
Somatic CAG Repeat Stability in a Transgenic Sheep Model of Huntington’s Disease
Abstract
Somatic instability of the huntingtin (HTT) CAG repeat mutation modifies age-at-onset of Huntington’s disease (HD). Understanding the mechanism and pathogenic consequences of instability may reveal therapeutic targets. Using small-pool PCR we analyzed CAG instability in the OVT73 sheep model which expresses a full-length human cDNA HTT transgene. Analyses of five- and ten-year old sheep revealed the transgene (CAG)69 repeat was remarkably stable in liver, striatum, and other brain tissues. As OVT73 sheep at ten years old have minimal cell death and behavioral changes, our findings support instability of the HTT expanded-CAG repeat as being required for the progression of HD.
Variation in inherited length of an expanded (>35) CAG repeat in the Huntington’s disease (HD) gene HTT explains the majority (∼60%) of variation in the age-at-onset (AAO) of motor symptoms of HD, with longer repeats resulting in earlier onset [1]. The residual variation in AAO is also highly heritable [1, 2], with evidence that expansion-biased somatic instability of the expanded CAG repeat drives the rate of pathogenesis [3, 4]. Somatic instability occurs in the HD brain [5–10] with greatest instability observed in the striatum and cortex, and peripheral tissues to a lesser extent [5, 10–12]. Striatal expansions are evident even in tissue from Vonsattel Grade 0 brains (minimal pathology) implying that instability is an early molecular event [6]. Functional studies in mouse models [13–17] and HD cell lines [18–20] show that somatic instability of the CAG repeat is mediated by DNA repair genes. Variants in, or associated with, six such genes (FAN1, LIG1, MLH1, MSH3, PMS1, and PMS2) have been identified as modifiers of AAO through genome wide association [4,21,22] and candidate genotype studies [11], and several have been found to modify repeat instability in HD patient blood samples [4, 11].
Current data support a two-step theory of pathogenesis in HD where the rate of somatic CAG repeat expansion controls the timing/rate of disease onset [3]. This theory proposes that the inherited expanded HTT CAG repeat allele undergoes somatic expansion towards a critical threshold(s) in individual cells. When this threshold is reached within a cell a toxic mechanism is then triggered to cause dysfunction and cell death, with symptom onset and disease progression occurring as increasing numbers of cells pass the expansion threshold. Recent reviews [23, 24] suggest that studying pre-symptomatic and early symptomatic low-grade tissues will help to resolve the critical intracellular CAG repeat threshold and the consequent pathogenic mechanisms.
We quantified somatic variation in CAG repeat length in the OVT73 transgenic sheep model of HD in striatum and liver. These two tissues exhibit high instability both in HD knock-in and transgenic mouse models [25–27] and in patients [6, 7, 9–11]. OVT73 are a premanifest model expressing an 11,625 bp transgene consisting of full length human HTT cDNA with an exon 1 repeat (CAG)69(CAACAG)2, under the control of a minimal human HTT promoter (1.1 Kb genomic DNA immediately upstream of exon 1) [28]. The OVT73 model develops HTT positive aggregates, gene expression and metabolic changes in the brain but no discernible cell loss [28–40]. The transgene is integrated as multiple copies at a single locus in an intergenic region of ovine chromosome 10, with approximately 10 full length copies integrated head-to-tail, surrounded by several smaller transgene fragments that lack promoter sequence [30].
Transgene repeat instability was first assessed in six post-mortem OVT73 sheep aged 5-years (3 ewes, 3 rams; G1 and G2) (Supplementary Table 1). High resolution small pool-PCR (SP-PCR) was used to analyze CAG repeat length in single DNA molecules containing the OVT73 transgene, based on limiting dilution and Poisson analysis. All tissue samples reported in this manuscript were obtained from the South Australian Research and Development Institute (SARDI) and were sampled in accordance with approval of the Department of Primary Industries and Regions (PIRSA) Animal Ethics Committee (Approval number 19/02).The SP-PCR protocol was based on that previously described [8], with the use of sheep- (first round amplification) and transgene- (second round amplification) specific primers. The transgene-specific forward primer was 6-FAM labelled to enable resolution of the PCR product and quantification of repeat length using an automated ABI3130XL DNA sequencer with GeneScantrademark 600 LIZ® Size Standard (Applied Biosystems). Pure CAG repeat length was calculated from the tallest peak (modal repeat) of each trace compared against an OVT73 sheep standard with structure (CAG)69(CAACAG)2 (Supplementary Material). At least 50 single molecules were genotyped per sample.
For all samples, the modal repeat was determined to be 69 CAG units (43.1–71.1% of molecules per sample; Supplementary Dataset 1). This aligns with genotyping of bulk genomic DNA (1000 genome equivalents) from these animals and previous reports for the OVT73 line [28, 29]. A smaller peak was also detected at a lower frequency (6.6–20.8% of molecules per sample). Sanger sequencing confirmed this is a transgene copy with structure (CAG)26(CAACAG)2. This short transgene copy is also detected in genomic DNA from the OVT73 founder animal HD260 (Supplementary Figure 1). SP-PCR of fibroblast cells derived from founder HD260 (Supplementary Material) shows that both (CAG)26(CAACAG)2 and (CAG)69(CAACAG)2 transgenes are present in all cells (1:4 ratio). Previously reported capture sequencing estimates there are 10 full-length transgene copies at the single OVT73 locus [30]. It therefore appears that a fragment of a single copy of the transgene integrated, or that the construct DNA used in microinjection to create the founder OVT73 animal HD260 [28] inadvertently contained a short repeat clone in addition to the predominant cDNA which integrated along with the other copies at the OVT73 locus. The latter is most likely as other potential founder animals generated at the same time as HD260 also carry the short (CAG)26(CAACAG)2 transgene (Supplementary Figure 1). The absence of an mRNA corresponding to the (CAG)26(CAACAG)2 transgene in OVT73 indicates that it is not expressed.
As the (CAG)26(CAACAG)2 repeat is not expanded it was removed from the SP-PCR dataset for instability analysis. For the remaining products, a size distribution all alleles with >56 repeats, the modal repeat for each sample remained as 69 CAG units with modest variation (57 –80 CAG) observed (Fig. 1, Supplementary Dataset 1).
Fig. 1
Somatic variation of the expanded CAG repeat of the OVT73 sheep transgene. Distribution and frequency of somatic variation in pure CAG repeat length of the OVT73 transgene is shown for liver and striatum tissues from six OVT73 sheep (5 years old) with constitutive polyglutamine-coding repeat structure (CAG)69(CAACAG)2. Repeat lengths were assessed in single molecules by small pool PCR. Modal repeat length for all animals was 69 CAG.
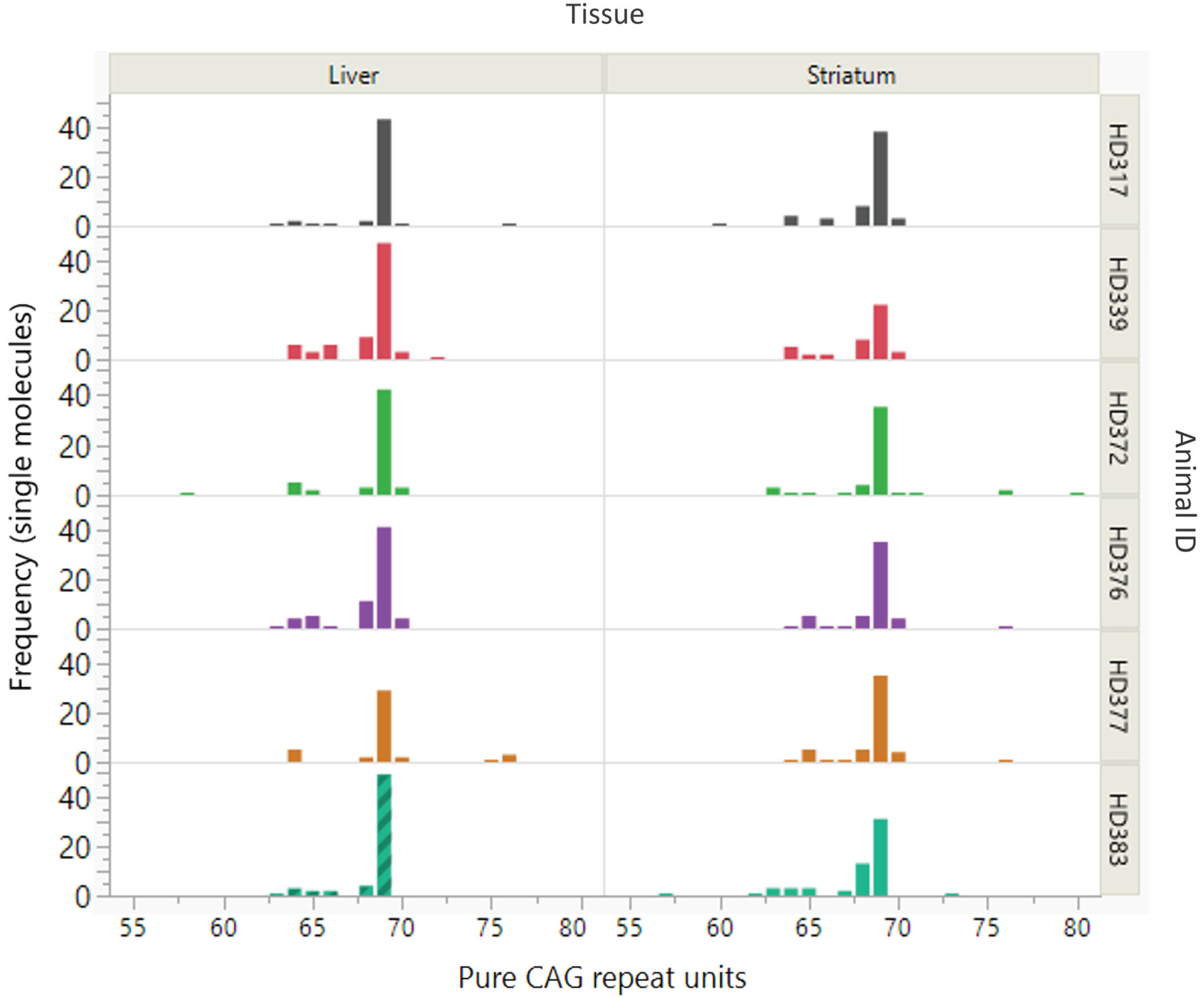
Interestingly, the SP-PCR data also revealed a cluster of alleles ∼65 CAGs as well as a few alleles between 75 and 80 CAGs. Further analyses, presented below, revealed that additional copies of the transgene at the multi-copy locus had repeat lengths in these ranges. Given this, it is likely that CAG-containing alleles around these sizes are products of other transgene copies, rather than somatic derivatives of the 69 CAG repeat-containing transgene. The presence of multiple transgene copies precludes accurate quantification of repeat instability, e.g., using Instability Index methods [8, 25]. This is because, for repeats that are closely spaced in length it is difficult to distinguish expansion peaks originating from shorter alleles from contraction peaks originating from longer alleles. Further, for transgenes with the same repeat length, it is impossible to discern the specific transgene(s) from which expansion or contraction peaks originate. The small sample number in this study also precluded meaningful assessment for effects of generation, age, or inherited CAG repeat length on instability, as has been observed in mouse models [6, 26, 27, 41, 42]. Regardless, the data clearly show that the polyglutamine-coding repeat in the dominant OVT73 sheep transgene (CAG)69(CAACAG)2 is remarkably stable. Notably, a repeat of this length is expected to exhibit significant levels of somatic expansion after 5 years, given the instability present in a knock-in HD mouse model with 72 CAG repeats [6].
To examine whether older sheep might show evidence for somatic expansion, we performed bulk PCR and MiSeq analysis in three OVT73 sheep of advanced age (10-year-old, G3 ewes). These animals were recently utilized in a non-invasive cohort study [39] where OVT73 sheep at 9 and 10 years of age were found to have elevated mHTT levels in CSF and changes in brain white matter structure as assessed by MRI. The microstructural white matter changes correlated with declining gait and mHTT levels of CSF, indicating measurable disease progression over the one-year period. To assess potential brain somatic expansion more broadly bulk PCR was performed here on seven brain regions (brainstem, caudate, cerebellum, motor cortex, piriform cortex, putamen, temporal lobe) as well as liver (Supplementary Material). Interestingly, at least four discrete peaks were observed in tissues from all three animals, corresponding to pure CAG repeat length estimates of 27, 65–66, 69–70 and 82–83 units (respectively), and a peak with 76-CAG evident only in animal HD909 (Fig. 2, Supplementary Figures 2 and 3).
Fig. 2
Alternative copies of the OVT73 transgene have different polyglutamine coding repeat lengths (Animal 909). Bulk PCR traces from striatal genomic DNA of three 10-year-old (G3) OVT73 sheep indicate presence of multiple copies of the OVT73 transgene with differing polyglutamine-coding repeat lengths, inserted at the ovine chromosome 10 locus. Bulk PCR traces from brain and liver tissues are shown for a single animal (animal 909). The dominant pure CAG repeat length was 70. Repeat lengths of 27 (short allele), 65, 76 and 82 are also observed. Traces were consistent across the eight tissues examined. GeneScan 500 LIZ internal size standard was used to determine product size. CAG repeat size was estimated against a knock-in mouse model standard, with adjustment for known differences in the polyglutamine-polyproline repeat sequence structure between the mouse and sheep models (refer to Supplementary Material).
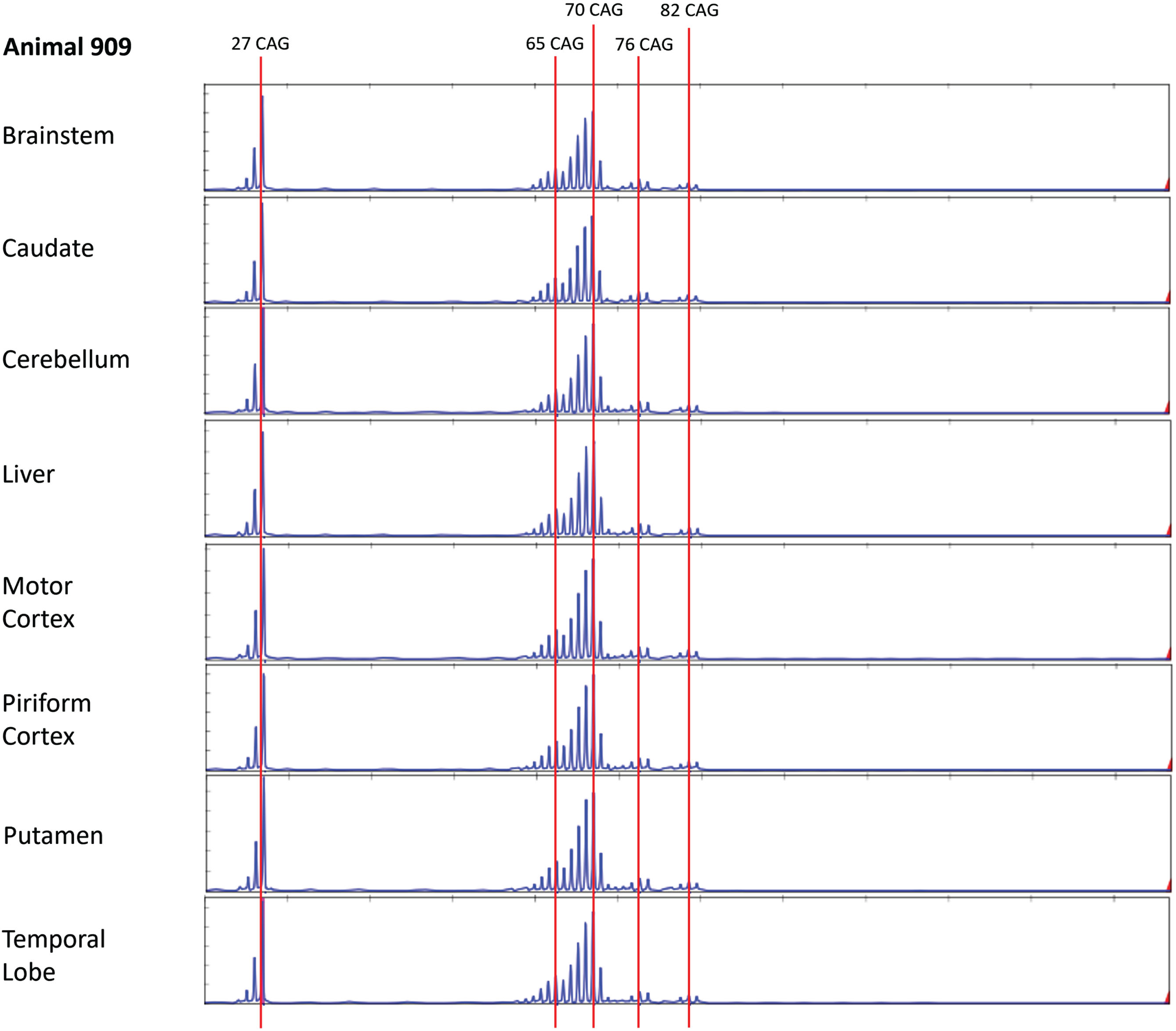
The bulk PCR electrophoretogram peaks correspond with the 5-year-old SP-PCR data and MiSeq data, although there is a discrepancy in the short transgene copy repeat length which was called as 26 CAGs in the SP-PCR data and 27 CAGs by MiSeq. CAG repeat length calls for both fragment sizing datasets were calculated based on the OVT73 transgene polyglutamine-polyproline coding repeat structure (CAG)n(CAACAG)2(CCG)9(CCT)3, previously determined by sanger sequencing of all OVT73 generations (data not shown) and validated here by MiSeq sequencing of bulk PCR products from the 10-year-old animals HD909 and HD913 (Supplementary Figure 4). Review of the shape of SP-PCR electrophoretograms (Supplementary Figure 1) supports that the predominant short transgene copy in the 5-year-old animals likely also contains 27 CAGs, as the height of the 27 CAG peak is almost equal to that of the 26 CAG peak. The modal call of 26 CAGs for the short transgene in the SP-PCR dataset may therefore reflect PCR slippage caused by overamplification of the short allele during SP-PCR. Other repeat lengths detected in the 10-year-old bulk PCR fragment analysis (65–66, 76 and 82–83 units) were present with lower abundance as evident by the electrophoretogram peak heights (Fig. 2, Supplementary Figures 2 and 3) consistent with constitutive CAG repeat lengths of lower abundance transgene copies, rather than instability of the predominant (CAG)69(CAACAG)2 transgene copies. The presence of transgene copies with these CAG repeat lengths was confirmed in the MiSeq data (Supplementary Figure 4). The electrophoretogram peak indicating a 76 CAG repeat length in animal HD909 (Fig. 2) may reflect germline expansion of a copy of the transgene. As explained above, CAG-containing alleles detected by SP-PCR in the 5-year sheep around the repeat sizes of these major repeat length clusters are most likely attributable to individual 65–66-, 76-, and 82–83-unit polyglutamine coding transgene copies. Analysis of many more single molecules by SP-PCR would confirm the presence of these alternative repeat-length transgene copies in the 5-year-old animals, although the narrow repeat range (65–87) prevents the accurate assignment of variation to specific transgene alleles.
It was notable that two of the three 10-year-old animals (all G3) had a 70-unit CAG repeat allele, compared to 69 units in all 5-year-old sheep (G1 and G2). This agrees with previously reported one-unit generational creep between G2 and G3 animals [29]. Identifying generational and/or individual differences in CAG repeat length is relevant for breeding of the OVT73 model. We also note that the shapes of the 69–70 CAG repeat distributions differed between animals, e.g., in animal 909, the peak to the right of the modal peak drops sharply in height relative to the modal 70-repeat peak, while in animal 912, the peak to the right of the modal peak is relatively close in height to the modal 69-repeat peak. This may reflect subtly different degrees of somatic expansion between these sheep; however, the shapes of these CAG length distributions might also suggest the presence of transgene copies with both 69 and 70 CAGs and overlapping PCR products generated from each (Fig. 2, Supplementary Figures 2 and 3). Regardless, these data reveal that at 10 years the OVT73 transgene CAG repeat is still remarkably stable in all tissues tested.
Taken together, these data provide new insight into OVT73 transgene locus composite sequence, where at least 4–5 different repeat lengths are apparent in different full-length copies of the transgene, with a majority of copies having (CAG)69-70(CAACAG)2 polyglutamine-coding repeats. As explained above, the presence of multiple transgene copies is a complicating factor for quantifying somatic variation in this model. However, minimal repeat length variation observed overall indicates that all transgene copies are quite stable, including between tissues, individuals, generations, and ages. As the OVT73 sheep are a prodromal model with no overt neurological symptoms or cell death, these findings provide further evidence that somatic instability of the HTT expanded CAG repeat beyond a threshold length is required for onset and progression of HD and suggest that ∼70 CAGs may be insufficient to trigger disease onset.
Cis- and/or trans-genetic factors may limit the instability of the OVT73 transgene. Cis-factors include the transgene sequence and genomic structure. The transgene polyglutamine-polyproline coding repeat structure is (CAG)n(CAACAG)2(CCG)9(CCT)3, confirmed by MiSeq sequencing data from the 10-year-old animals described here and Sanger sequencing of all generations (Supplementary Figure 4). Although it is a non-canonical human sequence including a duplicate polyglutamine-coding (CAACAG)2 sequence following the (CAG)n repeat, the (CAG)n repeat is uninterrupted and therefore should be permissive for instability [4].
Other cis-factors potentially limiting OVT73 transgene repeat instability include that it is cDNA rather than genomic DNA [43], or that the chromatin context surrounding the transgene may not be permissive for instability. Expanded CAG/CTG repeats flanked by genomic DNA are reported to be more unstable than those within cDNA transgenes (reviewed by [43]). The genomic locus of expanded CAG/CTG repeats also contributes to their ‘expandability’ as evidenced by differences in instability between CAG/CTG repeat expansion diseases [43]. The randomly integrated OVT73 cDNA transgene may therefore lack cis-elements that are important for instability in HD. There is also evidence that transcription is required for instability [44, 45]. Protein expression of the OVT73 transgene is relatively low (80–90% lower than levels seen in several transgenic rodent models [29]), potentially limiting repeat instability caused by transcriptional mechanisms [43, 46]. The low levels of transgene expression may be explained by genomic context, where the transgene is integrated in an intergenic region and is also methylated close to (∼2 kb) the CAG repeat [47]. We note however that in humans, hypermethylation of a CGG repeat expansion immediately upstream of the FMR1 gene (causative of Fragile X syndrome) does not appear to inhibit further somatic repeat expansion [48]. Regardless of somatic instability, low transgene expression alone may explain why the OVT73 model does not manifest overt pathology and neurological symptoms.
Sheep may also lack the appropriate complement of trans-factors that modify HTT CAG repeat instability. Human huntingtin has a longer polyglutamine coding tract than all other species, thought to confer an evolutionary advantage in neuronal function [49, 50]. Although direct comparison of HD patient and animal model data is difficult due to differences in lifespan, mouse models of HD appear to require very long repeat lengths relative to humans to achieve the frequency and size of mutations that is seen in humans [51–53]. Additionally, a knock-in minipig model with CAG150 shows less instability than a knock-in mouse with comparable age and repeat length [54]. The mechanism for the species difference is not fully understood but likely involves DNA repair proteins that influence instability [43, 49]. Key DNA repair genes that have been implicated in somatic repeat instability in HD (MSH3, FAN1, MLH1, LIG1, PMS1, PMS2) (reviewed by [43]) are expressed in multiple sheep tissues, according to the publicly available RNAseq database BioGPS; http://biogps.org/sheepatlas/#goto=welcome (Supplementary Table 2). Predicted amino acid sequences for these genes also demonstrate high conservation between human and sheep (Supplementary Table 2). Functional expression is therefore likely, as expected of a complex mammal. Investigating genetic variation in these DNA repair genes in the OVT73 model and expression levels may provide additional insight into their influence as trans-modifiers of repeat stability in sheep.
With limited availability of human tissue, characterizing the handful of animal models capturing prodromal and early HD is important to resolve the intracellular threshold CAG repeat length that triggers cellular dysfunction and the transition from pre-symptomatic to symptomatic [23]. Understanding the mechanism of instability may further reveal possible targets to suppress expansion. Mouse models of HD and other repeat disorders have demonstrated that DNA repair genes influence repeat instability [13–16, 43, 55]. The reason for the stability of the OVT73 sheep transgene expanded CAG repeat is not currently clear but may involve access by the DNA repair gene complement. Attempting to introduce instability via modulation/knock-out of DNA repair genes would test this idea and potentially enable study of the CAG length pathogenic threshold, with the complex brain and longer lifespan of sheep providing a realistic window to assess the mechanism of the prodromal-symptomatic transition. A humanized knock-in sheep would complement this by resolving complications of transgene composition and transgenesis.
ACKNOWLEDGMENTS
We thank the staff at the South Australian Research and Development Institute for all animal management and preparation of tissues. Marian DiFiglia and Ellen Sapp (Massachusetts General Hospital) kindly provided tissue samples for the analysis of 10-year-old OVT73 animals, originally donated from a longitudinal study cohort by Heather Grey-Edwards (University of Massachusetts). Kristine Boxen of the University of Auckland Centre for Genomics, Proteomics and Metabolomics (University of Auckland) performed Sanger sequencing and genescan services. We thank Emanuela Elezi of the MGH Mission Driven Service Core for DNA Fragment and MiSeq analyses.
FUNDING
This work was supported by the CHDI Foundation (A-2476, A-2690) and NIH grants R01 NS049206 and R01 NS091161.
CONFLICT OF INTEREST
V.C.W. was a founding scientific advisory board member with financial interest in Triplet Therapeutics Inc. Her financial interests were reviewed and are managed by Massachusetts General Hospital and Mass General Brigham in accordance with their conflict-of-interest policies. She is a scientific advisory board member of LoQus23 Therapeutics Ltd. and has provided paid consulting services to Acadia Pharmaceuticals Inc., Alnylam Inc., Biogen Inc., Passage Bio and Rgenta Therapeutics. She has received research support from Pfizer Inc.
DATA AVAILABILITY
The data supporting the findings of this study are available within the article and/or its supplementary material.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JHD-231516.
REFERENCES
[1] | Lee JM , Ramos EM , Lee JH , Gillis T , Mysore JS , Hayden MR , et al. CAG repeat expansion in Huntington disease determines age at onset in a fully dominant fashion. Neurology. (2012) ;78: (10):690–5. doi: 10.1212/WNL.0b013e318249f683. |
[2] | Wexler NS , Lorimer J , Porter J , Gomez F , Moskowitz C , Shackell E , et al. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington’s disease age of onset. Proc Natl Acad Sci U S A. (2004) ;101: (10):3498–503. doi: 10.1212/WNL.0b013e318249f683. |
[3] | Hong EP , MacDonald ME , Wheeler VC , Jones L , Holmans P , Orth M , et al. Huntington’s disease pathogenesis: Two sequential components. J Huntingtons Dis. (2021) ;10: (1):35–51. doi: 10.3233/JHD-200427. |
[4] | Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium CAG repeat not polyglutamine length determines timing of Huntington’s disease onset. Cell. (2019) ;178: (4):887–900s. doi: 10.1016/j.cell.2019.06.036. |
[5] | Telenius H , Kremer B , Goldberg YP , Theilmann J , Andrew SE , Zeisler J , et al. Somatic and gonadal mosaicism of the Huntington disease gene CAG repeat in brain and sperm. Nat Genet. (1994) ;6: (4):409–14. doi: 10.1038/ng0494-409. |
[6] | Kennedy L , Evans E , Chen CM , Craven L , Detloff PJ , Ennis M , et al. Dramatic tissue-specific mutation length increases are an early molecular event in Huntington disease pathogenesis. Hum Mol Genet. (2003) ;12: (24):3359–67. doi: 10.1093/hmg/ddg352. |
[7] | Swami M , Hendricks AE , Gillis T , Massood T , Mysore J , Myers RH , et al. Somatic expansion of the Huntington’s disease CAG repeat in the brain is associated with an earlier age of disease onset. Hum Mol Genet. (2009) ;18: (16):3039–47. doi: 10.1093/hmg/ddp242. |
[8] | Geraerts FCA , Snell RG , Faull RLM , Williams L , Jacobsen JC , Reid SJ . Comparison of Huntington’s disease CAG repeat length stability in human motor cortex and cingulate gyrus. J Huntingtons Dis. (2016) ;5: (3):297–301. doi: 10.3233/JHD-160203. |
[9] | Shelbourne PF , Keller-McGandy C , Bi WL , Yoon SR , Dubeau L , Veitch NJ , et al. Triplet repeat mutation length gains correlate with cell-type specific vulnerability in Huntington disease brain. Hum Mol Genet. (2007) ;16: (10):1133–42. doi: 10.1093/hmg/ddm054. |
[10] | Pinto RM , Arning L , Giordano J V , Razghandi P , Andrew MA , Gillis T , et al. Patterns of CAG repeat instability in the central nervous system and periphery in Huntington’s disease and in spinocerebellar ataxia type 1. Hum Mol Genet. (2020) ;29: (15):2551–67. doi: 10.1093/hmg/ddaa139. |
[11] | Ciosi M , Maxwell A , Cumming SA , Hensman Moss DJ , Alshammari AM , Flower MD , et al. A genetic association study of glutamine-encoding DNA sequence structures, somatic CAG expansion, and DNA repair gene variants, with Huntington disease clinical outcomes. EBioMedicine. (2019) ;48: :568–80. doi: 10.1016/j.ebiom.2019.09.020. |
[12] | Kacher R , Lejeune FX , Noël S , Cazeneuve C , Brice A , Humbert S , et al. Propensity for somatic expansion increases over the course of life in Huntington disease. Elife. (2021) ;10: :e64674. doi: 10.7554/eLife.64674. |
[13] | Pinto RM , Dragileva E , Kirby A , Lloret A , Lopez E , St. Claire J , et al. Mismatch repair genes Mlh1 and Mlh3 modify CAG instability in Huntington’s disease mice: Genome-wide and candidate approaches. PLoS Genet. (2013) ;9: (10):e1003930. doi: 10.1371/journal.pgen.1003930. |
[14] | Dragileva E , Hendricks A , Teed A , Gillis T , Lopez ET , Friedberg EC , et al. Intergenerational and striatal CAG repeat instability in Huntington’s disease knock-in mice involve different DNA repair genes. Neurobiol Dis. (2009) ;33: (1):37–47. doi: 10.1016/j.nbd.2008.09.014. |
[15] | Tomé S , Manley K , Simard JP , Clark GW , Slean MM , Swami M , et al. MSH3 polymorphisms and protein levels affect CAG repeat instability in Huntington’s disease mice. PLoS Genet. (2013) ;9: (2):e1003280. doi: 10.1371/journal.pgen.1003280. |
[16] | Loupe JM , Pinto RM , Kim KH , Gillis T , Mysore JS , Andrew MA , et al. Promotion of somatic CAG repeat expansion by Fan1 knock-out in Huntington’s disease knock-in mice is blocked by Mlh1 knock-out. Hum Mol Genet. (2020) ;29: (18):3044–53. doi: 10.1093/hmg/ddaa196. |
[17] | Kovalenko M , Dragileva E , St. Claire J Gillis T , Guide JR , New J , et al. Msh2 acts in medium-spiny striatal neurons as an enhancer of cag instability and mutant huntingtin phenotypes in Huntington’s disease knock-in mice. PLoS One. (2012) ;7: (9):e44273. doi: 10.1371/journal.pone.0044273. |
[18] | Goold R , Flower M , Moss DH , Medway C , Wood-Kaczmar A , Andre R , et al. FAN1 modifies Huntington’s disease progression by stabilizing the expanded HTT CAG repeat. Hum Mol Genet. (2019) ;28: (4):650–61. doi: 10.1093/hmg/ddy375. |
[19] | Kim KH , Hong EP , Shin JW , Chao MJ , Loupe J , Gillis T , et al. Genetic and functional analyses point to FAN1 as the source of multiple Huntington disease modifier effects. Am J Hum Genet. (2020) ;107: (1):96–110. doi: 10.1016/j.ajhg.2020.05.012. |
[20] | Roy JCL , Vitalo A , Andrew MA , Mota-Silva E , Kovalenko M , Burch Z , et al. Somatic CAG expansion in Huntington’s disease is dependent on the MLH3 endonuclease domain, which can be excluded via splice redirection. Nucleic Acids Res. (2021) ;49: (7):3907–18. doi: 10.1093/nar/gkab152. |
[21] | Lee JM , Chao MJ , Harold D , Elneel KA , Gillis T , Holmans P , et al. A modifier of Huntington’s disease onset at the MLH1 locus. Hum Mol Genet. (2017) ;26: (19):3859–67. doi: 10.1093/hmg/ddx286. |
[22] | Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell. (2015) ;162: (3):516–26. doi: 10.1016/j.cell.2015.07.003. |
[23] | Monckton DG . The contribution of somatic expansion of the CAG repeat to symptomatic development in Huntington’s disease: A historical perspective. J Huntingtons Dis. (2021) ;10: (1):7–33. doi: 10.3233/JHD-200429. |
[24] | Donaldson J , Powell S , Rickards N , Holmans P , Jones L . What is the pathogenic CAG expansion length in Huntington’s disease. J Huntingtons Dis. (2021) ;10: (1):175–202. doi: 10.3233/JHD-200445. |
[25] | Lee JM , Zhang J , Su AI , Walker JR , Wiltshire T , Kang K , et al. A novel approach to investigate tissue-specific trinucleotide repeat instability. BMC Syst Biol. (2010) ;4: :29. doi: 10.1186/1752-0509-4-29. |
[26] | Larson E , Fyfe I , Morton AJ , Monckton DG . Age-, tissue- and length-dependent bidirectional somatic CAG•CTG repeat instability in an allelic series of R6/2 Huntington disease mice. Neurobiol Dis. (2015) ;76: :98–111. doi: 10.1016/j.nbd.2015.01.004. |
[27] | Ishiguro H , Yamada K , Sawada H , Nishii K , Ichino N , Sawada M , et al. Age-dependent and tissue-specific CAG repeat instability occurs in mouse knock-in for a mutant Huntington’s disease gene. J Neurosci Res. (2001) ;65: (4):289–97. doi: 10.1002/jnr.1153. |
[28] | Jacobsen JC , Bawden CS , Rudiger SR , McLaughlan CJ , Reid SJ , Waldvogel HJ , et al. An ovine transgenic Huntington’s disease model. Hum Mol Genet. (2010) ;19: (10):1873–82. doi: 10.1093/hmg/ddq063. |
[29] | Reid SJ , Patassini S , Handley RR , Rudiger SR , McLaughlan CJ , Osmand A , et al. Further molecular characterisation of the OVT73 transgenic sheep model of Huntington’s disease identifies cortical aggregates. J Huntingtons Dis. (2013) ;2: (3):279–95. doi: 10.3233/JHD-130067. |
[30] | Chiang C , Jacobsen JC , Ernst C , Hanscom C , Heilbut A , Blumenthal I , et al. Complex reorganization and predominant non-homologous repair following chromosomal breakage in karyotypically balanced germline rearrangements and transgenic integration. Nat Genet. (2012) ;44: (4):390–7. doi: 10.1038/ng.2202. |
[31] | Handley RR , Reid SJ , Patassini S , Rudiger SR , Obolonkin V , McLaughlan CJ , et al. Metabolic disruption identified in the Huntington’s disease transgenic sheep model. Sci Rep. (2016) ;6: :20681. doi: 10.1038/srep20681. |
[32] | Handley RR , Reid SJ , Brauning R , MacLean P , Mears ER , Fourie I , et al. Brain urea increase is an early Huntington’s disease pathogenic event observed in a prodromal transgenic sheep model and HD cases. Proc Natl Acad Sci U S A. (2017) ;114: (52):E11293–302. doi: 10.1073/pnas.1711243115. |
[33] | Mears ER , Handley RR , Grant MJ , Reid SJ , Day BT , Rudiger SR , et al. A multi-omic Huntington’s disease transgenic sheep-model database for investigating disease pathogenesis. J Huntingtons Dis. (2021) ;10: (4):423–34. doi: 10.3233/JHD-210482. |
[34] | Morton AJ , Rudiger SR , Wood NI , Sawiak SJ , Brown GC , Mclaughlan CJ , et al. Early and progressive circadian abnormalities in Huntington’s disease sheep are unmasked by social environment. Hum Mol Genet. (2014) ;23: (13):3375–83. doi: 10.1093/hmg/ddu047. |
[35] | Skene DJ , Middleton B , Fraser CK , Pennings JLA , Kuchel TR , Rudiger SR , et al. Metabolic profiling of presymptomatic Huntington’s disease sheep reveals novel biomarkers. Sci Rep. (2017) ;7: :43030. doi: 10.1038/srep43030. |
[36] | Morton AJ , Middleton B , Rudiger S , Bawden CS , Kuchel TR , Skene DJ . Increased plasma melatonin in presymptomatic Huntington disease sheep (Ovis aries): Compensatory neuroprotection in a neurodegenerative disease? J Pineal Res. (2020) ;68: (2):12624. doi: 10.1111/jpi.12624. |
[37] | Schneider WT , Vas S , Nicol AU , Morton AJ . Abnormally abrupt transitions from sleep-to-wake in Huntington’s disease sheep (Ovis aries) are revealed by automated analysis of sleep/wake transition dynamics. PLoS One. (2021) ;16: (5):e0251767. doi: 10.1371/journal.pone.0251767. |
[38] | Spick M , Hancox TPM , Chowdhury NR , Middleton B , Skene DJ , Morton AJ . Metabolomic analysis of plasma in Huntington’s disease transgenic sheep (Ovis aries) reveals progressive circadian rhythm dysregulation. J Huntingtons Dis. (2023) ;12: (1):31–42. doi: 10.3233/jhd-220552. |
[39] | Taghian T , Gallagher J , Batcho E , Pullan C , Kuchel T , Denney T , et al. Brain alterations in aged OVT73 sheep model of Huntington’s disease: An MRI based approach. J Huntingtons Dis. (2022) ;11: (4):391–406. doi: 10.3233/JHD-220526. |
[40] | Sapp E , Boudi A , Reid SJ , Trombetta BA , Kivisäkk P , Taghian T , et al. Levels of synaptic proteins in brain and neurofilament light chain in cerebrospinal fluid and plasma of OVT73 Huntington’s disease sheep support a prodromal disease state. J Huntingtons Dis. (2023) ;12: (3):201–13. doi: 10.3233/JHD-230590. |
[41] | Lee JM , Pinto RM , Gillis T , St. Claire JC Wheeler VC . Quantification of age-dependent somatic CAG repeat instability in Hdh CAG knock-in mice reveals different expansion dynamics in striatum and liver. PLoS One. (2011) ;6: (8):e23647. doi: 10.1371/journal.pone.0023647. |
[42] | Wheeler VC , Lebel LA , Vrbanac V , Teed A , te Riele H , MacDonald ME . Mismatch repair gene Msh2 modifies the timing of early disease in HdhQ111 striatum. Hum Mol Genet. (2003) ;12: (3):273–83. doi: 10.1093/hmg/ddg056. |
[43] | Wheeler VC , Dion V . Modifiers of CAG/CTG repeat instability: Insights from mammalian models. J Huntingtons Dis. (2021) ;10: (1):123–48. doi: 10.3233/JHD-200426. |
[44] | Lin Y , Hubert L , Wilson JH . Transcription destabilizes triplet repeats. Mol Carcinog. (2009) ;48: (4):350–61. doi: 10.1002/mc.20488. |
[45] | Lin Y , Dion V , Wilson JH Transcription and triplet repeat instability. In: Wells RD, Ashizawa T, editors. Genetic Instabilities and Neurological Diseases. Elsevier; 2006. p. 691-704. |
[46] | Dion V , Wilson JH . Instability and chromatin structure of expanded trinucleotide repeats. Trends in Genetics. (2009) ;25: (7):288–97. doi: 10.1016/j.tig.2009.04.007. |
[47] | Lu AT , Narayan P , Grant MJ , Langfelder P , Wang N , Kwak S , et al. DNA methylation study of Huntington’s disease and motor progression in patients and in animal models. Nat Commun. (2020) ;11: (1):4529. doi: 10.1038/s41467-020-18255-5. |
[48] | Tabolacci E , Nobile V , Pucci C , Chiurazzi P . Mechanisms of the FMR1 repeat instability: How does the CGG sequence expand? Int J Mol Sci. (2022) ;23: (10):5425. doi: 10.3390/ijms23105425. |
[49] | Khristich AN , Mirkin SM . On the wrong DNA track: Molecular mechanisms of repeat-mediated genome instability. J Biol Chem. (2020) ;295: (13):4134–70. doi: 10.1074/jbc.REV119.007678. |
[50] | Iennaco R , Formenti G , Trovesi C , Rossi RL , Zuccato C , Lischetti T , et al. The evolutionary history of the polyQ tract in huntingtin sheds light on its functional pro-neural activities. Cell Death Differ. (2022) ;29: (2):293–305. doi: 10.1038/s41418-021-00914-9. |
[51] | Neto JL , Lee JM , Afridi A , Gillis T , Guide JR , Dempsey S , et al. Genetic contributors to intergenerational CAG repeat instability in Huntington’s disease knock-in mice. Genetics. (2017) ;205: (2):503–16. doi: 10.1534/genetics.116.195578. |
[52] | Gomes-Pereira M , Foiry L , Nicole A , Huguet A , Junien C , Munnich A , et al. CTG trinucleotide repeat ‘big jumps’: Large expansions, small mice. PLoS Genet. (2007) ;3: (4):e52. doi: 10.1371/journal.pgen.0030052. |
[53] | Seznec H , Lia-Baldini AS , Duros C , Fouquet C , Lacroix C , Hofmann-Radvanyi H , et al. Transgenic mice carrying large human genomic sequences with expanded CTG repeat mimic closely the DM CTG repeat intergenerational and somatic instability. Hum Mol Genet. (2021) ;9: (8):1185–94. doi: 10.1093/hmg/9.8.1185. |
[54] | Bai D , Yin P , Zhang Y , Sun F , Chen L , Lin L , et al. Lack of association of somatic CAG repeat expansion with striatal neurodegeneration in HD knock-in animal models. Hum Mol Genet. (2021) ;30: (16):1497–508. doi: 10.1093/hmg/ddab129. |
[55] | Zhao XN , Usdin K . FAN1 protects against repeat expansions in a Fragile X mouse model. DNA Repair (Amst). (2018) ;69: :1–5. doi: 10.1016/j.dnare2018.07.001. |

