Excitation-Contraction Coupling Alterations in Myopathies
Abstract
During the complex series of events leading to muscle contraction, the initial electric signal coming from motor neurons is transformed into an increase in calcium concentration that triggers sliding of myofibrils. This process, referred to as excitation–contraction coupling, is reliant upon the calcium-release complex, which is restricted spatially to a sub-compartment of muscle cells (“the triad”) and regulated precisely. Any dysfunction in the calcium-release complex leads to muscle impairment and myopathy. Various causes can lead to alterations in excitation–contraction coupling and to muscle diseases. The latter are reviewed and classified into four categories: (i) mutation in a protein of the calcium-release complex; (ii) alteration in triad structure; (iii) modification of regulation of channels; (iv) modification in calcium stores within the muscle. Current knowledge of the pathophysiologic mechanisms in each category is described and discussed.
THE CALCIUM-RELEASE COMPLEX (CRC) IN MUSCLE EXCITATION–CONTRACTION (EC) COUPLING
Activation of skeletal muscle at the neuromuscular junction induces a membrane depolarization that propagates along the plasma membrane to invaginations called “transverse tubules” (T-tubules). Depolarization in the latter activates L-type calcium channel dihydropyridine receptors (DHPRs). DHPRs stimulate opening of the sarcoplasmic reticulum (SR) calcium channel and type-1 ryanodine receptors (RyR1s). Opening of RyR1s results in massive release of calcium from the SR to the cytosol, and an increase in intracellular calcium concentration that will be responsible for the gliding of myofibrils and muscle contraction. Muscle relaxation occurs after closing of RyR1s, and calcium uptake into the SR by sarcoplasmic reticulum calcium ATPase (SERCA).
This process of EC coupling is done in the triads of skeletal muscle. Triads are formed by tight apposition of two terminal cisternae of SR on both sides of a T-tubule, and are localized specifically at the transition of A-I bands in mammalian skeletal muscle. The two main players of EC coupling (DHPRs and RyR1s) are associated together and form the core of a supramolecular complex, the calcium release complex (CRC), anchored in T-tubules and SR membrane, and localized exclusively in triads. Additional regulatory proteins are associated with these two calcium channels in the CRC (triadin, junctin, calsequestrin, FKBP12, STAC3) (Fig. 1). Efficacy of EC coupling is reliant on close physical contact between RyR1s and DHPRs. Very specific organization of triads allows close apposition of T-tubules and SR membranes that are separated by a space of only 15 nm, filled almost entirely by the huge cytosolic part of RyR1 detected as a dense structure by electron microscopy [1].
Several myopathies are related to default in the EC coupling process. The genetic alterations leading to these myopathies affect mainly RyR1 and DHPR genes. Mutations in several genes involved in regulation of both channels are also linked directly to myopathies as well as mutations in genes involved in organization of triad structure. However, whereas the number of RyR1 mutations probably exceeds 300, mutations in the DHPR are rarely reported, and very few mutations in other genes that regulate EC coupling have been revealed.
This review focuses on the major functional effects related to these different genetic modifications, and also touches upon alterations in EC coupling as a secondary effect to genetic alterations affecting other pathways.
MYOPATHIES RELATED TO RYR1
RyR1 is the major skeletal muscle isoform of the ryanodine receptor. The 150 kb sequence of its gene (RYR1) is located on chromosome 19q13.2. It is composed of 106 exons, with two alternatively spliced exons (70 and 83), and gives rise to a protein of 5038 amino acids (560 kDa). The functional RyR1 channel is a massive 2.5 MDa homotetramer whose structure at near atomic resolution has been shown recently using cryo-electron microscopy [2–4]. The major part (first 4000 amino acids) of the RyR1 sequence is cytosolic, and forms the “feet” of the molecule observed in electron microscopy. The last 1000 C-terminal amino acids of the protein contain the transmembrane helices responsible for anchoring in the SR membrane and form the pore domain of the channel.
Initially associated only with the dominant form of central core disease (CCD), the RYR1 gene has now also been linked to multiminicore disease (MmD), core myopathies with rods, centronuclear myopathy and congenital fiber type disproportion [5, 6]. Altogether, the diseases involving the RYR1 gene are classified as RyR1-related congenital myopathies (RyR1-RCM).
In its initial clinical description, CCD is a muscle disease associated with congenital hypotonia, delayed motor acquisition during childhood, and a slow evolution towards proximal muscle weakness in the legs [7]. Histologic characterization has shown the presence in central part of the fiber of zones (named “cores”) without oxidative and phosphorylative activities. Other histologic presentations have been described (cores and rods, multiple cores, minicores) [8]. The RYR1 mutations associated with dominant CCD are localized mainly in the C-terminal pore-forming region of the protein [9–11]. The description of recessive forms of congenital myopathies called Multimini core Disease (MmD) and linked to RyR1 is more recent [12–14]. Initially, the selenoprotein N gene (SEPN1) was associated with 50% of the severe so called “classical” forms of MmD [15]. However, RYR1 mutations were further identified in families with MmD forms associated with external ophtamloplegia [13].
In addition to these congenital myopathies, RYR1 has been associated with triggered myopathies: anesthesia-induced hyperthermia (malignant hyperthermia (MH)) [16, 17]; exercise-induced hyperthermia (exertional heat stroke) [18, 19]. Both diseases share uncontrolled release of calcium in muscle and massive metabolic alterations upon exposure to triggering conditions. In these induced myopathies however, no alteration of muscle structure has been described, and outside of the hyperthermia crisis, muscle function is normal.
PATHOPHYSIOLOGIC MECHANISMS ASSOCIATED WITH RYR1 MUTATIONS
Functional studies have been undertaken using different tools to understand the effect of RyR1 mutations, and resulted in several possible pathophysiologic mechanisms [20–22]. These studies were carried out with different cell models, among which cells produced from patients (differentiated primary myoblasts, primary fibroblasts pushed towards myogenic differentiation by MyoD expression or immortalized lymphocytes); non-muscle cell lines expressing transfected mutants of RyR1 (mainly HEK293 cells); RYR1 knockout (KO) mouse muscle cultures expressing RyR1 variants. Few mutations have been studied simultaneously with the different expression systems leading to different putative mechanisms, suggesting that each expression system has its drawback. Therefore, the actual pathophysiologic mechanisms resulting from a precise mutation are probably more complex than the simplified and schematic mechanisms described thereafter.
RyR1 gain of function
Some mutations have been shown to induce a gain of function (i.e., an increased amount of calcium released by the channel upon stimulation). RYR1 gain-of-function mutations could have two consequences. First, they can result in hypersensitivity of the channel to specific triggers, which is characteristic of the so called “MH mutation” inducing hypersensitivity to volatile anesthetics. In the presence of the triggering agent, MH mutations could lower the threshold of RyR1 activation by SR luminal calcium, thus inducing massive and unregulated calcium efflux [23]. This calcium overload would then be responsible for the generalized muscle contraction and hypermetabolic state typical for MH crisis. The second mode of action of a RYR1 gain-of-function mutation is calcium leak in physiologic situations when the channel should be closed: at very low (< μM) or very high (>mM) calcium concentrations. If the calcium leak is not compensated by an increased reverse calcium influx toward the SR, the net result is reduction in the basal SR calcium concentration. In this condition, the number of calcium ions released upon stimulation is reduced and the efficiency of muscle contraction altered [24]. Depending on the cell model used to assay such mutations (muscle cells equipped with all the calcium channels and pumps, or non-muscle cells expressing none or only few of them) and on the type of ectopic RyR1 expression undertaken (transient or long term) a compensation of the calcium leak may occur. Increase in calcium flux toward the SR by overexpression of SERCA pumps or of the membrane calcium channel involved in the store-operated calcium entry (SOCE) could compensate for such a calcium leak when chronic expression of the RyR1 mutant is carried out. This phenomenon could explain why discordant results have been reported for the same mutation, such as the RyR1 p.R2435L mutation, for which increase in the basal cytosolic calcium concentration has been observed in some expression systems but not in others [25, 26]. Nevertheless, if presence of a permanently leaky calcium channel does not always lead to a detectable increase in the resting cytosolic calcium concentration, it always induces a reduction in the amount of SR calcium, explaining why a gain-of-function mutation in RyR1 could be also associated with congenital myopathy [26].
RyR1 loss of function
For some missense RyR1 mutations associated with congenital myopathies, an alteration in the coupling between DHPR and RyR1 has been proposed as the origin of the disease [27]. With those mutations, the calcium release induced by membrane depolarization is reduced but the calcium content in the SR is normal, excluding the presence of a leaky RyR1. The mutation p.I4898T in RyR1, for instance, cannot mediate calcium efflux if stimulated by the DHPR, though it is correctly targeted to the triad [28]. A mouse model reproducing the p.I4898T mutation clearly demonstrated an alteration in RyR1 permeability to calcium [29]. This mutation, lethal in homozygous mice but also at a heterozygous state in humans [30], results in a muscle weakness which most probably reflects the inefficient coupling between the excitation stimulus mediated by DHPR and the RyR1 channels opening. Until now, a physical EC uncoupling (i.e., the dissociation of the RyR1–DHPR interaction) has never been observed in patients because it would be too damaging for the muscle or because this interaction relies on multiple contact points, thereby involving different regions of RyR1 that cannot be altered by a single point mutation.
Reduction in RyR1 content
Descriptions of patients with mutations affecting RyR1 expression level have led to the most recent hypothesis of the pathophysiologic mechanism [31, 32]. Disruption of one of the RYR1 alleles is not sufficient in itself to induce muscle weakness or damageable reduction in the amount of protein as demonstrated in heterozygous RyR1 KO mice [33]. However, association of such an allele with a second point mutation is often observed in recessive forms of RYR1-related congenital myopathies. In this case, a “null” mutation (resulting in the complete or almost complete absence of protein production from one allele) leads to expression of the second RYR1 variation in a hemizygous context. Disease severity in such a case is dependent on the nature of the expressed second mutation as well as on the residual expression level of the first-hypomorphic allele. If the second mutation also affects allele expression, overall RyR1 expression may be under a threshold limit, leading to worsening of the phenotype. Overall severe reduction in RyR1 expression affects the amount of calcium release, but could also have an impact on the stoichiometry of proteins of the CRC and its function. If the expression level of regulatory proteins (triadin, junctin, calmodulin, STAC3) is unchanged, reduced amounts of RyR1 could lead to an excess of these free regulatory proteins, which could have a negative effect on CRC function.
Tetrameric organization of RyR1 and its physical interaction with the DHPR add complexity to interpretation of the effect of mutations. The balance of combinations between normal or mutant monomers of RyR1 leading to dominant forms of myopathies, or between mutant monomers in recessive forms, may influence the apparent discrepancy of clinical outcome and histologic presentation for identical genotypes [22].
MUTATIONS IN THE DHPR
The main trigger for calcium release by RyR1 is DHPR activation. The α1-subunit of the DHPR forms the pore domain, and is encoded by the CACNA1S gene mutated in hypokalemic periodic paralysis (HPPK) and malignant hyperthermia susceptibility [34].
Hypokalemic periodic paralysis is characterized by episodic crisis of muscle paralysis associated with a reduction of potassium concentration in blood. The severity of the crisis may vary from weakness to actual paralysis, and last from hours to days. Outside this crisis, muscle function is normal, but its histologic analysis may reveal alterations (e.g., centralized nuclei or apparition of vacuoles).
Strikingly, mutations responsible for hypokalemic paralysis or malignant hyperthermia in the CACNA1S gene cluster in separate regions of the gene. Theα1-subunit pore structure is composed of four almost identical membrane domains each containing six transmembrane helices. One of these helices (S4) is highly charged and has the role of a “sensor” of membrane potential because its physical positioning within the lipid bilayer is modified upon membrane depolarization. Canonical mutation of CACNA1S associated with hypokalemic paralysis affects an arginine residue of the S4 helix, altering the sensitivity of the helix movement to depolarization. Overall, it is not the conductance of the channel that is affected by hypokalemic paralysis mutation but rather the sensitivity of its gating to membrane potential [35].
On the other hand, malignant hyperthermia mutations were found first in sequences of the gene encoding interaction domains with RyR1. A recent functional study showed that a DHPR channel mutated in an extracellular loop had increased sensitivity to caffeine and faster activation, suggesting that excessive calcium entry via the DHPR might by itself confer MHS [36]. Whether this calcium triggers muscle contraction directly or more probably modifies the threshold of RyR1 activation is not known.
MUTATIONS IN OTHER PROTEINS INVOLVED IN EC COUPLING
Calsequestrin
Two calsequestrin isoforms with similar functions are expressed in skeletal muscle, and are encoded by CASQ1 and CASQ2 genes. Calsequestrin 2 is the unique cardiac isoform and expressed in varying proportions in skeletal muscle. Calsequestrin 1 is expressed only in skeletal muscle. Calsequestrin is the main calcium buffer of sarcoplasmic reticulum. It is a luminal protein of the SR, anchored at the skeletal muscle triad through its interaction with two SR membrane proteins: triadin and junctin. It can bind loosely high concentrations of calcium, which enhances local calcium concentration close to RyR1 pore, contributing to regulation of the channel and massive release of calcium once the channel is activated [37]. It was long suspected that a mutation in the CASQ1 gene could account for skeletal-muscle defects because mutations in CASQ2 are responsible for a severe form of cardiac arrhythmia without skeletal muscle abnormalities, named catecholaminergic polymorphic ventricular tachycardia (CPVT). Abnormal release of calcium in CPVT cardiomyocytes [38] is considered to be the cardiac equivalent of the calcium-release defects of skeletal muscle in malignant hyperthermia (i.e., excessive release of calcium upon specific triggering). Confirmation of the hypothesis of CASQ1 involvement in muscle disease came first with a mouse model with KO of the CASQ1 gene, in which muscle weakness is associated with ultrastructural alterations resembling cores and hypersensitivity to halothane [39]. Recently, the first mutation (p.N244G) in human CASQ1 was identified in patients with a recessive myopathy characterized by weakness, fatigue, and large vacuoles containing characteristic inclusions resulting from aggregation of SR proteins [40]. Physiologically, calsequestrin forms a polymer after binding calcium within the lumen of SR. The effect of the p.N244G mutation is to lower the calcium concentration at which the protein forms inactive aggregates [41]. The N244G mutation triggers a loss of calsequestrin function, and most probably the inclusions detected in the muscles of patients, who can be considered as having functional KO of the CASQ1 gene. The MH status of individuals who possess the N244G mutation in CASQ1 is not known.
Triadin
Triadin is a transmembrane protein of sarcoplasmic reticulum, associated with RyR1 in the calcium release complex and is involved in anchoring of calsequestrin in triads. Deletion of triadin in mice results in alteration of triad shape, with flattening of the terminal cisternae, alteration of calcium release and muscle weakness, and is associated with cardiac arrhythmia [42–44]. The connection between triadin deletion and alteration of triads is under investigations, but can be related to its ability to link the reticulum and microtubules network [45]. In addition, part of the defects (alteration of calcium release and cardiac arrhythmia) is probably due to diffusion of calsequestrin out of the triad. The triadin gene (TRDN) encodes, via alternative splicing, four isoforms of the protein expressed in skeletal muscle and cardiac muscle [46]. Four mutations in the TRDN gene have been identified in patients with a similar recessive phenotype: a severe cardiac arrhythmia, which can be lethal, associated with moderate muscle weakness [47–49]. Molecular investigations have suggested that TRDN mutations lead to a null allele, and that complete loss of the protein is responsible for the phenotypes of cardiac muscle and skeletal muscle.
SERCA
Mutation in ATP2A1 encoding the Ca2+-ATPase of fast-twitch muscle (SERCA1) results in a rare recessive myopathy: Brody disease. The latter is characterized by muscle stiffness and impairment in muscle relaxation during exercise [50]. Mutations in the ATP2A1 gene are responsible for the absence of SERCA1 protein (non-sense or missense mutations that destabilize the protein) or affect calcium reuptake by SERCA1 into SR after contraction, altering muscle relaxation and, therefore, impacting subsequent muscle contractions. In the presence of two null alleles, it has been shown that upregulation of the SERCA2 isoform could explain the mild phenotype with regard to the central role of calcium reuptake in SR during the excitation-contraction coupling [51]. In addition, an increase in cytosolic calcium has been detected in the muscles of the patient, which could explain positive testing for malignant hyperthermia [51].
STAC3
Recently, STAC3 has been identified as a new protein involved in EC coupling that could link the DHPR and RyR1. KO of the STAC3 gene in mice is lethal in the perinatal period and cells without STAC3 have impaired Ca2+ release after membrane depolarization [52]. The p.W284S STAC3 mutation has been identified as being responsible for the Native American myopathy [53], a recessive myopathy with congenital muscle weakness, susceptibility to malignant hyperthermia, multiple joint contractures, and dysmorphic facial features with ptosis. The p.W284S is so far the only mutation of the gene, and has been shown to decrease EC coupling in a zebrafish model [53]. Recently, it has been proposed that STAC3 could be involved in the trafficking of the alpha-1 subunit of the DHPR to T-tubules [54], thereby explaining the phenotype of KO animals (i.e., impairment of electrically evoked skeletal-muscle contraction) and of the EC coupling due to the p.W284S mutation. This mutation is localized in an SH3 adaptor domain that could link the alpha-1 subunit of the DHPR or a component of the trafficking machinery required for its transport directly.
STIM1 and ORAI1
A specific form of calcium influx, so called the “Store Operated Calcium Entry” (SOCE), is responsible for the refilling of sarcoplasmic reticulum upon calcium depletion. This mechanism is performed by a channel that relies on the close interaction of two proteins: STIM1 and ORAI1. STIM1, a calcium sensor of the SR membrane multimerizes upon calcium depletion in the lumen of reticulum and interacts with ORAI1, a calcium channel of the plasma membrane, to activate calcium influx. Any modification in this SOCE, through a loss of function or gain of function, results in disease [55], as observed for RyR1 mutations. The close proximity of STIM1 and ORAI is needed to generate SOCE. The membranes of triads, as contact points between reticulum and plasma membranes, are ideally arranged to support SOCE [56]. Moreover, it was suggested recently that a muscle-specific isoform of STIM1 is pre-activated constitutively to generate fast SOCE in response to SR calcium depletion [57]. SOCE is present in many tissues, so the outcome of mutations of STIM1 or ORAI1 is often multi-systemic disease, as observed for mutations leading to absence of protein expression [58–59]. Conversely, mutations in STIM1 or ORAI1 resulting in a constitutively active channel have been shown as responsible for tubular aggregate myopathy [60–62]. In all of these situations, the result of the mutation and cause of the disease are abnormal SR calcium content.
EC COUPLING ALTERATIONS AS A SECONDARY EFFECT
Channel regulation
Oxidation/nitrosation
Dysfunction of the RyR1 channel is not only the consequence of RYR1 mutations, but can also arise from post-translational modifications of the protein. For many years, RyR1 has been known to be sensitive to oxidizing agents [63], and was proposed as a redox sensor in skeletal muscle cells [64]. The amino acids involved in this sensitivity have been identified [65], and it has been shown that oxidative modification such as S-nitrosylation (addition of a nitric oxide moiety to a reactive cysteine thiol [66]) results in reduction in the association of RyR1 with FKBP12 (a small protein that stabilizes the closed state of the RyR1 homotetramer [67–69]). Accordingly,S-nitrosylation has been proposed to create a leaky calcium channel, which would increase production of oxygen or nitrogen reactive species by mitochondria and, in turn, increase oxidative modifications of RyR1 and calcium leak in a deleterious circle. This mechanism has been observed in Duchenne muscular dystrophy [70] and aging [71], where it amplified muscle weakness but was also demonstrated to be the direct consequence of a RyR1 mutation such as the p.Y522S mutation [72]. Increase in oxidative modifications in RyR1 can also be the indirect result of mutations in selenoprotein N (SelN), which is responsible for Multiminicore Disease (MmD) [15]. SelN is a reticulum protein involved in protection against oxidative/nitrosative stress. Alteration of RyR1 function as a result of alteration of SelN function is a major pathophysiologic mechanism hypothesized in MmD [73], but it has been demonstrated recently that SERCA can also be altered by SelN absence [74]. Therefore, alteration in calcium handling seems to be a major pathophysiologic mechanism in SelN-related myopathy due to uncontrolled RyR1 and other oxidation-driven modifications in calcium-handling proteins.
This pathophysiologic mechanism is the first one to be the target of therapeutic intervention. In fact, the antioxidant agent N-acetylcysteine (NAC) has been demonstrated to be effective in reducing the amount of oxidized RyR1 and in increasing muscle strength in a zebrafish model of RyR1-related myopathy as well as in cultured myotubes from human CCD patients [75]. NAC could, therefore, be a promising pharmacologic treatment in these myopathies, and is under evaluation.
Splicing modifications
In several diseases with genetic instability in trinucleotides repeats (such as myotonic dystrophy, DM1), increased trinucleotides CTG stretches result in alteration in the level of RNA-binding proteins (muscleblind or CUG-BP proteins), that can be involved in the splicing of many pre-mRNA molecules. Muscle is not the only affected organ in these diseases, and it has been shown that some muscle alterations could be explained by abnormalities in the splicing of proteins involved in EC coupling, such as RyR1, DHPR, SERCA [76] and the amphiphysin 2/BIN1 [77].
Triad alterations
For many years, it has been difficult to understand the pathophysiologic mechanisms in centronuclear myopathies, as mutations in very different genes lead to a similar phenotype. Centronuclear myopathies can be caused by mutations in: myotubularin-MTM1, a lipid phosphatase the mutations of which lead to X-Linked Myotubular Myopathy; dynamin-DNM2, a GTPase involved in membrane scission; amphiphysin 2-BIN1, a protein involved in sensing and remodeling of membrane curvature; RyR1, a calcium channel. This genetic heterogeneity has been reconciled by identification of common pathophysiologic mechanisms and of triad alterations after mutations of MTM1, DNM2 and BIN1, thereby resulting in alterations in EC coupling and RyR1 function ([78–80] and for review [81]). Triad alterations can be due to alterations of lipid content (MTM1 mutations), T-tubule formation (DNM2 and BIN1 mutations) or in modification of the trafficking of triad proteins to the triad. More recently, mutations have been identified in titin (a protein of sarcomeres) but a different mechanism probably operates in this case [82].
CONCLUSIONS
Alterations in EC coupling can have numerous origins because of the complex structure of the triad and of the many proteins involved in the EC coupling and calcium homeostasis within muscle. The different types of alterations are summarized in Fig. 2 and can be clustered schematically into four groups: (1) direct mutation in a protein of the CRC; (2) alteration in triad structure; (3) alteration in regulation of calcium channels; (4) alteration in calcium stores.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Franzini-Armstrong C and Nunzi G . Junctional feet and particles in the triads of a fast-twitch muscle fibre. J Muscle Res Cell Motil. (1983) ;4: :233–52. |
[2] | Efremov RG , Leitner A , Aebersold R and Raunser S . Architecture and conformational switch mechanism of the ryanodine receptor. Nature. (2015) ;517: :39–43. doi: 10.1038/nature13916 |
[3] | Yan Z , Bai XC , Yan C , Wu J , Li Z , Xie T , et al. Structure of the rabbit ryanodine receptor RyR1 at near-atomic resolution. Nature. (2015) ;517: :50–5. doi: 10.1038/nature14063 |
[4] | Zalk R , Clarke OB , des Georges A , Grassucci RA , Reiken S , Mancia F , et al. Structure of a mammalian ryanodine receptor. Nature. (2015) ;517: :44–9. doi: 10.1038/nature13950 |
[5] | Clarke NF , Waddell LB , Cooper ST , Perry M , Smith RL , Kornberg AJ , et al. Recessive mutations in RYR1 are a common cause of congenital fiber type disproportion. Hum Mutat. (2010) ;31: :E1544–50. doi: 10.1002/humu.21278 |
[6] | Wilmshurst JM , Lillis S , Zhou H , Pillay K , Henderson H , Kress W , et al. RYR1 mutations are a common cause of congenital myopathies with central nuclei. Ann Neurol. (2010) ;68: :717–26. |
[7] | Shy GM and Magee KR . A new congenital non-progressive myopathy. Brain. (1956) ;79: :610–21. |
[8] | Romero NB , Herasse M , Monnier N , Leroy JP , Fischer D , Ferreiro AM , et al. Clinical and histopathological aspects of central core disease associated and non-associated with RYR1 locus. Acta Myol. (2005) ;24: :70–3. |
[9] | Lynch PJ , Tong J , Lehane M , Mallet A , Giblin L , Heffron JJ , et al. A mutation in the transmembrane/luminal domain of the ryanodine receptor is associated with abnormal Ca2+release channel function and severe central core disease. Proc Natl Acad Sci U.S.A. (1999) ;96: :4164–9. |
[10] | Monnier N , Romero NB , Lerale J , Landrieu P , Nivoche Y , Fardeau M , et al. Familial and sporadic forms of central core disease are associated with mutations in the C-terminal domain of the skeletal muscle ryanodine receptor. Hum Mol Genet. (2001) ;10: :2581–92. |
[11] | Scacheri PC , Hoffman EP , Fratkin JD , Semino-Mora C , Senchak A , Davis MR , et al. A novel ryanodine receptor gene mutation causing both cores and rods in congenital myopathy. Neurology. (2000) ;55: :1689–96. |
[12] | Ferreiro A , Monnier N , Romero NB , Leroy JP , Bonnemann C , Haenggeli CA , et al. A recessive form of central core disease, transiently presenting as multi-minicore disease, is associated with a homozygous mutation in the ryanodine receptor type 1 gene. Ann Neurol. (2002) ;51: :750–9. |
[13] | Monnier N , Ferreiro A , Marty I , Labarre-Vila A , Mezin P and Lunardi J . A homozygous splicing mutation causing a depletion of skeletal muscle RYR1 is associated with multi-minicore disease congenital myopathy with ophthalmoplegia. Hum Mol Genet. (2003) ;12: :1171–8. |
[14] | Romero NB , Monnier N , Viollet L , Cortey A , Chevallay M , Leroy JP , et al. Dominant and recessive central core disease associated with RYR1 mutations and fetal akinesia. Brain. (2003) ;126: :2341–9. |
[15] | Ferreiro A , Quijano-Roy S , Pichereau C , Moghadaszadeh B , Goemans N , Bonnemann C , et al. Mutations of the selenoprotein N gene, which is implicated in rigid spine muscular dystrophy, cause the classical phenotype of multiminicore disease: Reassessing the nosology of early-onset myopathies. Am J Hum Genet. (2002) ;71: :739–49. |
[16] | Monnier N , Kozak-Ribbens G , Krivosic-Horber R , Nivoche Y , Qi D , Kraev N , et al. Correlations between genotype and pharmacological, histological, functional and clinical phenotypes in malignant hyperthermia susceptibility. Hum Mutat. (2005) ;26: :413–25. |
[17] | Sambuughin N , Holley H , Muldoon S , Brandom BW , de Bantel AM , Tobin JR , et al. Screening of the entire ryanodine receptor type 1 coding region for sequence variations associated with malignant hyperthermia susceptibility in the North American population. Anesthesiology. (2005) ;102: :515–21. |
[18] | Carpenter D , Robinson RL , Quinnell RJ , Ringrose C , Hogg M , Casson F , et al. Genetic variations in RYR1 and malignant hyperthermia phenotypes. Br J Anaesth. (2009) ;103: :538–48. doi: 10.1093/bja/aep204 |
[19] | Roux-Buisson N , Monnier N , Sagui E , Abriat A , Brosset C , Bendahan D , et al. Identification of variants of the ryanodine receptor type 1 in patients with exertional heat stroke and positive response to the malignant hyperthermia in vitro contracture test. Br J Anaesth. (2016) ;116: :566–8. doi: 10.1093/bja/aew047 |
[20] | Betzenhauser MJ and Marks AR . Ryanodine receptor channelopathies. Pflugers Arch. (2010) ;460: :467–80. doi: 10.1007/s00424-010-0794-4 |
[21] | Lanner JT , Georgiou DK , Joshi AD and Hamilton SL . Ryanodine receptors: Structure, expression, molecular details, and function in calcium release. Cold Spring Harb Perspect Biol. (2010) ;2: :a960039. doi: 10.1101/cshperspect.a003996 |
[22] | MacLennan DH and Zvaritch E . Mechanistic models for muscle diseases and disorders originating in the sarcoplasmic reticulum. Biochim Biophys Acta. (2011) ;1813: :948–64. doi: 10.1016/j.bbamcr.2010.11.009 |
[23] | Tong J , Oyamada H , Demaurex N , Grinstein S , McCarthy TV and MacLennan DH . Caffeine and halothane sensitivity of intracellular Ca2+ release is altered by 15 calcium release channel (ryanodine receptor) mutations associated with malignant hyperthermia and/or central core disease. J Biol Chem. (1997) ;272: :26332–9. |
[24] | Tong J , McCarthy TV and MacLennan DH . Measurement of resting cytosolic Ca2+ concentrations and Ca2+ store size in HEK-293 cells transfected with malignant hyperthermia or central core disease mutant Ca2+ release channels. J Biol Chem. (1999) ;274: :693–702. |
[25] | Dirksen RT and Avila G . Distinct effects on Ca2+ handling caused by malignant hyperthermia and central core disease mutations in RyR1. Biophys J. (2004) ;87: :3193–204. |
[26] | Ghassemi F , Vukcevic M , Xu L , Zhou H , Meissner G , Muntoni F , et al. A recessive ryanodine receptor 1 mutation in a CCD patient increases channel activity. Cell Calcium. (2009) ;45: :192–7. doi: 10.1016/j.ceca.2008.10.001 |
[27] | Dirksen RT and Avila G . Altered ryanodine receptor function in central core disease: Leaky or uncoupled Ca2+ release channels? Trends Cardiovasc Med. (2002) ;12: :189–97. |
[28] | Avila G , O’Brien JJ and Dirksen RT . Excitation–contraction uncoupling by a human central core disease mutation in the ryanodine receptor. Proc Natl Acad Sci U.S.A. (2001) ;98: :4215–20. |
[29] | Loy RE , Orynbayev M , Xu L , Andronache Z , Apostol S , Zvaritch E , et al. Muscle weakness in Ryr1IT/WT knock-in mice as a result of reduced ryanodine receptor Ca2+ion permeation and release from the sarcoplasmic reticulum. J Gen Physiol. (2011) ;137: :43–57. doi: 10.1085/jg201010523 |
[30] | Hernandez-Lain A , Husson I , Monnier N , Farnoux C , Brochier G , Lacene E , et al. De novo RYR1 heterozygous mutation (IT) causing lethal core-rod myopathy in twins. Eur J Med Genet. (2011) ;54: :29–33. doi: 10.1016/j.ejmg.2010.09.009 |
[31] | Monnier N , Marty I , Faure J , Castiglioni C , Desnuelle C , Sacconi S , et al. Null mutations causing depletion of the type 1 ryanodine receptor (RYR1) are commonly associated with recessive structural congenital myopathies with cores. Hum Mutat. (2008) ;29: :670–8. doi: 10.1002/humu.20696 |
[32] | Zhou H , Yamaguchi N , Xu L , Wang Y , Sewry C , Jungbluth H , et al. Characterization of recessive RYR1 mutations in core myopathies. Hum Mol Genet. (2006) ;15: :2791–803. |
[33] | Cacheux M , Blum A , Sébastien M , Wozny AS , Brocard J , Mamchaoui K , et al. Functional characterization of a central core disease RyR1 mutation (YH) associated with quantitative defect in RyR1 protein. J Neuromusc Dis. (2015) ;2: :421–32. doi: 10.3233/JND-150073 |
[34] | Monnier N , Stieglitz P , Procaccio V and Lunardi J . Malignant hyperthermia susceptibility is associated with a mutation of the alpha1-subunit of the human dihydropyridine sensitive L-type voltage dependent calcium channel receptor in skeletal muscle. Am J Hum Genet. (1997) ;60: :1316–25. |
[35] | Moreau A , Gosselin-Badaroudine P and Chahine M . Mutations in the voltage sensors of domains I and II of Nav1. 5 that are associated with arrhythmias and dilated cardiomyopathy generate gating pore currents. Front Pharmacol. (2015) ;6: :301. doi: 10.3389/fphar.2015.00301 |
[36] | Pirone A , Schredelseker J , Tuluc P , Gravino E , Fortunato G , Flucher BE , et al. Identification and functional characterization of malignant hyperthermia mutation TS in the outer pore of the Cav-alpha1S-subunit. Am J Physiol Cell Physiol. (2010) ;299: :C1345–54. doi: 10.1152/ajpcell.00008.2010 |
[37] | Beard NA , Sakowska MM , Dulhunty AF and Laver DR . Calsequestrin is an inhibitor of skeletal muscle ryanodine receptor calcium release channels. Biophys J. (2002) ;82: (1 pt 1):310–20. doi: 10.1016/S0006-3495(02)75396-4 |
[38] | Priori SG and Chen SR . Inherited dysfunction of sarcoplasmicreticulum Ca2+ handling and arrhythmogenesis. Circ Res. (2011) ;108: :871–83. doi: 10.1161/CIRCRESAHA.110.226845 |
[39] | Paolini C , Quarta M , Wei-LaPierre L , Michelucci A , Nori A , Reggiani C , et al. Oxidative stress, mitochondrial damage, and cores in muscle from calsequestrin-1 knockout mice. Skelet Musc.. (2015) ;5: :10. doi: 10.1186/s13395-015-0035-9 |
[40] | Rossi D , Vezzani B , Galli L , Paolini C , Toniolo L , Pierantozzi E , et al. A mutation in the CASQ1 gene causes a vacuolar myopathy with accumulation of sarcoplasmic reticulum protein aggregates. Hum Mutat. (2014) ;35: :1163–70. doi: 10.1002/humu.22631 |
[41] | Lewis KM , Ronish LA , Ríos E and Kang C . Characterization of two human skeletal calsequestrin mutants implicated in malignant hyperthermia and vacuolar aggregate myopathy. J Biol Chem. (2015) ;290: :28665–74. doi: 10.1074/jbc.M115.686261 |
[42] | Shen X , Franzini-Armstrong C , Lopez JR , Jones LR , Kobayashi YM , Wang Y , et al. Triadins modulate intracellular Ca2+ homeostasis but are not essential for excitation-contraction coupling in skeletal muscle. J Biol Chem. (2007) ;282: :37864–74. |
[43] | Chopra N , Yang T , Asghari P , Moore ED , Huke S , Akin B , et al. Ablation of triadin causes loss of cardiac Ca2+ release units, impaired excitation-contraction coupling, and cardiac arrhythmias. Proc Natl Acad Sci U.S.A. (2009) ;106: :7636–41. doi: 10.1073/pnas.0902919106 |
[44] | Oddoux S , Brocard J , Schweitzer A , Szentesi P , Giannesini B , Brocard J , et al. Triadin deletion induces impaired skeletal muscle function. J Biol Chem. (2009) ;284: :34918–29. doi: 10.1074/jbc.M109.022442 |
[45] | Fourest-Lieuvin A , Rendu J , Osseni A , Pernet-Gallay K , Rossi D , Oddoux S , et al. Role of triadin in the organization of reticulum membrane at the muscle triad. J Cell Sci. (2012) ;125: (pt 14):3443–53. doi: 10.1242/jcs.100958 |
[46] | Thevenon D , Smida-Rezgui S , Chevessier F , Groh S , Henry-Berger J , Romero NB , et al. Human skeletal muscle triadin: Gene organization and cloning of the major isoform, Trisk 51. Biochem Biophys Res Comm. (2003) ;303: :669–75 . |
[47] | Roux-Buisson N , Cacheux M , Fourest-Lieuvin A , Fauconnier J , Brocard J , Denjoy I , et al. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Hum Mol Genet. (2012) ;21: :2759–67. doi: 10.1093/hmg/dds104 |
[48] | Altmann HM , Tester DJ , Will ML , Middha S , Evans JM , Eckloff BW , et al. Homozygous/compound heterozygous triadin mutations associated with autosomal-recessive long-QT syndrome and pediatric sudden cardiac arrest: Elucidation of the triadin knockout syndrome. Circulation. (2015) ;131: :2051–60. doi: 10.1161/CIRCULATIONAHA.115.015397 |
[49] | Walsh MA , Stuart AG , Schlecht HB , James AF , Hancox JC and Newbury-Ecob RA . Compound heterozygous triadin mutation causing cardiac arrest in two siblings. Pacing Clin Electrophysiol. (2016) ;39: :497–501. doi: 10.1111/pace.12813 |
[50] | Brody IA . Muscle contracture induced by exercise. A syndrome attributable to decreased relaxing factor. N Engl J Med. (1969) ;281: :187–92. |
[51] | Sambuughin N , Zvaritch E , Kraeva N , Sizova O , Sivak E , Dickson K , et al. Exome analysis identifies Brody myopathy in a family diagnosed with malignant hyperthermia susceptibility. Mol Genet Genomic Med. (2014) ;2: :472–83. doi: 10.1002/mgg3.91 |
[52] | Nelson BR , Wu F , Liu Y , Anderson DM , McAnally J , Lin W , et al. Skeletal muscle-specific T-tubule protein STAC3 mediates voltage-induced Ca2+ release and contractility. Proc Natl Acad Sci U.S.A. (2013) ;110: :11881–6. doi: 10.1073/pnas.1310571110 |
[53] | Horstick EJ , Linsley JW , Dowling JJ , Hauser MA , McDonald KK , Ashley-Koch A , et al. Stac3 is a component of the excitation–contraction coupling machinery and mutated in Native American myopathy. Nat Commun. (2013) ;4: :1952. doi: 10.1038/ncomms2952 |
[54] | Polster A , Perni S , Bichraoui H and Beam KG . Stac adaptor proteins regulate trafficking and function of muscle and neuronal L-type Ca2+ channels. Proc Natl Acad Sci U.S.A.. (2015) ;112: :602–6. doi: 10.1073/pnas.1423113112 |
[55] | Lacruz RS and Feske S . Diseases caused by mutations in ORAI1 and STIM1. Ann N Y Acad Sci. (2015) ;1356: :45–79. doi: 10.1111/nyas.12938 |
[56] | Kiviluoto S , Decuypere JP , De Smedt H , Missiaen L , Parys JB and Bultynck G . STIM1 as a key regulator for Ca2+ homeostasis in skeletal-muscle development and function. Skelet Musc. (2011) ;1: :16. doi: 10.1186/2044-5040-1-16 |
[57] | Darbellay B , Arnaudeau S , Bader CR , Konig S and Bernheim L . STIM1L is a new actin-binding splice variant involved in fast repetitive Ca2+ release. J Cell Biol. (2011) ;194: :335–46. doi: 10.1083/jcb.201012157 |
[58] | McCarl CA , Picard C , Khalil S , Kawasaki T , Röther J , Papolos A , et al. ORAI1 deficiency and lack of store-operated Ca2+ entry cause immunodeficiency, myopathy, and ectodermal dysplasia. J Allergy Clin Immunol. (2009) ;124: :1311–1318.e7. doi: 10.1016/j.jaci.2009.10.007 |
[59] | Picard C , McCarl CA , Papolos A , Khalil S , Lüthy K , Hivroz C , et al. STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. N Engl J Med. (2009) ;360: :1971–80. doi: 10.1056/NEJMoa0900082 |
[60] | Böhm J , Chevessier F , Koch C , Peche GA , Mora M , Morandi L , et al. Clinical, histological and genetic Characterization of patients with tubular aggregate myopathy caused by mutations in STIM1. J Med Genet. (2014) ;51: :824–33. doi: 10.1136/jmedgenet-2014-102623 |
[61] | Nesin V , Wiley G , Kousi M , Ong EC , Lehmann T , Nicholl DJ , et al. Activating mutations in STIM1 and ORAI1 cause overlapping syndromes of tubular myopathy and congenital miosis. Proc Natl Acad Sci U.S.A. (2014) ;111: :4197–202. doi: 10.1073/pnas.1312520111 |
[62] | Endo Y , Noguchi S , Hara Y , Hayashi YK , Motomura K , Miyatake S , et al. Dominant mutations in ORAI1 cause tubular aggregate myopathy with hypocalcemia via constitutive activation of store-operated Ca²+ channels. Hum Mol Genet. (2015) ;24: :637–48. doi: 10.1093/hmg/ddu477 |
[63] | Eu JP , Sun J , Xu L , Stamler JS and Meissner G . The skeletal muscle calcium release channel: Coupled O2 sensor and NO signaling functions. Cell. (2000) ;102: :499–509. |
[64] | Hidalgo C , Donoso P and Carrasco MA . The ryanodine receptors Ca2+ release channels: Cellular redox sensors? IUBMB Life (2005) ;57: :315–22. |
[65] | Aracena-Parks P , Goonasekera SA , Gilman CP , Dirksen RT , Hidalgo C and Hamilton SL . Identification of cysteines involved in S-nitrosylation, S-glutathionylation, and oxidation to disulfides in ryanodine receptor type 1. J Biol Chem. (2006) ;281: :40354–68. |
[66] | Hess DT , Matsumoto A , Kim SO , Marshall HE and Stamler JS . Protein S-nitrosylation: Purview and parameters. Nat Rev Mol Cell Biol. (2005) ;6: :150–66. |
[67] | Brillantes AB , Ondrias K , Scott A , Kobrinsky E , Ondriasová E , Moschella MC , et al. Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell. (1994) ;77: :513–23. |
[68] | Aracena P , Tang W , Hamilton SL and Hidalgo C . Effects of S-glutathionylation and S-nitrosylation on calmodulin binding to triads and FKBP12 binding to type 1 calcium release channels. Antioxid Redox Signal. (2005) ;7: :870–81. |
[69] | Zissimopoulos S , Docrat N and Lai FA . Redox sensitivity of the ryanodine receptor interaction with FK506-binding protein. J Biol Chem. (2007) ;282: :6976–83. |
[70] | Bellinger AM , Reiken S , Carlson C , Mongillo M , Liu X , Rothman L , et al. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat Med. (2009) ;15: :325–30. doi: 10.1038/nm.1916 |
[71] | Andersson DC , Betzenhauser MJ , Reiken S , Meli AC , Umanskaya A , Xie W , et al. Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab. (2011) ;14: :196–207. doi: 10.1016/j.cmet.2011.05.014 |
[72] | Durham WJ , Aracena-Parks P , Long C , Rossi AE , Goonasekera SA , Boncompagni S , et al. RyR1S-nitrosylation underlies environmental heat stroke and sudden death in Y522S RyR1 knockin mice. Cell. (2008) ;133: :53–65. doi: 10.1016/j.cell.2008.02.042 |
[73] | Arbogast S , Beuvin M , Fraysse B , Zhou H , Muntoni F and Ferreiro A . Oxidative stress in SEPN1-related myopathy: From pathophysiology to treatment. Ann Neurol. (2009) ;65: :677–86. doi: 10.1002/ana.21644 |
[74] | Marino M , Stoilova T , Giorgi C , Bachi A , Cattaneo A , Auricchio A , et al. SEPN1, an endoplasmic reticulum-localized selenoprotein linked to skeletal muscle pathology, counteracts hyperoxidation by means of redox-regulating SERCA2 pump activity. Hum Mol Genet. (2015) ;24: :1843–55. doi: 10.1093/hmg/ddu602 |
[75] | Dowling JJ , Arbogast S , Hur J , Nelson DD , McEvoy A , Waugh T , et al. Oxidative stress and successful antioxidant treatment in models of RYR1-related myopathy. Brain. (2012) ;135: :1115–27. doi: 10.1093/brain/aws036 |
[76] | Santoro M , Piacentini R , Masciullo M , Bianchi ML , Modoni A , Podda MV , et al. Alternative splicing alterations of Ca2+ handling genes are associated with Ca2+ signal dysregulation in myotonic dystrophy type 1 (DM1) and type 2 (DM2) myotubes. Neuropathol Appl Neurobiol. (2014) ;40: :464–76. doi: 10.1111/nan.12076 |
[77] | Fugier C , Klein AF , Hammer C , Vassilopoulos S , Ivarsson Y , Toussaint A , et al. Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat Med. (2011) ;17: :720–5. doi: 10.1038/nm.2374 |
[78] | Al-Qusairi L , Weiss N , Toussaint A , Berbey C , Messaddeq N , Kretz C , et al. T-tubule disorganization and defective excitation-contraction coupling in muscle fibers lacking myotubularin lipid phosphatase. Proc Natl Acad Sci U.S.A. (2009) ;106: :18763–8. doi: 10.1073/pnas.0900705106 |
[79] | Dowling JJ , Vreede AP , Low SE , Gibbs EM , Kuwada JY , Bonnemann CG , et al. Loss of myotubularin function results in T-tubule disorganization in zebrafish and human myotubular myopathy. PLoS Genet. (2009) ;5: :e1000372. doi: 10.1371/journal.pgen.1000372 |
[80] | Bohm J , Biancalana V , Malfatti E , Dondaine N , Koch C , Vasli N , et al. Adult-onset autosomal dominant centronuclear myopathy due to BIN1 mutations. Brain. (2014) ;137: :3160–70. doi: 10.1093/brain/awu272 |
[81] | Jungbluth H and Gautel M . Pathogenic mechanisms in centronuclear myopathies. Front Aging Neurosci. (2014) ;6: :339. doi: 10.3389/fnagi.2014.00339 |
[82] | Ceyhan-Birsoy O , Agrawal PB , Hidalgo C , Schmitz-Abe K , DeChene ET , Swanson LC , et al. Recessive truncating titin gene, TTN, mutations presenting as centronuclear myopathy. Neurology. (2013) ;81: :1205–14. doi: 10.1212/WNL.0b013e3182a6ca62 |
Figures and Tables
Fig.1
Excitation–contraction coupling is done at the triads of skeletal muscle, which are localized regularly at the interface of band A and I. The major player is the calcium-release complex, which is composed of two calcium channels, the DHPR (with five subunits α1, α2, β, γ and δ) and RyR1, anchored in two membranes (T-tubule and sarcoplasmic reticulum (SR)), and numerous associated proteins, among which triadin, junctin, calsequestrin, FKBP12 and STAC3 are represented. The alpha 1 subunit of DHPR is in direct interaction with RyR1, allowing the coupling between the two proteins. The blue squares on calsequestrin represent the calcium ions.
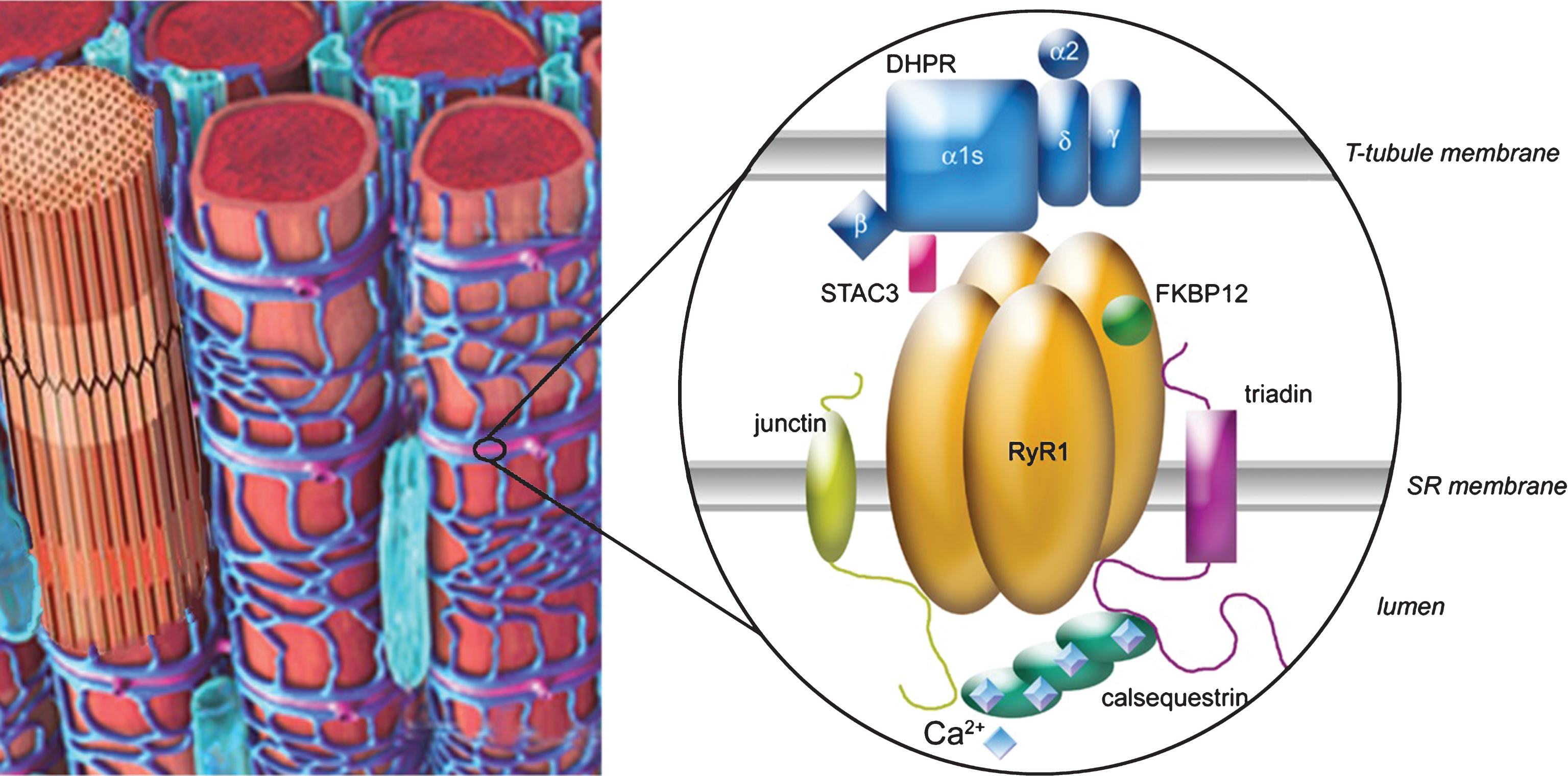
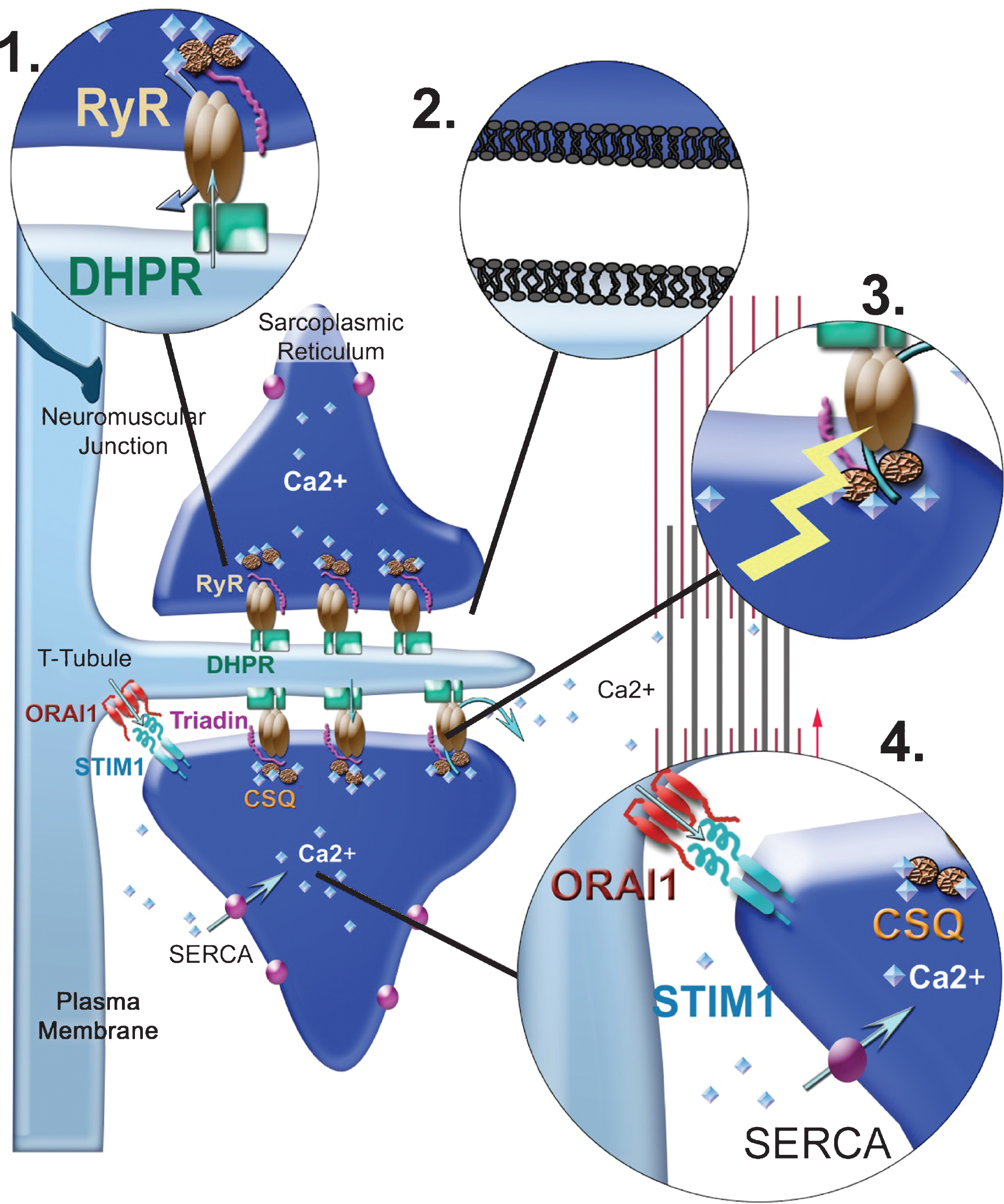
Fig.2
Possible origins for alterations in EC coupling. (1) Mutations can occur in one of the proteins of the calcium-release complex (CRC), mainly RyR1 and the DHPR. The arrows represent the calcium fluxes, which can therefore be modified by mutations in the channels. (2) Mutations can occur in proteins involved in triad formation/maintenance resulting in modification of triads’ shape, or in the triad lipid composition or in the triad targeting of CRC proteins (mutations in MTM1, DNM2, BIN1). (3) Alterations in the redox state (ROS/RNS) of the muscle fiber can alter RyR1 regulation by direct mutation in a protein involved in redox regulation (SelN1) or by indirect increase in the oxidative stress (Duchenne muscular dystrophy). RyR1 regulation can also be altered by modification in its splicing, for example, as a result of sequestration of Muscleblind proteins or in splicing regulators (in DM1). (4) Calcium stores can be altered as a result of mutations in the proteins involved in their refilling (SERCA, STIM1, ORAI1).