
Pathological (Dis)Similarities in Neuronal Exosome-Derived Synaptic and Organellar Marker Levels Between Alzheimer’s Disease and Frontotemporal Dementia
Abstract
Background:
Alzheimer’s disease (AD) and frontotemporal dementia (FTD) are pathologically distinct neurodegenerative disorders with certain overlap in cognitive and behavioral symptoms. Both AD and FTD are characterized by synaptic loss and accumulation of misfolded proteins, albeit, in different regions of the brain.
Objective:
To investigate the synaptic and organellar markers in AD and FTD through assessment of the levels of synaptic protein, neurogranin (Ng) and organellar proteins, mitofusin-2 (MFN-2), lysosomal associated membrane protein-2 (LAMP-2), and golgin A4 from neuronal exosomes.
Methods:
Exosomes isolated from the plasma of healthy controls (HC), AD and FTD subjects were characterized using transmission electron microscopy. Neurodegenerative status was assessed by measurement of neurofilament light chain (NfL) using Simoa. The pooled exosomal extracts from each group were analyzed for Ng, MFN-2, LAMP-2, and golgin A4 by western blot analysis using enhanced chemiluminescence method of detection.
Results:
The densitometric analysis of immunoreactive bands demonstrated a 65% reduction of Ng in AD and 53% in FTD. Mitochondrial protein MFN-2 showed a significant reduction by 32% in AD and 46% in FTD. Lysosomal LAMP-2 and Golgi complex associated golgin A4 were considerably increased in both AD and FTD.
Conclusion:
Changes in Ng may reflect the ongoing synaptic degeneration that are linked to cognitive disturbances in AD and FTD. Importantly, the rate of synaptic degeneration was more pronounced in AD. Changes to a similar extent in both the dementia groups in organellar proteins indicates shared mechanisms of protein accumulation/degradation common to both AD and FTD.
INTRODUCTION
Exosomes are a subset of extracellular vesicles of endocytic origin released by cells whose ability to cross the blood-brain barrier and peripheral detection make them interesting candidates in the field of brain disorders [1]. Exosomes reflect the heterogeneous biological changes of the parental cell, and their cargo can serve as potential biomarker source for early diagnosis and clinical assessment of neurodegenerative disorders [2].
Alzheimer’s disease (AD) and frontotemporal dementia (FTD) are two neurodegenerative diseases that cause cognitive impairment and functional disability in the elderly. The overlapping clinical symptoms and pathological features make it challenging to differentiate between these conditions [3]. Abnormal protein conformations in neurodegenerative disorders and their cellular and neuroanatomical distribution constitute the major histological features essential in making a specific neuropathological diagnosis. The characteristic defining features of AD are the extraneuronal deposition of insoluble fibrous amyloid proteins and the neuronal tau inclusions called neurofibrillary tangles. FTD is characterized by TDP-43 cytoplasmic/nuclear inclusions or tau depositions in the frontal and temporal regions of the brain [4].
At the cellular level, dementia syndrome is characterized by a progressive synaptic failure. Changes in synaptic function are usually reflected by alterations in the concentration of proteins in the presynaptic or at the post synaptic density [5]. Among them, neurogranin (Ng) is a 78 amino acid post synaptic protein highly enriched in the neuronal dendritic spines in cerebral cortex, amygdala, caudate, putamen, and the hippocampus, has been widely investigated in the context of AD [6]. A decrease in Ng concentration in the brain and an elevation in cerebrospinal fluid (CSF) are related to CNS dysfunction in AD. It is associated with tau and amyloid pathology and poorer cognitive performance, cortical glucose metabolism, and hippocampal volumes in individuals with mild cognitive impairment and AD [7]. However, reports on the alterations in synaptic homeostasis in other neurodegenerative disorders are poorly available [8].
With respect to disease-associated intracellular dynamics, studying the organelles provide new insights for understanding the pathobiology of neurodegenerative disorders. Mitochondrial dysfunction is an early feature of AD which occurs prior to the development of Aβ plaque formation [9, 10]. Mitofusin-2 (MFN-2), a transmembrane GTPase located in the outer mitochondrial membrane is a key player of mitochondrial activities such as fusion, trafficking, turnover, and contacts with other organelles. Impaired activity of MFN-2 affects mitochondrial shape, function, and distribution within the cell. Fusion is important for the health and physiological functions of mitochondria, including complementation of damaged mitochondrial DNA, and maintenance of membrane potential [11].
Lysosomal-associated membrane glycoprotein-2 (LAMP-2) is required for the fusion of autophagosomes with the lysosomes which is the final stage of macroautophagy, and it serves as a receptor in the lysosomal membrane for the substrate proteins of chaperone-mediated autophagy [12]. Neurons especially rely on lysosomal degradation since they live extremely long without cell division. Therefore, the defects in autophagy and endocytosis are relevant to neurodegenerative diseases. Altered expression and activity of a number of lysosomal proteins in CSF have been reported in association with AD [13].
Morphological defects such as Golgi fragmentation have been observed in both animal and cell models of AD and is considered as an early irreversible process [14]. In particular, golgin A4 is a protein associated with the trans-Golgi network and functions in transport of golgin specific cargo proteins. An elevation of golgin A4 in the CSF of AD patients has recently been reported [15]. Gradual failure of these organelles coupled with synaptic loss may contribute to age-associated neurodegenerative disorders [16].
Based on this premise, the objective of our study is to investigate the levels of synaptic protein, Ng and organellar proteins, MFN-2, LAMP-2, and golgin A4 from neuronal exosomes as indicators of underlying pathological changes in AD and FTD brains. The selected proteins have been analyzed by immunoblotting method. The extent of neurodegeneration was assessed by the quantification of NfL in exosomal extracts by using single molecule array (Simoa). Extent of alterations in these candidate proteins in AD and FTD are discussed.
MATERIALS AND METHODS
Subjects
The participants were selected from the Geriatric Psychiatry Unit of NIMHANS, Bengaluru, India. The study was approved by the Institutional Human Ethics committee, NIMHANS (IEC Number NIMHANS/ IEC (BS & NS DIV.)/14th MEETING/2018 dated September 17, 2019). Written informed consent was obtained from all participants. The socio-demographic details of the participants and the criteria followed for their selection and clinical assessment scores are described earlier [17].
Isolation and characterization of exosomes
Exosomes were isolated from 1 mL of plasma by using total exosome isolation kit (Thermofisher Scientific) following the manufacturer’s instructions. The isolated exosomes were suspended in 1 mL of 0.01 M phosphate buffered saline (PBS), pH 7.4. Exosomes were characterized by using transmission electron microscopy (TEM). The exosomes were processed by negative staining method using freshly prepared 1% phosphotungstic acid. 20μL of the exosome suspension was placed onto collodion-coated 400 mesh copper grids. After 5 min, excess suspension was removed with filter paper and allowed to dry. The grids were then dipped in the stain and allowed to dry for 15 min. All the grid handling practices were performed under sterile conditions. The exosome grids were viewed under Jeol plus 1200 TEM, Japan; at magnification x15000.
Exosomal NfL measurement by Single molecule array (Simoa) technique
The exosomal proteins were extracted by sonication (3 cycles, 10 s each) in 0.5% triton X-100 in 0.1 M PBS. The mixture was then centrifuged at 4000 rpm for 5 min at 4°C. The clear supernatant was aliquoted and stored at –80°C for further analysis. Concentration of NfL in the total exosomal protein extracts was measured by Simoa using HD-X Analyser (Quanterix, USA) with Simoa NF-light Advantage Kit following the manufacturer’s instructions and standard procedures. Lower limit of quantification of the assay was 0.174 pg/mL and the lower limit of detection was 0.038 pg/mL.
Generation of antibodies to N-terminal region of neurogranin and epitope analysis
The N terminal peptide corresponding to the primary amino acid sequence KPDDDILDIPLDDPGANAAAAKIQASFRGHMARKK (residues 11-45) used as antigen for polyclonal antibody production was custom synthesized by Merrifield’s solid phase synthesis (Biotech Desk Pvt Ltd, Hyderabad, India). The purity of the synthetic peptide was ascertained by HPLC using Phenomenex Luna C18 column (4.6 x 250 mm) on LC-2010 HT system (Shimadzu). Mobile phase contained 0.1% trifluoroacetic acid in water and acetonitrile (0–100% gradient). The fractions were eluted at a flow rate of 1 ml/min with detector wavelength set at 220 nm. The analyses revealed that the synthetic peptide was homogeneous to an extent of > 90%. For production of polyclonal anti-peptide antibodies, albino rabbits were procured from Institute’s Central Animal Research Facility. For primary injection, 1 mg of peptide in 500μl of distilled water emulsified with equal volume of Freund’s complete adjuvant was subcutaneously administered to rabbits. This was followed by administration of three boosters at 3-week intervals with half the dose in Freund’s incomplete adjuvant for each booster. Animal experiments involved in this study were approved by the Institutional Animal Ethics Committee (IAEC number AEC/66/395/N. C dated November 13, 2017). Analysis of the anti-peptide antiserum collected 10 days after the final booster indicated a titer of 1 : 8000 against N-terminal 11-45 AA peptide for 50% Bmax binding. Epitope mapping analysis of these antibodies was carried out against overlapping tridecapeptides spanning the entire sequence of neurogranin (1-78) wherein, cysteine residues at the N-terminal end were substituted with alanine, to avoid potential disulphide bridge formation and related conformational alterations that may interfere with linear epitope analysis.
SDS-poly acrylamide gel electrophoresis of total exosomal protein extract
Electrophoresis was carried out in a vertical slab electrophoresis system (Mini-PROTEAN Tetra cell, Bio-Rad) using 5% stacking gel and 6% or 10 % resolving gel. 100μg of the exosomal protein extract was mixed with 5X gel loading dye in the presence of β-mercaptoethanol, heated in a boiling water bath for 5 min and loaded into wells. A broad range of SDS-PAGE molecular weight marker (Cytiva, UK) was used and electrophoresed at 120 V.
Immunoblotting
The proteins were transferred to a 0.45μm pore-sized PVDF membrane (Sigma, P2938) using the semi-dry blotting technique at 100 mA, 1 h for proteins < 150 kDa and 200 mA, 2 h for proteins > 150 kDa. Blocking was performed overnight at 4°C using 3% BSA in 0.01 M PBS, pH 7.4. Incubations with the in house generated polyclonal rabbit Ng antibody, mouse monoclonal-L1CAM (sc-514360, Santa Cruz Biotechnology), LAMP-2 (sc-18822, Santa Cruz Biotechnology), and MFN-2 (sc-515647, Santa Cruz Biotechnology) antibodies (1 : 100 dilution) and rabbit monoclonal Golgin A4 antibodies (#MBS8291539, Mybiosource) (1 : 500 dilution) in 0.3% BSA in 0.01 M PBS were performed at 37°C for 2 h. The membranes were washed for 8×5 min each in wash buffer [50 mM phosphate, pH 7.4 containing 0.25% gelatin, 1.0 M NaCl, 5 mM EDTA and 0.05% Triton X-100] and then incubated with appropriate HRP-conjugated secondary antibody (Sigma) (1 : 2500 dilution) for 1.5 h at room temperature. Following 10 x 10 min washes each, the membranes were developed for 1 min with ECL western blotting detection reagent (Sigma) in BioRad ChemiDoc MP imaging system. The densitometric analysis of the protein bands was performed using Image lab version 5.2.
Statistical analysis
The data were analyzed using Graphpad version 8.4.3 (Prism; Graphpad Software Inc, San Diego, CA, USA). Prior to analysis the outliers were removed by the software. Exosomal NfL measurements and the signal intensities from the immunoblots followed a non-normal distribution and hence analyzed by non-parametric Kruskal Wallis test followed by Dunn’s post hoc test for multiple comparisons. Data were reported as median and interquartile ranges (IQR). Statistical significance was defined for p-values less than or equal to 0.05 (*), 0.01 (**), and 0.001(***).
RESULTS
Participants selected for the study included HC (n = 10), AD (n = 9), and FTD (n = 10) subjects with their age in the range of 52–88, 52–65, and 56–62 years respectively. With respect to gender distribution, 6 males and 4 females were included in each HC and FTD group whereas 5 males and 4 females participated in AD group. In FTD and AD groups, the duration from the reported onset of cognitive disturbances till the time point of sample collection ranged from 0–2 years. Clinical Dementia Rating (CDR) score of HC group was 0, and that of AD and FTD groups were > 1. The Hindi Mental State Examination (HMSE) scores of the participants in the HC, AD and FTD groups were 29.6±1.43, 5.89±1.76, and 15±7.56 respectively.
Characterization of circulating exosomes in AD and FTD by transmission electron microscopy
Exosomes fractionated from the plasma of HC, AD, and FTD subjects were analyzed by Jeol plus 1200 TEM, Japan. The results confirmed round/oval shaped vesicles of diameter ranging from 60–90 nm in all the study groups. The representative images of exosomes from each group are shown in Fig. 1 (Supplementary Figure 1).
Fig. 1
Electron micrographs of plasma exosomes. Representative images of samples from two independent subjects in each group are shown. Panel A and B correspond to exosomes from healthy controls; Panel C and D represent exosomes purified from AD subjects; exosomes fractionated from plasma samples of frontotemporal dementia subjects are shown in panel E and F. [Magnification = x15000].
![Electron micrographs of plasma exosomes. Representative images of samples from two independent subjects in each group are shown. Panel A and B correspond to exosomes from healthy controls; Panel C and D represent exosomes purified from AD subjects; exosomes fractionated from plasma samples of frontotemporal dementia subjects are shown in panel E and F. [Magnification = x15000].](https://ip.ios.semcs.net:443/media/jad/2023/94-s1/jad-94-s1-jad220829/jad-94-jad220829-g001.jpg)
Quantitation of NfL as a measure of neurodegeneration
The quantification of NfL present in the exosomes was performed by using Simoa. The results showed that the median concentrations of NfL in the AD group [3.04 (1.62–6.620) pg/mL] and the FTD group [3.29 (1.92–4.46) pg/mL] were significantly higher than that of the HC group [0.60 (0.39–0.71) pg/mL] with p < 0.001 (Fig. 2).
Fig. 2
Comparison of exosomal NfL concentrations in HC, AD, and FTD subjects. Data represented as median (interquartile ranges [IQR]); HC [0.60 (0.39–0.71) pg/mL], AD [3.04 (1.62–6.620) pg/mL], and FTD [3.29 (1.92–4.46) pg/mL]. Boxes encompass IQR with median line and whiskers designate 10th–90th percentiles. Statistically significant difference of ***p < 0.001 recorded between groups.
![Comparison of exosomal NfL concentrations in HC, AD, and FTD subjects. Data represented as median (interquartile ranges [IQR]); HC [0.60 (0.39–0.71) pg/mL], AD [3.04 (1.62–6.620) pg/mL], and FTD [3.29 (1.92–4.46) pg/mL]. Boxes encompass IQR with median line and whiskers designate 10th–90th percentiles. Statistically significant difference of ***p < 0.001 recorded between groups.](https://ip.ios.semcs.net:443/media/jad/2023/94-s1/jad-94-s1-jad220829/jad-94-jad220829-g002.jpg)
Epitope mapping analysis of Ng antibodies
Prior to conducting western blotting analysis with Ng antibodies, as a first step, epitope mapping studies were carried out. Binding of protein-A Sepharose affinity purified antibodies to tridecapeptides spanning the entire sequence of neurogranin with 8 amino acid overlap between the consecutive 13-mer peptides was measured. Epitope scan analysis results revealed that the minimal sequence recognized by these antibodies spanned residues 11-23 (Table 1).
Table 1
Epitope scan analysis of Ng peptides with rabbit polyclonal antibodies
| Sequence of tridecapeptide | Immunoreactivity with Ng |
| peptide (11-45) antibodies | |
| MDAATENAASKPD (1-13) | – |
| ENAASKPDDDILD (6-18) | – |
| KPDDDILDIPLDD (11-23) | ++++ |
| ILDIPLDDPGANA (16-28) | – |
| LDDPGANAAAAKI (21-33) | – |
| ANAAAAKIQASFR (26-38) | – |
| AKIQASFRGHMAR (31-43) | – |
| SFRGHMARKKIKS (36-48) | – |
| MARKKIKSGERGR (41-53) | – |
| IKSGERGRKGPGP (46-58) | – |
| RGRKGPGPGGPGG (51-63) | – |
| PGPGGPGGAGVAR (56-68) | – |
| PGGAGVARGGAGG (61-73) | – |
| VARGGAGGGPSGD (66-78) | – |
(Amino acid number in the primary sequence of neurogranin is indicated in parentheses. Cysteine residues in the N-terminal peptide (1–13) substituted with alanine are shown underlined. Epitope peptide is shown in bold). ++++, very strong binding with optical density (OD) values > 3.0 at 450 nm. –, no binding, with OD values < 0.1 at 450 nm.
Immunoblotting analysis of synaptic and organellar markers in exosomal extracts from AD and FTD
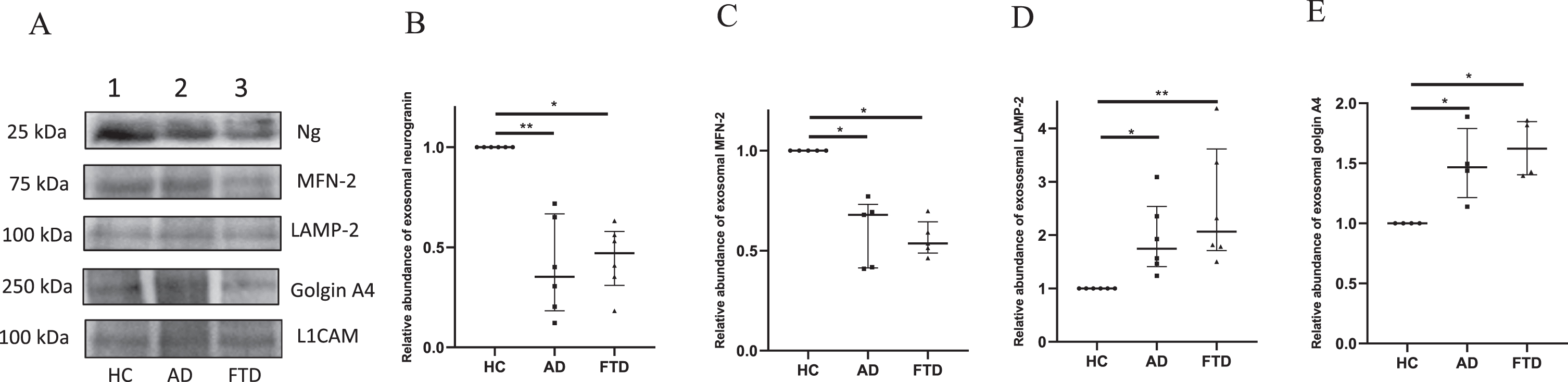
After ascertaining the binding pattern, Ng peptide antibodies were used for western blotting experiments. In addition, immunoblotting analyses were carried out for the organellar proteins MFN-2, LAMP-2, and golgin A4 in the pooled exosomal extracts of HC, AD, and FTD groups (Fig. 3, Supplementary Figure 2). The levels of neuron-specific exosomal marker L1CAM in the extracts were also analyzed by western blotting. All the western blot data were normalized with the signal intensity of L1CAM protein migrated at 100 kDa in the respective groups and the signal intensities have been compared with that of the HC group. The immunoblots and the densitometric analysis data are presented in Fig. 3 and Supplementary Figure 2. The analysis of Ng immunoreactive band migrated at 25 kDa showed a significant reduction in signal intensity in both AD and FTD by 65% and 53% respectively when compared with HC group. The median intensities in the AD [0.35 (0.18–0.67)] and FTD [0.47 (0.31–0.58)] groups were significantly lower than that of the HC group with p < 0.01 and p < 0.05 respectively. The median signal intensity of the bands corresponding to MFN-2 at 75 kDa in AD and FTD groups were [0.68 (0.41–0.73)], (p < 0.05) and [0.54 (0.49–0.64)], (p < 0.05) respectively, with a significant reduction in the signal intensity by 32% in AD and 46% in FTD when compared to HC. The median intensities of LAMP-2 immunoreactive band observed at 100 kDa in AD group [1.74 (1.41–2.54)] and FTD group [2.07 (1.71–3.62)] was significantly higher than that of the HC group with p < 0.05 and p < 0.01 respectively i.e., the relative abundance of LAMP-2 was 1.7 times higher in AD and 2 times higher in FTD than that of HC. The analysis of golgin A4 band observed at 250 kDa had an intensity 1.5 times higher in AD and 1.6 times higher in FTD than that of the controls. The median signal intensities of golgin immunoreactive band in AD and FTD groups were 1.47 (1.21–1.79) and 1.62 (1.4–1.85) respectively and both the intensities were significantly higher in comparison to HC with p < 0.05.
Fig. 3
Western blot analysis for immunodetection of L1 CAM, Ng, MFN-2, LAMP-2, and golgin A4 in exosomal protein extracts from HC, AD, and FTD subjects. In Panel A, representative immunoblots of the synaptic and organellar markers are shown with corresponding molecular weights indicated. In panel B to E, densitometric analyses of the signal intensities corresponding to synaptic and organellar proteins in the exosomes of HC, AD, and FTD groups are shown. Data presented as scatter plot with median value and interquartile ranges of signal intensities obtained from independent western blot experiments of neurogranin (n = 6), MFN-2 (n = 5), LAMP-2 (n = 6), and golgin A4 (n = 4). Statistical difference of p < 0.05 was considered significant (*p < 0.05 and **p < 0.01).
DISCUSSION
The shape and size of the exosomes in HC and AD groups were similar to that of the previously reported study from our laboratory [18] wherein the TEM characterization of exosomes was performed using Tecnai G2 Spirit Biotwin, The Netherlands. It was observed that the inter-instrument variation has not affected the quality of images. In addition to the HC and AD samples, the present study also included exosomes from FTD samples. The characteristics of the exosomes from this group showed no apparent difference in the appearance and size range. The negative staining study helped to confirm the typical morphology of the exosomes. Treatment with phosphotungstic acid resulted in the deposition of heavy metal salts on the surface of exosomes and allowed the visualization of exosomes [19].
NfL, an intermediate filament protein expressed exclusively in neurons, has emerged as a promising biomarker of neurodegeneration. CSF NfL concentration has reproducibly shown to reflect neurodegeneration in brain disorders [20]. In AD, plasma NfL level correlates strongly with the CSF NfL level and associates with cognitive decline and disease related structural brain changes [21]. While CSF and blood levels of NfL reflect the amount that is released into the interstitial fluid, the exosomal NfL is directly associated with the neuronal cell of origin. In our study, the concentration of exosomal NfL as measured by Simoa demonstrated significant increase in AD and FTD compared to HC, thus confirming the neurodegenerative status in disease groups. Current findings are one among the first few studies that has quantified exososmal NfL in AD and FTD.
Through the immunoblotting experiments we have assessed the levels of Ng, MFN-2, LAMP-2, and golgin A4 in the neuronal exosomes of study groups. The immunoblots showed that these proteins were present in detectable levels in the exosomes. Signal intensity of the L1CAM immunoreactive band at 100 kDa has been used to normalize the levels of other proteins under study. L1CAM is a member of cell adhesion molecules primarily expressed in nervous system and has been proven as a marker of exosomes of neuronal origin with a molecular weight of 100 kDa [22, 23].
Ng is a 7.6 kDa protein highly enriched in the dendritic spines of neurons. Ng functions in postsynaptic calcium signaling and memory formation via binding to calmodulin in a calcium dependent manner [24]. For the detection of Ng in the exosomal extracts by immunoblotting, we have used an in-house generated polyclonal antibody, with epitope mapped to N-terminal region (residues 11-23) in Ng. An immunoreactive band at 25 kDa was observed in the blot. In literature, multiple forms of neurogranin have been reported. For example, Ng from CSF and brain homogenates has been reported to migrate as 12 kDa species in gel electrophoresis whereas the same species in size exclusion chromatography, has eluted as 25 kDa fraction [25]. Bereczki et al. have reported 15 kDa Ng species in AD brain homogenates by electrophoresis [8]. Ng is a protein which is subjected to various post translational modifications such as the addition of N-terminal acetyl group, disulfide bridge at C3-C4/C4-C9, glutathione additions at C3/C9, phosphorylation at the Serine, etc. which may contribute to the differences in the reported molecular forms of Ng [26]. Alternatively, association of Ng with binding partners such as calmodulin can also give rise to higher molecular form(s) [27] which awaits experimental confirmation.
The immunoreactivity produced by Ng antibody has shown a significant reduction in signal intensity in dementia groups compared to HC with the extent of decrease in the order of AD > FTD>HC, which was proportionately related to HMSE scores. Multiple studies have reported that CSF Ng is increased in AD correlating with total tau, phospho tau [28], and cognitive decline [29]. Current results are suggestive of an inverse relationship in the distribution of Ng between CSF and exosomes. Further, our current findings are in agreement with a recent study that reported a reduction in the concentration of synaptic proteins including Ng in the exosomes of AD as well as FTD when measured by ELISA [30]. Extensive synaptic loss and decrease in synaptic density in the autopsy analysis of FTD cortex tissues have also been documented [31]. The impaired intellectual and behavioral symptoms manifested in FTD has also been attributed to the decrease in post synaptic density [28]. Tau pathology also has been shown to cause synaptic damage through inflammatory processes which could be probably a late event in FTD syndrome [32]. Synaptic degeneration occurs as a part of any neurodegenerative process. But the main brain regions affected in AD such as parietal, temporal cortices, amygdala, and hippocampus are the regions with highest Ng expression [33]. In addition, AD characteristically exhibits a remarkable alteration in the memory impairment at the early stages. This may begin with the subtle alterations that take place at the synaptic level [34]. In FTD, the distribution of neuropathology differs from that in AD, i.e., the FTD-related pathologies are more abundantly found in the frontal and temporal lobes. FTD is characterized by a variety of dysexecutive, language, and behavioral disturbances and cognitive impairment may occur at a later stage [35]. Altogether our data strongly supports that synaptic degeneration/dysfunction is an early event in AD indicating the role of Ng as a potential clinical marker of AD.
In the present study, the levels of MFN-2 was found to be reduced in AD and FTD groups to a similar extent. Mitochondria are dynamic organelles that undergo constant fission and fusion. In AD, Aβ overproduction increases mitochondrial fission and simultaneously decreases mitochondrial fusion activity causing mitochondrial fragmentation and neuronal death [36]. Increased levels of mitochondrial fission proteins and decreased levels of fusion proteins in the frontal cortical tissues of AD patients have been reported [37]. Similar to AD, mitochondrial alterations are also observed in FTD, like deregulation of mitochondrial genes, significant reduction in the enzymatic activity of the mitochondrial complexes, etc. [38]. It is known that mitophagy is a key pathway of mitochondrial quality control mechanisms which is involved in the sequestration of defective mitochondria into autophagosomes for subsequent lysosomal degradation [39]. Upon the loss of inner mitochondrial membrane potential, PINK 1, a mitochondrial kinase is stabilized to the surface of the dysfunctional mitochondria, which then recruits Parkin, a ubiquitin ligase that ubiquitinates mitochondrial outer membrane proteins including MFN-2. This Parkin-mediated turnover of MFN-2 may lead to a shift in the balance of mitochondrial dynamics towards decreased fusion and increased fission [40, 41].
Mitochondria can make contact with ER to regulate vital cellular homeostatic functions including mitochondrial quality control, lipid metabolism, calcium homeostasis, the unfolded protein response, and ER stress. There are several mitochondria-ER contact sites (MERCS) tethering proteins that maintain these contacts and facilitate their function. MFN2 acts as a physical tether and maintains Ca2 + homeostasis and regulates mitochondrial morphological changes. Dysfunctional MERCS have been implicated in neurodegenerative diseases including AD and FTD [42]. Mitochondrial fusion process has not been thoroughly investigated in frontotemporal dementia, but a few reports have demonstrated damage to ER-mitochondria signaling in response to the TDP-43 inclusions associated with FTD [43]. Any minor alteration in mitochondrial metabolism accelerates production of reactive oxygen species, reduced ATP production and causes cellular damage. Our results suggest an imbalance in mitochondrial fusion-fission process in the neurodegenerative diseases under study.
Our immunoblotting studies with LAMP-2 has shown an immunoreactive band at 100 kDa [44]. There was a significant upregulation in the LAMP-2 levels in both AD and FTD in comparison to HC in the order of FTD > AD>HC which was inversely related to HMSE scores. These findings concur with our previously reported elevated LAMP-2 levels in exosomal protein extracts of AD subjects quantified by using ELISA [18]. Insufficient clearance of neurotoxic proteins has been implicated in several neurodegenerative disorders. Chaperone mediated autophagy (CMA) plays a crucial role in cellular proteostasis under physiological as well as pathological conditions. LAMP-2 acts as a receptor for the cytosolic cargo in CMA. CMA activity is upregulated in neurodegeneration [45]. Postmortem of AD brain has shown accumulation of large autophagic vacuoles with undegraded aggregates including Aβ. Neuronal endosomal enlargement and upregulation of genes related to endocytosis and associated proteins are early events observed in AD. These changes are followed by an increase in lysosome biogenesis, autophagy impairment, alterations in genes and proteins related to lysosomal network suggesting a failure of lysosomal protein degradation in AD [46]. In FTD, the major genetic causes like GRN mutations, C9orf72 expansions play a critical role in lysosome function and autophagy [47]. Our findings from LAMP-2 immunoblotting suggest that the protein clearance mechanisms might be compromised in AD, FTD, and related protein aggregation disorders.
Densitometric analysis of the golgin A4 bands has shown an overall increase in the signal intensity in both the dementia groups. Golgin A4 concentration has been reported to be significantly higher in CSF from patients with AD than in CSF of controls. Immunohistochemical studies of AD brains have also demonstrated the accumulation of golgin A4 granules within the cytoplasm of neuronal cells. Elevated golgin A4 levels may indicate an early response due to either Golgi alterations or cellular stress associated with elevated levels of misfolded proteins [16]. Golgin A4 also plays a critical role during the initial steps of autophagy because it has been detected in the autophagosomes during the biogenesis [15]. Golgi fragmentation has been observed in the neurons of AD patients but the molecular mechanism leading to Golgi fragmentation in AD remains elusive. It has been reported that the Golgi fragmentation enhances vesicle budding from the Golgi membranes and accelerates protein trafficking. In response to protein aggregation, unstacking and fragmentation of Golgi accelerates the protein trafficking which in turn upregulates the proteins associated with the transport machinery [48]. It has also been proposed that Aβ accumulation accelerates the activity of kinases which phosphorylates Golgi structural proteins and causes Golgi fragmentation. The phosphorylation of Golgi structural proteins can also contribute to the fragmentation in AD. The inhibition of kinases has also resulted in the reduction of Golgi fragmentation and rescue of the organelle in cell culture studies [49]. The Golgi alterations in other neurodegenerative disorders have not been thoroughly investigated, although, Golgi fragmentation as a phenomenon has been observed in many protein aggregation disorders [50].
In summary, this study has explored the analysis of synaptic and organellar-specific proteins in the neuronal exosomes of HC, AD, and FTD. The immunoblotting studies of Ng from the exosomes showed significant synaptic degeneration in the dementia groups. Although there was an overlap in Ng levels in AD and FTD, relatively lower Ng levels in AD than FTD may contribute to greater cognitive deficits seen early on in AD. It was observed that, changes in the structural and functional dynamics of organelles may be common pathological features in AD and FTD. Our findings highlight the clinical utility of plasma exosomes as a less invasive mode of assessment of the changes in neurons from which they are derived from. Large-scale studies with longitudinal monitoring of these synaptic and organellar markers may support evaluation of cellular and molecular changes in the course of neurodegenerative disease progression/treatment response. While simultaneous analysis of markers for three different organelles which are crucial in cellular protein homeostasis forms the strength of the present study, smaller sample size and lack of inclusion of other neurodegenerative dementias form limitations to the present study. Scope for future research includes efforts in reducing Golgi fragmentation may help improve trafficking of proteins involved in fast axonal transport [51] including tau, thus reducing pathogenesis of protein aggregation disorders. Further, means to improve lysosomal functions through upregulation of lysosomal proteins/enzymes and pH regulation [52], adapting strategies to induce mitochondrial biogenesis through improving mitochondrial-nuclear communication [53] may potentially offer advantage to disease modifying therapies for proteinopathic neurodegenerative dementias.
ACKNOWLEDGMENTS
Financial assistance from SERB, New Delhi, India [Grant no. EMR/2017/ 000849] and ICMR, New Delhi, India [Grant no.5/4-5/181/Neuro/2018-NCD-I] are gratefully acknowledged. GK is a recipient of Junior Research Fellowship from Council of Scientific and Industrial Research, New Delhi, India.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/22-0829r1).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-220829.
REFERENCES
[1] | Saeedi S , Israel S , Nagy C , Turecki G ((2019) ) The emerging role of exosomes in mental disorders. Transl Psychiatry 9: , 122. |
[2] | Kalluri R , LeBleu VS ((2020) ) The biology, function, and biomedical applications of exosomes. Science 367: , eaau6977. |
[3] | Simrée J , Ashton NJ , Blennow K , Zetterberg H ((2020) ) An update on fluid biomarkers for neurodegenerative diseases: recent success and challenges ahead. Curr Opin Neurobiol 61: , 29–39. |
[4] | Dugger BN , Dickson DW ((2017) ) Pathology of neurodegenerative diseases. Cold Spring Harb Perspect Biol 9: , a028035. |
[5] | Becker B , Nazir FH , Brinkmalm G , Camporesi E , Kvartsberg H , Portelius E , Boström M , Kalm M , Höglund K , Olsson M , Zetterberg H , Blennow K ((2018) ) Alzheimer-associated cerebrospinal fluid fragments of neurogranin are generated by Calpain-1 and prolyl endopeptidase. Mol Neurodegener 13: , 47. |
[6] | Blennow K , Zetterberg H ((2018) ) Biomarkers for Alzheimer’s disease: current status and prospects for the future. J Intern Med 284: , 643–663. |
[7] | Portelius E , Olsson B , Höglund K , Cullen NC , Kvartsberg H , Andreasson U , Zetterberg H , Sandelius Å , Shaw LM , Lee V , Irwin DJ , Grossman M , Weintraub D , Chen-Plotkin A , Wolk DA , McCluskey L , Elman L , McBride J , Toledo JB , Trojanowski JQ , Blennow K ((2018) ) Cerebrospinal fluid neurogranin concentration in neurodegeneration: relation to clinical phenotypes and neuropathology. Acta Neuropathol 136: , 363–376. |
[8] | Bereczki E , Francis PT , Howlett D , Pereira JB , Höglund K , Bogstedt A , Cedazo-Minguez A , Baek JH , Hortobáagyi T , Attems J , Ballard C , Aarsland D ((2016) ) Synaptic proteins predict cognitive decline in Alzheimer’s disease and Lewy body dementia. Alzheimers Dement 12: , 1149–1158. |
[9] | Filadi R , Pendin D , Pizzo P ((2018) ) Mitofusin 2: from functions to disease. Cell Death Dis 9: , 330. |
[10] | Sita G , Hrelia P , Graziosi A , Morroni F ((2020) ) Back to the fusion: mitofusin-2 in Alzheimer’s disease. J Clin Med 9: , 126. |
[11] | Li YJ , Cao YL , Feng JX , Qi Y , Meng S , Yang JF , Zhong YT , Kang S , Chen X , Lan L , Luo L , Yu B , Chen S , Chan DC , Hu J , Gao S ((2019) ) Structural insights of human mitofusin-2 into mitochondrial fusion and CMT2A onset. Nat Commun 10: , 4914. |
[12] | Van Acker ZP , Bretou M , Annaert W ((2019) ) Endo-lysosomal dysregulations and late-onset Alzheimer’s disease: impact of genetic risk factors. Mol Neurodegener 14: , 20. |
[13] | Armstrong A , Mattsso N , Appelqvist H , Janefjord C , Sandin L , Agholme L , Olsson B , Svensson S , Blennow K , Zetterberg H , Kågedal K ((2014) ) Lysosomal network proteins as potential novel CSF biomarkers for Alzheimer’s disease. Neuromolecular Med 16: , 150–160. |
[14] | Asik RM , Suganthy N , Aarifa MA , Kumar A , Szigeti K , Mathe D , Gulyáas B , Archunan G , Padmanabhan P ((2021) ) Alzheimer’s disease: A molecular view of β-amyloid induced morbific events. Biomedicines 9: , 1126. |
[15] | Kork F , Jankowski J , Goswami A , Weis J , Brook G , Yamoah A , Anink J , Aronica E , Fritz S , Huck C , Schipke C , Peters O , Tepel M , Noels H , Jankowski V ((2018) ) Golgin A4 in CSF and granulovacuolar degenerations of patients with Alzheimer disease. Neurology 91: , e1799–e1808. |
[16] | Petkovic M , O’Brien CE , Jan YN ((2021) ) Interorganelle communication, aging, and neurodegeneration. Genes Dev 35: , 449–469. |
[17] | Subramanian S , Krishna G , Sivakumar PT , Dahale AB , Kumar JS , Sinha P , Varghese M ((2021) ) Plasma neurofilament L to amyloid β42 ratio in differentiating Alzheimer’s type from non-Alzheimer’s dementia: A cross-sectional pilot study from India. Asian J Psychiatr 66: , 102914. |
[18] | Krishna G , Anu KN , Kumar RS , Sagar BC , Philip M , Dahale AB , Issac TG , Mukku S , Sivakumar PT , Subramanian S ((2020) ) Higher levels of lysosomal associated membrane protein-2 (LAMP-2) in plasma exosomes from Alzheimer’s disease: An exploratory study from South India. Asian J Psychiatr 48: , 101898. |
[19] | Corso G , Mäger , Lee Y , Görgens A , Bultema J , Giebel B , Wood M , Nordin JZ , Andaloussi SE ((2017) ) Reproducible and scalable purification of extracellular vesicles using combined bind-elute and size exclusion chromatography. Sci Rep 7: , 11561. |
[20] | Andersson E , Janelidze S , Lampinen B , Nilsson M , Leuzy A , Stomrud E , Blennow K , Zetterberg H , Hansson O ((2020) ) Blood and cerebrospinal fluid neurofilament light differentially detect neurodegeneration in early Alzheimer’s disease. Neurobiol Aging 95: , 143–153. |
[21] | Zetterberg H , Skillbäck T , Mattsson N , Trojanowski JQ , Portelius E , Shaw LM , Weiner MW , Blennow K , Alzheimer’s Disease Neuroimaging Initiative ((2016) ) Association of cerebrospinal fluid neurofilament light concentration with Alzheimer disease progression. JAMA Neurol 73: , 60–67. |
[22] | Zhao A , Li Y , Yan Y , Qiu Y , Li B , Xu W , Wang Y , Liu J , Deng Y ((2020) ) Increased prediction value of biomarker combinations for the conversion of mild cognitive impairment to Alzheimer’s dementia. Transl Neurodegener 9: , 30. |
[23] | Mustapic M , Eitan E , Werner JK Jr , Berkowitz ST , Lazaropoulos MP , Tran J , Goetzl EJ , Kapogiannis D ((2017) ) Plasma extracellular vesicles enriched for neuronal origin: a potential window into brain pathologic processes. Front Neurosci 11: , 278. |
[24] | Petersen A , Gerges NZ ((2015) ) Neurogranin regulates CaM dynamics at dendritic spines. Sci Rep 5: , 11135. |
[25] | Nazir FH , Camporesi E , Brinkmalm G , Lashley T , Toomey CE , Kvartsberg H , Zetterberg H , Blennow K , Becker B ((2021) ) Molecular forms of neurogranin in cerebrospinal fluid. J Neurochem 157: , 816–833. |
[26] | Kvartsberg H , Lashley T , Murray CE , Brinkmalm G , Cullen NC , Höglund K , Zetterberg H , Blennow K , Portelius E ((2019) ) The intact postsynaptic protein neurogranin is reduced in brain tissue from patients with familial and sporadic Alzheimer’s disease. Acta Neuropathol 137: , 89–102. |
[27] | Willemse EAJ , De Vos A , Herries EM , Andreasson U , Engelborghs S , van der Flier WM , Scheltens P , Crimmins D , Ladenson JH , Vanmechelen E , Zetterberg H , Fagan AM , Blennow K , Bjerke M , Teunissen CE ((2018) ) Neurogranin as cerebrospinal fluid biomarker for Alzheimer disease: an assay comparison study. Clin Chem 64: , 927–937. |
[28] | Kester MI , Teunissen CE , Crimmins DL , Herries EM , Ladenson JH , Scheltens P , Van der Flier WM , Morris JC , Holtzman DM , Fagan AM ((2015) ) Neurogranin as a cerebrospinal fluid biomarker for synaptic loss in symptomatic Alzheimer disease. JAMA Neurol 72: , 1275–1280. |
[29] | Xue M , Sun FR , Ou YN , Shen XN , Li HQ , Huang YY , Dong Q , Tan L , Yu JT , Alzheimer’s Disease Neuroimaging Initiative ((2020) ) Association of cerebrospinal fluid neurogranin levels with cognition and neurodegeneration in Alzheimer’s disease. Aging (Albany NY) 12: , 9365–9379. |
[30] | Goetzl EJ , Kapogiannis D , Schwartz JB , Lobach IV , Goetzl L , Abner EL , Jicha GA , Karydas AM , Boxer A , Miller BL ((2016) ) Decreased synaptic proteins in neuronal exosomes of frontotemporal dementia and Alzheimer’s disease. FASEB J 30: , 4141–4148. |
[31] | Salmon E , Bahri MA , Plenevaux A , Becker G , Seret A , Delhaye E , Degueldre C , Balteau E , Lemaire C , Luxen A , Bastin C ((2021) ) In vivo exploration of synaptic projections in frontotemporal dementia. Sci Rep 11: , 16092. |
[32] | Ferrer I ((1999) ) Neurons and their dendrites in frontotemporal dementia. Dement Geriatr Cogn Disord 10: Suppl 1, 55–60. |
[33] | Brunello CA , Merezhko M , Uronen RL , Huttunen H ((2020) ) Mechanisms of secretion and spreading of pathological tau protein. Cell Mol Life Sci 77: , 1721–1744. |
[34] | Nilsson J , Gobom J , Sjödin S , Brinkmalm G , Ashton NJ , Svensson J , Johansson P , Portelius E , Zetterberg H , Blennow K , Brinkmalm A ((2021) ) Cerebrospinal fluid biomarker panel for synaptic dysfunction in Alzheimer’s disease. Alzheimers Dement (Amst) 13: , e12179. |
[35] | Park LQ , Harvey D , Johnson J , Farias ST ((2015) ) Deficits in everyday function differ in AD and FTD. Alzheimer Dis Assoc Disord 29: , 301–306. |
[36] | Shields LY , Li H , Nguyen K , Kim H , Doric Z , Garcia JH , Gill TM , Haddad D , Vossel K , Calvert M , Nakamura K ((2021) ) Mitochondrial fission is a critical modulator of mutant APP-induced neural toxicity. J Biol Chem 296: , 100469. |
[37] | Manczak M , Calkins MJ , Reddy PH ((2011) ) Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Hum Mol Genet 20: , 2495–2509. |
[38] | Leskelä S , Huber N , Hoffmann D , Rostalski H , Remes AM , Takalo M , Hiltunen M , Haapasalo A ((2021) ) Expression of C9orf72 hexanucleotide repeat expansion leads to formation of RNA foci and dipeptide repeat proteins but does not influence autophagy or proteasomal function in neuronal cells. Biochim Biophys Acta Mol Cell Res 1868: , 119021. |
[39] | Cai Q , Jeong YY ((2020) ) Mitophagy in Alzheimer’s disease and other age-related neurodegenerative diseases. Cells 9: , 150. |
[40] | McLelland GL , Goiran T , Yi W , Dorval G , Chen CX , Lauinger ND , Krahn AI , Valimehr S , Rakovic A , Rouiller I , Durcan TM , Trempe JF , Fon EA ((2018) ) Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent release of ER from mitochondria to drive mitophagy. ELife 7: , e32866. |
[41] | Rana A , Oliveira MP , Khamoui AV , Aparicio R , Rera M , Rossiter HB , Walker DW ((2017) ) Promoting Drp1-mediated mitochondrial fission in midlife prolongs healthy lifespan of Drosophila melanogaster. Nat Commun 8: , 448. |
[42] | Wilson EL , Metzakopian E ((2021) ) ER-mitochondria contact sites in neurodegeneration: genetic screening approaches to investigate novel disease mechanisms. Cell Death Differ 28: , 1804–1821. |
[43] | Lau D , Hartopp N , Welsh NJ , Mueller S , Glennon EB , Mórotz GM , Annibali A , Gomez-Suaga P , Stoica R , Paillusson S , Miller C ((2018) ) Disruption of ER-mitochondria signalling in fronto-temporal dementia and related amyotrophic lateral sclerosis. Cell Death Dis 9: , 327. |
[44] | Ferreira JV , Soares AR , Ramalho J , Carvalho CM , Cardoso MH , Pintado P , Carvalho AS , Beck HC , Matthiesen R , Zuzarte M , Girão H , van Niel G , Pereira P ((2022) ) LAMP2A regulates the loading of proteins into exosomes. Sci Adv 8: , eabm1140. |
[45] | Kanno H , Handa K , Murakami T , Aizawa T , Ozawa H ((2022) ) Chaperone-mediated autophagy in neurodegenerative diseases and acute neurological insults in the central nervous system. Cells 11: , 1205. |
[46] | Uddin MS , Stachowiak A , Mamun AA , Tzvetkov NT , Takeda S , Atanasov AG , Bergantin LB , Abdel-Daim MM , Stankiewicz AM ((2018) ) Autophagy and Alzheimer’s disease: from molecular mechanisms to therapeutic implications. Front Aging Neurosci 10: , 04. |
[47] | Root J , Merino P , Nuckols A , Johnson M , Kukar T ((2021) ) Lysosome dysfunction as a cause of neurodegenerative diseases: Lessons from frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol Dis 154: , 105360. |
[48] | Thayer DA , Jan YN , Jan LY ((2013) ) Increased neuronal activity fragments the Golgi complex. Proc Natl Acad Sci U S A 110: , 1482–1487. |
[49] | Joshi G , Chi Y , Huang Z , Wang Y ((2014) ) Aβ-induced Golgi fragmentation in Alzheimer’s disease enhances Aβ production. Proc Natl Acad Sci U S A 111: , E1230–E1239. |
[50] | Martínez-Menáarguez JÁ , Tomáas M , Martínez-Martínez N , Martínez-Alonso E ((2019) ) Golgi fragmentation in neurodegenerative diseases: is there a common cause? Cells 8: , 748. |
[51] | Hammerschlag R , Stone GC , Bolen FA , Lindsey JD , Ellisman MH ((1982) ) Evidence that all newly synthesized proteins destined for fast axonal transport pass through the Golgi apparatus. J Cell Biol 93: , 568–575. |
[52] | Koh JY , Kim HN , Hwang JJ , Kim YH , Park SE ((2019) ) Lysosomal dysfunction in proteinopathic neurodegenerative disorders: possible therapeutic roles of cAMP and zinc. Mol Brain 12: , 18. |
[53] | Zhu D , Li X , Tian Y ((2022) ) Mitochondrial-to-nuclear communication in aging: an epigenetic perspective. Trends Biochem Sci 47: , 645–659. |


