
Harnessing Passive Pulsatile Shear Stress for Alzheimer’s Disease Prevention and Intervention
Abstract
Alzheimer’s disease (AD) affects more than 40 million people worldwide and is the leading cause of dementia. This disease is a challenge for both patients and caregivers and puts a significant strain on the global healthcare system. To address this issue, the Lancet Commission recommends focusing on reducing modifiable lifestyle risk factors such as hypertension, diabetes, and physical inactivity. Passive pulsatile shear stress (PPSS) interventions, which use devices like whole-body periodic acceleration, periodic acceleration along the Z-axis (pGz), and the Jogging Device, have shown significant systemic and cellular effects in preclinical and clinical models which address these modifiable risks factors. Based on this, we propose that PPSS could be a potential non-pharmacological and non-invasive preventive or therapeutic strategy for AD. We perform a comprehensive review of the biological basis based on all publications of PPSS using these devices and demonstrate their effects on the various aspects of AD. We draw from this comprehensive analysis to support our hypothesis. We then delve into the possible application of PPSS as an innovative intervention. We discuss how PPSS holds promise in ameliorating hypertension and diabetes while mitigating physical inactivity, potentially offering a holistic approach to AD prevention and management.
INTRODUCTION
Alzheimer’s disease (AD) is the leading cause of dementia and one of the most expensive, lethal, and burdensome diseases. In 2020, it was estimated that more than 6 million people live with clinical AD in the United States (US), and based on population increases, this number will more than double to nearly 14 million by 2060 [1]. Globally, it has been estimated that between 1990 and 2016 the number of people living with dementia doubled from 20.2 to 43.8 million worldwide and is expected to surpass 150 million people by 2050 [2, 3]. The global economic burden of AD is estimated to be at least $9.1 trillion by 2050 [4]. There is no doubt that AD represents a significant challenge in healthcare for both patients and caregivers. In 2019 the World Health Organization (WHO) released guidelines for the reduction of the risk of cognitive decline and dementia, which include lifestyle interventions such as physical activity, diet, and reduction in obesity, tobacco and alcohol use, hypertension, and diabetes. Physical activity should be recommended to adults with normal cognition to reduce the risk of cognitive decline, and a conditional recommendation that physical activity may be recommended to adults with mild cognitive impairment (MCI) to reduce the risk of further cognitive decline [5]. Recently, the Lancet Commission published 12 steps to enhance cognitive reserves (CR), which refer to individual differences in how tasks are performed that may allow some people to be more resilient than others. CR indicates resilience to neuropathological damage and the way the brain uses its damaged resources. It could be defined as the ability to optimize or maximize performance through differential recruitment of brain networks and/or alternative cognitive strategies and risk reduction for cognitive decline; minimize diabetes, treat hypertension, prevent head injury, stop smoking, reduce air pollution, reduce midlife obesity, maintain frequent exercises, reduce the occurrence of depression, avoid excessive alcohol, treat hearing impairment, maintain frequent social contact, attain high levels of education [6]. The Lancet Commission estimates that 40% of dementia cases could be prevented or delayed by addressing 12 known lifestyle risk factors [6].
Unfortunately, the challenge of physical inactivity persists on a global scale, with an estimated cost of nearly INT$54 billion worldwide and is classified as a pandemic [7, 8]. Diabetes, another global concern, currently affects 537 million adults as of 2021 and is projected to rise to 783 million by 2045 [9]. Hypertension, a major risk factor for coronary heart disease and stroke, is prevalent among adults in the US, affecting approximately 43.7% of those aged 20 and older in 2019, with an associated annual cost of $51 billion [10]. Reducing physical inactivity, diabetes, and hypertension represents a vital step toward preventing overall cognitive decline, as emphasized by the Lancet Commission [6].
An intervention that can concurrently address physical inactivity, diabetes, and hypertension while having a cellular and physiological basis to reduce inflammation, enhance cerebral blood flow (CBF), improve calcium homeostasis, promote neurotrophin expression, and increase endothelial nitric oxide production would be a valuable addition to the arsenal against AD.
AD is characterized by hallmark pathological features, namely ‘plaques’ and ‘tangles.’ Plaques comprise clumps of amyloid protein, which leads to the death of brain cells, while tangles result from aberrant polymerization of tau protein, culminating in the destruction of neural connections. The precise pathophysiology of AD remains incompletely elucidated. Current understanding of the disease’s preclinical phase highlights neuroinflammation, vascular changes, aging, compromised neuronal calcium handling, glymphatic system dysfunction, and amyloid-β (Aβ) accumulation as contributing factors [3]. Notably, the multifaceted nature of AD’s pathophysiological pathways makes it challenging to rely solely on a single pharmacological approach for effective treatment or reversal, leading to numerous failed clinical trials [11, 12]. The current consensus supports a multimodal and multifactorial therapeutic approach, emphasizing the importance of reducing diabetes, hypertension, and increasing physical activity [13].
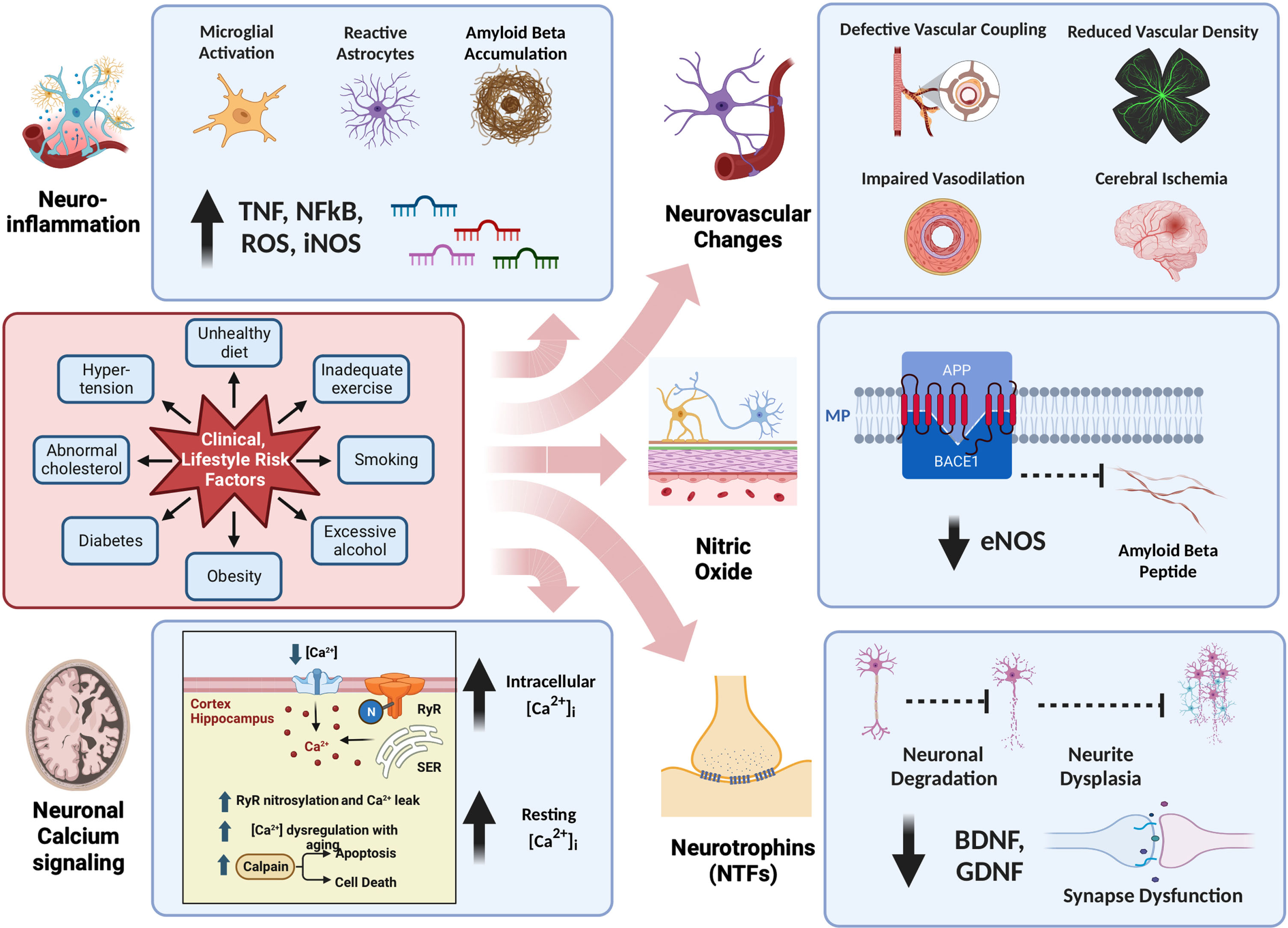
In this review, we will delve into crucial pathophysiological aspects of AD and subsequently explore a novel non-pharmacologic intervention, passive pulsatile shear stress (PPSS), as a potential preventive and therapeutic strategy for AD. Figure 1 provides a simplified and current summary of the pathophysiological aspects of AD, as discussed in this review.
Fig. 1
Pathophysiology of Alzheimer’s Disease. This figure offers a comprehensive overview of the intricate pathophysiology of Alzheimer’s disease, organized into five major categories: a) Neuroinflammation, b) Neuronal Calcium Signaling, c) Neurovascular Changes, d) Nitric Oxide, and e) Neurotrophins. Notably, Lifestyle Risk Factors exert a significant influence on all or some of these five categories, underscoring the multifaceted nature of their impact on Alzheimer’s disease.
INFLAMMATION AND NEUROINFLAMMATION IN AD
Neuroinflammation is a complex process, intended acutely as a host defense mechanism to protect and restore structural and functional integrity of the brain against infections, injury, or toxic metabolites with activation of the innate immune system via glial cells and blood-born immune cells. Chronic inflammation disrupts the equilibrium of brain repair and the pro-inflammatory phenotype, leading to neuronal damage. Cellular involvement in neuroinflammation includes microglia, astrocytes, T cells, and mast cells, which release pro-inflammatory molecules. The main source of cytokines in AD are microglia and astrocytes. Additionally, these two cells are also responsible for the production of high concentrations of nitric oxide (NO) via inducible nitric oxide synthase (iNOS) and a source of reactive oxygen species (ROS) and peroxynitrites, which are toxic to neurons. An important orchestrator of the inflammatory response is nuclear factor κ β (NF-κ β) which promotes the transcription of many proinflammatory target genes. Furthermore, in AD brain Toll-like receptors (TLRs) are overexpressed in microglia and neurons, and TLRs activate NF-κ β signaling pathways, with subsequent release of cytokines and chemokines [14]. Neuroinflammation appears to be present in the early stages of AD [15–17]. The relationship between neuroinflammation and accumulation of Aβ has been reviewed by others, but summarized in a model in which chronic background inflammation provides an initial stimulus resulting in microglial priming followed by a protective wave of microglial activation and Aβ deposition. The ineffective clearance of Aβ and tau aggregation alters microglial defense and produces an exaggerated microglial response in the later stages of AD [18]. Reviews of neuroinflammation related to AD have recently been published [15–19].
CEREBRAL BLOOD FLOW AND NEUROVASCULAR ALTERATIONS IN AD
Vascular aging, defined as age-related changes in the vasculature, plays an important role in AD. Vascular alterations are present in more than 50% of clinically diagnosed AD cases, with reduced microvascular density (vascular rarefaction) present in 90% of AD brains. Furthermore, decreased CBF has been described in AD [20–23] and observed in the preclinical stages of AD. In addition, vascular dysfunction is an early manifestation of Aβ accumulation [21].
The brain receives 1/5 of cardiac output and consumes 1/5 of the body’s oxygen and glucose, delivered by CBF [22, 23]. The coupling of this metabolic demand and CBF is known as neurovascular coupling, which ensures a rapid increase in CBF to activated areas of the brain [20]. The neurovascular unit includes astrocytes, vascular smooth muscle cells, pericytes, and endothelial cells that contribute to this neurovascular coupling, and NO has been shown to play an important role in neurovascular coupling [24, 25]. Neurovascular coupling dysfunction in AD has previously been reviewed [22, 26]. In addition to dysfunction of the neurovascular coupling in AD, concomitant factors such as aging, hypertension, obesity, metabolic syndrome, diabetes, obstructive sleep apnea (with intermittent hypoxemia), and depression also contribute to cerebral hypoperfusion (decreased CBF) and glucose hypometabolism to promote AD [27]. A reduction in baseline CBF of <10–20% is reported in various regions of the brain in patients with AD, with studies showing a correlation between the severity of hypoperfusion and cognitive impairment [28].
CALCIUM SIGNALING IN AD
Calcium modulates many neural processes, including synaptic plasticity, intracellular cell signaling, and apoptosis. Neurons modulate intracellular [Ca2+] ([Ca2+]i) by regulating Ca2+ influx/efflux through ion channels, ATPase pumps, and ion exchangers in the cellular membrane and its release from internal sources such as the endoplasmic reticulum [29]. The pathogenesis of Alzheimer’s disease is complex and involves many alterations in molecular and cellular signaling pathways. However, there is growing evidence that sustained disturbances in intracellular Ca2+ homeostasis are a critical factor for neurodegenerative diseases, including AD, giving rise to the calcium hypothesis of AD [29–32].
In support of the Ca2+ hypothesis of AD, we demonstrated for the first time in adult neurons from 3xTg-AD mice and single transgenic mice expressing the Swedish mutation of human amyloid precursor protein a significant elevation of resting [Ca2+]i compared to control mice of the same age. Intracellular Ca2+ dysfunction that appears to be mediated by a Ca2+ influx through a nifedipine and SKF-sensitive Ca2+ pathways, as well as Ca2+ efflux from the endoplasmic reticulum through the inositol trisphosphate receptors [33]. A review of Ca2+ signaling and regulation in AD has recently been published [34, 35].
NEUROTROPHINS IN AD
Neurotrophic factors (NTFs) are endogenous proteins that promote or activate neuronal repair genes. NTFs control neuronal survival, migration, neurite outgrowth, synapse formation, and neuronal plasticity [36]. Brain-derived neurotrophic factor (BDNF) and glial-derived neurotrophic factor (GDNF). Each belongs to one of the two families of NTFs. The altered expression of NTFs (BDNF, GDNF, and nerve growth factor, NGF) correlates with the neurodegeneration process and cognitive impairment in AD [37–39].
BDNF is highly expressed in the central nervous system in both neurons and glia, in the hippocampus, cerebral cortex, hypothalamus, and amygdala, all regions of the brain involved in learning and memory processes. BDNF regulates long-term potentiation, synaptic plasticity, sprouting, and neuronal differentiation, and is critical in learning and memory processes. Some studies have reported reduced levels of BDNF mRNA and protein in postmortem brains of AD patients [40, 41], serum and brain of AD patients and a transgenic mouse model of tauopathy [42]. Gene transfer of BDNF into the cortex in APP transgenic mice and aged rats produced an increase in BDNF protein levels in the hippocampus and improved spatial learning [42–45].
GDNF is also highly expressed in the striatum, substantia nigra, and skeletal muscle, as well as other regions of the brain and peripheral tissue. GDNF is a promoter of neuronal survival in both the central and peripheral nervous system and has effects on hippocampal and cerebellar neurons, noradrenergic, serotoninergic and cholinergic neurons [46]. GDNF was suggested to be negatively regulated in a transgenic mice model of AD, and physical exercise and overexpression of GDNF improved learning and memory in this model [47, 48].
Recently, Nasrolahi et al. have thoroughly reviewed preclinical and clinical studies of NTFs to treat AD, concluding that NTFs are promising candidates for the treatment of AD. Limitations for clinical application of NTFs such, as delivery across the blood-brain barrier, short half-lives, and possible side effects, can potentially be resolved in the future with gene therapy vectors and biomaterial-based drug delivery systems [49].
Physical exercise is a method that has been shown to increase BDNF levels [50–54]. Ribeiro et al. reviewed circulating levels of BDNF in animal and human studies that performed training protocols from 4 to 64 weeks. They concluded that while animal model data on the relationship between long-term aerobic exercise and BDNF is unquestionable, human data on long-term aerobic exercise and circulating BDNF are more ambiguous [53].
NITRIC OXIDE IN AD
NO is a gas involved in a multitude of functions, such as vascular dilation, cytoprotection or cytotoxicity, second messenger, pro- and anti-inflammatory effects among some of the essential functions. NO is synthesized by the conversion of l-arginine and oxygen to citrulline using one of the three NO synthases (NOS). Endothelial nitric oxide synthase (eNOS) and neuronal nitric oxide synthase (nNOS) are constitutively expressed enzymes that require NADPH, calcium, and tetrahydrobiopterin (BH4). eNOS is found mainly in endothelial cells, whereas nNOS is found mainly in neuronal tissue. nNOS and eNOS both produce NO at low nanomolar concentrations. In contrast, iNOS is not constitutively expressed or calcium-calmodulin dependent, produced primarily by neutrophils, and macrophages, in very large micromolar quantities. The overall effect of NO depends on the concentration of NO. While the nanomolar concentration provides a prosurvival and anti-inflammatory effect, large concentrations, as produced by iNOS, are pro-death due to the radical activity of NO. Thus, NO at low nanomolar amounts is neuroprotective, and at high micromolar quantities, it is cytodestructive. NO signaling occurs via soluble guanylyl cyclase signaling or S-nitrosylation of proteins. Under pathological conditions, high concentrations of NO produce reactive nitrogen and oxygen species (RNS, ROS) with damaging peroxynitrites [55, 56].
The vascular endothelium participates in local control of blood flow and is critical for maintaining homeostasis and cytoprotection. NO produced via eNOS (eNO) is the most important vasodilator mechanism for control of blood flow, while decreasing platelet aggregation, inflammation, cell adhesion, and endothelial cell activation. The proper subcellular localization of eNOS ensures optimal regulation by mechanical forces (shear stress or pressure gradients), calcium ions, and kinases. Therefore, any agonist that mobilizes intracellular calcium (VEGF, bradykinin, histamine, etc.) or alterations in intracellular signaling pathways that lead to enhanced calmodulin binding or reduced calmodulin dissociation has the potential to promote eNOS activity and NO release. Furthermore, eNOS control occurs at both the transcriptional and post-transcriptional levels [57].
The endothelial cell experiences a tangential frictional force created by blood flow (laminar shear); the latter produces a quiescent endothelium phenotype, which maintains non-inflammatory, cytoprotective, and vasoprotective properties. Pulsatile shear stress (PSS) is produced by the addition of a heartbeat to normal laminar blood flow. Pulsatile shear stress is cytoprotective and upregulates genes responsible for enhanced mitochondrial biogenesis and function, increase in eNOS-derived NO bioavailability, reduction in endothelial cell proliferation, inflammation, and cytoprotection [58–61]. Furthermore, both laminar and pulsatile shear stress induce the expression of eNOS and are critical for maintaining vasodilatation through eNO.
Nitric oxide has been shown to play an important role in AD [62–65]. eNO participates in the control of amyloid-β protein precursor (AβPP) metabolism [66]. The β-site amyloid precursor protein cleaving enzyme 1 (BACE1), is a membrane-bound protease that cleaves various substrates, particularly AβPP, to generate Aβ. BACE1 and Aβ levels in the brain and plasma Aβ are involved in AD [67], and eNO inhibits AβPP expression and the processing of AβPP to Aβ [68–71]. Furthermore, eNO has been shown to have neurovascular protective functions that have previously been reviewed [66]. In mouse models, partial deficiency of eNOS exacerbates cognitive deficit and amyloid pathology [72], and has also been shown to increase a pro-inflammatory phenotype in the brain [70]. In contrast, high concentrations of NO produced by iNOS, produce a massive generation of peroxynitrites, oxidative stress, and NOS uncoupling with nitration of Aβ and toxicity to neurons, producing synaptic transmission impairment [19]. Furthermore, elevated levels of NO, as occurs with iNOS, result in post-translational modification of proteins associated with mitochondrial dysfunction and bioenergetic compromise [73]. In subjects with mild AD, treatment with medications that augment the eNOS pathway are associated with increased CBF and improved cognitive performance [74]. Furthermore, loss of eNO could significantly contribute to the initiation of the progression of cognitive decline, and genetic inactivation of eNOS causes microglial activation and promotes an inflammatory phenotype of the brain [69, 70, 75]. Jeynes et al. showed a negative correlation between positive eNOS staining capillaries and neurofibrillary tangles and senile plaques in histological specimens of brain cortices from AD subjects [76].
PASSIVE PULSATILE SHEAR STRESS AS PRODUCED BY WHOLE BODY PERIODIC ACCELERATION AND NON-INVASIVE PASSIVE JOGGING DEVICE
Pulsations to the vasculature occur with each heartbeat. In elegant experiments carried out by Hutchenson et al., they found that EC produced NO as a function of pulsatility with an optimal frequency of 2–8 Hz (120–480 cpm) [77]. The addition of pulsations beyond the basal heart rate can be produced by active or passive means. The prototype of active PSS is walking/jogging or running. The typical cadence of running, which adds pulses to the circulation in male and female recreational runners, is between 163 and 169 steps per min [78]. This pulsation frequency occurs as the foot hits the ambulating surface and is not timed with the individual’s heart rate. PPSS can be performed by adding external pulsations to the circulation, such as occurs with Whole Body Periodic Acceleration (WBPA, a.k.a. pGz). WBPA is the motion of the supine body in the head to the caudal direction that produces external motion of the abdomen and adds pulses to the circulation. The typical frequency of WBPA in humans is 100–150 cycles per min (1.6 to 2.5 Hz), and the acceleration forces in the Z plane (head to toe) of Gz±0.3 mt/s2 [79]. This motion adds pulsations to the circulation that are not timed to the cardiac cycle. WBPA is a nonportable device designed as a bed with dimensions of approximately 91 cm wide and 182 cm long (36×72 in), weighing around 120 kilograms (300 lbs). Its nonportability and weight necessitate a dedicated space. Most individuals, ranging from 1 to 90 years old, tolerate the bed’s movement well and can stay on it for more than 1 h, often falling asleep during the process. However, a small percentage (less than 5%) may find the movement intolerable due to anxiety or perceived dizziness. Subjects who use WBPA should refrain from eating or drinking 1–2 h before use to avoid discomfort caused by abdominal fluid movement. After at least 30 min of WBPA, subjects commonly report the need to urinate (which is likely the result of increased renal blood flow, as we have shown in our microvascular flow studies) [80]. Unlike traditional exercise, WBPA is a passive activity that requires minimal assistance to access the bed. It has been used in various health conditions, including Parkinson’s disease (PD), AD, cardiovascular diseases, diabetes, and postoperative recovery. The cost of a WBPA device typically ranges from $3,000 to $10,000 (€ 2,777 to € 9,259). Because the WBPA is a large heavy, nonportable bed-like device, a portable predicate device was engineered, which also adds external pulses to the circulation by the alternating motion of the feet on a ‘jogging device’ (JD, Gentle Jogger, Movewell Technologies).
The JD produces passive upward alternating motion of the feet on a pedal, and with each downward motion, the forefoot gently strikes a bumper simulating jogging or running. It achieves passive foot movement simulating walking or jogging, providing pulsatile shear stress at a rate of 120 to 190 steps per min, and can be performed in a seated or supine posture. JD is simple to operate, portable, and can be performed by anyone regardless of physical or cognitive limitations [79]. The device is significantly lighter, weighing less than 10 lbs, with dimensions of 34× 35×10 cm (13× 14× 4 in). It is more affordable, costing less than $1,000 (€ 925). Although generally well tolerated, hyperactive individuals may find it difficult to remain seated or supine for extended periods. The device is applied locally to the feet, but its effects are systemic [81]. Like WBPA, JD has been applied across various health conditions. Its portability allows usage under a desk, during work, while watching television, or even during meals. In a recent study, home use compliance of JD was high, with only one out of 21 subjects being noncompliant. Most subjects used the device for more than the recommended 90 min per day [82].
To provide context on the status of WBPA and JD in human subjects, a summary of relevant clinical data is essential. Fourteen clinical trials have been reported involving WBPA, involving more than 250 adult subjects aged 18 to 90 years. No adverse events have been reported in any of these trials. Similarly, JD has been examined in seven clinical trials with more than 140 subjects, and no adverse events were observed.
In particular, three trials focused on WBPA in patients with PD. In one instance, WBPA was administered for 10 sessions of 45 min each, with pre- and post-assessments of motor symptoms. Sixty-six percent of the subjects exhibited significant improvements in motor activity. Another study involving a single 45-min WBPA session in patients with PD revealed positive outcomes, including increased flexibility, reduced shuffling gait, decreased or eliminated freezing of gait, improved control of turning and balance, clearer speech articulation, and improved facial appearance [83]. In a separate investigation, 13 patients with PD underwent 12 45-min sessions of WBPA, resulting in improved sleep quality, decreased depressive symptoms, increased daily activity and well-being, and a reduction in systolic blood pressure [84]. Although clinical trials specifically for JD and PD are lacking, anecdotal reports suggest that JD may produce similar results.
We hypothesize that passive pulsatile shear stress may be a preventive or therapeutic strategy for AD. In support of the hypothesis, we review the beneficial effects of WBPA or JD based on published data on each of the aforementioned and point to its clinical utility as a multimodal approach to AD.
CEREBRAL BLOOD FLOW AND WBPA
In an animal model, regional microvascular blood flow has been measured in anesthetized swine under non-stressed conditions and during and after whole body ischemia-reperfusion injury. Under unstressed conditions, 30 min of pGz increased regional cortical blood flow by 200% of baseline values, and brain stem blood flow by 200% of baseline values [80]. Whole body ischemia-reperfusion injury reduces both cortical and brain stem regional blood flows to 13 and 10% respectively from baseline levels. pGz performed during reperfusion of whole-body ischemia (cardiac arrest), significantly increased cortical blood and regional blood flows from the brain stem by 167% and 94% of baseline values [85]. WBPA has been performed as preconditioning stimuli 1 h prior to reperfusion injury from total body ischemia reperfusion injury, 2 h after reperfusion, regional cortical blood flow remained higher than the values for non-preconditioned animals (184±12 versus 130±8 ml/min/100 g of tissue, preconditioned WBPA versus non preconditioned) [86]. Additionally, WBPA performed as a delayed post-conditioning strategy (beginning 30 min after reperfusion) also significantly increased regional blood flow in the cortex and brainstem by 40% compared to non-postconditioned animals [87]. Collectively, these animal studies confirm the increase in cerebral microvascular blood flow induced by WBPA.
CALCIUM SIGNALING IN THE BRAIN AND WBPA
We have reported an age-dependent increase in neuronal [Ca2+]i in mice. We examined the regulation of [Ca2+]i in cortical (in vivo) and hippocampal (in vitro) neurons from young, middle-aged, and aged mice. We found a progressive age-related elevation of [Ca2+]i in cortical (in vivo) and hippocampal (in vitro) neurons associated with increased hippocampal neuronal calpain activity and reduced cell viability, which was associated with age-related cognitive decline [88]. Our laboratory has also described an age-dependent elevation of [Ca2+]i, in both cortical and hippocampal neurons from the mouse model of Duchenne Muscular Dystrophy (mdx), associated with cognitive deficits [89]. Treatment with WBPA/pGz in this mdx mouse model for 8 days (1 h per day), reduced cortical [Ca2+]i overload, reduced oxidative stress, improved neuronal viability, and cognitive decline [90]. These findings support the hypothesis that intracellular Ca2+ dyshomeostasis is a major mechanism underlying cognitive deficits seen in both normal aging and degenerative neurologic diseases, and the reduction in [Ca2+]I by WBPA/pGz.
INFLAMMATION AND WBPA
The whole body inflammatory response to WBPA has been studied in a mouse model of lipopolysaccharide E. coli endotoxin-induced sepsis like (LPS) [91]. This model uses a lethal dose of LPS (40 mg/kg) with 100% mortality at 24 h in control mice. WBPA performed as a pretreatment intervention (3 days before LPS) or after treatment (30 min after LPS for 1 h) significantly improves survival at 24 h from 0% to 75% and 100% before and after WBPA treatments, respectively. Furthermore, the expression of pro-inflammatory cytokines (TNFα, NF-κ β-p65, IL-1β, IL-6) is significantly reduced before and after WBPA treatment and the anti-inflammatory cytokine (IL-10) increased [79]. In addition, eNOS, p-eNOS, and nNOS expression were restored from LPS levels with both pre- and post-treatment with WBPA. Both pre and post treatment with WBPA markedly decreased LPS induced elevation of iNOS expression induced by LPS by 40 and 50%, respectively from LPS levels. These data support an anti-inflammatory phenotype induced by WBPA when used as a pre- or post-treatment intervention in a severe model of LPS.
NEUTROPHINS AND WBPA
Neurotropins BDNF and GDNF have been measured in whole brain homogenates in mice. Two weeks of WBPA (1 h daily) significantly increases the expression of the BDNF and GDNF proteins in the brain by 30% of the control values [92]. This increase is in the range of those reported for 2 weeks of voluntary wheel running in rats [93]. However, it was modest compared to other studies of specific brain regions and exercise strategies of longer duration [94–100].
ENDOTHELIAL DERIVED NITRIC OXIDE, ANTIOXIDANTS, AND WBPA AND JD
Pulsatile shear stress produced by WBPA and JD has been shown to increase eNOS genomic expression and NO bioavailability [81, 101].
WBPA increases the expression of eNOS in endothelial cells and cardiomyocytes. The latter has been shown at both the individual cellular level and whole animal models. The initial response includes acute phosphorylation (activation) of eNOS through the phosphoinositol 3-kinase pathway (PI3K AKT), followed by genomic upregulation. Genomic upregulation remains effective for at least 3 days after WBPA usage in mice. Furthermore, antioxidant defense mechanisms (superoxide dismutase 1 [SOD1], Catalase [CAT], glutathioneperoxidase [GPX1], and total antioxidant capacity) are also activated with the use of WBPA, with a concomitant decrease in oxidative stress [102].
The ability of WBPA to decrease oxidative stress has been tested in animal models with a marked burden of oxidative stress, such as Duchenne muscular dystrophy (mdx) and type 1 and 2 diabetes (TD1, TD2). In both mdx and diabetics, the production of ROS was markedly reduced in cardiomyocytes after WBPA [102].
The JD has also been shown to increase NO bioavailability after short (30 min) or longer periods (1 h per day for 7 days) in human subjects [81]. PPSS is a powerful intervention to increase the expression of beneficial eNOS and increase its phosphorylation, with increased NO bioavailability, and reduction of oxidative stress.
PHYSICAL INACTIVITY, HYPERTENSION, DIABETES, AND JD
The reduction of cardiovascular risk factors, especially hypertension, diabetes, and decreased physical inactivity are at the forefront of reducing risk for AD [6]. The use of JD decreases physical inactivity; it can be used during work in the sitting position, while performing other tasks, and in the supine position for those who cannot sit or are hospitalized. In recent human studies of both hypertensive and non-hypertensive subjects, JD acutely reduced the elevation of blood pressure induced by sedentary behavior both in supine and seated postures [103]. Furthermore, the longer-term use of JD also decreased systolic and diastolic blood pressure in a cohort of subjects who used JD for 7 days. The effect on blood pressure was sustained for at least 3 days after discontinuation of JD, supporting the use of JD as a non-pharmacologic aid in the management of high blood pressure.
Glycemic control and reduction in diabetes have also been shown to be important risk reduction factors for AD. JD performed in both diabetic and non-diabetic subjects for 1 week, significantly decreased glucose variability, measured by continuous subcutaneous glucose monitoring, during JD. Furthermore, JD reduced the area under the curve of an oral glucose challenge in non-diabetic subjects. These data support the use of JD as an adjunct to the management of diabetes [104]. Figures 2 and 3 provide an overview of the published effects of PPSS and a mechanistic summary of its effects on AD, respectively.
Fig. 2
Overview of the published effects of Passive Pulsatile Shear Stress. The figure shows the effects of PPSS produced by whole body periodic acceleration (WBPA/pGz) and Jogging Device (JD) on the modifiable risk factors and other pathophysiological mechanisms involved in AD.
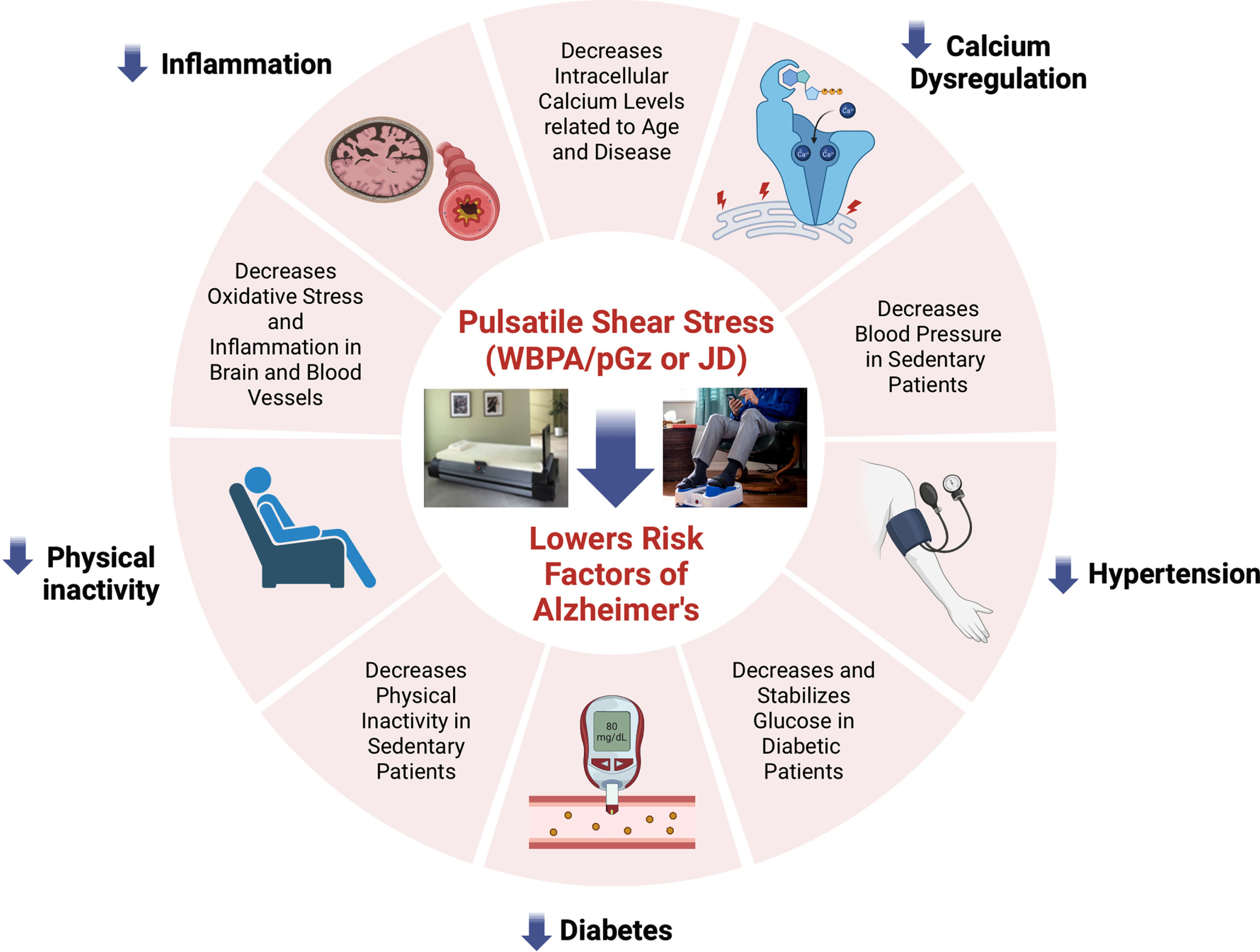
Fig. 3
Mechanistic Summary of the Effects of Passive Pulsatile Shear Stress (WBPA/pGz or JD) in Alzheimer’s Disease. PPSS as produced by WBPA/pGz or JD, elicits a wide variety of hemodynamic and protein expression changes which include: increase regional cortical and microvascular blood flow at rest, during ischemia and during reperfusion; improved [Ca2+]i homeostasis and dysregulation, with a decrease in Ca2+ leak and RyR nitrosilation, a decrease in Ca2+ influx, and a decrease in Calpain (A); decrease in the inflammatory phenotype with decrease in pro-inflammatory cytokines (TNFα, NF-κβ-p65, IL-1β, IL-6) and increase in anti-inflammatory cytokine (IL-10); increase in protein expression of eNOS, p-eNOS; and antioxidants (GPX1, CAT, SOD); decrease in reactive oxygen species (ROS) production and increase NO bioavailability (B); increase in brain neurotrophins; BDNF and GDNF (C).
![Mechanistic Summary of the Effects of Passive Pulsatile Shear Stress (WBPA/pGz or JD) in Alzheimer’s Disease. PPSS as produced by WBPA/pGz or JD, elicits a wide variety of hemodynamic and protein expression changes which include: increase regional cortical and microvascular blood flow at rest, during ischemia and during reperfusion; improved [Ca2+]i homeostasis and dysregulation, with a decrease in Ca2+ leak and RyR nitrosilation, a decrease in Ca2+ influx, and a decrease in Calpain (A); decrease in the inflammatory phenotype with decrease in pro-inflammatory cytokines (TNFα, NF-κβ-p65, IL-1β, IL-6) and increase in anti-inflammatory cytokine (IL-10); increase in protein expression of eNOS, p-eNOS; and antioxidants (GPX1, CAT, SOD); decrease in reactive oxygen species (ROS) production and increase NO bioavailability (B); increase in brain neurotrophins; BDNF and GDNF (C).](https://ip.ios.semcs.net:443/media/jad/2024/98-2/jad-98-2-jad231010/jad-98-jad231010-g003.jpg)
LIMITATIONS
There are limitations which must be acknowledged to this review. The data presented relate to PPSS; however, active pulsatile shear stress (as occurs with active aerobic exercise) was not discussed in detail. The current knowledge and molecular mechanisms of exercise induced improvements in cognitive dysfunction, have been recently reviewed, focusing on exercise enhancement of neural plasticity, structure, function, mitochondrial health, and modification of the muscle brain axis and others [105]. Furthermore, a recent summary of the impact of physical exercise on AD in animal models and humans has been published [106]. Active exercise has been shown in various systematic reviews and meta-analysis to be beneficial. An umbrella review of systematic reviews and meta-analysis concluded that the largest effect of exercise on AD was on cognition. They concluded that exercise is an effective way to treat AD symptoms with a low incidence of related adverse effects [107]. Other meta-analysis of randomized controlled trials (RCTs) showed that aerobic exercise improved cognitive function in AD patients, and a worse baseline cognitive status contributed to a more significant improvement in cognitive function [108]. Recently, an umbrella review of the existing meta-analysis showed strong evidence for a protective effect of regular physical activity against AD, but the dose response remained unclear [109]. Zeng et al. in a meta-analysis of RCT on the effects of various physical activity interventions on executive function in older adults with dementia, showed that executive function benefited from physical activity and the benefit was affected by the type, intensity, total duration, and frequency of exercise [110]. A systematic review and meta-analysis that examined whether mid-life physical activity is protective for all causes of dementia, AD, and vascular dementia, in subjects with follow-up more than 1 year, concluded that physical activity was associated with a lower incidence of all causes of dementia and AD, even at longer follow-up (20 years) [111]. In an interesting systematic review by Ayari et al. of exercise modalities that reduce pro-inflammatory cytokines in humans and animal models with MCI or dementia, they concluded that in rodents with the AD phenotype various exercise interventions delay the mechanisms of dementia progression, and in humans aerobic exercise is effective in subjects with mild AD and MCI, decreasing pro-inflammatory cytokine markers [112]. These findings are also consistent with a recent review by Wang et al. [113]. In a recent large-scale portrait analysis of AD gene expression using 22 large-scale AD gene expression datasets, the authors, found that exercise reversed the expression patterns of hundreds of AD genes in multiple categories including cytoskeleton, blood vessel development, mitochondrion, and interferon-stimulated related genes, and ranked as the best treatment across in most of the individual region-specific AD datasets and meta-analysis dataset [114]. Although the thresholds, dose, and frequency of physical activity as a preventive or therapeutic intervention are unknown at this time, most studies support physical activity as part of a multimodal approach to AD and MCI.
We also did not discuss other potential applications of PPSS. Recent reviews on spaceflight, microgravity, and simulated microgravity have shown that multiple organ systems are involved in the response of the organism to these stressors [115–120]. The response to these stressors converges on the inflammatory response, which is a driver of aging [117]. Countermeasures for spaceflight and microgravity in the form of active pulsatile shear stress (aerobic exercise) have been reviewed by others [121–124]. PPSS has been used in the form of plantar mechanical stimulation in animal models that show promise in these preclinical models [125], dynamic foot stimulation [126] and repeated horizontal jumping [127] in humans. Furthermore, these exercise countermeasures for microgravity are being adapted for use in bedrest studies in older adults [121, 128, 129]. The application of PPSS in the latter conditions deserves future studies.
Finally, we did not discuss pharmacologic interventions that can improve vascular function or noninvasive transcranial Doppler ultrasound, which has been used to assess pharmacological interventions and as a potential therapeutic intervention for AD. These topics have been explored in various review articles [130–134].
CONCLUSIONS
Passive pulsatile shear stress as produced by WBPA/pGz and JD can be considered an ‘exercise mimetic’, which produces many of the beneficial effects of exercise, a concept that has been reviewed by Gubert et al. in the context of pharmacological interventions [135]. Data on PPSS as produced by WBPA or JD provide compelling rationale that these interventions may be of clinical utility in AD (Fig. 3). To prove our hypothesis that PPSS may be a preventive or therapeutic strategy or adjunct for AD, clinical trials with long-term use of JD or WBPA, and appropriate outcome measures are needed. PPSS is poised to be a disruptive approach to prevention/therapy and an adjunct to AD [136]. PPSS may play a therapeutic role in AD; however, it is unlikely to treat or eradicate the disease solely, and a comprehensive therapeutic approach is needed that incorporates a reduction in diabetes, hypertension, and increased physical activity. Long-term use of PPSS interventions along with a current multimodal approach to AD management may prove to be valuable for the management of such a devastating disease for patients and their caregivers.
AUTHOR CONTRIBUTIONS
Jose Antonio Adams (Conceptualization; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Writing – original draft; Writing – review & editing); Arkady Uryash (Conceptualization; Investigation; Writing – original draft; Writing – review & editing; Graphics); Jose R. Lopez (Conceptualization; Investigation; Writing – original draft; Writing – review & editing).
ACKNOWLEDGMENTS
The figures were created using BioRender.com
FUNDING
This project was funded in part by a grant to JAA from the Florida Heart Research Institute.
CONFLICT OF INTEREST
Jose A. Adams was a part patent owner for the jogging device. The patent was sold in 2020 to Movewell Technology (Hollywood, FL, USA). He does not consult, receive honoraria or have ownership of Movewell Technology. The remainder of the authors have no conflict of interest to report.
DATA AVAILABILITY
Data sharing is not applicable to this article as no datasets were generated or analyzed during this study.
REFERENCES
[1] | Rajan KB , Weuve J , Barnes LL , McAninch EA , Wilson RS , Evans DA ((2021) ) Population estimate of people with clinical Alzheimer’s disease and mild cognitive impairment in the United States (2020–2060). Alzheimers Dement 17: , 1966–1975. |
[2] | Collaborators GBDD ((2019) ) Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 18: , 88–106. |
[3] | Scheltens P , De Strooper B , Kivipelto M , Holstege H , Chetelat G , Teunissen CE , Cummings J , van der Flier WM ((2021) ) Alzheimer’s disease. Lancet 397: , 1577–1590. |
[4] | Nandi A , Counts N , Chen S , Seligman B , Tortorice D , Vigo D , Bloom DE ((2022) ) Global and regional projections of the economic burden of Alzheimer’s disease and related dementias from 2019 to 2050: A value of statistical life approach. EClinicalMedicine 51: , 101580. |
[5] | World Health Organization (2019) Risk reduction of cognitive decline and dementia. World Health Organization. |
[6] | Livingston G , Huntley J , Sommerlad A , Ames D , Ballard C , Banerjee S , Brayne C , Burns A , Cohen-Mansfield J , Cooper C , Costafreda SG , Dias A , Fox N , Gitlin LN , Howard R , Kales HC , Kivimaki M , Larson EB , Ogunniyi A , Orgeta V , Ritchie K , Rockwood K , Sampson EL , Samus Q , Schneider LS , Selbaek G , Teri L , Mukadam N ((2020) ) Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 396: , 413–446. |
[7] | Amini H , Habibi S , Islamoglu AH , Isanejad E , Uz C , Daniyari H ((2021) ) COVID-19 pandemic-induced physical inactivity: The necessity of updating the Global Action Plan on Physical Activity 2018–2030. Environ Health Prev Med 26: , 1–3. |
[8] | Andersen LB , Mota J , Di Pietro L ((2016) ) Update on the global pandemic of physical inactivity. Lancet 388: , 1255–1256. |
[9] | Forouhi NG , Wareham NJ ((2022) ) Epidemiology of diabetes. Medicine 50: , 638–643. |
[10] | Tsao CW , Aday AW , Almarzooq ZI , Alonso A , Beaton AZ , Bittencourt MS , Boehme AK , Buxton AE , Carson AP , Commodore-Mensah Y , Elkind MSV , Evenson KR , Eze-Nliam C , Ferguson JF , Generoso G , Ho JE , Kalani R , Khan SS , Kissela BM , Knutson KL , Levine DA , Lewis TT , Liu J , Loop MS , Ma J , Mussolino ME , Navaneethan SD , Perak AM , Poudel R , Rezk-Hanna M , Roth GA , Schroeder EB , Shah SH , Thacker EL , VanWagner LB , Virani SS , Voecks JH , Wang NY , Yaffe K , Martin SS ((2022) ) Heart Disease and Stroke Statistics-2022 Update: A Report From the American Heart Association. Circulation 145: , e153–e639. |
[11] | Asher S , Priefer R ((2022) ) Alzheimer’s disease failed clinical trials. Life Sci 306: , 120861. |
[12] | Yiannopoulou KG , Anastasiou AI , Zachariou V , Pelidou SH ((2019) ) Reasons for failed trials of disease-modifying treatments for Alzheimer disease and their contribution in recent research. Biomedicines 7: , 97. |
[13] | Rao RV , Subramaniam KG , Gregory J , Bredesen AL , Coward C , Okada S , Kelly L , Bredesen DE ((2023) ) Rationale for a multi-factorial approach for the reversal of cognitive decline in Alzheimer’s disease and MCI: A review. Int J Mol Sci 24: , 1659. |
[14] | Sun E , Motolani A , Campos L , Lu T ((2022) ) The pivotal role of NF-kB in the pathogenesis and therapeutics of Alzheimer’s disease. Int J Mol Sci 23: , 8972. |
[15] | Ghosh P , Singh R , Ganeshpurkar A , Pokle AV , Singh RB , Singh SK , Kumar A ((2021) ) Cellular and molecular influencers of neuroinflammation in Alzheimer’s disease: Recent concepts & roles. Neurochem Int 151: , 105212. |
[16] | Heneka MT , Carson MJ , El Khoury J , Landreth GE , Brosseron F , Feinstein DL , Jacobs AH , Wyss-Coray T , Vitorica J , Ransohoff RM , Herrup K , Frautschy SA , Finsen B , Brown GC , Verkhratsky A , Yamanaka K , Koistinaho J , Latz E , Halle A , Petzold GC , Town T , Morgan D , Shinohara ML , Perry VH , Holmes C , Bazan NG , Brooks DJ , Hunot S , Joseph B , Deigendesch N , Garaschuk O , Boddeke E , Dinarello CA , Breitner JC , Cole GM , Golenbock DT , Kummer MP ((2015) ) Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14: , 388–405. |
[17] | Thakur S , Dhapola R , Sarma P , Medhi B , Reddy DH ((2023) ) Neuroinflammation in Alzheimer’s disease: Current progress in molecular signaling and therapeutics. Inflammation 46: , 1–17. |
[18] | Leng F , Edison P ((2021) ) Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? . Nat Rev Neurol 17: , 157–172. |
[19] | Justo AFO , Suemoto CK ((2022) ) The modulation of neuroinflammation by inducible nitric oxide synthase. J Cell Commun Signal 16: , 155–158. |
[20] | Iadecola C ((2004) ) Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci 5: , 347–360. |
[21] | Iturria-Medina Y , Sotero RC , Toussaint PJ , Mateos-Perez JM , Evans AC , Alzheimer’s Disease Neuroimaging Initiative ((2016) ) Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat Commun 7: , 11934. |
[22] | Kisler K , Nelson AR , Montagne A , Zlokovic BV ((2017) ) Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat Rev Neurosci 18: , 419–434. |
[23] | Zlokovic BV ((2011) ) Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci 12: , 723–738. |
[24] | Dormanns K , Brown RG , David T ((2016) ) The role of nitric oxide in neurovascular coupling. J Theor Biol 394: , 1–17. |
[25] | Lourenco CF , Laranjinha J ((2021) ) Nitric oxide pathways in neurovascular coupling under normal and stress conditions in the brain: Strategies to rescue aberrant coupling and improve cerebral blood flow. Front Physiol 12: , 729201. |
[26] | Solis E Jr. , Hascup KN , Hascup ER ((2020) ) Alzheimer’s disease: The link between amyloid-beta and neurovascular dysfunction. J Alzheimers Dis 76: , 1179–1198. |
[27] | Daulatzai MA ((2017) ) Cerebral hypoperfusion and glucose hypometabolism: Key pathophysiological modulators promote neurodegeneration, cognitive impairment, and Alzheimer’s disease. J Neurosci Res 95: , 943–972. |
[28] | Bracko O , Cruz Hernandez JC , Park L , Nishimura N , Schaffer CB ((2021) ) Causes and consequences of baseline cerebral blood flow reductions in Alzheimer’s disease. J Cereb Blood Flow Metab 41: , 1501–1516. |
[29] | LaFerla FM ((2002) ) Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat Rev Neurosci 3: , 862–872. |
[30] | Khachaturian ZS ((1989) ) Calcium, membranes, aging, and Alzheimer’s disease. Introduction and overview. Ann N Y Acad Sci 568: , 1–4. |
[31] | Landfield PW ((1987) ) ‘Increased calcium-current’ hypothesis of brain aging. Neurobiol Aging 8: , 346–347. |
[32] | Landfield PW , Pitler TA ((1984) ) Prolonged Ca2+-dependent afterhyperpolarizations in hippocampal neurons of aged rats. Science 226: , 1089–1092. |
[33] | Lopez JR , Lyckman A , Oddo S , Laferla FM , Querfurth HW , Shtifman A ((2008) ) Increased intraneuronal resting [Ca2+] in adult Alzheimer’s disease mice. J Neurochem 105: , 262–271. |
[34] | Cascella R , Cecchi C ((2021) ) Calcium dyshomeostasis in Alzheimer’s disease pathogenesis. Int J Mol Sci 22: , 4914. |
[35] | Huang DX , Yu X , Yu WJ , Zhang XM , Liu C , Liu HP , Sun Y , Jiang ZP ((2022) ) Calcium signaling regulated by cellular membrane systems and calcium homeostasis perturbed in Alzheimer’s disease. Front Cell Dev Biol 10: , 834962. |
[36] | Lubke JH , Idoon F , Mohasel-Roodi M , Alipour F , Hami J , Ehteshampour A , Mostafaee H , Sadeghi A ((2021) ) Neurotrophic factors in Alzheimer’s disease: Pathogenesis and therapy. Acta Neurobiol Exp (Wars) 81: , 314–327. |
[37] | Sopova K , Gatsiou K , Stellos K , Laske C ((2014) ) Dysregulation of neurotrophic and haematopoietic growth factors in Alzheimer’s disease: From pathophysiology to novel treatment strategies. Curr Alzheimer Res 11: , 27–39. |
[38] | Du Y , Wu HT , Qin XY , Cao C , Liu Y , Cao ZZ , Cheng Y ((2018) ) Postmortem brain, cerebrospinal fluid, and blood neurotrophic factor levels in Alzheimer’s disease: A systematic review and meta-analysis. J Mol Neurosci 65: , 289–300. |
[39] | Qin XY , Cao C , Cawley NX , Liu TT , Yuan J , Loh YP , Cheng Y ((2017) ) Decreased peripheral brain-derived neurotrophic factor levels in Alzheimer’s disease: A meta-analysis study (N=7277). Mol Psychiatry 22: , 312–320. |
[40] | Meng C , He Z , Xing D ((2013) ) Low-level laser therapy rescues dendrite atrophy via upregulating BDNF expression: Implications for Alzheimer’s disease. J Neurosci 33: , 13505–13517. |
[41] | Michalski B , Fahnestock M ((2003) ) Pro-brain-derived neurotrophic factor is decreased in parietal cortex in Alzheimer’s disease. Brain Res Mol Brain Res 111: , 148–154. |
[42] | Jiao SS , Shen LL , Zhu C , Bu XL , Liu YH , Liu CH , Yao XQ , Zhang LL , Zhou HD , Walker DG , Tan J , Gotz J , Zhou XF , Wang YJ ((2016) ) Brain-derived neurotrophic factor protects against tau-related neurodegeneration of Alzheimer’s disease. Transl Psychiatry 6: , e907. |
[43] | Blurton-Jones M , Kitazawa M , Martinez-Coria H , Castello NA , Muller FJ , Loring JF , Yamasaki TR , Poon WW , Green KN , LaFerla FM ((2009) ) Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc Natl Acad Sci U S A 106: , 13594–13599. |
[44] | Lattanzio F , Carboni L , Carretta D , Rimondini R , Candeletti S , Romualdi P ((2014) ) Human apolipoprotein E4 modulates the expression of Pin1, Sirtuin 1, and Presenilin 1 in brain regions of targeted replacement apoE mice. Neuroscience 256: , 360–369. |
[45] | Nagahara AH , Merrill DA , Coppola G , Tsukada S , Schroeder BE , Shaked GM , Wang L , Blesch A , Kim A , Conner JM , Rockenstein E , Chao MV , Koo EH , Geschwind D , Masliah E , Chiba AA , Tuszynski MH ((2009) ) Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat Med 15: , 331–337. |
[46] | Allen SJ , Watson JJ , Shoemark DK , Barua NU , Patel NK ((2013) ) GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol Ther 138: , 155–175. |
[47] | Revilla S , Sunol C , Garcia-Mesa Y , Gimenez-Llort L , Sanfeliu C , Cristofol R ((2014) ) Physical exercise improves synaptic dysfunction and recovers the loss of survival factors in 3xTg-AD mouse brain. Neuropharmacology 81: , 55–63. |
[48] | Revilla S , Ursulet S , Alvarez-Lopez MJ , Castro-Freire M , Perpina U , Garcia-Mesa Y , Bortolozzi A , Gimenez-Llort L , Kaliman P , Cristofol R , Sarkis C , Sanfeliu C ((2014) ) Lenti-GDNF gene therapy protects against Alzheimer’s disease-like neuropathology in 3xTg-AD mice and MC65 cells. CNS Neurosci Ther 20: , 961–972. |
[49] | Nasrolahi A , Javaherforooshzadeh F , Jafarzadeh-Gharehziaaddin M , Mahmoudi J , Asl KD , Shabani Z ((2022) ) Therapeutic potential of neurotrophic factors in Alzheimer’s Disease. Mol Biol Rep 49: , 2345–2357. |
[50] | Chou W , Liu YF , Lin CH , Lin MT , Chen CC , Liu WP , Chang CP , Chio CC ((2018) ) Exercise rehabilitation attenuates cognitive deficits in rats with traumatic brain injury by stimulating the cerebral HSP20/BDNF/TrkB signalling axis. Mol Neurobiol 55: , 8602–8611. |
[51] | Coulson EJ , Bartlett PF ((2017) ) An exercise path to preventing Alzheimer’s disease: An Editorial Highlight on ‘Exercise and BDNF reduce Ab production by enhancing alpha-secretase processing of APP’. J Neurochem 142: , 191–193. |
[52] | Nigam SM , Xu S , Kritikou JS , Marosi K , Brodin L , Mattson MP ((2017) ) Exercise and BDNF reduce Abeta production by enhancing alpha-secretase processing of APP. J Neurochem 142: , 286–296. |
[53] | Ribeiro D , Petrigna L , Pereira FC , Muscella A , Bianco A , Tavares P ((2021) ) The impact of physical exercise on the circulating levels of BDNF and NT 4/5: A review. Int J Mol Sci 22: , 8814. |
[54] | Mazo CE , Miranda ER , Shadiow J , Vesia M , Haus JM ((2022) ) High intensity acute aerobic exercise elicits alterations in circulating and skeletal muscle tissue expression of neuroprotective exerkines. Brain Plast 8: , 5–18. |
[55] | Dyer RR , Ford KI , Robinson RAS ((2019) ) The roles of S-nitrosylation and S-glutathionylation in Alzheimer’s disease. Methods Enzymol 626: , 499–538. |
[56] | Forstermann U , Sessa WC (2012) Nitric oxide synthases: Regulation and function. Eur Heart J 33, 829-837, 837a- 837d. |
[57] | Garcia V , Sessa WC ((2019) ) Endothelial NOS: Perspective and recent developments. Br J Pharmacol 176: , 189–196. |
[58] | Chien S ((2007) ) Mechanotransduction and endothelial cell homeostasis: The wisdom of the cell. Am J Physiol Heart Circ Physiol 292: , H1209–1224. |
[59] | He M , Martin M , Marin T , Chen Z , Gongol B ((2020) ) Endothelial mechanobiology. APL Bioeng 4: , 010904. |
[60] | Zhou J , Li YS , Chien S ((2014) ) Shear stress-initiated signaling and its regulation of endothelial function. Arterioscler Thromb Vasc Biol 34: , 2191–2198. |
[61] | Trinity JD , Richardson RS ((2019) ) Physiological impact and clinical relevance of passive exercise/movement. Sports Med 49: , 1365–1381. |
[62] | Dubey H , Gulati K , Ray A ((2020) ) Alzheimer’s disease: A contextual link with nitric oxide synthase. Curr Mol Med 20: , 505–515. |
[63] | Azargoonjahromi A ((2023) ) Dual role of nitric oxide in Alzheimer’s disease. Nitric Oxide 134-135: , 23–37. |
[64] | Iova OM , Marin GE , Lazar I , Stanescu I , Dogaru G , Nicula CA , Bulboaca AE ((2023) ) Nitric oxide/nitric oxide synthase system in the pathogenesis of neurodegenerative disorders-an overview. Antioxidants (Basel) 12: , 1–23. |
[65] | Katusic ZS , d’Uscio LV , He T ((2023) ) Emerging roles of endothelial nitric oxide in preservation of cognitive health. Stroke 54: , 686–696. |
[66] | Katusic ZS , Austin SA ((2016) ) Neurovascular protective function of endothelial nitric oxide - recent advances. Circ J 80: , 1499–1503. |
[67] | Zhou H , Gao F , Yang X , Lin T , Li Z , Wang Q , Yao Y , Li L , Ding X , Shi K , Liu Q , Bao H , Long Z , Wu Z , Vassar R , Cheng X , Li R , Shen Y ((2022) ) Endothelial BACE1 impairs cerebral small vessels via tight junctions and eNOS. Circ Res 130: , 1321–1341. |
[68] | Austin SA , Santhanam AV , Katusic ZS ((2010) ) Endothelial nitric oxide modulates expression and processing of amyloid precursor protein. Circ Res 107: , 1498–1502. |
[69] | Austin SA , Katusic ZS ((2020) ) Partial loss of endothelial nitric oxide leads to increased cerebrovascular beta amyloid. J Cereb Blood Flow Metab 40: , 392–403. |
[70] | Austin SA , Santhanam AV , Hinton DJ , Choi DS , Katusic ZS ((2013) ) Endothelial nitric oxide deficiency promotes Alzheimer’s disease pathology. J Neurochem 127: , 691–700. |
[71] | Chu Y , Heistad DD ((2010) ) No answer to Alzheimer’s disease? . Circ Res 107: , 1400–1402. |
[72] | Ahmed S , Jing Y , Mockett BG , Zhang H , Abraham WC , Liu P ((2022) ) Partial endothelial nitric oxide synthase deficiency exacerbates cognitive deficit and amyloid pathology in the APPswe/PS1DeltaE9 mouse model of Alzheimer’s disease. Int J Mol Sci 23: , 7316. |
[73] | Nakamura T , Lipton SA ((2017) ) ‘SNO’-storms compromise protein activity and mitochondrial metabolism in neurodegenerative disorders. Trends Endocrinol Metab 28: , 879–892. |
[74] | Degrush E , Shazeeb MS , Drachman D , Vardar Z , Lindsay C , Gounis MJ , Henninger N ((2022) ) Cumulative effect of simvastatin, L-arginine, and tetrahydrobiopterin on cerebral blood flow and cognitive function in Alzheimer’s disease. Alzheimers Res Ther 14: , 134. |
[75] | Katusic ZS , Austin SA ((2014) ) Endothelial nitric oxide: Protector of a healthy mind. Eur Heart J 35: , 888–894. |
[76] | Jeynes B , Provias J ((2009) ) Significant negative correlations between capillary expressed eNOS and Alzheimer lesion burden. Neurosci Lett 463: , 244–248. |
[77] | Hutcheson IR , Griffith TM ((1991) ) Release of endothelium-derived relaxing factor is modulated both by frequency and amplitude of pulsatile flow. Am J Physiol 261: , H257–H262. |
[78] | Clermont CA , Benson LC , Osis ST , Kobsar D , Ferber R ((2019) ) Running patterns for male and female competitive and recreational runners based on accelerometer data. J Sports Sci 37: , 204–211. |
[79] | Adams JA , Uryash A , Lopez JR ((2022) ) Non-invasive pulsatile shear stress modifies endothelial activation; a narrative review. Biomedicines 10: , 3050. |
[80] | Adams JA , Mangino MJ , Bassuk J , Kurlansky P , Sackner MA ((2001) ) Regional blood flow during periodic acceleration. Crit Care Med 29: , 1983–1988. |
[81] | Adams JA , Lopez JR , Nadkarni V , Zolkipli-Cunningham Z , Ischiropoulos H , Sackner MA ((2022) ) The effects of a motorized passive simulated jogging device on descent of the arterial pulse waveform dicrotic notch: A single arm placebo-controlled cross-over trial. Physiol Rep 10: , e15418. |
[82] | Adams JA , Banderas V , Lopez JR , Sackner MA ((2020) ) Portable gentle jogger improves glycemic indices in type 2 diabetic and healthy subjects living at home: A pilot study. J Diabetes Res 2020: , 8317973. |
[83] | Sackner MA ((2012) ) Whole body periodic acceleration: “passive exercise” for Parkinson’s disease. J Parkinsonism Restless Legs Syndrome 2: , 1–5. |
[84] | Southard V , Donoghue J , Belmonte J , Liboreiro M , Musa M ((2018) ) The effects of whole body periodic acceleration on non-motor symptoms in people with mild to moderate Parkinson’s disease. J Adv Med Med Res 27: , 1–9. |
[85] | Wu D , Bassuk J , Arias J , Peschiera I , Lamet A , Kurlansky P , Adams JA ((2006) ) Post-resuscitation reperfusion injury: Comparison of periodic Gz acceleration versus Thumper CPR. Resuscitation 70: , 454–462. |
[86] | Adams JA , Wu H , Bassuk JA , Arias J , Uryash A , Jorapur V , Lamas GA , Kurlansky P ((2010) ) Periodic acceleration (pGz) prior to whole body ischemia reperfusion injury provides early cardioprotective preconditioning. Life Sci 86: , 707–715. |
[87] | Adams JA , Uryash A , Wu H , Bassuk JA , Nadkarni V , Berg R , Jorapur V , Kurlansky P ((2011) ) Microcirculatory and therapeutic effects of whole body periodic acceleration (pGz) applied after cardiac arrest in pigs. Resuscitation 82: , 767–775. |
[88] | Uryash A , Flores V , Adams JA , Allen PD , Lopez JR ((2020) ) Memory and learning deficits are associated with Ca(2+) dyshomeostasis in normal aging. Front Aging Neurosci 12: , 224. |
[89] | Lopez JR , Kolster J , Uryash A , Esteve E , Altamirano F , Adams JA ((2018) ) Dysregulation of intracellular Ca(2+) in dystrophic cortical and hippocampal neurons. Mol Neurobiol 55: , 603–618. |
[90] | Lopez JR , Uryash A , Kolster J , Esteve E , Zhang R , Adams JA ((2018) ) Enhancing endogenous nitric oxide by whole body periodic acceleration elicits neuroprotective effects in dystrophic neurons. Mol Neurobiol 55: , 8680–8694. |
[91] | Adams JA , Lopez JR , Uryash A , Sackner MA ((2021) ) Whole body periodic acceleration (pGz) improves endotoxin induced cardiomyocyte contractile dysfunction and attenuates the inflammatory response in mice. Heliyon 7: , e06444. |
[92] | Adams JA , Uryash A , Bassuk J , Sackner MA , Kurlansky P ((2014) ) Biological basis of neuroprotection and neurotherapeutic effects of Whole Body Periodic Acceleration (pGz). Med Hypotheses 82: , 681–687. |
[93] | Hall JM , Vetreno RP , Savage LM ((2014) ) Differential cortical neurotrophin and cytogenetic adaptation after voluntary exercise in normal and amnestic rats. Neuroscience 258: , 131–146. |
[94] | Alomari MA , Khabour OF , Alzoubi KH , Alzubi MA ((2013) ) Forced and voluntary exercises equally improve spatial learning and memory and hippocampal BDNF levels. Behav Brain Res 247: , 34–39. |
[95] | Chen MJ , Russo-Neustadt AA ((2009) ) Running exercise-induced up-regulation of hippocampal brain-derived neurotrophic factor is CREB-dependent. Hippocampus 19: , 962–972. |
[96] | Gomez-Pinilla F , Ying Z , Zhuang Y ((2012) ) Brain and spinal cord interaction: Protective effects of exercise prior to spinal cord injury. PLoS One 7: , e32298. |
[97] | Kobilo T , Liu QR , Gandhi K , Mughal M , Shaham Y , van Praag H ((2011) ) Running is the neurogenic and neurotrophic stimulus in environmental enrichment. Learn Mem 18: , 605–609. |
[98] | Zielinski MR , Davis JM , Fadel JR , Youngstedt SD ((2013) ) Influence of chronic moderate sleep restriction and exercise training on anxiety, spatial memory, and associated neurobiological measures in mice. Behav Brain Res 250: , 74–80. |
[99] | Griesbach GS , Gomez-Pinilla F , Hovda DA ((2007) ) Time window for voluntary exercise-induced increases in hippocampal neuroplasticity molecules after traumatic brain injury is severity dependent. J Neurotrauma 24: , 1161–1171. |
[100] | Lee MC , Okamoto M , Liu YF , Inoue K , Matsui T , Nogami H , Soya H ((2012) ) Voluntary resistance running with short distance enhances spatial memory related to hippocampal BDNF signaling. J Appl Physiol (1985) 113: , 1260–1266. |
[101] | Rokutanda T , Izumiya Y , Miura M , Fukuda S , Shimada K , Izumi Y , Nakamura Y , Araki S , Hanatani S , Matsubara J , Nakamura T , Kataoka K , Yasuda O , Kaikita K , Sugiyama S , Kim-Mitsuyama S , Yoshikawa J , Fujita M , Yoshiyama M , Ogawa H ((2011) ) Passive exercise using whole-body periodic acceleration enhances blood supply to ischemic hindlimb. Arterioscler Thromb Vasc Biol 31: , 2872–2880. |
[102] | Uryash A , Bassuk J , Kurlansky P , Altamirano F , Lopez JR , Adams JA ((2015) ) Antioxidant properties of whole body periodic acceleration (pGz). PLoS One 10: , e0131392. |
[103] | Sackner MA , Patel S , Adams JA ((2019) ) Changes of blood pressure following initiation of physical inactivity and after external addition of pulses to circulation. Eur J Appl Physiol 119: , 201–211. |
[104] | Adams JA , Uryash A , Lopez JR , Sackner MA ((2021) ) The endothelium as a therapeutic target in diabetes: A narrative review and perspective. Front Physiol 12: , 638491. |
[105] | Lu Y , Bu FQ , Wang F , Liu L , Zhang S , Wang G , Hu XY ((2023) ) Recent advances on the molecular mechanisms of exercise-induced improvements of cognitive dysfunction. Transl Neurodegener 12: , 1–29. |
[106] | Andrade-Guerrero J , Rodriguez-Arellano P , Barron-Leon N , Orta-Salazar E , Ledesma-Alonso C , Diaz-Cintra S , Soto-Rojas LO ((2023) ) Advancing Alzheimer’s therapeutics: Exploring the impact of physical exercise in animal models and patients. Cells 12: , 2531. |
[107] | Andrade A , Siqueira TC , D’Oliveira A , Dominski FH ((2022) ) Effects of exercise in the treatment of Alzheimer’s disease: An umbrella review of systematic reviews and meta-analyses. J Aging Phys Act 30: , 535–551. |
[108] | Zhang S , Zhen K , Su Q , Chen Y , Lv Y , Yu L ((2022) ) The effect of aerobic exercise on cognitive function in people with Alzheimer’s disease: A systematic review and meta-analysis of randomized controlled trials. Int J Environ Res Public Health 19: , 15700. |
[109] | Lopez-Ortiz S , Lista S , Valenzuela PL , Pinto-Fraga J , Carmona R , Caraci F , Caruso G , Toschi N , Emanuele E , Gabelle A , Nistico R , Garaci F , Lucia A , Santos-Lozano A ((2023) ) Effects of physical activity and exercise interventions on Alzheimer’s disease: An umbrella review of existing meta-analyses. J Neurol 270: , 711–725. |
[110] | Zeng Y , Wang J , Cai X , Zhang X , Zhang J , Peng M , Xiao D , Ouyang H , Yan F ((2023) ) Effects of physical activity interventions on executive function in older adults with dementia: A meta-analysis of randomized controlled trials. Geriatr Nurs 51: , 369–377. |
[111] | Iso-Markku P , Kujala UM , Knittle K , Polet J , Vuoksimaa E , Waller K ((2022) ) Physical activity as a protective factor for dementia and Alzheimer’s disease: Systematic review, meta-analysis and quality assessment of cohort and case-control studies. Br J Sports Med 56: , 701–709. |
[112] | Ayari S , Abellard A , Carayol M , Guedj E , Gavarry O ((2023) ) A systematic review of exercise modalities that reduce pro-inflammatory cytokines in humans and animals’ models with mild cognitive impairment or dementia. Exp Gerontol 175: , 112141. |
[113] | Wang Z , Emmerich A , Pillon NJ , Moore T , Hemerich D , Cornelis MC , Mazzaferro E , Broos S , Ahluwalia TS , Bartz TM , et al ((2022) ) Genome-wide association analyses of physical activity and sedentary behavior provide insights into underlying mechanisms and roles in disease prevention. Nat Genet 54: , 1332–1344. |
[114] | Hill MA , Gammie SC ((2022) ) Alzheimer’s disease large-scale gene expression portrait identifies exercise as the top theoretical treatment. Sci Rep 12: , 17189. |
[115] | Baran R , Marchal S , Garcia Campos S , Rehnberg E , Tabury K , Baselet B , Wehland M , Grimm D , Baatout S ((2021) ) The cardiovascular system in space: Focus on in vivo and in vitro studies. Biomedicines 10: , 59. |
[116] | Bizzarri M , Fedeli V , Piombarolo A , Angeloni A ((2022) ) Space biomedicine: A unique opportunity to rethink the relationships between physics and biology. Biomedicines 10: , 2633. |
[117] | Capri M , Conte M , Ciurca E , Pirazzini C , Garagnani P , Santoro A , Longo F , Salvioli S , Lau P , Moeller R , Jordan J , Illig T , Villanueva MM , Gruber M , Burkle A , Franceschi C , Rittweger J ((2023) ) Long-term human spaceflight and inflammaging: Does it promote aging? . Ageing Res Rev 87: , 101909. |
[118] | Chaloulakou S , Poulia KA , Karayiannis D ((2022) ) Physiological alterations in relation to space flight: The role of nutrition. Nutrients 14: , 4896. |
[119] | Grimm D , Hemmersbach R ((2022) ) Translation from microgravity research to earth application. Int J Mol Sci 23: , 10995. |
[120] | Hart DA ((2023) ) Homo sapiens-A species not designed for space flight: Health risks in low earth orbit and beyond, including potential risks when traveling beyond the geomagnetic field of earth. Life (Basel) 13: , 757. |
[121] | Hedge ET , Patterson CA , Mastrandrea CJ , Sonjak V , Hajj-Boutros G , Faust A , Morais JA , Hughson RL ((2022) ) Implementation of exercise countermeasures during spaceflight and microgravity analogue studies: Developing countermeasure protocols for bedrest in older adults (BROA). Front Physiol 13: , 928313. |
[122] | Moosavi D , Wolovsky D , Depompeis A , Uher D , Lennington D , Bodden R , Garber CE ((2021) ) The effects of spaceflight microgravity on the musculoskeletal system of humans and animals, with an emphasis on exercise as a countermeasure: A systematic scoping review. Physiol Res 70: , 119–151. |
[123] | Scott JM , Feiveson AH , English KL , Spector ER , Sibonga JD , Dillon EL , Ploutz-Snyder L , Everett ME ((2023) ) Effects of exercise countermeasures on multisystem function in long duration spaceflight astronauts. NPJ Microgravity 9: , 11. |
[124] | Gao T , Huang J , Zhang X , Gao F ((2023) ) Exercise counteracts vascular aging in long-term spaceflight: Challenges and perspective. Curr Opin Physiol 31: , 100628. |
[125] | Tyganov SA , Mochalova E , Belova S , Sharlo K , Rozhkov S , Kalashnikov V , Turtikova O , Mirzoev T , Shenkman B ((2021) ) Plantar mechanical stimulation attenuates protein synthesis decline in disused skeletal muscle via modulation of nitric oxide level. Sci Rep 11: , 9806. |
[126] | Layne CS , Forth KE ((2008) ) Plantar stimulation as a possible countermeasure to microgravity-induced neuromotor degradation. Aviat Space Environ Med 79: , 787–794. |
[127] | Cleather DJ , Price PDB , Kennett JE ((2022) ) Repeated horizontal jumping is a feasible exercise countermeasure for microgravity. Micrograv Sci Technol 34: , 1–10. |
[128] | Konda NN , Karri RS , Winnard A , Nasser M , Evetts S , Boudreau E , Caplan N , Gradwell D , Velho RM ((2019) ) A comparison of exercise interventions from bed rest studies for the prevention of musculoskeletal loss. NPJ Microgravity 5: , 12. |
[129] | Robin A , Wang L , Custaud MA , Liu J , Yuan M , Li Z , Lloret JC , Liu S , Dai X , Zhang J , Lv K , Li W , Gauquelin-Koch G , Wang H , Li K , Li X , Qu L , Navasiolava N , Li Y ((2022) ) Running vs. resistance exercise to counteract deconditioning induced by 90-day head-down bedrest. Front Physiol 13: , 902983. |
[130] | Chen L , Jiao J , Zhang Y ((2022) ) Therapeutic approaches for improving cognitive function in the aging brain. Front Neurosci 16: , 1060556. |
[131] | Linh TTD , Hsieh YC , Huang LK , Hu CJ ((2022) ) Clinical trials of new drugs for vascular cognitive impairment and vascular dementia. Int J Mol Sci 23: , 11067. |
[132] | Nicodemus NE , Becerra S , Kuhn TP , Packham HR , Duncan J , Mahdavi K , Iovine J , Kesari S , Pereles S , Whitney M , Mamoun M , Franc D , Bystritsky A , Jordan S ((2019) ) Focused transcranial ultrasound for treatment of neurodegenerative dementia. Alzheimers Dement (N Y) 5: , 374–381. |
[133] | Pan Y , Wan W , Xiang M , Guan Y ((2022) ) Transcranial doppler ultrasonography as a diagnostic tool for cerebrovascular disorders. Front Hum Neurosci 16: , 841809. |
[134] | Wan Y , Teng X , Li S , Yang Y ((2022) ) Application of transcranial Doppler in cerebrovascular diseases. Front Aging Neurosci 14: , 1035086. |
[135] | Gubert C , Hannan AJ ((2021) ) Exercise mimetics: Harnessing the therapeutic effects of physical activity. Nat Rev Drug Discov 20: , 862–879. |
[136] | Grossman JH (2007) Disruptive Innovation in Health Care: Challenges for Engineering. InNAE Annual Meeting Technical Symposium, The Bridge, pp. 10-16. |


