
Cancer: From Wild-Type to Mutant Huntingtin
Abstract
Huntingtin (HTT) is a scaffold protein mostly known because it gives rise to the severe and incurable inherited neurological disorder Huntington’s disease (HD) when mutated. The Huntingtin gene (HTT) carries a polymorphic trinucleotide expansion of CAGs in exon 1 that ranges from 9 to 35 in the non-HD affected population. However, if it exceeds 35 CAG repeats, the altered protein is referred to as mutant HTT and leads to the development of HD. Given the wide spectrum of severe symptoms developed by HD individuals, wild-type and mutant HTT have been mostly studied in the context of this disorder. However, HTT expression is ubiquitous and several peripheral symptoms in HD have been described, suggesting that HTT is of importance, not only in the central nervous system (CNS), but also in peripheral organs. Accordingly, HTT and mutant HTT may interfere with non-brain-related diseases. Correlative studies have highlighted a decreased cancer incidence in the HD population and both wild-type and mutant HTT have been implicated in tumor progression. In this review, we describe the current evidence linking wild-type and mutant HTT to cancer and discuss how CAG polymorphism, HTT function, and partners may influence carcinogenesis and metastatic progression.
INTRODUCTION
Huntingtin (HTT) is a large scaffold protein of 350 kDa, conserved from flies to mammals, that has been mostly studied in the context of Huntington’s disease (HD), a rare inherited neurological disorder. HTT carries a polymorphic expansion of glutamine in its N-terminal sequence encoded by CAG repeats in its gene, HTT [1]. CAG repeats range from 9 to 35 CAGs in the non-HD population, with an average between 17 and 20 repeats (Fig. 1) [2]. When above 35 CAGs, it leads to the development of HD, generally during adulthood, with a lower penetrance observed in carriers of 36 to 39 repeats [3–5]. Individuals carrying alleles with an intermediate number (between 27 and 35 CAGs) may display some aspects of the disease [6, 7].
Fig. 1
The CAG polymorphism in HTT is associated with Huntington disease and cancer risk and evolution.
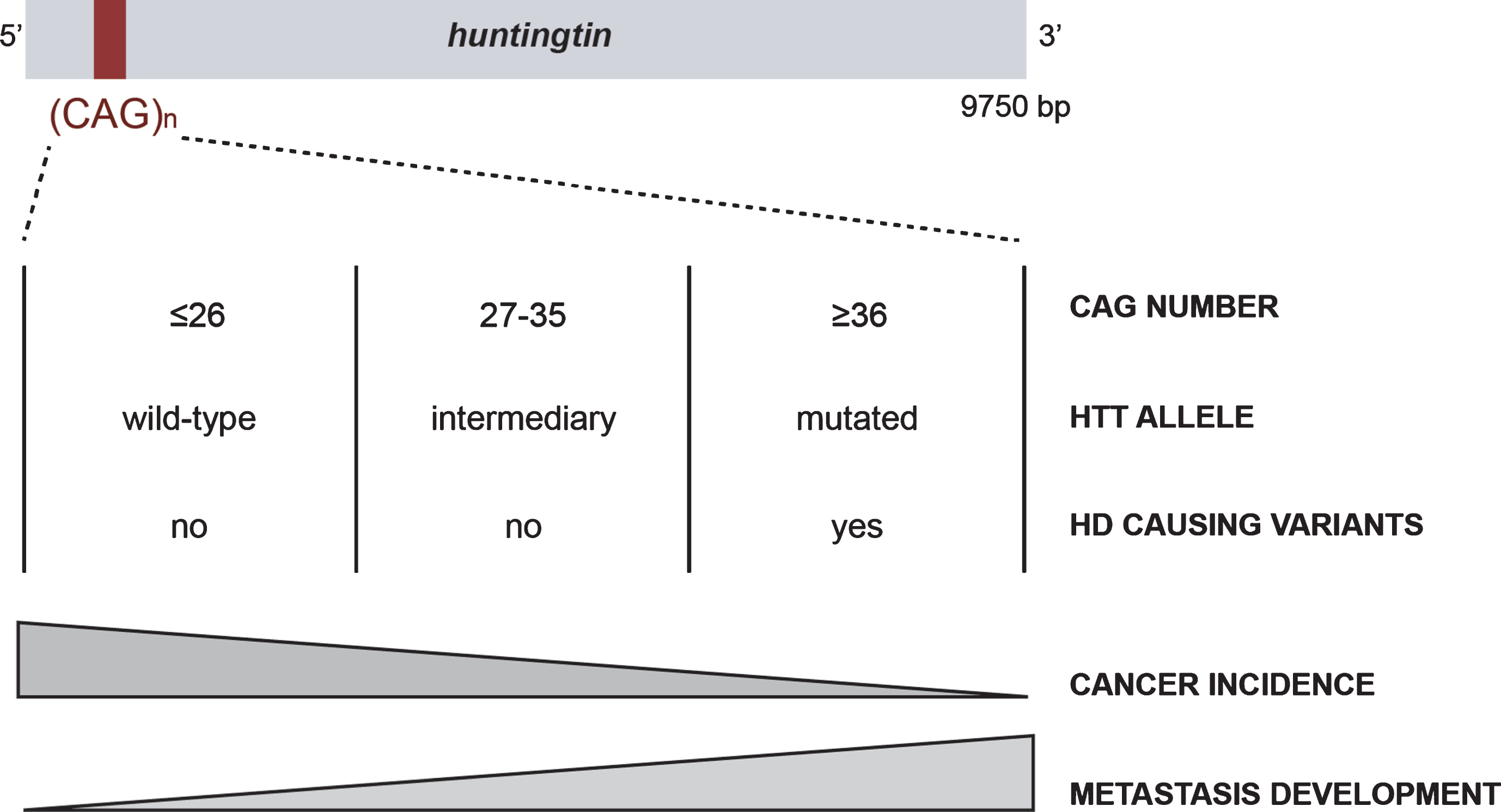
HD symptoms, including motor, cognitive, and psychiatric disturbances, are severe and ultimately lead to dementia [8]. Thus, much effort has been focused on understanding the molecular mechanisms leading to HD in the central nervous system (CNS) and the function of HTT has been mostly described in cells of neuronal origin. HTT participates in numerous cellular functions essential for the physiology of the CNS, such as cell division, transcriptional regulation, and intracellular transport [9]. It is subjected to multiple post-translational modifications including acetylation, sumoylation, ubiquitination, palmitoylation, and phosphorylation, which regulate its function and toxicity in HD [9]. However, HTT is widely expressed in the body and accumulating evidence suggests that it has a function in organs other than the brain [10, 11]. HD patients display peripheral symptoms, such as weight loss, muscular atrophy, cardiac failure, and osteoporosis [11], consistent with a role of HTT outside the CNS. Furthermore, some of these symptoms develop early during the disease and contribute to the mortality of HD patients. Several lines of evidence suggest that these peripheral manifestations are not dependent on cerebral dysfunction, but may be the consequence of mutant HTT expression in peripheral organs [11].
Our laboratory has shown that HTT is expressed in mammary tissue and regulates its morphogenesis and tumorigenesis [12–15]. We demonstrated that loss of HTT or the expression of mutant HTT is pro-metastatic in breast cancer. Additionally, we showed that wild-type HTT is involved in normal mammary gland development [12, 14, 15]. Moreover, we described an association between the size of CAG repeats in wild-type HTT and cancer prognosis [16], as well as decreased cancer incidence in the HD population [17]. In this review, we discuss the function of HTT during carcinogenesis, as well as the relationship between CAG repeats size and cancer incidence and prognosis. We also discuss several protein partners of HTT and signaling pathways in which HTT is engaged that may mediate the effects of HTT during cancer progression.
PARADOXICAL ASSOCIATION BETWEEN HD AND CANCER
The first study to examine the association between mutant HTT and cancer was published at the end of the last century [18]. Sorensen and Fenger investigated the leading causes of death in HD individuals and reported that only 5% were attributable to cancer. A few years later, Sorensen et al. reported a lower incidence of all cancers in HD individuals, except for those of the buccal cavity and the pharynx [19]. In 2012, Ji et al. described a similar decrease in the incidence of most cancer types in individuals with HD or other polyglutamine disorders [20]. In both studies, the authors did not have information concerning the length of CAG repeats or the clinicopathological characteristics of the cancer (grade, survival, metastasis) and suggested that HD and other polyglutamine disorders share common mechanisms leading to protection against cancer [20]. They proposed that HTT can act as a tumor suppressor by modulating the apoptosis of neoplastic cells. More recently, we confirmed the decreased risk of cancer in HD and spinocerebellar ataxia (SCA) patients [17].
Thus, long repeats could be protective against cancer, which may explain the advantage of germinal and somatic instability in HTT that leads to increased CAG repeats size, even though it is detrimental with respect to HD [21, 22]. However, there are exceptions depending on the type of cancer. For example, decreased incidence of cancer in HD patients does not apply to digestive system related cancers [20] or skin cancers [17]. The patterns of expression, the functions and interactors of each protein, and/or the nature of the signaling pathways involved in the neurodegenerative processes may differentially influence the specificity of action in cancer.
Finally, in sharp contrast to the low risk of cancer in HD, one of our studies paradoxically suggests that pathological HD CAG expansions increase cancer severity once it develops (Fig. 1) [15]. Indeed, mutant HTT increases cancer growth and metastasis development in mouse and cellular models of HD through the accumulation of Erb-B2 Receptor Tyrosine Kinase 2 (ERBB2) (HER2 in human) at the cell membrane. Mutant HTT impairs HER2 recycling by interacting with dynamin 2, leading to its accumulation at the membrane and subsequent overactivation of signaling pathways involved in proliferation and metastasis. Thus, even though long CAG repeats correlate with a lower incidence of cancer, they may enhance the severity of specific subtypes of cancer once it is initiated.
NON HD-PATHOLOGICAL CAG POLYMORPHISMS AND CANCER
The link between pathological HD CAG expansions and cancer risk and severity raises the question of whether CAG polymorphism is associated with cancer features and outcomes in a non-HD situation (in which the HTT CAG repeats is strictly below 36). We investigated the length of CAG repeats in the HTT gene in cohorts of cancer patients and found that it negatively correlated with ovarian cancer incidence in individuals that carry the cancer predisposition mutation in the BRCA2 gene [16], consistent with the lower incidence of cancer in HD patients [17, 18, 20]. Moreover, cancer onset occurred earlier in individuals with HTT alleles with an intermediate number of CAG repeats than in those with <27 CAG repeats in BRCA1 mutation carriers who developed breast cancer. Finally, the length of the CAG repeats in HTT appears to be an independent prognostic factor of metastasis development, specifically of the HER2 subtype, in good agreement with increased cancer aggressiveness by mutant HTT and the described molecular mechanism. This suggests that there may be a linear relationship between the length of CAG repeats in HTT and cancer incidence and metastasis development rather than a threshold effect (Fig. 1). Long CAG repeats may protect against cancer whereas it enhances cancer progression once tumorigenesis has been initiated, even below the pathological threshold that results in HD.
The relationship between CAG polymorphism and disease has also been described for the androgen receptor. The androgen receptor gene carries a polymorphic CAG repeats in exon 1 and, as HTT, is associated with cancer risk and evolution, as well as with neurodegeneration [23]. Depending on its size, the CAG expansion in the androgen receptor is linked to an increased risk of prostate (≤21 CAG repeats) or of breast cancers (≥21 CAG) or to the development of spinal and bulbar muscular atrophy (>40 CAG repeats) [23–25].
Finally, a functional relationship between CAG repeats and cancer cell death was recently described [26]. The authors identified a family of siRNA against CAG/CUG repeats which bind the open reading frames of several genes containing long complementary repeats. siCAG/CUG are highly toxic to cancer cells in vitro and efficient in vivo to decrease xenografted ovarian tumor progression in mice.
WILD-TYPE HTT IS ANTI-METASTATIC IN BREAST CANCER
Breast cancer is a highly heterogeneous disease with many different histological and molecular subtypes showing different levels of differentiation, proliferation profiles, and outcomes [27–29]. We have used cellular and murine models, as well as the analysis of HTT expression in a large cohort of breast cancer patients, to show that HTT influences breast cancer differentiation and progression [14, 30]. HTT expression increases in human in situ carcinomas as compared to healthy tissue. In contrast, it decreases in invasive cells as compared to in situ carcinomas (Fig. 2). Indeed, loss of HTT is itself pro-metastatic in mice. We focused our attention on the phosphorylation of HTT at serine 421 (S421-P-HTT), which regulates its normal function and is neuroprotective in HD [31–36]. We found that it colocalizes at cell-cell junctions with zonula occludens 1 (ZO-1), a marker of tight junctions, and regulates its expression and localization. S421-P-HTT is much less abundant in human in situ tumors than healthy tissue and nearly absent from invasive cancer cells and colocalize at cell-cell junctions with ZO-1 (Fig. 2). HTT and ZO-1 expression positively correlate with the glandular differentiation stage of human carcinomas and are concomitantly downregulated in low-grade, poorly differentiated carcinomas associated with a poor prognosis.
Fig. 2
HTT and S421-P-HTT during tumor progression. Schematic representation of HTT cellular localization and S421-P-HTT during mammary gland tumor progression. (from [30]).
![HTT and S421-P-HTT during tumor progression. Schematic representation of HTT cellular localization and S421-P-HTT during mammary gland tumor progression. (from [30]).](https://ip.ios.semcs.net:443/media/jhd/2018/7-3/jhd-7-3-jhd180290/jhd-7-jhd180290-g002.jpg)
Other studies have highlighted ZO-1 dysregulation in cancer. As for S421-P-HTT, the expression of ZO-1 is lower in tumoral tissue than in healthy mammary glands [37] and correlates with glandular differentiation of the carcinomas [38]. Furthermore, the loss of ZO-1 expression is linked to a poor prognosis and metastasis development [39]. Thus, HTT may modulate tumor differentiation, intercellular adhesion, and therefore metastasis progression, by regulating ZO-1. ZO-1 shares sequence homology with the tumor suppressor discs-large DLG protein in drosophila [40], suggesting that it may act as a tumor suppressor in mammals as well. Moreover, we observed nuclear localization of both S421-P-HTT and ZO-1 in vitro in mammary cell lines but we have been unable to detect S421-P-HTT within the cell nuclei in human biopsies. However, we observed ZO-1 localization in the nuclei of poorly differentiated basal carcinomas [14]. Nuclear ZO-1 localization has been previously shown to regulate cell proliferation through its interactor, ZONAB [41, 42]. HTT could therefore potentially act on ZO-1 through regulation of ZONAB.
HUNTINGTIN MODULATES CELLULAR ADHESION AND DIFFERENTIATION
Several pathways are regulated by HTT, disregulated in HD, and known to contribute to cancer progression. Among these, several lines of evidence suggest that HTT modulates cellular adhesion and polarity, with an impact on differentiation. For example, HTT regulates the differentiation of mammary cells during development in healthy mammary glands [12]. In contrast, the differentiation process is affected in human HD-derived pluripotent stem cell lines [43, 44], consistent with the reported role of HTT as a positive transcriptional regulator of genes involved in neuronal maintenance, a function that is lost when HTT is mutated [45]. Thus, mutant HTT may have lost the ability to regulate cellular differentiation.
At the molecular level, HTT regulates proteins involved in cell adhesion and differentiation. It interacts with the metalloprotease ADAM10, thus allowing the proper cleavage of the neuronal cadherin, N-cadherin [46, 47]. The absence of HTT leads to decreased expression of N-cadherin and ZO-1 in zebrafish, altering homotypic interactions between neuroepithelial cells and neurulation [47]. Additionally, HTT favors N-cadherin recycling through RAB11, thus influencing the attachment of newly generated neurons to the fiber of radial glial cells during cortical development [48].
HTT has also been implicated in the regulation of components of adherence junctions. The mRNA levels for such proteins are reduced in embryonic stem cells and neurons with lower HTT levels [49]. In neurons, HTT interacts with the F-box protein β-TrcP and axin, which are part of the multisubunit β-catenin destruction complex [50–52]. By permitting β-catenin degradation through this interaction, HTT regulates the WNT signaling pathway, which itself controls crucial aspects of cellular differentiation. Furthermore, loss of HTT from basal cells in healthy mammary tissue leads to the disruption of ZO-1 transport from the plasma membrane to the cytoplasm of luminal cells, leading to altered polarity [12]. In healthy and tumoral tissue, S421-P-HTT colocalizes with ZO-1 at the cell membrane and modulates its localization, as already discussed. As HTT phosphorylation at serine 421 is involved in microtubule-mediated vesicular transport to the cell membrane [34], it may regulate adhesion by transporting various components of intercellular complexes. Mutant HTT appears to act as a loss of function mutant, leading to decreased expression of ZO-1 and E-cadherin and increased metastatic capacity of tumoral cells [15]. The function of HTT in cellular adhesion may play a key role during embryonic neurogenesis, as neuronal adhesion and polarity are essential for the establishment of a functional network.
On a global scale, HTT expression appears to correlate with the stage of differentiation of normal and pathological tissues. HTT levels are higher in epithelial cell types, such as skin, squamous, and mammary cells, than in mesenchymal cell types, such as skin fibroblasts, mammary adipocytes, and stromal cells from the uterus [10]. HTT expression in breast cancer correlates with the differentiation stages of the tumoral tissue [14], being highly expressed in well-differentiated breast carcinomas and poorly expressed in poorly differentiated breast cancers, which are associated with a poor prognosis.
HTT-INTERACTING PROTEINS: PARTNERS IN CANCER?
Since 2003, more than 350 interactors of HTT have been identified, whereas only few have been further validated by binding assays and co-localization experiments [53–58]. At the molecular level, most of these partners participate in one of the following pathways: intracellular dynamics, protein degradation, cytoskeletal dynamics, and gene expression. Obviously, proteins participating in such pathways could be involved in HTT-mediated effects during tumorigenesis. However, little is known on these interactions, except for the above-mentioned role of HTT on HER2 recycling and the maintenance of ZO-1 at junctions.
One pathway of particular interest is autophagy, a mechanism in which HTT acts as a scaffold protein [59, 60]. Autophagy prevents cancer development and conversely, once cancer is established, increased autophagic flux often enables tumor cell survival and growth [61]. In fact, a role for autophagy at nearly every phase of the metastatic cascade has been identified [62]. Mutant HTT exon 1 protein is expressed in patients and knock-in HD mouse models as a result of incomplete HTT gene transcription [63, 64]. Interestingly, mutant HTT exon 1 protein expression causes cellular stress that activates autophagy [65, 66]. Thus, the activation of autophagy by mutant HTT expression in addition to a direct function for HTT in autophagy itself may be germane to a role for wild-type and mutant HTT in carcinogenesis and metastatic progression.
Also, studies have described a direct role for several HTT-interacting proteins in carcinogenesis. For example, the tumor suppressor P53 is mutated in many breast carcinomas [67]. Mutant HTT increases P53 expression, which has been shown to accumulate in murine and cellular HD models and postmortem brains of HD patients [68]. Thus, if this accumulation occurs as well in other cell types that do express mutant HTT, it may participate in the lower cancer incidence in the HD population [17, 19, 20], although this is yet to be established.
The HTT interacting protein 1, HIP1, an endocytic protein, is overexpressed in adenocarcinomas, such as breast, ovarian, colon, prostate, and lung cancers [69]. HIP1 acts as a pro-survival factor and its expression has been associated with metastatic progression and a poor prognosis for prostate cancer patients [69]. HIP1 is also associated with poor overall survival of acute myeloid leukemia patients [70]. Surprisingly, others found that HIP1 acts to suppress metastasis and regulates the epithelial-mesenchymal transition in lung cancer [71]. Moreover, HTT interacting protein 14, HIP14, is an oncogene that can induce tumor formation in mice [72]. Finally, expression of HAP1 (HTT associated protein 1) is lower in breast cancer than in healthy tissue and its overexpression decreases the migration velocity and invasive capacities of mammary tumoral cells [27, 73].
HTT may thus participate in the tumorigenic or anti-tumorigenic effects induced by its partners. This hypothesis could be tested by investigating the effect of HTT partners on tumorigenesis in the absence of HTT or in the presence of mutant HTT.
CONCLUSION
Cancer and neurodegenerative disorders, which represent important health public issues, are two research topics rarely associated. However, they clearly share dysregulated signaling pathways, as shown by the example of HTT that is a key regulator of the balance between cell survival and cell death under normal, neoplastic, and neurodegenerative conditions. The specificity of the cancer types that are regulated by HTT may depend on its expression levels which are heterogeneous across cell types as well as on the signaling pathways involved in the tumorigenic cascade. Unraveling the molecular mechanisms involved in both diseases will lead to a better comprehension of the function of a central scaffold protein such as HTT and of the association between CAG repeats length and cancers in polyglutamine disorders.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
This review is dedicated to HD and breast cancer patients and their families. We thank F. Saudou and laboratory members for fruitful discussions. This work was supported by grants from the Agence Nationale pour la Recherche - Maladies Rares (ANR-09-BLAN-0080, S.H.), Association pour la Recherche sur le Cancer (ARC subvention libre n°3188, S.H.), Fondation pour la Recherche Médicale (FRM, équipe labellisée, S.H.), Cancéropôle Ile-de France (INCA_6517, S.H.), CNRS, INSERM and the Institut Curie. S.H. is an INSERM investigator. M.S.T. was supported by a MENRT doctoral fellowship and an ARC fellowship.
REFERENCES
[1] | The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. (1993) ;72: (6):971–83. |
[2] | Kremer B , Goldberg P , Andrew SE , Theilmann J , Telenius H , Zeisler J , et al. A worldwide study of the Huntington’s disease mutation, The sensitivity and specificity of measuring CAG repeats. N Engl J Med. (1994) ;330: (20):1401–6. |
[3] | Rubinsztein DC , Leggo J , Coles R , Almqvist E , Biancalana V , Cassiman JJ , et al. Phenotypic characterization of individuals with 30-40 CAG repeats in the Huntington disease (HD) gene reveals HD cases with 36 repeats and apparently normal elderly individuals with 36-39 repeats. Am J Hum Genet, (1996) ;59: (1):16–22. |
[4] | Andrew SE , Goldberg YP , Kremer B , Telenius H , Theilmann J , Adam S , et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nat Genet, (1993) ;4: (4):398–403. |
[5] | Snell RG , MacMillan JC , Cheadle JP , Fenton I , Lazarou LP , Davies P , et al. Relationship between trinucleotide repeat expansion and phenotypic variation in Huntington’s disease. Nat Genet, (1993) ;4: (4):393–7. |
[6] | Killoran A , Biglan KM , Jankovic J , Eberly S , Kayson E , Oakes D , et al. Characterization of the Huntington intermediate CAG repeat expansion phenotype in PHAROS. Neurology, (2013) ;80: (22):2022–7. |
[7] | Semaka A , Hayden MR . Evidence-based genetic counselling implications for Huntington disease intermediate allele predictive test results. Clin Genet, (2014) ;85: (4):303–11. |
[8] | Ross CA , Tabrizi SJ . Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol, (2011) ;10: (1):83–98. |
[9] | Saudou F , Humbert S . The biology of huntingtin. Neuron, (2016) ;89: (5):910–26. |
[10] | Marques Sousa C , Humbert S . Huntingtin: Here, there, everywhere!. J Huntingtons Dis. (2013) ;2: (4):395–403. |
[11] | van der Burg JM , Bjorkqvist M , Brundin P . Beyond the bra: Widespread pathology in Huntington’s disease. Lancet Neurol. (2009) ;8: (8):765–74. |
[12] | Elias S , Thion MS , Yu H , Sousa CM , Lasgi C , Morin X , et al. Huntingtin regulates mammary stem cell division and differentiation. Stem Cell Reports, (2014) ;2: (4):491–506. |
[13] | Elias S , McGuire JR , Yu H , Humbert S . Huntingtin Is required for epithelial polarity through RAB11A-mediated apical trafficking of PAR3-aPKC. PLoS Biol. (2015) ;13: (5):1002142. |
[14] | Thion MS , McGuire JR , Sousa CM , Fuhrmann L , Fitamant J , Leboucher S , et al. Unraveling the role of huntingtin in breast cancer metastasis. J Natl Cancer Inst. (2015) ;107: (10):208. |
[15] | Moreira Sousa C , McGuire JR , Thion MS , Gentien D , de la Grange P , Tezenas du Montcel S , et al. The Huntington disease protein accelerates breast tumour development and metastasis through ErbB2/HER2 signalling. EMBO Mol Med, (2013) ;5: (2):309–25. |
[16] | Thion MS , Tezenas du Montcel S , Golmard JL , Vacher S , Barjhoux L , Sornin V , et al. CAG repeat size in Huntingtin alleles is associated with cancer prognosis. Eur J Hum Genet, (2016) ;24: (9):1310–5. |
[17] | Coarelli G , Diallo A , Thion MS , Rinaldi D , Calvas F , Boukbiza OL , et al. Low cancer prevalence in polyglutamine expansion diseases. Neurology, (2017) ;88: (12):1114–9. |
[18] | Sorensen SA , Fenger K . Causes of death in patients with Huntington’s disease and in unaffected first degree relatives. J Med Genet, (1992) ;29: (12):911–4. |
[19] | Sorensen SA , Fenger K , Olsen JH . Significantly lower incidence of cancer among patients with Huntington disease: An apoptotic effect of an expanded polyglutamine tract? Cancer. (1999) ;86: (7):1342–6. |
[20] | Ji J , Sundquist K , Sundquist J . Cancer incidence in patients with polyglutamine diseases: A population-based study in Sweden. Lancet Oncol, (2012) ;13: (6):642–8. |
[21] | Duyao M , Ambrose C , Myers R , Novelletto A , Persichetti F , Frontali M , et al. Trinucleotide repeat length instability and age of onset in Huntington’s disease. Nat Genet, (1993) ;4: (4):387–92. |
[22] | Telenius H , Kremer B , Goldberg YP , Theilmann J , Andrew SE , Zeisler J , et al. Somatic and gonadal mosaicism of the Huntington disease gene CAG repeat in brain and sperm. Nat Genet, (1994) ;6: (4):409–14. |
[23] | Kumar R , Atamna H , Zakharov MN , Bhasin S , Khan SH , Jasuja R . Role of the androgen receptor CAG repeat polymorphism in prostate cancer, and spinal and bulbar muscular atrophy. Life Sci, (2011) ;88: (13-14):565–71. |
[24] | Song YN , Geng JS , Liu T , Zhong ZB , Liu Y , Xia BS , et al. Long CAG repeat sequence and protein expression of androgen receptor considered as prognostic indicators in male breast carcinoma. PLoS One. (2012) ;7: (12):52271. |
[25] | Paz YMC , Robles P , Salazar C , Leone PE , Garcia-Cardenas JM , Naranjo M , et al. Positive association of the androgen receptor CAG repeat length polymorphism with the risk of prostate cancer. Mol Med Re, (2016) ;14: (2):1791–8. |
[26] | Murmann AE , Gao QQ , Putzbach WE , Patel M , Bartom ET , Law CY , et al. Small interfering RNAs based on huntingtin trinucleotide repeats are highly toxic to cancer cells. EMBO Ree, (2018) ;19: (3):45336. |
[27] | Perou CM , Sorlie T , Eisen MB , van de Rijn M , Jeffrey SS , Rees CA , et al. Molecular portraits of human breast tumours. Nature. (2000) ;406: (6797):747–52. |
[28] | Sorlie T , Perou CM , Tibshirani R , Aas T , Geisler S , Johnsen H , et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA, (2001) ;98: (19):10869–74. |
[29] | Sorlie T , Tibshirani R , Parker J , Hastie T , Marron JS , Nobel A , et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA, (2003) ;100: (14):8418–23. |
[30] | Thion MS , Humbert S . Beyond the bra: Huntingtin in breast cancers. Med Sci (Paris). (2016) ;32: (8–9):674–7. |
[31] | Humbert S , Bryson EA , Cordelieres FP , Connors NC , Datta SR , Finkbeiner S , et al. The IGF-1/Akt pathway is neuroprotective in Huntington’s disease and involves Huntingtin phosphorylation by Akt. Dev Cell, (2002) ;2: (6):831–7. |
[32] | Pardo R , Colin E , Regulier E , Aebischer P , Deglon N , Humbert S , et al. Inhibition of calcineurin by FK506 protects against polyglutamine-huntingtin toxicity through an increase of huntingtin phosphorylation at S421. J Neurosci, (2006) ;26: (5):1635–45. |
[33] | Rangone H , Poizat G , Troncoso J , Ross CA , MacDonald ME , Saudou F , et al. The serum- and glucocorticoid-induced kinase SGK inhibits mutant huntingtin-induced toxicity by phosphorylating serine 421 of huntingtin. Eur J Neurosci, (2004) ;19: (2):273–9. |
[34] | Colin E , Zala D , Liot G , Rangone H , Borrell-Pages M , Li XJ , et al. Huntingtin phosphorylation acts as a molecular switch for anterograde/retrograde transport in neurons. EMBO J, (2008) ;27: (15):2124–34. |
[35] | Zala D , Colin E , Rangone H , Liot G , Humbert S , Saudou F . Phosphorylation of mutant huntingtin at S421 restores anterograde and retrograde transport in neurons. Hum Mol Genet, (2008) ;17: (24):3837–46. |
[36] | Schilling B , Gafni J , Torcassi C , Cong X , Row RH , LaFevre-Bernt MA , et al. Huntingtin phosphorylation sites mapped by mass spectrometry, Modulation of cleavage and toxicity. J Biol Chem. (2006) ;281: (33):23686–97. |
[37] | Tokes AM , Szasz AM , Juhasz E , Schaff Z , Harsanyi L , Molnar IA , et al. Expression of tight junction molecules in breast carcinomas analysed by array PCR and immunohistochemistry. Pathol Oncol Res, (2012) ;18: (3):593–606. |
[38] | Hoover KB , Liao SY , Bryant PJ . Loss of the tight junction MAGUK ZO-1 in breast cancer: Relationship to glandular differentiation and loss of heterozygosity. Am J Pathol, (1998) ;153: (6):1767–73. |
[39] | Martin TA , Watkins G , Mansel RE , Jiang WG . Loss of tight junction plaque molecules in breast cancer tissues is associated with a poor prognosis in patients with breast cancer. Eur J Cancer, (2004) ;40: (18):2717–25. |
[40] | Willott E , Balda MS , Fanning AS , Jameson B , Van Itallie C , Anderson JM . The tight junction protein ZO-1 is homologous to the Drosophila discs-large tumor suppressor protein of septate junctions. Proc Natl Acad Sci U S A, (1993) ;90: (16):7834–8. |
[41] | Balda MS , Garrett MD , Matter K . The ZO-1-associated Y-box factor ZONAB regulates epithelial cell proliferation and cell density. J Cell Biol, (2003) ;160: (3):423–32. |
[42] | Balda MS , Matter K . The tight junction protein ZO-1 and an interacting transcription factor regulate ErbB-2 expression. EMBO J, (2000) ;19: (9):2024–33. |
[43] | Conforti P , Besusso D , Bocchi VD , Faedo A , Cesana E , Rossetti G , et al. Faulty neuronal determination and cell polarization are reverted by modulating HD early phenotypes. Proc Natl Acad Sci U S A., (2018) ;115: (4):71. |
[44] | Consortium HDi. Developmental alterations in Huntington’s disease neural cells and pharmacological rescue in cells and mice. Nat Neurosci, (2017) ;20: (5):648–60. |
[45] | Zuccato C , Tartari M , Crotti A , Goffredo D , Valenza M , Conti L , et al. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet, (2003) ;35: (1):76–83. |
[46] | Reis SA , Thompson MN , Lee JM , Fossale E , Kim HH , Liao JK , et al. Striatal neurons expressing full-length mutant huntingtin exhibit decreased N-cadherin and altered neuritogenesis. Hum Mol Genet, (2011) ;20: (12):2344–55. |
[47] | Lo Sardo V , Zuccato C , Gaudenzi G , Vitali B , Ramos C , Tartari M , et al. An evolutionary recent neuroepithelial cell adhesion function of huntingtin implicates ADAM10-Ncadherin. Nat Neurosci, (2012) ;15: (5):713–21. |
[48] | Barnat M , Le Friec J , Benstaali C , Humbert S . Huntingtin-mediated multipolar-bipolar transition of newborn cortical neurons is critical for their postnatal neuronal morphology. Neuron, (2017) ;93: (1):99–114. |
[49] | Strehlow AN , Li JZ , Myers RM . Wild-type huntingtin participates in protein trafficking between the Golgi and the extracellular space. Hum Mol Genet, (2007) ;16: (4):391–409. |
[50] | Gines S , Ivanova E , Seong IS , Saura CA , MacDonald ME . Enhanced Akt signaling is an early pro-survival response that reflects N-methyl-D-aspartate receptor activation in Huntington’s disease knock-in striatal cells. J Biol Chem, (2003) ;278: (50):50514–22. |
[51] | Godin JD , Poizat G , Hickey MA , Maschat F , Humbert S . Mutant huntingtin-impaired degradation of beta-catenin causes neurotoxicity in Huntington’s disease. EMBO J, (2010) ;29: (14):2433–45. |
[52] | Dupont P , Besson MT , Devaux J , Lievens JC . Reducing canonical Wingless/Wnt signaling pathway confers protection against mutant Huntingtin toxicity in Drosophila. Neurobiol Dis, (2012) ;47: (2):237–47. |
[53] | Harjes P , Wanker EE . The hunt for huntingtin function: Interaction partners tell many different stories. Trends Biochem Sci, (2003) ;28: (8):425–33. |
[54] | Goehler H , Lalowski M , Stelzl U , Waelter S , Stroedicke M , Worm U , et al. A protein interaction network links GIT1, an enhancer of huntingtin aggregation, to Huntington’s disease. Mol Cell, (2004) ;15: (6):853–65. |
[55] | Kaltenbach LS , Romero E , Becklin RR , Chettier R , Bell R , Phansalkar A , et al. Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS Genet. (2007) ;3: (5):82. |
[56] | Culver BP , Savas JN , Park SK , Choi JH , Zheng S , Zeitlin SO , et al. Proteomic analysis of wild-type and mutant huntingtin-associated proteins in mouse brains identifies unique interactions and involvement in protein synthesis. J Biol Chem, (2012) ;287: (26):21599–614. |
[57] | Ratovitski T , Chighladze E , Arbez N , Boronina T , Herbrich S , Cole RN , et al. Huntingtin protein interactions altered by polyglutamine expansion as determined by quantitative proteomic analysis. Cell Cycle, (2012) ;11: (10):2006–21. |
[58] | Shirasaki DI , Greiner ER , Al-Ramahi I , Gray M , Boontheung P , Geschwind DH , et al. Network organization of the huntingtin proteomic interactome in mammalian brain. Neuron, (2012) ;75: (1):41–57. |
[59] | Ochaba J , Lukacsovich T , Csikos G , Zheng S , Margulis J , Salazar L , et al. Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc Natl Acad Sci U S A, (2014) ;111: (47):16889–94. |
[60] | Rui YN , Xu Z , Patel B , Chen Z , Chen D , Tito A , et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat Cell Biol, (2015) ;17: (3):262–75. |
[61] | Levy JMM , Towers CG , Thorburn A . Targeting autophagy in cancer. Nat Rev Cancer, (2017) ;17: (9):528–42. |
[62] | Mowers EE , Sharifi MN , Macleod KF . Autophagy in cancer metastasis. Oncogene, (2017) ;36: (12):1619–30. |
[63] | Neueder A , Landles C , Ghosh R , Howland D , Myers RH , Faull RLM , et al. The pathogenic exon 1 HTT protein is produced by incomplete splicing in Huntington’s disease patients. Sci Rep. (2017) ;7: (1):1307. |
[64] | Sathasivam K , Neueder A , Gipson TA , Landles C , Benjamin AC , Bondulich MK , et al. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc Natl Acad Sci U S A, (2013) ;110: (6):2366–70. |
[65] | Ravikumar B , Vacher C , Berger Z , Davies JE , Luo S , Oroz LG , et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet, (2004) ;36: (6):585–95. |
[66] | Yamamoto A , Cremona ML , Rothman JE . Autophagy-mediated clearance of huntingtin aggregates triggered by the insulin-signaling pathway. J Cell Biol, (2006) ;172: (5):719–31. |
[67] | Petitjean A , Mathe E , Kato S , Ishioka C , Tavtigian SV , Hainaut P , et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: Lessons from recent developments in the IARC TP53 database. Hum Mutat, (2007) ;28: (6):622–9. |
[68] | Bae BI , Xu H , Igarashi S , Fujimuro M , Agrawal N , Taya Y , et al. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington’s disease. Neuron, (2005) ;47: (1):29–41. |
[69] | Rao DS , Hyun TS , Kumar PD , Mizukami IF , Rubin MA , Lucas PC , et al. Huntingtin-interacting protein 1 is overexpressed in prostate and colon cancer and is critical for cellular survival. J Clin Invest, (2002) ;110: (3):351–60. |
[70] | Wang J , Yu M , Guo Q , Ma Q , Hu C , Ma Z , et al. Prognostic significance of huntingtin interacting protein 1 expression on patients with acute myeloid leukemia. Sci Re. (2017) ;7: :45960. |
[71] | Hsu CY , Lin CH , Jan YH , Su CY , Yao YC , Cheng HC , et al. Huntingtin-interacting protein-1 is an early-stage prognostic biomarker of lung adenocarcinoma and suppresses metastasis via Akt-mediated epithelial-mesenchymal transition. Am J Respir Crit Care Med, (2016) ;193: (8):869–80. |
[72] | Ducker CE , Stettler EM , French KJ , Upson JJ , Smith CD . Huntingtin interacting protein 14 is an oncogenic human protein: Palmitoyl acyltransferase. Oncogene. (2004) ;23: (57):9230–7. |
[73] | Zhu L , Song X , Tang J , Wu J , Ma R , Cao H , et al. Huntingtin-associated protein A potential biomarker of breast cancer. Oncol Re, (2013) ;29: (5):1881–7. |

