–67. doi: 10.1038/nrg2091 |
[25] | Howe K , Clark MD , Torroja CF , Torrance J , Berthelot C , Muffato M , et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature. (2013) ;496: :498–503. doi: 10.1038/nature12111 |
[26] | Steffen LS , Guyon JR , Vogel ED , Howell MH , Zhou Y , Weber GJ , et al. The zebrafish Runzel muscular dystrophy is linked to the titin gene. Dev Biol. (2007) ;309: :180–92. doi: 10.1016/j.ydbio.2007.06.015 |
[27] | Li M , Hromowyk KJ , Amacher SL , Currie PD . Muscular dystrophy modeling in zebrafish. Methods Cell Biol.. (2017) ;138: :347–80. doi: 10.1016/bs.mcb.2016.11.004 |
[28] | Gibbs EM , Horstick EJ , Dowling JJ . Swimming into prominence: The zebrafish as a valuable tool for studying human myopathies and muscular dystrophies. FEBS J. (2013) ;280: :4187–97. doi: 10.1111/febs.12412 |
[29] | Goody MF , Carter EV , Kilroy EA , Maves L , Henry CA . “Muscling” throughout life: Integrating studies of muscle development, homeostasis, and disease in zebrafish. Curr Top Dev Biol.. (2017) ;124: :197–234. doi: 10.1016/bs.ctdb.2016.11.002 |
[30] | Murphey RD , Stern HM , Straub CT , Zon LI . A chemical genetic screen for cell cycle inhibitors in zebrafish embryos. Chem Biol Drug Des. (2006) ;68: :213–9. doi: 10.1111/j.1747-0285.2006.00439.x |
[31] | Kokel D , Bryan J , Laggner C , White R , Cheung CYJ , Mateus R , et al. Rapid behavior-based identification of neuroactive small molecules in the zebrafish. Nat Chem Biol. (2010) ;6: :231–7. doi: 10.1038/nchembio.307 |
[32] | Rihel J , Prober DA , Arvanites A , Lam K , Zimmerman S , Jang S , et al. Zebrafish behavioral profiling links drugs to biological targets and rest/wake regulation. Science. (2010) ;327: :348–51. doi: 10.1126/science.1183090 |
[33] | Milan DJ , Peterson TA , Ruskin JN , Peterson RT , MacRae CA . Drugs that induce repolarization abnormalities cause bradycardia in zebrafish. Circulation. (2003) ;107: :1355–8. |
[34] | Basu S , Sachidanandan C . Zebrafish: A multifaceted tool for chemical biologists. Chem Rev. (2013) ;113: :7952–80. doi: 10.1021/cr4000013 |
[35] | Granato M , van Eeden FJ , Schach U , Trowe T , Brand M , Furutani-Seiki M , et al. Genes controlling and mediating locomotion behavior of the zebrafish embryo and larva. Development. (1996) ;123: :399–413. |
[36] | Steffen LS , Guyon JR , Vogel ED , Beltre R , Pusack TJ , Zhou Y , et al. Zebrafish orthologs of human muscular dystrophy genes. BMC Genomics. (2007) ;8: :79. doi: 10.1186/1471-2164-8-79 |
[37] | Bassett DI , Bryson-Richardson RJ , Daggett DF , Gautier P , Keenan DG , Currie PD . Dystrophin is required for the formation of stable muscle attachments in the zebrafish embryo. Development. (2003) ;130: :5851–60. doi: 10.1242/dev.00799 |
[38] | Hall TE , Bryson-Richardson RJ , Berger S , Jacoby AS , Cole NJ , Hollway GE , et al. The zebrafish candyfloss mutant implicates extracellular matrix adhesion failure in laminin alpha2-deficient congenital muscular dystrophy. Proc Natl Acad Sci USA. (2007) ;104: :7092–7. doi: 10.1073/pnas.0700942104 |
[39] | Guyon JR , Mosley AN , Zhou Y , O’Brien KF , Sheng X , Chiang K , et al. The dystrophin associated protein complex in zebrafish. Hum Mol Genet. (2003) ;12: :601–15. |
[40] | Doyon Y , McCammon JM , Miller JC , Faraji F , Ngo C , Katibah GE , et al. Heritable targeted gene disruption in zebrafish using designed zinc-finger nucleases. Nat Biotechnol.. (2008) ;26: :702–8. doi: 10.1038/nbt1409 |
[41] | Meng X , Noyes MB , Zhu LJ , Lawson ND , Wolfe SA . Targeted gene inactivation in zebrafish using engineered zinc-finger nucleases. Nat Biotechnol. (2008) ;26: :695–701. doi: 10.1038/nbt1398 |
[42] | Huang S-H , Hsiao C-D , Lin D-S , Chow C-Y , Chang C-J , Liau I . Imaging of zebrafish in Vivo with second-harmonic generation reveals shortened sarcomeres associated with myopathy induced by statin. PLoS ONE. (2011) ;6: :e24764. doi: 10.1371/journal.pone.0024764 |
[43] | Sander JD , Cade L , Khayter C , Reyon D , Peterson RT , Joung JK , et al. Targeted gene disruption in somatic zebrafish cells using engineered TALENs. Nat Biotechnol. (2011) ;29: :697–8. doi: 10.1038/nbt.1934 |
[44] | Hwang WY , Fu Y , Reyon D , Maeder ML , Tsai SQ , Sander JD , et al. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol. (2013) ;31: :227–9. doi: 10.1038/nbt.2501 |
[45] | Amores A , Force A , Yan YL , Joly L , Amemiya C , Fritz A , et al. Zebrafish hox clusters and vertebrate genome evolution. Science. (1998) ;282: :1711–4. |
[46] | Dehal P , Boore JL . Two rounds of whole genome duplication in the ancestral vertebrate. PLOS Biology. (2005) ;3: :e314. doi: 10.1371/journal.pbio.0030314 |
[47] | Blomme T , Vandepoele K , De Bodt S , Simillion C , Maere S , Van de Peer Y . The gain and loss of genes during 600 million years of vertebrate evolution. Genome Biology. (2006) ;7: :R43. doi: 10.1186/gb-2006-7-5-r43 |
[48] | Brady CA , Rennekamp AJ , Peterson RT . Chemical screening in zebrafish. Methods Mol Biol. (2016) ;1451: :3–16. doi: 10.1007/978-1-4939-3771-4_1 |
[49] | Kaufman CK , White RM , Zon L . Chemical genetic screening in the zebrafish embryo. Nat Protoc. (2009) ;4: :1422–32. doi: 10.1038/nprot.2009.144 |
[50] | Pardo-Martin C , Chang T-Y , Koo BK , Gilleland CL , Wasserman SC , Yanik MF . High-throughput in Vivo vertebrate screening. Nat Methods. (2010) ;7: :634–6. doi: 10.1038/nmeth.1481 |
[51] | Bischel LL , Mader BR , Green JM , Huttenlocher A , Beebe DJ . Zebrafish Entrapment By Restriction Array (ZEBRA) device: A low-cost, agarose-free zebrafish mounting technique for automated imaging. Lab Chi. (2013) ;13: :1732–6. doi: 10.1039/c3lc50099c |
[52] | Petzold AM , Bedell VM , Boczek NJ , Essner JJ , Balciunas D , Clark KJ , et al. SCORE imaging: Specimen in a corrected optical rotational enclosure. Zebrafish. (2010) ;7: :149–54. doi: 10.1089/zeb.2010.0660 |
[53] | Masselink W , Wong JC , Liu B , Fu J , Currie PD . Low-cost silicone imaging casts for zebrafish embryos and larvae. Zebrafish. (2014) ;11: :26–31. doi: 10.1089/zeb.2013.0897 |
[54] | Street SF . Lateral transmission of tension in frog myofibers: A myofibrillar network and transverse cytoskeletal connections are possible transmitters. J Cell Physiol. (1983) ;114: :346–64. doi: 10.1002/jc1041140314 |
[55] | Rybakova IN , Patel JR , Ervasti JM . The dystrophin complex forms a mechanically strong link between the sarcolemma and costameric actin. J Cell Biol. (2000) ;150: :1209–14. |
[56] | Hoffman EP , Brown RH , Kunkel LM . Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell. (1987) ;51: :919–28. |
[57] | Bladen CL , Salgado D , Monges S , Foncuberta ME , Kekou K , Kosma K , et al. The TREAT-NMD DMD Global Database: Analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat. (2015) ;36: :395–402. doi: 10.1002/humu.22758 |
[58] | Monaco AP , Bertelson CJ , Liechti-Gallati S , Moser H , Kunkel LM . An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics. (1988) ;2: :90–5. |
[59] | Ramaswamy KS , Palmer ML , van der Meulen JH , Renoux A , Kostrominova TY , Michele DE , et al. Lateral transmission of force is impaired in skeletal muscles of dystrophic mice and very old rats. J Physiol (Lond).. (2011) ;589: :1195–208. doi: 10.1113/jphysiol.2010.201921 |
[60] | Petrof BJ , Stedman HH , Shrager JB , Eby J , Sweeney HL , Kelly AM . Adaptations in myosin heavy chain expression and contractile function in dystrophic mouse diaphragm. Am J Physiol. (1993) ;265: :C834–41. doi: 10.1152/ajpcell.1993.265.3.C834 |
[61] | Claflin DR , Brooks SV . Direct observation of failing fibers in muscles of dystrophic mice provides mechanistic insight into muscular dystrophy. Am J Physiol, Cell Physiol. (2008) ;294: :C651–8. doi: 10.1152/ajpcell.00244.2007 |
[62] | McNeil PL , Khakee R . Disruptions of muscle fiber plasma membranes. Role in exercise-induced damage. Am J Pathol. (1992) ;140: :1097–109. |
[63] | Yeung EW , Whitehead NP , Suchyna TM , Gottlieb PA , Sachs F , Allen DG . Effects of stretch-activated channel blockers on [Ca2+]i and muscle damage in the Mdx mouse. J Physiol (Lond). (2005) ;562: :367–80. doi: 10.1113/jphysiol.2004.075275 |
[64] | Whitehead NP , Streamer M , Lusambili LI , Sachs F , Allen DG . Streptomycin reduces stretch-induced membrane permeability in muscles from Mdx mice. Neuromuscul Disord. (2006) ;16: :845–54. doi: 10.1016/j.nmd.2006.07.024 |
[65] | Spencer MJ , Croall DE , Tidball JG . Calpains are activated in necrotic fibers from Mdx Dystrophic Mice . J Biol Chem. (1995) ;270: :10909–14. |
[66] | Lindahl M , Bäckman E , Henriksson KG , Gorospe JR , Hoffman EP . Phospholipase A2 activity in dystrophinopathies. Neuromuscul Disord. (1995) ;5: :193–9. |
[67] | Prins KW , Humston JL , Mehta A , Tate V , Ralston E , Ervasti JM . Dystrophin is a microtubule-associated protein. J Cell Biol. (2009) ;186: :363–9. doi: 10.1083/jcb.200905048 |
[68] | Khairallah RJ , Shi G , Sbrana F , Prosser BL , Borroto C , Mazaitis MJ , et al. Microtubules underlie dysfunction in Duchenne muscular dystrophy. Sci Signal. (2012) ;5: :ra56. 10.1126/scisignal.2002829 |
[69] | Gervásio OL , Whitehead NP , Yeung EW , Phillips WD , Allen DG . TRPC1 binds to caveolin-3 and is regulated by Src kinase - role in Duchenne muscular dystrophy. J Cell Sci. (2008) ;121: :2246–55. doi: 10.1242/jcs.032003 |
[70] | Kumar A , Boriek AM . Mechanical stress activates the nuclear factor-kappaB pathway in skeletal muscle fibers: A possible role in Duchenne muscular dystrophy. FASEB J. (2003) ;17: :386–96. doi: 10.1096/fj.02-0542com |
[71] | Brenman JE , Chao DS , Xia H , Aldape K , Bredt DS . Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell. (1995) ;82: :743–52. |
[72] | Sander M , Chavoshan B , Harris SA , Iannaccone ST , Stull JT , Thomas GD , et al. Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with Duchenne muscular dystrophy. Proc Natl Acad Sci USA. (2000) ;97: :13818–23. doi: 10.1073/pnas.250379497 |
[73] | Kobayashi YM , Rader EP , Crawford RW , Iyengar NK , Thedens DR , Faulkner JA , et al. Sarcolemma-localized nNOS is required to maintain activity after mild exercise. Nature. (2008) ;456: :511–5. doi: 10.1038/nature07414 |
[74] | Serrano AL , Mann CJ , Vidal B , Ardite E , Perdiguero E , Muñoz-Cánoves P . Cellular and molecular mechanisms regulating fibrosis in skeletal muscle repair and disease. Curr Top Dev Biol. (2011) ;96: :167–201. doi: 10.1016/B978-0-12-385940-2.00007-3 |
[75] | Bassett D , Currie PD . Identification of a zebrafish model of muscular dystrophy. Clin Exp Pharmacol Physiol. (2004) ;31: :537–40. doi: 10.1111/j.1440-1681.2004.04030.x |
[76] | Guyon JR , Goswami J , Jun SJ , Thorne M , Howell M , Pusack T , et al. Genetic isolation and characterization of a splicing mutant of zebrafish dystrophin. Hum Mol Genet. (2009) ;18: :202–11. doi: 10.1093/hmg/ddn337 |
[77] | Kawahara G , Karpf JA , Myers JA , Alexander MS , Guyon JR , Kunkel LM . Drug screening in a zebrafish model of Duchenne muscular dystrophy. Proc Natl Acad Sci USA. (2011) ;108: :5331–6. doi: 10.1073/pnas.1102116108 |
[78] | Smith LL , Beggs AH , Gupta VA . Analysis of skeletal muscle defects in larval zebrafish by birefringence and touch-evoke escape response assays. J Vis Exp. (2013) :e50925. 10.3791/50925 |
[79] | Berger J , Sztal T , Currie PD . Quantification of birefringence readily measures the level of muscle damage in zebrafish. Biochem Biophys Res Commun. (2012) ;423: :785–8. doi: 10.1016/j.bbrc.2012.06.040 |
[80] | Berger J , Berger S , Hall TE , Lieschke GJ , Currie PD . Dystrophin-deficient zebrafish feature aspects of the Duchenne muscular dystrophy pathology. Neuromuscul Disord. (2010) ;20: :826–32. doi: 10.1016/j.nmd.2010.08.004 |
[81] | Smith LL , Gupta VA , Beggs AH . Bridging integrator 1 (Bin1) deficiency in zebrafish results in centronuclear myopathy. Hum Mol Genet.-78. (2014) ;23: :3566–78. doi: 10.1093/hmg/ddu067 |
[82] | Gupta VA , Ravenscroft G , Shaheen R , Todd EJ , Swanson LC , Shiina M , et al. Identification of KLHL41 mutations implicates BTB-Kelch-mediated ubiquitination as an alternate pathway to myofibrillar disruption in nemaline myopathy. Am J Hum Genet. (2013) ;93: :1108–17. doi: 10.1016/j.ajhg.2013.10.020 |
[83] | Li M , Arner A . Immobilization of dystrophin and laminin A2-chain deficient zebrafish larvae in Vivo prevents the development of muscular dystrophy. PLoS ONE. (2015) ;10: :e0139483. 10.1371/journal.pone.0139483 |
[84] | Petrof BJ , Shrager JB , Stedman HH , Kelly AM , Sweeney HL . Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci USA. (1993) ;90: :3710–4. |
[85] | Widrick JJ , Alexander MS , Sanchez B , Gibbs DE , Kawahara G , Beggs AH , et al. Muscle dysfunction in a zebrafish model of Duchenne muscular dystrophy. Physiol Genomics. (2016) ;48: :850–60. doi: 10.1152/physiolgenomics.00088.2016 |
[86] | Li M , Andersson-Lendahl M , Sejersen T , Arner A . Muscle dysfunction and structural defects of dystrophin-null sapje mutant zebrafish larvae are rescued by ataluren treatment. FASEB J. (2014) ;28: :1593–9. doi: 10.1096/fj.13-240044 |
[87] | Alexander MS , Casar JC , Motohashi N , Vieira NM , Eisenberg I , Marshall JL , et al. MicroRNA-486-dependent modulation of DOCK3/PTEN/AKT signaling pathways improves muscular dystrophy-associated symptoms. J Clin Invest.-67. (2014) ;124: :2651–67. doi: 10.1172/JCI73579 |
[88] | Lynch GS , Hinkle RT , Chamberlain JS , Brooks SV , Faulkner JA . Force and power output of fast and slow skeletal muscles from Mdx mice 6-28 months old. J Physiol (Lond). (2001) ;535: :591–600. |
[89] | Lowe DA , Williams BO , Thomas DD , Grange RW . Molecular and cellular contractile dysfunction of dystrophic muscle from young mice. Muscle Nerve. (2006) ;34: :92–100. doi: 10.1002/mus.20562 |
[90] | Kornegay JN , Bogan DJ , Bogan JR , Childers MK , Cundiff DD , Petroski GF , et al. Contraction force generated by tarsal joint flexion and extension in dogs with golden retriever muscular dystrophy. J Neurol Sci. (1999) ;166: :115–21. |
[91] | Tegeler CJ , Grange RW , Bogan DJ , Markert CD , Case D , Kornegay JN , et al. Eccentric contractions induce rapid isometric torque drop in dystrophin-deficient dogs. Muscle Nerve. (2010) ;42: :130–2. doi: 10.1002/mus.21699 |
[92] | Deconinck N , Rafael JA , Beckers-Bleukx G , Kahn D , Deconinck AE , Davies KE , et al. Consequences of the combined deficiency in dystrophin and utrophin on the mechanical properties and myosin composition of some limb and respiratory muscles of the mouse. Neuromuscul Disord. (1998) ;8: :362–70. |
[93] | Moens P , Baatsen PH , Maréchal G . Increased susceptibility of EDL muscles from Mdx mice to damage induced by contractions with stretch. J Muscle Res Cell Motil. (1993) ;14: :446–51. |
[94] | Widrick JJ , Jiang S , Choi SJ , Knuth ST , Morcos PA . An octaguanidine-morpholino oligo conjugate improves muscle function of Mdx mice. Muscle Nerve. (2011) ;44: :563–70. doi: 10.1002/mus.22126 |
[95] | Dellorusso C , Crawford RW , Chamberlain JS , Brooks SV . Tibialis anterior muscles in Mdx mice are highly susceptible to contraction-induced injury. J Muscle Res Cell Motil. (2001) ;22: :467–75. |
[96] | Yang HT , Shin J-H , Hakim CH , Pan X , Terjung RL , Duan D . Dystrophin deficiency compromises force production of the extensor carpi ulnaris muscle in the canine model of Duchenne muscular dystrophy. PLoS ONE. (2012) ;7: :e44438. 10.1371/journal.pone.0044438 |
[97] | Consolino CM , Brooks SV . Susceptibility to sarcomere injury induced by single stretches of maximally activated muscles of Mdx mice. J Appl Physiol. (2004) ;96: :633–8. doi: 10.1152/japplphysiol.00587.2003 |
[98] | Kawahara G , Gasperini MJ , Myers JA , Widrick JJ , Eran A , Serafini PR , et al. Dystrophic muscle improvement in zebrafish via increased heme oxygenase signaling. Hum Mol Genet.. (2014) ;23: :1869–78. doi: 10.1093/hmg/ddt579 |
[99] | Waugh TA , Horstick E , Hur J , Jackson SW , Davidson AE , Li X , et al. Fluoxetine prevents dystrophic changes in a zebrafish model of Duchenne muscular dystrophy. Hum Mol Genet. (2014) ;23: :4651–62. doi: 10.1093/hmg/ddu185 |
[100] | Percival JM , Whitehead NP , Adams ME , Adamo CM , Beavo JA , Froehner SC . Sildenafil reduces respiratory muscle weakness and fibrosis in the Mdx mouse model of Duchenne muscular dystrophy. J Pathol. (2012) ;228: :77–87. doi: 10.1002/path.4054 |
[101] | Asai A , Sahani N , Kaneki M , Ouchi Y , Martyn JAJ , Yasuhara SE . Primary role of functional ischemia, quantitative evidence for the two-hit mechanism, and phosphodiesterase-5 inhibitor therapy in mouse muscular dystrophy. PLoS ONE. (2007) ;2: :e806. 10.1371/journal.pone.0000806 |
[102] | Adamo CM , Dai D-F , Percival JM , Minami E , Willis MS , Patrucco E , et al. Sildenafil reverses cardiac dysfunction in the Mdx mouse model of Duchenne muscular dystrophy. Proc Natl Acad Sci USA. (2010) ;107: :19079–83. doi: 10.1073/pnas.1013077107 |
[103] | Hammers DW , Sleeper MM , Forbes SC , Shima A , Walter GA , Sweeney HL . Tadalafil treatment delays the onset of cardiomyopathy in dystrophin-deficient hearts. J Am Heart Assoc. (2016) ;5: . 10.1161/JAHA.116.003911 |
[104] | Carre-Pierrat M , Mariol M-C , Chambonnier L , Laugraud A , Heskia F , Giacomotto J , et al. Blocking of striated muscle degeneration by serotonin in C. Elegans. J Muscle Res Cell Motil. (2006) ;27: :253–8. doi: 10.1007/s10974-006-9070-9 |
[105] | Carre-Pierrat M , Lafoux A , Tanniou G , Chambonnier L , Divet A , Fougerousse F , et al. Pre-clinical study of 21 approved drugs in the Mdx mouse. Neuromuscul Disord. (2011) ;21: :313–27. doi: 10.1016/j.nmd.2011.01.005 |
[106] | Dick DJ , Gardner-Medwin D , Gates PG , Gibson M , Simpson JM , Walls TJ . A trial of flunarizine in the treatment of Duchenne muscular dystrophy. Muscle Nerve. (1986) ;9: :349–54. doi: 10.1002/mus.880090412 |
[107] | Michele DE , Barresi R , Kanagawa M , Saito F , Cohn RD , Satz JS , et al. Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature. (2002) ;418: :417–22. doi: 10.1038/nature00837 |
[108] | Thornhill P , Bassett D , Lochmüller H , Bushby K , Straub V . Developmental defects in a zebrafish model for muscular dystrophies associated with the loss of fukutin-related protein (FKRP). Brain. (2008) ;131: :1551–61. doi: 10.1093/brain/awn078 |
[109] | Kawahara G , Guyon JR , Nakamura Y , Kunkel LM . Zebrafish models for human FKRP muscular dystrophies. Hum Mol Genet. (2010) ;19: :623–33. doi: 10.1093/hmg/ddp528 |
[110] | Brockington M , Blake DJ , Prandini P , Brown SC , Torelli S , Benson MA , et al. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin alpha2 deficiency and abnormal glycosylation of alpha-dystroglycan. Am J Hum Genet. (2001) ;69: :1198–209. doi: 10.1086/324412 |
[111] | Brockington M , Yuva Y , Prandini P , Brown SC , Torelli S , Benson MA , et al. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum Mol Genet. (2001) ;10: :2851–9. |
[112] | Poppe M , Cree L , Bourke J , Eagle M , Anderson LVB , Birchall D , et al. The phenotype of limb-girdle muscular dystrophy type 2I. Neurology. (2003) ;60: :1246–51. |
[113] | Quijano-Roy S , Martí-Carrera I , Makri S , Mayer M , Maugenre S , Richard P , et al. Brain MRI abnormalities in muscular dystrophy due to FKRP mutations. Brain Dev. (2006) ;28: :232–42. doi: 10.1016/j.braindev.2005.08.003 |
[114] | Darin N , Kroksmark A-K , Ahlander A-C , Moslemi A-R , Oldfors A , Tulinius M . Inflammation and response to steroid treatment in limb-girdle muscular dystrophy 2I. Eur J Paediatr Neurol. (2007) ;11: :353–7. doi: 10.1016/j.ejpn.2007.02.018 |
[115] | Wu B , Shah SN , Lu P , Richardson SM , Bollinger LE , Blaeser A , et al. Glucocorticoid steroid and alendronate treatment alleviates dystrophic phenotype with enhanced functional glycosylation of α-dystroglycan in mouse model of limb-girdle muscular dystrophy with FKRPP448L mutation. Am J Pathol. (2016) ;186: :1635–48. doi: 10.1016/j.ajpath.2016.02.015 |
[116] | Frattini P , Villa C , De Santis F , Meregalli M , Belicchi M , Erratico S , et al. Autologous intramuscular transplantation of engineered satellite cells induces exosome-mediated systemic expression of Fukutin-related protein and rescues disease phenotype in a murine model of limb-girdle muscular dystrophy type 2I. Hum Mol Genet. (2017) ;26: :3682–98. doi: 10.1093/hmg/ddx252 |
[117] | Xu L , Lu PJ , Wang C-H , Keramaris E , Qiao C , Xiao B , et al. Adeno-associated virus 9 mediated FKRP gene therapy restores functional glycosylation of α-dystroglycan and improves muscle functions. Mol Ther. (2013) ;21: :1832–40. doi: 10.1038/mt.2013.156 |
[118] | Gicquel E , Maizonnier N , Foltz SJ , Martin WJ , Bourg N , Svinartchouk F , et al. AAV-mediated transfer of FKRP shows therapeutic efficacy in a murine model but requires control of gene expression. Hum Mol Genet. (2017) ;26: :1952–65. doi: 10.1093/hmg/ddx066 |
[119] | Vannoy CH , Xiao W , Lu P , Xiao X , Lu QL . Efficacy of gene therapy is dependent on disease progression in dystrophic mice with mutations in the FKRP gene. Mol Ther Methods Clin Dev. (2017) ;5: :31–42. doi: 10.1016/j.omtm.2017.02.002 |
[120] | Serafini PR , Feyder MJ , Hightower RM , Garcia-Perez D , Vieira NM , Lek A , et al. A limb-girdle muscular dystrophy 2I model of muscular dystrophy identifies corrective drug compounds for dystroglycanopathies. JCI Insight. (2018) ;3: . 10.1172/jci.insight.120493 |
[121] | Kawahara G , Kunkel LM . Zebrafish based small molecule screens for novel DMD drugs. Drug Discov Today Technol. (2013) ;10: :e91–6. doi: 10.1016/j.ddtec.2012.03.001 |
[122] | Chen RJY , Chung T-Y , Li F-Y , Yang W-H , Jinn T-R , Tzen JTC . Steroid-like compounds in Chinese medicines promote blood circulation via inhibition of Na+/K+-ATPase. Acta Pharmacol Sin. (2010) ;31: :696–702. doi: 10.1038/aps.2010.61 |
[123] | Kunkel SD , Suneja M , Ebert SM , Bongers KS , Fox DK , Malmberg SE , et al. mRNA expression signatures of human skeletal muscle atrophy identify a natural compound that increases muscle mass. Cell Metab. (2011) ;13: :627–38. doi: 10.1016/j.cmet.2011.03.020 |
[124] | Dowling JJ , Arbogast S , Hur J , Nelson DD , McEvoy A , Waugh T , et al. Oxidative stress and successful antioxidant treatment in models of RYR1-related myopathy. Brain. (2012) ;135: :1115–27. doi: 10.1093/brain/aws036 |
[125] | Dou Y , Andersson-Lendahl M , Arner A . Structure and function of skeletal muscle in zebrafish early larvae. J Gen Physiol. (2008) ;131: :445–53. doi: 10.1085/jg200809982 |
[126] | Sloboda DD , Claflin DR , Dowling JJ , Brooks SV . Force measurement during contraction to assess muscle function in zebrafish larvae. J Vis Ex. (2013) . 10.3791/50539 |


