
Improving Cognition Without Clearing Amyloid: Effects of Tau and Ultrasound Neuromodulation
Abstract
Alzheimer’s disease is characterized by progressive impairment of neuronal functions culminating in neuronal loss and dementia. A universal feature of dementia is protein aggregation, a process by which a monomer forms intermediate oligomeric assembly states and filaments that develop into end-stage hallmark lesions. In Alzheimer’s disease, this is exemplified by extracellular amyloid-β (Aβ) plaques which have been placed upstream of tau, found in intracellular neurofibrillary tangles and dystrophic neurites. This implies causality that can be modeled as a linear activation cascade. When Aβ load is reduced, for example, in response to an anti-Aβ immunotherapy, cognitive functions improve in plaque-forming mice. They also deteriorate less in clinical trial cohorts although real-world clinical benefits remain to be demonstrated. Given the existence of aged humans with unimpaired cognition despite a high plaque load, the central role of Aβ has been challenged. A counter argument has been that clinical symptoms would eventually develop if these aged individuals were to live long enough. Alternatively, intrinsic mechanisms that protect the brain in the presence of pathology may exist. In fact, Aβ toxicity can be abolished by either reducing or manipulating tau (through which Aβ signals), at least in preclinical models. In addition to manipulating steps in this linear pathocascade model, mechanisms of restoring brain reserve can also counteract Aβ toxicity. Low-intensity ultrasound is a neuromodulatory modality that can improve cognitive functions in Aβ-depositing mice without the need for removing Aβ. Together, this highlights a dissociation of Aβ and cognition, with important implications for therapeutic interventions.
INTRODUCTION
The 100th volume of the Journal of Alzheimer’s Disease (JAD) is a special issue dedicated to Mark A. Smith, who passed away prematurely and tragically in December 2010. Mark served as co-editor and chief of JAD, a journal founded by George Perry in 1998. Mark regularly challenged the amyloid cascade hypothesis and instead proposed oxidative stress and mitogenic dysregulation (i.e., aberrant cell cycle reentry) as the primary drivers of Alzheimer’s disease (AD), attributing not only tau but also amyloid-β (Aβ) subordinate roles in disease etiology.1,2 A major focus of his work was to understand how mitochondrial impairment and oxidative stress might initiate and/or contribute to AD pathogenesis.3 Here, our aim is not to challenge whether Aβ is a primary or secondary etiological agent, but, in light of the ongoing approval of anti-Aβ antibodies as a treatment for AD,4 to explore whether cognitive improvements rely on Aβ clearance.
When Auguste Deter began showing signs of presenile AD before reaching the age of 50, she started to have trouble with episodic memory, noticed shifts in her behavior, and encountered difficulties with both speaking and writing. Eventually, she was institutionalized by Alois Alzheimer in 1901, further deteriorated and then passed away in 1906. Upon examining her brain, Alzheimer observed not only that it had shrunk in certain areas, but he also identified two types of abundant histological features, the two hallmarks of AD, extracellular plaques and intracellular neurofibrillary tangles (NFTs).5 Independently, Oskar Fischer also discovered these lesions in senile dementia cases.6 The microscopically visible hallmarks contain molecules that had undergone a process of post-translational modifications of the monomer and subsequent oligomerization and fibrillization, with the peptide Aβ forming the plaques and the microtubule-associated protein tau the NFTs. With the identification of autosomal dominant mutations in familial AD in the gene that encodes the amyloid-β protein precursor (AβPP) from which Aβ is derived, the belief that Aβ triggers the disease, particularly in familial AD cases, and fuels its progression was further reinforced. This understanding has since influenced research efforts globally, both in basic science and translational explorations. Eventually, the amyloid hypothesis was formulated in 1991. While the AD brain, other than plaques and NFTs, also presents with tau-positive neuropil threads and dystrophic neurites, eosinophilic actin-containing Hirano bodies, activated microglia and reactive astrocytes, granulovacuolar degeneration as well as cerebral amyloid angiopathy as common features,7 Aβ has been posited as the major culprit.8 A direct connection was established between the deposition of Aβ and AD, a progressive dementia impacting memory, cognition, and behavior. The assumption, therefore, was that by tailoring mice to express sufficiently high levels of Aβ, as a consequence, their neurons would degenerate and this would lead to cognitive impairment. Consequently, it was assumed that a therapy that reduces Aβ in these mice to sufficiently low levels would restore the mouse’s cognitive functions.
When the first-generation transgenic mouse models were developed for AD (and for primary tauopathies, such as frontotemporal lobar degeneration, FTLD), the primary objective was to reproduce at a biochemical and histological, albeit not at an anatomical level, the fibril formation of Aβ and tau as well as the formation of argyrophilic and/or congophilic plaques and NFTs. After several iterations, this objective was attained, facilitated by the appropriate choice of expression vectors and the introduction of pathogenic mutations in AD- and FTLD-related genes into the protein-coding sequence of the respective transgenes.9,10 Despite the widespread use of these transgenic mouse models to test therapeutic interventions, determining the toxic forms of Aβ and tau at the post-translational modification and assembly levels has remained challenging to date, especially regarding tau.11 When the various mouse models are analyzed behaviorally to assess treatment outcomes, a regularly encountered challenge is that the test battery needs to reflect the anatomical distribution and severity of the Aβ and tau pathology, often yielding subtle and, in part, contradictory improvements of behavioral outcomes.12 This makes a comparative and integrative analysis of data obtained across preclinical models challenging.13 When translating therapeutic strategies like anti-Aβ vaccination, which originated from animal studies,14 to clinical trials, the main hurdle over the years has been the failure to clear Aβ at sufficiently high levels or to produce significant cognitive benefits. Interestingly, in 2018, the AT(N) conceptual framework was established, which redefined AD from a syndromal to a biological construct by categorizing the biomarkers into those of Aβ, tau, and neurodegeneration [AT(N)].15 This framework uses three traditional syndromal categories and a six-stage numeric scheme. The guideline’s authors emphasize ‘that this framework seeks to create a common language with which investigators can generate and test hypotheses about the interactions among different pathologic processes (denoted by biomarkers) and cognitive symptoms’.15
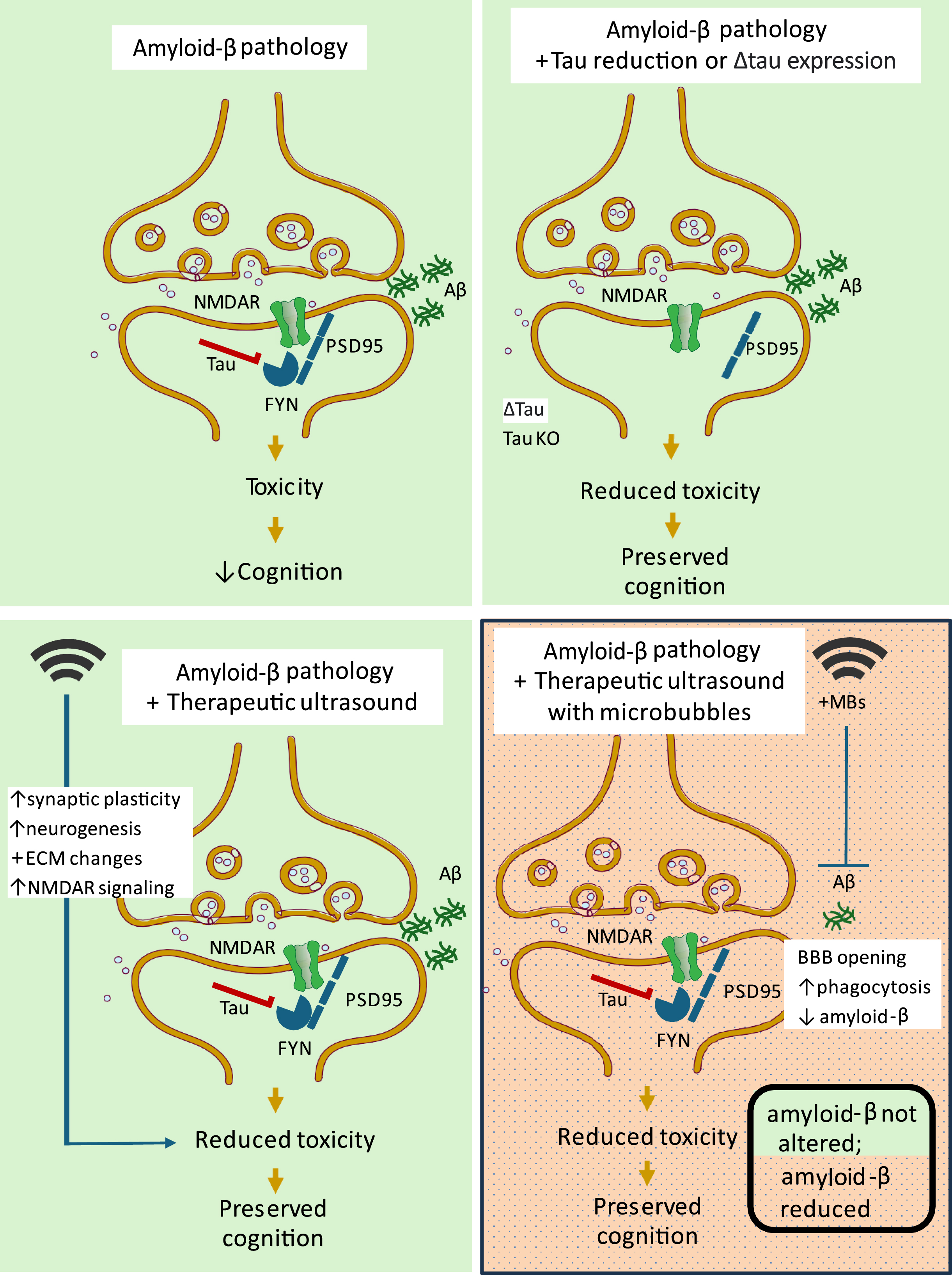
A question that arises in a disease setting is whether cognition can be improved or even restored without lowering Aβ levels. As discussed here, this may be possible, provided data in animal models with amyloidosis can be translated to the human situation. The amyloid cascade hypothesis places Aβ upstream of tau in the AD pathocascade.8 By assuming such a linear activation model with the peptide eliciting toxic downstream outcomes via a signaling cascade that involves tau and other components, one can envisage that by manipulating the components downstream of Aβ, cognitive functions may be restored without altering Aβ levels (Fig. 1). Another strategy to counteract Aβ toxicity is by activating brain or cognitive resilience mechanisms. Examples of such a strategy are neuromodulatory modalities such as low-intensity ultrasound. In the following, we will discuss the underlying mechanisms of these two complementary strategies and how they can potentially be combined in the clinic with the more classical approaches involving lowering Aβ.
Fig. 1
Improving cognition without clearing amyloid - effects of tau and ultrasound neuromodulation. Tau reduction as well as therapeutic ultrasound preserve cognition by abrogating amyloid-β (Aβ) toxicity in mouse models of Alzheimer’s disease. Tau is present in the post-synapse where it mediates Aβ toxicity through a PSD95-FYN kinase-NMDAR interaction. Depletion of tau or overexpression of a truncated form of tau (Δtau) protects APP transgenic mice from Aβ toxicity by limiting the excitotoxic signaling that occurs when FYN phosphorylates NMDARs, leading to improved cognition. Therapeutic ultrasound with microbubbles to achieve blood-brain barrier opening (BBBO) reduces Aβ levels by microglial activation and phagocytosis of Aβ. Therapeutic ultrasound without microbubbles has multiple stimulatory effects on the brain that lead to abrogated toxicity of Aβ and preserved cognition.
FROM TRANSGENIC MODELS TO HUMAN VACCINES
Animal models are widely used to study the etiology of AD and the underlying pathological mechanisms, as well as to design and validate therapeutic strategies. However, given the challenges in developing effective disease-modifying treatments for AD, as a fundamental problem, the lack of translatability of preclinical work using animal models of AD has been raised. We had recently argued that the existing models are useful within the limits of their design. We suggested that although they do not capture the entire complexity of human AD, animal models, in particular mice, will be indispensable for AD and other dementia research for the foreseeable future.13
The field has come a long way since the first transgenic animal models for AD were established more than two decades ago. In these mouse models, a major approach was to overexpress genes implicated in AD, based on the reasonable assumption that such a strategy was needed to recapitulate the key histopathological lesions, i.e., plaques and NFTs, that are found in diseased human brains. In recent years, this approach has come under scrutiny, and gene editing has been used to humanize endogenous loci and to introduce pathogenic mutations to establish more relevant models without the artificial overexpression of AβPP.16 However, these more refined approaches also have their inherent limitations, in part reflecting the fact that the course of a disease that in humans requires decades to develop has to be squeezed into the very short lifespan of a mouse. In modeling AD in mice, the question therefore arises as discussed recently, what actually constitutes a suitable AD model.10 In our view, this very much depends on the types of questions being asked, i.e., whether the key objective is to reproduce the biochemical and histological changes of AD at a cellular level and thereby induce functional impairments, including those at the level of cognition in the brain areas where the transgene-encoded proteins form protein aggregates, or whether the intention is to reproduce the clinical symptoms of AD as much as this is achievable in mice, recapitulating the temporal and spatial order of neurodegeneration observed in the human disease.
For practical reasons (which include financial considerations), a major objective of a suitable AD model for basic and translational research is that disease manifests at a reasonably young age and not necessarily only towards the end of a mouse’s lifespan. A downside of such a strategy is that even though the pathology may develop progressively, the contribution of aging to late-onset AD may not be appropriately modelled. A second aspect is the need for a reasonably large therapeutic window to facilitate drug testing. A third issue is that findings obtained in animal models need to be reproducible across laboratories. This is often difficult to accomplish given differences in mouse models, the genetic background on which they are maintained,17 and in methods and analysis tools, including behavioral test batteries, as well as the fact that mouse colonies experience genetic drift with time, which either augments, reduces or abrogates some of the pathological features.18 Despite these considerations, animal studies are essential for delineating the molecular and cellular processes specifically associated with AD pathology. They can identify and validate molecular targets pivotal to the disease process and investigate whether the modulation of such targets elicits a therapeutic effect.
The first robust Aβ plaque-developing mouse model was generated by expressing the familial APP mutation V717F (Indiana) under the neuron-directed platelet-derived growth factor-β promoter.19 Shortly thereafter, another strain was reported that expressed the 695 amino acid isoform of the human APP gene together with the Swedish double mutation K670N/M671L (APPswe).20 The mice developed numerous Congo red-positive plaques indicative of fibrillar amyloid in their core, and displayed impaired learning and memory in spatial reference and alternation tasks at 9-10 months but not three months of age. The Tg2576 mice were subsequently shown to have deficits in the Morris water maze, radial arm maze, object recognition, Y-maze spontaneous alternation, and contextual fear conditioning, and to display increased locomotor activity in the open field and the home cage environment (discussed in)21. The Tg2576 strain had to be maintained on a mixed C57BL/6×SJL genetic background to prevent lethality without concomitantly losing the amyloid pathology. Several years after the development of the Tg2576 mice on this mixed background, a separate line was created by systematically (>16 times) backcrossing hemizygous Tg2576 mice to the 129S6/SvEv (129) strain. Interestingly, when the mice were assessed in multiple behavioral tests, the behavioral impairments were more pronounced and occurred earlier on the 129 than the mixed background. Still, the Aβ pathology was similar between the two strains.21 While the assessment of Aβ pathology was somewhat cursory, this observation nonetheless indicates a potential decoupling between Aβ pathology and cognition.
In our own preclinical studies, we mainly use APP23 mice that express APPswe under the control of the murine Thy1 promoter, conferring a twofold neuronal overexpression of the transgene compared to the endogenous APP gene.22 The mice develop congophilic plaques surrounded by distorted neurites containing hyperphosphorylated Tau, implying, together with other corroborating evidence, a role for Aβ in the pathogenesis of AD (Fig. 1). The APP23 mice were maintained on a C57Bl/6 background and at least for a decade since they had been generated, they displayed, different from Tg2576 mice, a normal mortality. However, interestingly, with time, the strain began to drift such that in 2010, we reported a premature lethality of above 40% which reached a plateau at around the age of 4–6 months.23 This was the case for both an animal cohort bred and maintained in Sydney, Australia as well as in Basel, Switzerland.23 A 60% mortality was subsequently reported by a Spanish team for this strain, with mortality in female mice exceeding that in males.24 In our facility, the APP23 strain has since reached a stable mortality rate of around 60%.
The identification of Aβ and tau as key players in AD pathophysiology, together with the expansion of antibody development technologies at the turn of the century, led to substantial efforts in developing active and passive immunotherapies for AD, largely targeting Aβ and tau25,26 Our intension is not to provide a comprehensive overview of studies in transgenic mice but to convey that improvements in amyloid pathology generally led to cognitive improvements (in studies in which behavioral tests have been performed): Active immunizations with the Aβ42 peptide were shown to reduce amyloid plaque pathology and behavioral impairment,27,28 without negatively impacting sensorimotor or cognitive abilities long-term.29 Similar findings were obtained by passive immunization with a range of anti-Aβ antibodies,30,31 arguing for a therapeutic effect due to the restoration of synaptic deficits.32 One group described the development of aducanumab (BIIB037), a human monoclonal antibody that selectively interacts with Aβ aggregates, including soluble oligomers and insoluble fibrils, and a dose-escalation phase 1b randomized trial (PRIME; ClinicalTrials.gov identifier NCT01677572) in AD patients, reporting a slowing of clinical decline.33 In this study, reduction of brain amyloid was achieved in Tg2576 mice using a murinized version of aducanumab, but cognitive functions were not assessed.
After two phase 3 clinical trials (EMERGE and ENGAGE),34 the US Food and Drug Administration (FDA) approved aducanumab (AduhelmTM) in 2021, despite concerns relating to clinical evidence of its stated cognitive benefits. This marked the first approval for a drug aimed at reducing Aβ levels, a decision met with criticism from the scientific community. More recently, the FDA granted accelerated approval to another anti-Aβ antibody, lecanemab (LeqembiTM), following an 18-month phase 3 trial involving 1,795 early AD patients. This humanized IgG1 monoclonal antibody targets large, soluble Aβ protofibrils, demonstrating reduced Aβ levels and reduced cognitive and functional decline, albeit with modest effect sizes. Eventually, in July 2024, the FDA approved donanemab (KisunlaTM), an antibody targeting a pyroglutamylated form of Aβ. The regulatory approval process for anti-Aβ antibodies has been drawn-out and contentious, sparking extensive debate. Currently, the European Medicines Agency (EMA) has not approved any disease-modifying therapy for AD. Notably, Biogen, the manufacturer of aducanumab, announced its discontinuation in 2024 of aducanumab to prioritize marketing of lecanemab. This raises questions about whether anti-Aβ antibodies will deliver cognitive benefits in real-world settings and whether these benefits justify their high costs.
MANIPULATING TAU TO MITIGATE OR NULLIFY Aβ TOXICITY
An important step in AD pathophysiology is the interaction of Aβ in its oligomeric form with the neuronal plasma membrane surface. This serves as a triggering event that initiates a cascade of altered signal transduction leading to synaptic dysfunction and other functional impairments including neurodegeneration.35 The amyloid cascade hypothesis places tau downstream of Aβ; however, tau is by no means an innocent bystander, given that the protein has an essential role in mediating Aβ toxicity, at least in preclinical models. By crossing amyloid-depositing hAPP mice onto a tau knockout background, several learning and memory deficits were ameliorated and premature lethality was rescued, importantly without altering AβPP expression, Aβ42 levels, or Aβ plaque load.36 The question arises about the underlying mechanism in conferring this protection from Aβ toxicity when tau is lacking. It turns out that tau, perceived at the time as an axonal protein, has an important scaffolding function in dendritic spines by targeting Fyn to the post-synapse, where this tyrosine-directed kinase phosphorylates the NR2b subunit of the NMDA receptor (NMDAR), an ion-channel receptor found at most excitatory synapses and being a target of the AD drug memantine. Y1472-phosphorylated NR2b-containing NMDARs then recruit the scaffolding protein PSD-95 to form an excitotoxic signaling complex (Fig. 1).23 Not different from hAPP mice, the APP23 strain is also characterized by premature lethality, memory impairment and an increased susceptibility to experimentally induced seizures. By crossing APP23 mice with mice that either lack tau or overexpress only its so-called projection domain (Δtau strain), all three deficits were rescued, because Fyn was not targeted to the post-synapse and could therefore not phosphorylate NR2b (Fig. 1). Again, the improvements of cognitive functions in APP23 mice, when crossed onto a tau knock-out or Δtau-expressing background, occurred in the absence of any changes to APP mRNA levels, Aβ40 or Aβ42 levels, or Aβ plaque load.23 The broad implications of this are twofold, firstly of uncovering a linear model of a signal transduction cascade with Aβ being the primary ‘cause’ leading to ‘down-stream toxicity’ as its ‘consequence’, and secondly the dissociation of Aβ and cognition which has important implications for developing treatment strategies for AD.
We had referred to the interplay of tau and Aβ as a ‘toxic pas de deux’,37 and not surprisingly, the role of tau in mediating toxicity extends to clinical conditions other than AD, such as stroke and Dravet syndrome, the latter being a severe form of epilepsy characterized by frequent and prolonged seizures,38,39 Interestingly, tau ablation in mice has diverse effects on different neuronal sub-populations; it reduces the action potential firing of excitatory neurons, while increasing the excitability of inhibitory neurons.40 Also, in APPPS1 mice, tau deficiency was shown to decelerate the formation of new plaques.41 The differential role of tau and Aβ has been nicely summed up by Dr. George Bloom who aptly described Aβ as the trigger and tau as the bullet in AD pathogenesis, suggesting pathways mediating toxicity including excitotoxicity.42 With Fyn being a kinase that interlinks tau and Aβ, these three players have been referred to as a ‘toxic triad’.43
We subsequently referred to tau and Aβ as ‘brake and accelerator’44 in order to convey yet another point of difference between the two hallmark molecules, namely that pathological tau reduces protein translation more generally, whereas Aβ boosts tau translation more specifically.45,46 Tau and Fyn share the property of forming nanoclusters that may serve as potential subcellular platforms allowing for the integration of signaling pathways under both physiological and pathological conditions,47–51 with Fyn also being (independently of Aβ) a key regulator of tau pathology and its aggregation process.52–54 Treatment strategies for AD are therefore much broader than Aβ-targeting vaccines, including Fyn kinase inhibitors35,55 (NCT01864655) or antisense oligonucleotides (ASOs) targeting tau56 (NCT03186989). Ionis has developed an investigational anti-tau ASO by monthly intrathecal injections, demonstrating dose-dependent reductions in total tau as well as phospho-tau proteins in the cerebrospinal fluid by 30–50% in clinical phase I/II trials, in the absence of serious adverse events56 (NCT NCT03186989). While these investigational therapies target events in a presumed linear signal transduction cascade with Aβ at the top and tau and Fyn more downstream, in the following we will discuss how low-intensity ultrasound can achieve cognitive improvements without manipulating either Aβ or tau.
LOW-INTENSITY ULTRASOUND IMPROVES COGNITION WITHOUT CLEARING Aβ
Ultrasound is a modality that is increasingly being explored in the AD field, both in preclinical models and in clinical trials.57,58 The common underlying principle is the generation and focusing of sound waves above the range of human hearing (i.e., >20 kHz) into a defined target volume. At high intensity (HIFU), the modality is being used as a focused, incisionless, FDA-approved surgical tool for treating essential tremor and Parkinson’s disease. At low intensities (relevant to the treatment of AD; LIFU), the modality is being investigated to achieve neuromodulation or blood-brain barrier opening (BBBO). To attain BBBO, preformed micrometer-sized microbubbles (MBs), employed in a clinical setting as ultrasound contrast agents, are administered intravenously. When ultrasound is directed through the skull into the brain, the pulsed sound waves interact with the circulating MBs causing them to cavitate in the pressure field.59 This achieves safe BBBO in the focal volume, lasting several hours, by separating the tight and adherens junctions that typically keep brain endothelial cells tightly tethered, thereby facilitating paracellular transport. Cavitation further facilitates transcytoplasmic transport by increasing caveolae-mediated endocytosis60 and through sonoporation.61 Brain endothelial cells generally display very low pinocytotic activity and express multidrug efflux transporters, which together decrease the level of endo- and transcytosis.
Ultrasound-mediated BBBO allows for the focal uptake of intravenously injected drugs which are either typically prevented from entering the brain or only achieve ineffective intracerebral levels.62–64 A distinctive advantage of LIFU is that, unlike systemic drug administration, it allows for localized brain treatment by directing an ultrasound beam to specific brain areas; unlike radiation therapy, it exerts its effects only within the effective focal zone and not in tissue through which the sound waves travel, improving its safety. For diseases such as AD, however, which are characterized by the widespread distribution of Aβ and tau pathology, large parts of the brain may require sequential treatment in an approach we have termed scanning ultrasound (SUS).65,66
Operationally, we categorize scanning ultrasound (SUS) or focused ultrasound (FUS) into three categories: SUSonly, SUS+MB, and SUS+MB +drug.58 The first application uses ultrasound without the exogenous injection of MBs and/or drugs; the second combines SUS with MBs to transiently open the BBB, relying on the therapeutic effects of endogenous, unidentified blood-borne factors taken up by the brain; and the third incorporates BBBO with the delivery of drugs (such as monoclonal antibodies). For typical low-intensity SUSonly parameters, the bioeffects are largely caused by the acoustic radiation force67 and its influence on mechanosensitive ion channels.68 The underlying mechanisms for the SUS+MB and SUS+MB +drug paradigms include cavitation plus radiation forces. Interestingly, for a given biological read-out, their effects can be antagonistic, additive or synergistic,69 with different cell-types in the brain responding differently to particular ultrasound parametercombinations.70,71
When we first applied ultrasound to APP23 mice with an amyloid pathology, we applied a SUS+MB paradigm with weekly treatments over up to eight weeks. This achieved microglia-mediated clearance of Aβ and improved memory performance in three complementary tasks, including the active place avoidance (APA) paradigm (Fig. 1).65,72 That SUS+MB achieved massive amyloid reductions was demonstrated in several ways: by Campbell-Switzer stains to separately visualize compact, mature plaques and diffuse aggregates, by western blotting of different fractions, and by ELISAs, all showing SUS+MB-mediated significant reductions. Interestingly, memory functions in the APA test were restored to wild-type levels. As a control, sham-treated APP23 mice were included which were anaesthetized and injected with MBs but ultrasound was not delivered. Subsequently, we demonstrated that Aβ clearance requires BBBO and microglial activation, which was not achieved by using SUSonly.73 We therefore suspected initially that SUSonly would not improve memory functions in impaired APP23 mice.
In parallel, we also assessed wild-type (C57Bl/6) mice. Originally planned as an extended safety study following up on earlier ultrasound safety assessments,74,75 we treated senescent (20–22-month-old) wild-type mice with SUS+MB and SUSonly compared with sham. Senescent mice are massively impaired in the APA paradigm and long-term potentiation (LTP) as an electrophysiological paradigm of memory. Surprisingly, we found that both sonication paradigms (but in particular the one without MBs), improved APA memory in a dose-dependent manner and restored LTP induction.69 As an underlying mechanism, we identified reductions in the density of perineuronal nets, increased neurogenesis and synaptic signaling, as well as changes to NMDARs conducive with the restoration of LTP.
We then applied these findings to APP23 mice, exploring two ultrasound frequencies (only the 1 MHz used in the above experiments will be discussed here) together with SUSonly compared with sham. Aligning with our findings in the aged wild-type mice,69 we found that SUSonly improved APA memory and other functions, without reducing Aβ (Fig. 1).76 We further revealed using quantitative SWATH proteomics and functional magnetic resonance imaging that SUSonly induced long-lasting functional changes that correlated with the observed improvement in memory. We argued that ultrasound brain stimulation could act on mechanisms responsible for resilience and neural compensation (such as cellular energy levels or improved connectivity), considering that in AD patients some neurons are spared from degeneration until advanced age and that some individuals are even remarkably resilient to AD, despite significant pathology.77,78 Given the findings that Aβ mediates toxicity through tau,23 we also assessed tau in the SUSonly-treated APP23 mice even though this strain does not present with NFT pathology.22 We quantified tau levels in hippocampal synaptosomes, finding increased levels in sham-treated APP23 mice, and reduced levels of synaptic tau in the SUSonly-treated mice,76 preliminary work which should be followed up on. Whether ultrasound treatment of APP23 mice affects synaptic tau accumulation has not been determined. Our study found changes to the neuronal proteome lasting for up to two weeks post-treatment, with the dysregulated clusters being tightly related to synaptic vesicle function, exocytosis, axonogenesis, and neuronal signaling. Furthermore, an elevation was found in Golgi vesicle transport and membrane dynamics-associated processes, and a marked decrease in histone methylation-associated processes. Together with the behavioral and magnetic resonance imaging data, the proteomics analysis provides mechanistic evidence that could explain why SUSonly improves cognition without affecting amyloid, namely through profound changes in chromatin organization and removal of transcriptional silencing, simultaneous with increased secretory activity, that could collectively result in increased network connectivity and improved cognitive functions.
WHAT HAVE WE LEARNED AND FUTURE STUDIES
What we have discussed here is that amyloid toxicity as manifested by behavioral/cognitive impairments can either be nullified in animal models by removing tau or altering tau in a way (e.g., by expressing Δtau) that it cannot target Fyn to the dendritic spines where amyloid exerts its excitotoxic effects, or by using neuromodulation to improve neuronal connectivity and activate plasticity as well as other mechanisms that mediate resilience and neural compensation. In support of this notion, a recent clinical study in eight AD patients treated the right hippocampus with FUS+MB, finding an increased regional cerebral metabolic rate of glucose by 18F-fluoro-2-deoxyglucose positron emission tomography (PET) that was correlated with improvement in recognition memory.79 These data suggest that FUS+MB to the hippocampus of AD patients may improve the metabolic glucose rate of the target area and memory in the short term, even without BBBO (and hence, we would add, without amyloid clearance).
An important aspect for therapeutic interventions is the brain volume and specific regions of brain that need to be treated. A general assumption is that with improved diagnostic tools, treatments can be initiated before the pathology has spread throughout the brain. The stereotypical pattern of tau pathology inspired a model, guided by prion research, whereby tau pathology, initiated in a discrete brain area, spreads to other brain regions via neuron-to-neuron transmission of misfolded tau seeds, a process facilitated by the trans-synaptic transfer of seeds.13 The pattern of spread, that occurs across anatomically connected neurons is thereby determined by connectivity and not, proximity.80 A natural extension of this concept is the interpretation of AD as a dysfunction of neural networks, rather than a cellular problem of how Aβ and tau impair neuronal functions which in due course lead to neurodegeneration. Indeed, mounting evidence supports the notion that aberrant changes in neural network activity occur early in AD, driven by neuronal hyperactivity, and that they contribute to pathological protein accumulation and cognitive dysfunction in AD.81 A ‘cascading network failure’ model was recently proposed hypothesizing that synaptic remodeling, associated with compensatory shifts in large-scale network configurations, is related to Aβ pathology in hubs of high connectivity during the preclinical disease phase and tau pathology within specific networks during clinical disease stages. The model thus predicts that large-scale network changes would be related to both Aβ and tau.82 This model would also suggest that pathology needs to be reduced predominantly in these hubs, a claim however that needs to be experimentally substantiated. Another open question is whether network activity can be restored to physiological levels (for example by using ultrasound as a neuromodulatory tool targeted to these hubs) and whether improved cognitive outcomes can be achieved without reducing levels of pathological Aβ and tau in these and outside of these hubs.
In the future, we foresee a combination of therapeutic strategies that combine Aβ reduction with strategies that are Aβ-independent and do target cognitive resilience. Demonstrating the translational value of preclinical research which found that a combination treatment with the antibody aducanumab and FUS+MB achieved increased Aβ clearance and improved cognition in mice,83,84 three AD patients who had received a FUS+MB treatment and escalating doses of aducanumab over six months showed massively enhanced amyloid plaque clearance as measured by fluorine-18 florbetaben PET.85 However, as discussed by us, the potential of such a combination strategy lies in the fact that here, the therapeutic benefits of ultrasound likely go beyond increased uptake of aducanumab (and amyloid clearance).86 In fact, each component of this combination approach has different biological effects, which suggests multiple routes to improved therapeutic outcomes. Ultrasound-induced BBBO facilitates uptake of not only the administered antibody but also a range of blood-borne factors, some of which could augment or suppress aducanumab-mediated Aβ clearance through effects on microglia. The net effect of blood-borne factors that enter the brain after BBBO on Aβ levels and other therapeutic bioeffects remain to be determined. Furthermore, ultrasound alone— without MBs— has neuromodulatory effects owing to the associated radiation force. Given that mechanosensitive receptors are present in all brain cell types as well as in the brain endothelial cells that form the BBB itself, one can envisage that the effects that ultrasound exerts on the brain are pleiotropic, whether or not BBBO occurs (i.e., amyloid is being cleared).86
In conclusion, these different studies implicate a dissociation of Aβ and cognition, with important implications for therapeutic interventions. They also illustrate the potential of combining pharmacological with non-pharmacological approaches and highlight the prospect of low-intensity ultrasound that is currently being explored in multiple clinical trials for the treatment of prevalent brain diseases including AD.
AUTHOR CONTRIBUTIONS
Jürgen Götz (Conceptualization; Writing – original draft); Gerhard Leinenga (Conceptualization; Writing – review & editing); Pranesh Padmanabhan (Conceptualization; Writing – review & editing).
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
We acknowledge support from the NHMRC (Investigator grant GNT1176326 and GNT2026929), the Alzheimer’s Association (Zenith, 22-AAIIA-965230), and the NHMRC/JPND (EU Joint Program in Neurodegeneration) AUTOFUS program.
CONFLICT OF INTEREST
Jürgen Götz is an Editorial Board Member of this journal but was not involved in the peer-review process of this article nor had access to any information regarding its peer-review.
All other authors have no conflict of interest to report.
REFERENCES
1. | Zhu X , Lee HG , Perry G and Smith MA . Alzheimer disease, the two-hit hypothesis: an update. Biochim Biophys Acta (2007) ; 1772: : 494–502. |
2. | Mondragon-Rodriguez S , Basurto-Islas G , Lee HG , et al. Causes versus effects: the increasing complexities of Alzheimer’s disease pathogenesis. Expert Rev Neurother (2010) ; 10: : 683–691. |
3. | Marlatt MW , Lucassen PJ , Perry G , et al. Alzheimer’s disease: cerebrovascular dysfunction, oxidative stress, and advanced clinical therapies. J Alzheimers Dis (2008) ; 15: : 199–210. |
4. | Terao I and Kodama W . Comparative efficacy, tolerability, and acceptability of donanemab, lecanemab, aducanumab, melatonin, and aerobic exercise for a short time on cognitive function in mild cognitive impairment and mild Alzheimer’s disease: a systematic review and network meta-analysis. J Alzheimers Dis (2024) ; 98: : 825–835. |
5. | Moller HJ and Graeber MB . The case described by Alois Alzheimer in 1911. Historical and conceptual perspectives based on the clinical record and neurohistological sections. Eur Arch Psychiatry Clin Neurosci (1998) ; 248: : 111–122. |
6. | Goedert M . Oskar Fischer and the study of dementia. Brain (2009) ; 132: : 1102–1111. |
7. | DeTure MA and Dickson DW . The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener (2019) ; 14: : 32. |
8. | Hardy J and Selkoe DJ . The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science (2002) ; 297: : 353–356. |
9. | Dujardin S , Colin M and Buee L . Invited review: Animal models of tauopathies and their implications for research/translation into the clinic. Neuropathol Appl Neurobiol (2015) ; 41: : 59–80. |
10. | Götz J , Bodea LG and Goedert M . Rodent models for Alzheimer disease. Nat Rev Neurosci (2018) ; 19: : 583–598. |
11. | Polanco JC , Li C , Bodea LG , et al. Amyloid-beta and tau complexity - towards improved biomarkers and targeted therapies. Nat Rev Neurol (2018) ; 14: : 22–39. |
12. | Shepherd A , Tyebji S , Hannan AJ , et al. Translational assays for assessment of cognition in rodent models of Alzheimer’s disease and dementia. J Mol Neurosci (2016) ; 60: : 371–382. |
13. | Padmanabhan P and Götz J . Clinical relevance of animal models in aging-related dementia research. Nat Aging (2023) ; 3: : 481–493. |
14. | Schenk D , Barbour R , Dunn W , et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature (1999) ; 400: : 173–177. |
15. | Jack CR Jr , Bennett DA , Blennow K , et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement (2018) ; 14: : 535–562. |
16. | Saito T , Matsuba Y , Mihira N , et al. Single App knock-in mouse models of Alzheimer’s disease. Nat Neurosci (2014) ; 17: : 661–663. |
17. | Zurita E , Chagoyen M , Cantero M , et al. Genetic polymorphisms among C57BL/6 mouse inbred strains. Transgenic Res (2011) ; 20: : 481–489. |
18. | Ji H , Pai AV , West CA , et al. Loss of resistance to angiotensin II-induced hypertension in the Jackson Laboratory recombination-activating gene null mouse on the C57BL/6J background. Hypertension (2017) ; 69: : 1121–1127. |
19. | Games D , Adams D , Alessandrini R , et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature (1995) ; 373: : 523–527. |
20. | Hsiao K , Chapman P , Nilsen S , et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science (1996) ; 274: : 99–102. |
21. | Rustay NR , Cronin EA , Curzon P , et al. Mice expressing the Swedish APP mutation on a 129 genetic background demonstrate consistent behavioral deficits and pathological markers of Alzheimer’s disease. Brain Res (2010) ; 1311: : 136–147. |
22. | Sturchler-Pierrat C , Abramowski D , Duke M , et al. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci U S A (1997) ; 94: : 13287–13292. |
23. | Ittner LM , Ke YD , Delerue F , et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell (2010) ; 142: : 387–397. |
24. | Gimenez-Llort L , Marin-Pardo D , Marazuela P , et al. Survival bias and crosstalk between chronological and behavioral age: age- and genotype-sensitivity tests define behavioral signatures in middle-aged, old, and long-lived mice with normal and AD-associated aging. Biomedicines (2021) ; 9: : 636. |
25. | Congdon EE and Sigurdsson EM . Tau-targeting therapies for Alzheimer disease. Nat Rev Neurol (2018) ; 14: : 399–415. |
26. | Chen L , Cruz E , Oikari LE , et al. Opportunities and challenges in delivering biologics for Alzheimer’s disease by low-intensity ultrasound. Adv Drug Deliv Rev (2022) ; 189: : 114517. |
27. | Morgan D , Diamond DM , Gottschall PE , et al. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature (2000) ; 408: : 982–985. |
28. | Janus C , Pearson J , McLaurin J , et al. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature (2000) ; 408: : 979–982. |
29. | Arendash GW , Gordon MN , Diamond DM , et al. Behavioral assessment of Alzheimer’s transgenic mice following long-term Abeta vaccination: task specificity and correlations between Abeta deposition and spatial memory. DNA Cell Biol (2001) ; 20: : 737–744. |
30. | Rasool S , Martinez-Coria H , Wu JW , et al. Systemic vaccination with anti-oligomeric monoclonal antibodies improves cognitive function by reducing Abeta deposition and tau pathology in 3xTg-AD mice. J Neurochem (2013) ; 126: : 473–482. |
31. | Crehan H , Liu B , Kleinschmidt M , et al. Effector function of anti-pyroglutamate-3 Abeta antibodies affects cognitive benefit, glial activation and amyloid clearance in Alzheimer’s-like mice. Alzheimers Res Ther (2020) ; 12: : 12. |
32. | Hillen H , Barghorn S , Striebinger A , et al. Generation and therapeutic efficacy of highly oligomer-specific beta-amyloid antibodies. J Neurosci (2010) ; 30: : 10369–10379. |
33. | Sevigny J , Chiao P , Bussiere T , et al. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature (2016) ; 537: : 50–56. |
34. | Budd Haeberlein S , Aisen PS , Barkhof F , et al. Two randomized phase 3 studies of aducanumab in early Alzheimer’s disease. J Prev Alzheimers Dis (2022) ; 9: : 197–210. |
35. | Salazar SV and Strittmatter SM . Cellular prion protein as a receptor for amyloid-beta oligomers in Alzheimer’s disease. Biochem Biophys Res Commun (2017) ; 483: : 1143–1147. |
36. | Roberson ED , Scearce-Levie K , Palop JJ , et al. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science (2007) ; 316: : 750–754. |
37. | Ittner LM , Götz J . Amyloid-beta and tau - a toxic pas de deux in Alzheimer’s disease. Nat Rev Neurosci (2011) ; 12: : 65–72. |
38. | Bi M , Gladbach A , van Eersel J , et al. Tau exacerbates excitotoxic brain damage in an animal model of stroke. Nat Commun (2017) ; 8: : 473. |
39. | Shao E , Chang CW , Li Z , et al. Tau ablation in excitatory neurons and postnatal tau knockdown reduce epilepsy, SUDEP, and autism behaviors in a Dravet syndrome model. Sci Transl Med (2022) ; 14: : eabm5527. |
40. | Chang CW , Evans MD , Yu X , et al. Tau reduction affects excitatory and inhibitory neurons differently, reduces excitation/inhibition ratios, and counteracts network hypersynchrony. Cell Rep (2021) ; 37: : 109855. |
41. | Peters F , Salihoglu H , Pratsch K , et al. Tau deletion reduces plaque-associated BACE1 accumulation and decelerates plaque formation in a mouse model of Alzheimer’s disease. EMBO J (2019) ; 38: : e102345. |
42. | Bloom GS . Amyloid-beta and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol (2014) ; 71: : 505–508. |
43. | Haass C and Mandelkow E . Fyn-tau-amyloid: a toxic triad. Cell (2010) ; 142: : 356–358. |
44. | Cruz E , Nisbet RM , Padmanabhan P , et al. Proteostasis as a fundamental principle of Tau immunotherapy. bioRxiv (2024) ; doi: https://doi.org/10.1101/2024.02.12.580007 [Preprint]. Posted February 14, 2024. |
45. | Li C and Götz J . Somatodendritic accumulation of tau in Alzheimer’s disease is promoted by Fyn-mediated local protein translation. EMBO J (2017) ; 36: : 3120–3138. |
46. | Evans HT , Benetatos J , van Roijen M , et al. Decreased synthesis of ribosomal proteins in tauopathy revealed by non-canonical amino acid labelling. EMBO J (2019) ; 38: : e101174. |
47. | Padmanabhan P , Martinez-Marmol R , Xia D , et al. Frontotemporal dementia mutant Tau promotes aberrant Fyn nanoclustering in hippocampal dendritic spines. Elife (2019) ; 8: : e45040. |
48. | Padmanabhan P , Kneynsberg A , Cruz E , et al. Single-molecule imaging reveals Tau trapping at nanometer-sized dynamic hot spots near the plasma membrane that persists after microtubule perturbation and cholesterol depletion. EMBO J (2022) ; 41: : e111265. |
49. | Martinez-Marmol R , Small C , Jiang A , et al. Fyn nanoclustering requires switching to an open conformation and is enhanced by FTLD-Tau biomolecular condensates. Mol Psychiatry (2023) ; 28: : 946–962. |
50. | Longfield SF , Mollazade M , Wallis TP , et al. Tau forms synaptic nano-biomolecular condensates controlling the dynamic clustering of recycling synaptic vesicles. Nat Commun (2023) ; 14: : 7277. |
51. | Santos N , Segura L , Lewis A , et al. Multiscale modeling of macromolecular interactions between tau-amylin oligomers and asymmetric lipid nanodomains that link Alzheimer’s and diabetic diseases. Molecules (2024) ; 29: : 740. |
52. | Briner A , Götz J and Polanco JC . Fyn kinase controls tau aggregation in vivo. Cell Rep (2020) ; 32: : 108045. |
53. | Liu G , Fiock KL , Levites Y , et al. Fyn depletion ameliorates tau(P301L)-induced neuropathology. Acta Neuropathol Commun (2020) ; 8: : 108. |
54. | Stancu IC , Ferraiolo M , Terwel D , et al. Tau interacting proteins: gaining insight into the roles of tau in health and disease. Adv Exp Med Biol (2019) ; 1184: : 145–166. |
55. | Nygaard HB . Targeting Fyn kinase in Alzheimer’s disease. Biol Psychiatry (2018) ; 83: : 369–376. |
56. | Mummery CJ , Borjesson-Hanson A , Blackburn DJ , et al. Tau-targeting antisense oligonucleotide MAPT(Rx) in mild Alzheimer’s disease: a phase 1b, randomized, placebo-controlled trial. Nat Med (2023) ; 29: : 1437–1447. |
57. | Meng Y , Hynynen K and Lipsman N . Applications of focused ultrasound in the brain: from thermoablation to drug delivery Nat Rev Neurol (2021) ; 17: : 7–22. |
58. | Blackmore DG , Razansky D and Götz J . Ultrasound as a versatile tool for short- and long-term improvement and monitoring of brain function. Neuron (2023) ; 111: : 1174–1190. |
59. | Hynynen K , McDannold N , Vykhodtseva N , et al. Noninvasive MR imaging-guided focal opening of the blood-brain barrier in rabbits. Radiology (2001) ; 220: : 640–646. |
60. | Pandit R , Koh WK , Sullivan RKP , et al. Role for caveolin-mediated transcytosis in facilitating transport of large cargoes into the brain via ultrasound. J Control Release (2020) ; 327: : 667–675. |
61. | Memari E , Hui F , Yusefi H , et al. Fluid flow influences ultrasound-assisted endothelial membrane permeabilization and calcium flux. J Control Release (2023) ; 358: : 333–344. |
62. | Nisbet RM , van der Jeugd A , Leinenga G , et al. Combined effects of scanning ultrasound and a tau-specific single chain antibody in a tau transgenic mouse model. Brain (2017) ; 140: : 1220–1230. |
63. | Uribe Cardenas R , Laramee M , Ray I , et al. Influence of focused ultrasound on locoregional drug delivery to the brain: Potential implications for brain tumor therapy. J Control Release (2023) ; 362: : 755–763. |
64. | Kofoed RH , Dibia CL , Noseworthy K , et al. Efficacy of gene delivery to the brain using AAV and ultrasound depends on serotypes and brain areas. J Control Release (2022) ; 351: : 667–680. |
65. | Leinenga G and Götz J . Scanning ultrasound removes amyloid-beta and restores memory in an Alzheimer’s disease mouse model. Sci Transl Med (2015) ; 7: : 278ra33. |
66. | Götz J , Richter-Stretton G and Cruz E . Therapeutic ultrasound as a treatment modality for physiological and pathological ageing including Alzheimer’s disease. Pharmaceutics (2021) ; 13: : 1002. |
67. | Menz MD , Ye P , Firouzi K , et al. Radiation force as a physical mechanism for ultrasonic neurostimulation of the ex vivo retina. J Neurosci (2019) ; 39: : 6251–6264. |
68. | Yoo S , Mittelstein DR , Hurt RC , et al. Focused ultrasound excites cortical neurons via mechanosensitive calcium accumulation and ion channel amplification. Nat Commun (2022) ; 13: : 493. |
69. | Blackmore DG , Turpin F , Palliyaguru T , et al. Low-intensity ultrasound restores long-term potentiation and memory in senescent mice through pleiotropic mechanisms including NMDAR signaling. Mol Psychiatry (2021) ; 26: : 6975–6991. |
70. | Yu K , Niu X , Krook-Magnuson E , et al. Intrinsic functional neuron-type selectivity of transcranial focused ultrasound neuromodulation. Nat Commun (2021) ; 12: : 2519. |
71. | Murphy KR , Farrell JS , Gomez JL , et al. A tool for monitoring cell type-specific focused ultrasound neuromodulation and control of chronic epilepsy. Proc Natl Acad Sci U S A (2022) ; 119: : e2206828119. |
72. | Leinenga G , Bodea LG , Schröder J , et al. Transcriptional signature in microglia isolated from an Alzheimer’s disease mouse model treated with scanning ultrasound Bioeng Transl Med (2022) ; 8: : e10329. |
73. | Leinenga G , Koh WK and Götz J . Scanning ultrasound in the absence of blood-brain barrier opening is not sufficient to clear beta-amyloid plaques in the APP23 mouse model of Alzheimer’s disease. Brain Res Bull (2019) ; 153: : 8–14. |
74. | Hatch RJ , Leinenga G and Götz J . Scanning ultrasound (SUS) causes no changes to neuronal excitability and prevents age-related reductions in hippocampal CA1 dendritic structure in wild-type mice. PloS One (2016) ; 11: : e0164278. |
75. | Blackmore DG , Turpin F , Mohamed AZ , et al. Multimodal analysis of aged wild-type mice exposed to repeated scanning ultrasound treatments demonstrates long-term safety. Theranostics (2018) ; 8: : 6233–6247. |
76. | Leinenga G , To XV , Bodea LG , et al. Scanning ultrasound-mediated memory and functional improvements do not require amyloid-beta reduction. Mol Psychiatry (2024) ; doi: 10.1038/s41380-024-02509-5. |
77. | Oh H , Razlighi QR and Stern Y . Multiple pathways of reserve simultaneously present in cognitively normal older adults. Neurology (2018) ; 90: : e197–e205. |
78. | Stern Y , Barnes CA , Grady C , et al. Brain reserve, cognitive reserve, compensation, and maintenance: operationalization, validity, and mechanisms of cognitive resilience. Neurobiol Aging (2019) ; 83: : 124–129. |
79. | Jeong H , Song IU , Chung YA , et al. Short-term efficacy of transcranial focused ultrasound to the hippocampus in Alzheimer’s disease: a preliminary study. J Pers Med (2022) ; 12: : 250. |
80. | Ahmed Z , Cooper J , Murray TK , et al. A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: the pattern of spread is determined by connectivity, not proximity. Acta Neuropathol (2014) ; 127: : 667–683. |
81. | Zott B , Busche MA , Sperling RA , et al. What happens with the circuit in Alzheimer’s disease in mice and humans? Annu Rev Neurosci (2018) ; 41: : 277–297. |
82. | Jones DT , Knopman DS , Gunter JL , et al. Cascading network failure across the Alzheimer’s disease spectrum. Brain (2016) ; 139: : 547–562. |
83. | Leinenga G , Koh WK and Götz J . A comparative study of the effects of Aducanumab and scanning ultrasound on amyloid plaques and behavior in the APP23 mouse model of Alzheimer disease. Alzheimers Res Ther (2021) ; 13: : 76. |
84. | Kong C , Yang EJ , Shin J , et al. Enhanced delivery of a low dose of aducanumab via FUS in 5xFAD mice, an AD model. Transl Neurodegener (2022) ; 11: : 57. |
85. | Rezai AR , D’Haese PF , Finomore V , et al. Ultrasound blood-brain barrier opening and aducanumab in Alzheimer’s disease. N Engl J Med (2024) ; 390: : 55–62. |
86. | Götz J and Padmanabhan P . Ultrasound and antibodies - a potentially powerful combination for Alzheimer disease therapy. Nat Rev Neurol (2024) ; 20: : 257–258. |


