
The Novel Role of Mitochondrial Citrate Synthase and Citrate in the Pathophysiology of Alzheimer’s Disease
Abstract
Citrate synthase is a key mitochondrial enzyme that utilizes acetyl-CoA and oxaloacetate to form citrate in the mitochondrial membrane, which participates in energy production in the TCA cycle and linked to the electron transport chain. Citrate transports through a citrate malate pump and synthesizes acetyl-CoA and acetylcholine (ACh) in neuronal cytoplasm. In a mature brain, acetyl-CoA is mainly utilized for ACh synthesis and is responsible for memory and cognition. Studies have shown low citrate synthase in different regions of brain in Alzheimer’s disease (AD) patients, which reduces mitochondrial citrate, cellular bioenergetics, neurocytoplasmic citrate, acetyl-CoA, and ACh synthesis. Reduced citrate mediated low energy favors amyloid-β (Aβ) aggregation. Citrate inhibits Aβ25–35 and Aβ1–40 aggregation in vitro. Hence, citrate can be a better therapeutic option for AD by improving cellular energy and ACh synthesis, and inhibiting Aβ aggregation, which prevents tau hyperphosphorylation and glycogen synthase kinase-3 beta. Therefore, we need clinical studies if citrate reverses Aβ deposition by balancing mitochondrial energy pathway and neurocytoplasmic ACh production. Furthermore, in AD’s silent phase pathophysiology, when neuronal cells are highly active, they shift ATP utilization from oxidative phosphorylation to glycolysis and prevent excessive generation of hydrogen peroxide and reactive oxygen species (oxidative stress) as neuroprotective action, which upregulates glucose transporter-3 (GLUT3) and pyruvate dehydrogenase kinase-3 (PDK3). PDK3 inhibits pyruvate dehydrogenase, which decreases mitochondrial-acetyl-CoA, citrate, and cellular bioenergetics, and decreases neurocytoplasmic citrate, acetyl-CoA, and ACh formation, thus initiating AD pathophysiology. Therefore, GLUT3 and PDK3 can be biomarkers for silent phase of AD.
INTRODUCTION
Alzheimer’s disease (AD) is a chronic progressive neurodegenerative disease that affects the elderly population and results in dementia [1]. It is the sixth leading cause of death in elderly people. AD leads to declines in cognitive function like memory, language problems, planning, execution, and visuospatial problems and noncognitive psychiatric symptoms like depression, anxiety, agitation, hallucination, and delusion, which result in difficulties in daily activities of living [2]. Currently, 6.2 million people ≥65 years of age are living with AD, which may rise to 12.7 million by 2050 [3].
The underlying pathomechanism of AD starts years before the appearance of the clinical symptoms [4]. In AD, a decrease in acetylcholine (ACh) and its degradation by acetylcholinesterase or butyrylcholinesterase causes cognitive symptoms [5, 6]. Furthermore, senile plaques due to amyloid-beta (Aβ) deposition and neurofibrillary tangles due to tau hyperphosphorylation are two diagnostic parameters in AD [7], which cause N-methyl-D-aspartate (NMDA) induced excitotoxicity. Current treatment strategies such as acetylcholinesterase inhibitors and NMDA antagonists slow down the disease progression but do not cure the disease. So, we need to search for other pathways interlinked with the production of Aβ, its deposition, metabolism, clearance, and involvement of peripheral organs, which influence cognitive function and start the progression of AD [8]. In this review article, we investigate the role of the citrate synthase (CS) and its product citrate in AD and in pathophysiology of the prodromal phase of AD.
THE ROLE OF CITRATE SYNTHASE AND CITRATE IN CELLULAR BIOENERGETICS
Neuronal cells depend on glucose as their energy source. Glucose metabolizes to generate ATPs by glycolysis, tricarboxylic acid (TCA), and electron transport chain (ETC) pathways which involve glucokinase, CS, and cytochrome oxidase enzymes, respectively. CS is an important mitochondrial enzyme encoded in nuclear DNA. It is synthesized in the cytoplasmic ribosome and migrates to the mitochondrial matrix, where CS plays a key role in the TCA cycle in aerobic energy production by interconversion of metabolites.
Glycolysis produces pyruvate, which generates acetate. Acetate is carried with co-factor (acetyl-CoA) in the TCA cycle [9]. In neuronal cells, mitochondrial acetyl-CoA is utilized by CS [10–12]. In the TCA cycle, CS catalyzes the condensation of acetate residue of acetyl-CoA and oxaloacetate to form citrate [9].
This citrate in the TCA cycle is utilized to form 2NADH, 1FADH, and 1 ATP, which are further used in the ETC cycle to generate more ATPs using cytochrome oxidase enzymes (mitochondrial complex IV) by oxidative phosphorylation [13].
Low CS activity impairs the aerobic ATP synthesis pathway and decreases citrate or decreases cytoplasmic acetyl-CoA, which causes a low energy environment inside the cells [14]. A low-energy environment favors the aggregation of Aβ16–22 [15]. Several neurodegenerative conditions occur due to the inhibition of pyruvate dehydrogenase (PDH) that ultimately inhibits the acetyl-CoA formation in the mitochondria, which reduces the metabolic flux via the TCA cycle, decreases cellular bioenergetics, and inhibits the synthetic acetylation reaction in the neuronal cells sub-compartments.
Changes in the mitochondrial and neurocytoplasmic acetyl-CoA can significantly contribute to neurotoxic pathomechanisms and other neurodegenerative disorders [16]. Neuronal cells require a higher source of glucose energy than the non-excitable cell to strengthen the functions of neurotransmitters. Acetyl-CoA also serves as a direct substrate of energy for the brain neuronal cells [16]. ETC produces higher energy, and mitochondria perform these tasks if they are supplied with enough energy. At a low ATP state, mitochondria are unable to pump-out calcium from the inner mitochondrial membrane (IMM), and accumulation of Ca+2 causes mitochondrial dysfunction. Ca+2 overload in mitochondria promotes apoptosis by release of pro-apoptotic factors which lead to impaired mitochondrial membrane potential, decreased ATP production, increased reactive oxygen species (ROS) production, alteration of Ca+2 homeostasis, and mitochondrial dysfunction [17]. Aβ was also seen to cause mitochondrial and cytosolic Ca+2 overload in both in vivo and in vitro studies [18]. The fibroblasts of patients with AD show disrupted mitochondrial dynamics, cellular bioenergetics, and Ca+2 homeostasis [19, 20]. In AD, altered mitochondrial Ca+2 homeostasis causes defective mitophagy and accumulation of the damaged mitochondria leading to neurodegeneration, hence resulting in cell death by Ca+2 overload [21–24].
CS is the main rate limiting enzyme in the TCA cycle [25, 26]. Furthermore, CS is the quantitative marker of mitochondrial integrity, function, mass [27–29], and mitochondrial respiratory chain enzymes [30], as CS and citrate maintain the TCA, ETC energy cycle, and mitochondrial health. Lower levels of CS lead to decreased citrate, cytoplasmic acetyl-CoA, and ACh neurotransmitter formation, release, and re-synthesis [31].
CITRATE SYNTHASE AND CITRATE IN ACETYLCHOLINE SYNTHESIS
Oxaloacetate and acetyl-CoA are impermeable through mitochondrial membranes, whereas pyruvate and citrate can cross the mitochondrial membranes [32]. Citrate moves from mitochondrial membrane to neuronal cytoplasm through citrate-malate antiporter and converted to acetyl-CoA by ATP citrate lyase [33–38].
In neuronal cytoplasm, acetyl-CoA combines with choline in the presence of choline acetyl transferase (CAT) to form ACh [39, 40]. ACh is the main neurotransmitter for memory and cognition, and it is found to be deficient in AD [41]. Therefore, citrate as a substrate forms acetyl-CoA, that is utilized for ACh synthesis [38, 42, 43].
The mitochondrial acetyl-CoA is responsible for the level of neuro-cytoplasmic acetyl-CoA and ACh synthesis. The decrease in mitochondrial acetyl-CoA in neuronal cytoplasm results in many neurodegenerative diseases [16]. Pyruvate dehydrogenase complex (PDHC) and CAT correlated well in different brain regions, such as the caudate nucleus, amygdala, and putamen, but not in the frontal cortex or hippocampus. In patients with C-21 trisomy Down syndrome, the total activation and activities of PDHC and CAT were below normal, which increases the risk of AD. PDHC deficiency may also play a vital role in AD and Huntington’s disease because decreased PDHC activity correlates with the loss in cholinergic neurons and decreased cognitive functions due to less mitochondrial and neuronal cytosolic acetyl-CoA and less ACh synthesis [44–48].
Studies show that acetyl unit (acetyl-CoA) is transported as citrate for ACh synthesis and lipogenesis for brain tissues [35, 38, 49, 50]. In the immature brain, Acetyl-CoA is utilized in large amounts for the lipid synthesis that is necessary for structural element and myelin formation of neuronal tissue, while in the mature brain, acetyl-CoA is more utilized in the formation of ACh. Both glucose and ketone bodies are precursors for acetyl-CoA in suckling animal brains, while glucose is an exclusive substrate in the adult brain [38, 51, 52]. Acetyl-CoA has a significant role in neurodegenerative disease for the survival and death of the cholinergic neurons [48] and also has a role in epigenetic regulation [53] (Fig. 1).
Fig. 1
Citrate synthase and citrate in cellular bioenergetics and ACh formation, PDH, pyruvate dehydrogenase; TCA cycle, tricarboxylic acid cycle; CAT, choline acetyltransferase; ACL, ATP citrate lyase; ACh, acetylcholine; AChE, acetylcholinesterase; COX, cytochrome c oxidase.
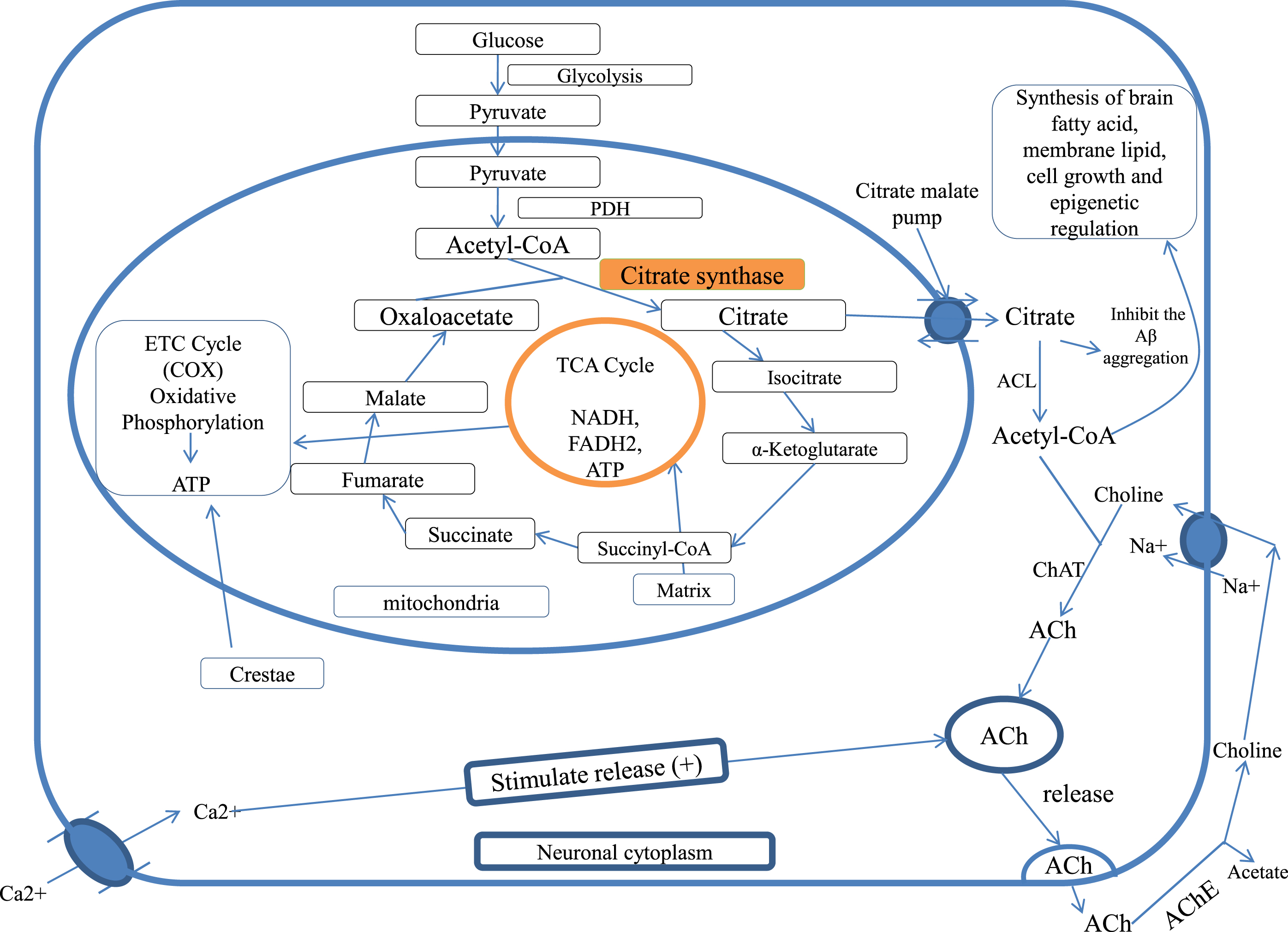
CITRATE SYNTHASE AND CITRATE IN ALZHEIMER’S DISEASE
Neuronal cells require higher amounts of overall energy for functioning neurotransmitters due to their continuous depolarization and repolarization. Acetyl-CoA is the direct source of energy for the brain cells. Neuronal cells mainly utilize the energy by oxidative phosphorylation [16].
The reduced activity of cytochrome c oxidase (COX) is found in neurodegenerative and non–degenerative AD tissue due to mitochondrial mutation [54]. COX maximizes mitochondrial energy production; therefore, when COX activity is reduced, it results in a lower energy production, which favors Aβ16 - 22 aggregation [15]. Aβ deposition in AD causes cytotoxicity by increasing oxidative stress, lipid peroxidation of the plasma membrane, ion motive ATPase impairment, GPCR uncoupling, glutamate uptake, and altering of calcium homeostasis intracellularly [55, 56].
Furthermore, Aβ generates tau protein kinase1 (TPK1) [57, 58]. TPK1 hyperphosphorylates tau [59] and inhibits PDH [60], which inhibits acetyl-CoA and citrate in mitochondria resulting in a decrease of ACh synthesis and causes memory and cognition impairment [15]. Aβ interacts with mitochondrial cyclophilin D and causes cell death [61]. Acetyl-CoA is the direct energy source for the brain cells [16]. Low CS activity impairs ATP synthesis aerobically [14] and decreases acetyl-CoA. Low energy favors Aβ16 - 22 aggregation [15] that further induces TPK1, which causes tau hyperphosphorylation and PDH inhibition. Thus, the same cycle continues with the inhibition of acetyl-CoA and ACh synthesis. Screening of patients with AD showed an overall reduction in brain COX activities. Hence, mitochondrial dysfunction in the cerebral or extracerebral contributes to AD pathology [54]. Therefore, instead of Aβ causing AD, low CS activity and citrate mediated low ACh production and bioenergetics could be the main reason for underlying AD symptoms.
Human teratocarcinoma cell line (NT2 cell) treated with Aβ25 - 35 and Aβ1 - 42 differentiates into neurons, secretes amyloid, and exhibits an excitotoxic response [62, 63]. Aβ1 - 42 is more toxic than Aβ25 - 35. Aβ25 - 35 leads to ATP depletion, reduces membrane potential of mitochondria, and inhibits mitochondrial respiratory chain complexes (complex 1-NADH ubiquinone oxidoreductase, complex 2/3-succinate cytochrome-c oxidoreductase, and complex 4-cytochrome–c oxidase) enzymes [56]. Pre-incubation of NT2 cells with antioxidants like vitamin E, melatonin, idebenone, and glutathione for 22 h prevents Aβ25 - 35 toxicity [64].
Interestingly, a lack of mitochondrial DNA prevents oxidative phosphorylation in NT2 cells. Furthermore, NT2 cells remained unaffected by Aβ1 - 42 or Aβ25 - 35. This proved that Aβ causes mitochondrial dysfunction and oxidative stress [56], promoting neuronal death and degeneration [65]. This shows that mitochondrial respiratory chain malfunction causes Aβ toxicity.
CITRATE SYNTHASE AND CITRATE IN TAU HYPERPHOSPHORYLATION
AD is caused by extracellular deposition of Aβ plaques and intracellular neurofibrillary tangles by abnormal paired helical filaments (PHF), leading to neuronal cell damage [57]. The abnormal PHF is composed by hyperphosphorylation of tau [58, 66] and in small portions by ubiquitin [67]. Tau is a microtubule-associated protein that provides strength and stability to axonal microtubules in neurons by tubulin-binding property and helps in intraneuronal transportation [68].
In AD, Aβ induces the synthesis of glycogen synthase kinase-3 beta [60] and tau protein kinase 1 and 2 (TPK1 and TPK2) in AD [58]. TPK1 hyperphosphorylates tau protein. It converts tau to PHF and forms neurofibrillary tangles, while TPK2 does not participate in PHF formation [59]. TPK1 was found to be positive in neurodegenerative Aβ plaques and neuropil threads in Down syndrome brains [69]. TPK1 also phosphorylates PDH and reduces the conversion of pyruvate to acetyl-CoA, which is needed for ACh synthesis [60], whereas TPK2 does not inactivate PDH [70]. In rat hippocampal cells, Aβ-mediated TPK1 activation causes inhibition of PDH and leads to accumulation of pyruvate, impaired glucose metabolism, decreased acetyl-CoA production, low CS activity, and citrate and ACh synthesis, resulting in AD [71].
Aβ AND IMPAIRMENT OF GLUCOSE TRANSPORTERS
The decrease in glucose uptake is observed in the silent phase of AD prior to neurodegeneration in the study of genetically at-risk AD patients [72–74]. By using brain imaging methods, neurotic plaques are detected in the brain regions where glucose uptake is impaired [75–77]. The mitochondrial enzyme activities of ketoglutarate dehydrogenase and PDHC also decreased [78]. In the impairment of neuronal glucose uptake, ATP production in mitochondria is reduced and increased neuroexcitotoxicity due to calcium overload is seen [79, 80]. In the brain, glucose is taken up by the special transporter GLUT3 in neuronal cells and by GLUT1 in endothelial cells, and the levels of these glucose transporters are reduced in the AD brain [81]. Aβ, the proteolytic fragment of the amyloid-β protein precursor (AβPP), is majorly responsible for neurotic plaques in AD [82]. Aβ toxicity leads to membrane peroxidation [83] and membrane transporter signaling impairment, along with impairment of ion-motive ATPases [84], ion homeostasis disruption [85], apoptosis [86], glutamate transporter [87], and muscarinic cholinergic receptor binding signaling [88]. The level of lipids [89], proteins by oxidative stress [90], and the endproducts of glycation are associated with neurofibrillary tangles [91], an aldehydic product of lipid peroxidation (4-hydroxynonenal), disruption in ion-motive ATPases, and calcium homeostasis [92]. According to data, Aβ also impairs the glucose uptake reduction in ATP levels in cultured cortical and hippocampal neurons mediated by 4-hydroxynonenal to GLUT3 conjugation and lipid peroxidation. So Aβ may result in a decrease in glucose uptake and neurodegeneration in AD [93]. Glucose transporter is impaired by Aβ deposition that ultimately affects the intra-mitochondrial acetyl–CoA synthesis and low substrate availability for CS that is responsible for low levels of citrate and neuronal cytoplasmic acetyl–CoA, which affects the synthesis of ACh.
Fluorodeoxyglucose-positron emission tomography (PET) studies in asymptomatic middle-aged APOE ɛ4 allele carriers show the reduction in glucose utilization and changes in the mitochondrial bioenergetics [94]. APOE ɛ4, an isoform of apolipoprotein E, forms a peptide of mitochondrial targeting sequence that inhibits COX and results in mitochondrial dysfunction and neurotoxicity [95–97]. Activities of the mitochondrial COX and CS were also reduced in the platelets of APOE ɛ4 carriers versus non-carriers [98].
ABNORMAL ENERGY METABOLISM IN ALZHEIMER’S DISEASE
Poly ADP ribose polymerase-1 (PARP-1) activation affects the protein levels of cytochrome oxidase-IV and causes a decline in functional bioenergetics, rate of oxygen consumption, and membrane potential that results in deficits in cellular bioenergetics and also increases the expression of pyruvate kinases and modulates the glycolytic pathway through PARP-1. PARP-1 overactivation is associated with neurodegenerative diseases [99]. In addition, the level of full-length AβPP and its β-secretase cleavage product C99 were reported to cause impairment in energy metabolism and mitochondrial functions in the mice [100–102].
Moreover, low levels of peroxisome proliferator-activated receptor-gamma co-activator (PGC-1α) might also result in mitochondrial dysfunction. There is convincing evidence of a link between AD and PGC-1α [103]. Postmortem brain tissue samples of patients with AD showed a decrease in PGC-1α levels [104]. Additionally, AβPPswe/PS1dE9 mice were also reported to show decreased PGC-1α levels [105]. PGC-1α is one of the important regulators of carbohydrates and proteins and initiates mitochondrial respiration and biogenesis. The proteins and the mRNA level of PGC-1α were significantly reduced in neurodegenerative diseases [106].
The low levels of PGC-1α may cause mitochondrial dysfunction [107] and impairment of mitochondrial biogenesis [108]. In different AD mouse models, CS activities were significantly decreased in the mitochondria of brain cells, which indicated a decrease in mitochondrial mass and reduction in the levels of PGC-1α, cytochrome c-oxidase (complex IV) activity, ATP, and mitochondrial membrane protein [109–111].
Mitochondrial function is improved by restoring the levels of ATP, COX, and mitochondrial membrane potential, mitochondrial mass, enhancement of CS activity, and the levels of oxidative phosphorylation. Physiological CS activity and citrate level helps in enhancing acetyl-CoA-mediated cellular bioenergetics and ACh synthesis [112].
REDUCTION IN COX AND ENERGY ALTER AβPP TO THE AMYLOIDOGENIC PATHWAY
The presence of distinct features outside of the AD brain and inside the many tissues suggested that AD is not a brain-limited event or process. Aβ oligomerization and aggregation processes in the brain can alter AβPP homeostasis and energy metabolism in multiple tissues. Gabuzda et al. reported that the inhibition of COX shifts the process of AβPP to the amyloidogenic pathway and results in Aβ deposition [113]. Mitochondrial ROS also shifts the AβPP process towards Aβ [114], and different cell culture studies suggest that interference with cellular bioenergetics shifts the AβPP process to the amyloidogenic pathway [115, 116]. The triple transgenic female mice model of reduced COX activity showed an increase in glycolytic activity prior to amyloidosis [117]. CS knock down cells also result in impairment of respiratory activity and significantly decrease ATP production [118] (Fig. 2).
Fig. 2
Low citrate synthase and citrate impairs cellular bioenergetics, acetylcholine synthesis, and loss of cognitive function in Alzheimer’s disease
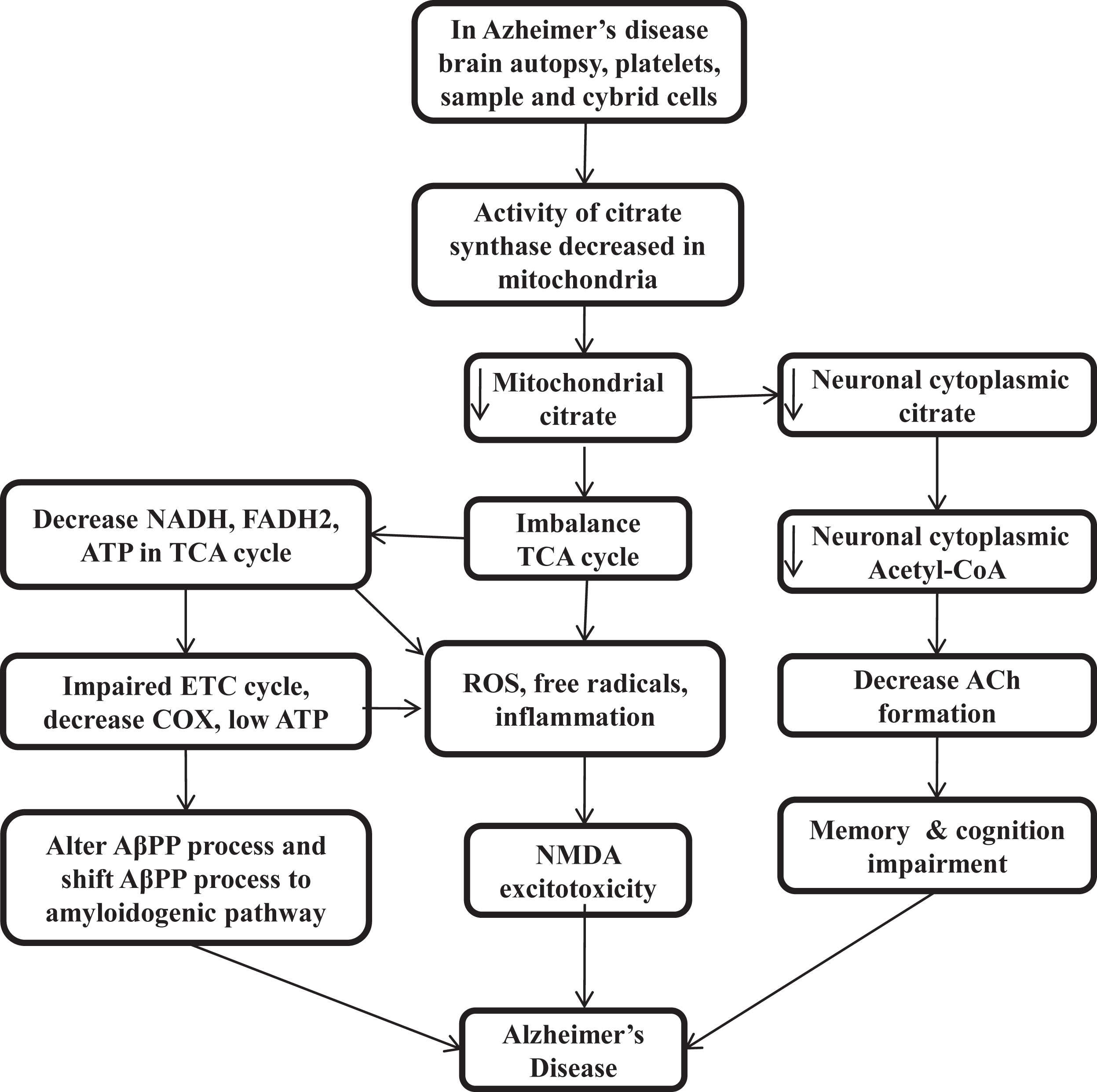
CITRATE SYNTHASE ACTIVITY IN THE CLINICAL STUDIES OF ALZHEIMER’S DISEASE PATIENTS
In patients with AD, low CS activity was found in brain homogenate [119, 120], platelets [121, 122], prefrontal cortex [123, 124], temporal cortex [125], parietal cortex [126, 127], cybrid cells [128], and APOE4 carrier [98]. Patients with low CS levels show low citrate production, low cellular bioenergetics, reduced acetyl-CoA, and decreased ACh formation. Then low energy and mitochondrial ROS alter the AβPP process and shift it to amyloidogenic pathways [113–116]. At low energy, proteins have the tendency to self-aggregation, while at physiological ATP level, it prevents self-aggregation. Low cellular energy induces free radicals, ROS, and NMDA excitotoxicity [129]. The low level of PDH and Acetyl-CoA also results in many neurodegenerative diseases [44–47] that decreases the level of citrate in mitochondria and in neuronal cytoplasm and decrease the ACh synthesis and impairs cellular bioenergetics homeostasis or calcium homeostasis. Therefore, maintaining the level of CS and citrate by external sources can be a promising therapeutic target for curing AD by maintaining the cellular energy homeostasis, acetyl-CoA, and ACh production and lipogenesis of brain tissue. Interestingly, citrate, as a surfactant, also inhibits Aβ aggregation [130–133] (Fig. 2).
CITRATE IN PREVENTION OF AβAGGREGATION
The Aβ molecule is an amphiphilic peptide in nature and has N-terminal hydrophilic and C-terminal hydrophobic regions. Aβ form self-assembled aggregates of various morphologies like fibrils, protofibrils, filaments, dimers, and oligomers [134–138] due to intermolecular electrostatic and hydrophobic interaction [139–143]. Studies show that in different forms of Aβ, like monomeric, oligomeric, and fibrillar Aβ, the soluble forms of Aβ oligomers are more toxic in comparison to fibrillar and monomeric forms [144]. Further, inter-molecular electrostatic and hydrophobic interaction results in the aggregation of Aβ [139–143]. The aggregation of Aβ depends on the hydrophobicity of Aβ [133], so the molecule which inhibits the formation of Aβ aggregation could be a therapeutic option. The aggregation of Aβ was found to be inhibited by small number of surfactants that bind to the hydrophobic region of Aβ, which is responsible for its self-assembly [145–148]. Here citrate has an important role due to its high anionic charge density and small size [130]. Citrate has been reported to inhibit the aggregation of Aβ25 - 35 [131, 132] and aggregation of Aβ1 - 40 in vitro [133].
Furthermore, cerebrospinal fluid (CSF) levels of citrate were significantly related to CSF total proteins levels and changes in CSF citrate can be a diagnostic parameter in patients with AD and other neurologic disorders [149]. Citrate also dissolves Aβ1 - 40 and β-2-macroglobulin, presumed to chelate calcium [150].
MITOCHONDRIAL FUNCTION IN THE NORMAL NEURONAL CELLS
Neuronal cells depend on the mitochondria at the synapse to get ATP and the concentration of buffer Ca++ ion to generate membrane potential for neurotransmission. Therefore, the amounts of the mitochondria are higher in the synaptic area as compared to the other parts of neuronal cells [151–153]. The correct transport of mitochondria to the synapse is an indicator of its efficient function. The synaptic and non-synaptic mitochondria are produced in the neuronal soma and transported to the other required area of the neuronal cells. This transport of mitochondria with axon towards synapse occurs through microtubules, motor proteins like dynein and kinesin, and outer mitochondrial protein Rho GTPase. The metabolic demand and Ca++ concentration at the synaptic level influence this axonal transport of the mitochondria [154–157].
Oxidative phosphorylation of mitochondrial respiratory chain complex causes two side effects; first, the generation of IMM-potential necessary for the import of nuclear-encoded proteins of the mitochondria and shows the health of cell and mitochondria itself. Second, electron leakage from the respiratory chain complex forms ROS, so ROS is the by-product of mitochondrial bioenergetics pathways [158]. But under normal physiological conditions, this ROS production is counter balanced by the mitochondrial antioxidant system. In neurodegenerative conditions, this equilibrium fails and leads to molecular damage in the native and surrounding areas. ROS targets the protein, carbohydrates, lipids, nucleic acids, and all the macromolecules of the cell [159].
In the brain, the hippocampus and cortex region is more affected by ROS due to their higher consumption of oxygen and dependence on the energy produced by mitochondria. Low levels of antioxidants and high levels of polyunsaturated fat trigger the vulnerability of the brain to ROS [160].
Mitochondria have a tubular network throughout the cytosol. The structural and morphological regulation of this mitochondrial network is made by the fusion and fission reaction [161]. Fission occurs when the mitochondrial tubule divides into the fragments and is controlled by proteins like dynamin-1-likeprotein (Drp1) with mitochondrial fission 1 protein (Fis1) and mitochondrial dynamics protein (MID49). Fusion occurs when two or more mitochondria are fused into one by the joint action of protein optic atrophy protein1 (OPA1) and Mitofusion1 protein (Mfn1 & 2). Through this fission and fusion mitochondrial biogenesis reaction, cells increase their mitochondrial mass [162].
PGC-1α is one of the key regulators of the mitochondrial biogenesis [163], which activates the chain of transcription factors and regulates the transcription, and replication of mitochondrial DNA [164] along with NFR-1, NFR-2 (Nuclear respiratory factor 1 & 2) and TFAM (Mitochondrial transcription factor A) and control the nuclear gene-encoded with mitochondrial proteins [163].
The cooperation between mitochondria and endoplasmic reticulum (ER) by forming a contact site mediates the intracellular buffer of calcium [165] that allows the uptake of calcium from cytosol and ion exchange between these two organelles [166]. Ca++ is an important regulator of mitochondrial metabolic enzymes [167], and mitochondria have two calcium transporter: 1) mitochondrial calcium uniporter, having high selectivity for calcium ions in the IMM [168], and 2) voltage dependent an ion channel (VDAC) in the outer mitochondrial membrane that regulates calcium release from the mitochondria [165].
VDAC works with IMM adenine nucleotide transporter and in the matrix with cyclophilin D after forming membrane permeability transition pore (mPTP) [169]. The opening of mPTP results in cell death by the activation of apoptosis [170], and the amount of calcium is regulated by the mitochondria for the neuro transmission and the execution of synaptic functions [171, 172].
Cellular homeostasis and mitochondrial functions are maintained by the quality control system of mitochondria. At the same time, the endogenous proteases and chaperons regulate the folding state and mitochondrial protein activity [173], and selective autophagy removes the damaged mitochondria, termed mitophagy [174]. That is primarily regulated by the signaling system of protein Pink1 (PTEN induced kinase1) and ubiquitin ligase parkin, which activate after losing the mitochondrial membrane potential [175]. Pink1accumulation at the damaged mitochondrial outer membrane recruits the ubiquitin ligase Parkin, which might label the mitochondria to the process of autophagy and engulfment of mitochondria by fusion with the lysosomes, and the material of mitochondria is digested [176].
MITOCHONDRIAL FUNCTION IN ALZHEIMER’S DISEASE
The Aβ oligomer is responsible for primary neurotoxicity [177] by numerous mechanisms like the production of ROS, inflammatory response, and altered Ca+2 homeostasis [178]. The accumulation of Aβ and mitochondrial dysfunction results in ROS production in AD [179]. In the human AD brain, different metabolic pathways and enzymes involved in glycolysis and ETC oxidative phosphorylation are affected at the transcriptional level and reduced their activity [180, 181]. In ETC, reduced oxidative phosphorylation protein expression of complex-1andcomplex-IVinpR5/APP/PS2 (triple transgenic mice model) leads to progressive reduction in cerebral aerobic glucose metabolism [182]. The bioenergetics deficiency of mitochondria is the main sign of AD pathophysiology in the AD triple transgenic female mouse model by decreasing the activity of PDH E1α and COX 4 and protein expression [117]. In mitochondrial deficiency, the cell may shift its energy requirements from mitochondria to glycolysis and other metabolic pathways. In the conversion of phosphoenolpyruvate to pyruvate, the pyruvate kinase is the main catalytic enzyme and rate-limiting step in glycolysis for energy production [183]. PKM2 slows the mitochondrial oxidative phosphorylation even in the oxygen presence like Warburg effects [184].
The variation in the energy pathway reduces glucose consumption in the AD disease brain [185]. According to PET studies in AD, glucose metabolism was found to be 20–30% lower in the area of memory processing like the hippocampus, parietal lobe, temporal lobe, and posterior cingulated area than in a healthy brain [186]. That shows the variation in metabolic signs occurs early than the appearance of changes in histopathological signs and symptoms [185]. In several animals models and human AD patients, mitochondrial functions are severely compromised [107] in terms of morphology [187] and numbers of mitochondria [188], oxidative phosphorylation, mitochondrial membrane potential, buffer calcium concentration, production of ROS [189], mutation and oxidation of mitochondrial DNA [190], contact sites of mitochondria and ER [191], biogenesis of mitochondria and transport to the neuronal axon [192], and mitophagy [172]. Dysfunction in these processes results in synaptic loss and other consequences in the brain [192].
The activity of mitochondrial enzymes involved in energy production like PDHC, citrate synthase isocitrate dehydrogenase, αKGDH (α-ketoglutarate dehydrogenase), complex-IVCOX, and ATP synthase were decreased in AD, while the activity of complex-II succinate dehydrogenase and the malate dehydrogenase were increased [120, 121]. That compromises the mitochondrial membrane potential and production of ATP [193].
The imbalance between the generation of ROS and antioxidants was found in the blood, CSF, and in the brain of AD patients [194]. Oxidative stress caused by ROS is considered a main contributing factor in the pathogenesis of AD [195] and mild cognitive impairment (MCI) in the early stage of AD by increasing protein oxidation and lipid peroxidation and decreasing the levels of antioxidants in the brain and the peripheral regions [196].
The sample of AD patients and transgenic mice models (mAPP mice) shows the association of mitochondrial dysfunction with oxidative stress by impaired oxidative phosphorylation, axonal transport of mitochondria, increased oxidative stress, and mPTP [197]. The triple transgenic mice model of AD shows the compromised bioenergetics, and increased oxidative stress level appears early than the development of Aβ plaque [107, 117]. The lymphocytes sample of peripheral blood of AD, AD brain, and MCI patients found the oxidation of the ATP synthase and the enzyme of oxidative phosphorylation [198] represents the reduction in the activities of ATP synthase, and the levels of the ATP in AD. Reduction in plasma antioxidant level and aconitase (ACO2) activity in the peripheral lymphocytes of MCI and AD patients also proves the correlation between mitochondrial dysfunction and oxidative stress [199].
In human-induced pluripotent stem cells (iPSCs) model of AD also shows that oxidative stress and mitochondrial dysfunction are the main causative factor in AD pathology by increasing ROS, respiratory chain complexes, and vulnerability to stressors in neurons and astrocytes of AD-iPSCs [200].
Mitochondrial dynamic processes of fusion and fission were imbalanced in AD patients, resulting in the distribution of compromised mitochondria in neurons [188] and fragmented mitochondria in fibroblast and the brain of AD patients [201]. In the hippocampus of AD, the mitochondrial protein expression is abnormal and finds an increase in fission protein Fis1 along with the downregulation of the Drp1, and the fusion proteins OPAI, Mfn1, and Mfn2 [202]. The same is also found in the cybrid cells model with shorter and bleb-like mitochondria compared to the control [203]. And the further increase in phosphorylation of Drp1 S-nitrosylation at the site of Ser-616 promotes the fission of mitochondria [204] which is higher in the AD brain than in control [202]. Protein Drp1 interacts with hyperphosphorylated tau and Aβ in brain homogenates of the AD patients [205]. A recent study of a sample of the healthy control group and AD subject shows the link between the polymorphism in AD and the MFN2 gene, suggests that polymorphism in the regulatory process of mitochondrial fusion might have a role in the pathogenesis of the AD [206]. Protein mfn2 work as a cord between ER and mitochondrial membrane [207]. Presenilin 2 (PS2) is influenced by mfn2 and its mutation is linked to familial AD in ER-mitochondrial contact site modulation [208]. Different AD experimental models of Aβ peptide treatment and over expression of AβPP are characterized as fragmentation of mitochondria and due to variation in protein levels of fusion and fission process leads to abnormal distribution of mitochondria along with neurons [209–211].
Another important mitochondrial function is bioenergetics which is impaired in AD and decreases the mitochondrial number in the hippocampus of the human AD brain or in other cell culture models due to compromised mitochondrial bioenergetics [188, 209]. The level of mitochondrial regulatory proteins like NRF1, NRF2, PGC1α, and TFAM was decreased in the hippocampus overexpressing cellular models of APP Swedish mutation [104, 212]. In other mouse models of AD, i.e., humans carrying mutant transgenes of the Presenilin-1 (PS1) and APP, the markers of mitochondrial bioenergetics were also found to decline in the hippocampus, and here melatonin shows some beneficial effects [213].
Another factor, mitophagy, was able to prevent cognitive deterioration, deposition of Aβ, tau hyperphosphorylation, and reverse memory impairment in many AD models [214]. But in AD, mitophagy is affected and causes neuronal dysfunctions by accumulations of mitochondrial damage. This may be due to fusion impairment between the lysosomes and the autophagosomes [215].
In AD, the somatic mutation found in mitochondrial DNA is higher than in the healthy brain, which triggers ROS production and promotes the amyloidogenic cascades and other neuropathological consequences [216].
The histopathological markers of AD, Aβ and tau, interact with mitochondria non-specifically [182] and adversely affect the mitochondrial axonal transport from soma to the synapse of the neurons. Overexpression Aβ mouse model of AD has damaged mitochondria, impaired mitochondrial axonal transport, decreased mitochondrial membrane potential, and inhibition of respiratory chain complexes with lower ATP production [217]. The AβPP inside the mitochondria interacts with the Aβ peptide and with the mitochondrial matrix component [218, 219]. Aβ impaired the import of mitochondrial nuclear-encoded proteins by the process of mitochondrial co-aggregation [220].
The tauopathies in AD also impaired the functions of mitochondria and their axonal transport at the presynaptic terminals [221]. In AD, hyperphosphorylated tau interacts with VDAC1 and leads to mitochondrial dysfunction [205]. Hyperphosphorylated tau decreases the activity of complex-1 and ATP production, increases ROS, lowers the mitochondrial membrane potential, stimulates the mitochondrial fission process, and the fragmentation of mitochondria in the postmortem brains of murine models and AD patients [222, 223], and oxidative stress stimulates tau hyperphosphorylation [224].
The outer membrane translocase homolog unit Tomm40 is a channel at the outer mitochondrial membrane responsible for the import of nuclear-encoded protein [225]. Aβ affected this import machinery and caused mitochondrial dysfunction in AD [226, 220]. TOMM40 has a closed gene cluster with the APOE gene at chromosome-19 and APOE is the most significant risk factor in sporadic genetic late-onset AD (LOAD) linked with high-risk isoform ɛ4. A variable-length and the polymorphism of deoxythymidine homo polymer of TOMM40 gene intron 6 shows genetic risk factors for LOAD [227–229]. In the Caucasian ethnic group, there are three variants of TOMM40 polymorphism. One of the variants, rs10524523 in APOE3/4 carriers, lowered the onset of LOAD by 7 years [230] which impaired the volume of the grey matter and cognition in the brain of AD [231]. Some other groups also influence TOMM40 “523” variant on APOE and TOMM40 gene transcription [232].
OXIDATIVE STRESS MEDIATED INFLUENCES ON CS AND CITRATE AND BEGINNING OF THE SILENT PHASE PATHOPHYSIOLOGY OF ALZHEIMER’S DISEASE
An accumulation of Aβ and hyperphosphorylation of tau proteins might not be the primary cause of neuronal degeneration in LOAD. Additionally, some other factors might be responsible for it [233, 234]. Abnormal cellular bioenergetics contributes to normal aging and pathogenesis of LOAD [235, 236]. The hippocampal culture or neuronal cells get oxygen consumption and energy utilization by oxidative phosphorylation to power the neuronal cells activities [237]. But, when neuronal cells are highly synaptically active or high in metabolic activity, the O2 consumption is increased multifold, which generate H2O2 and ROS in higher amounts. Therefore, the neuronal cell shifts ATP utilization from oxidative phosphorylation to aerobic glycolysis (pentose sugar pathway) to prevent this ROS and free radicals that damage the neuronal cell and start inflammatory pathway. This shift is called the Warburg effect. The loss of CS is directly linked with the Warburg effect. CS knockout cells severely decrease the respiratory activity and showed a marked decrease in ATP production but a huge increase in glycolytic metabolism [11]. This shift of neuronal cell energy utilization from the glycolysis pathway decreases ROS and decreases the activity of mitochondria [238–240]. Actually, it is the mechanism of acquired neuroprotection [241]. This transition helps in decreasing the accumulation of ROS. But, at the same time in order to prevent ROS and free radicals, the shift of energy utilization from glycolysis pathway by the hippocampal neurons that are highly synaptically activated, upregulate both GLUT3 (neuronal glucose transporter) for increasing glucose uptake and the pyruvate dehydrogenase kinase-3 (PDK3) enzyme [242–244]. In the hippocampus, PDHC activity is found to be highest [44], and PDK3 phosphorylates the PDH in mitochondria and inhibits the conversion of glycolysis endproduct pyruvate to acetyl-CoA or ultimately inhibit acetyl-CoA and citrate. Then less acetyl-CoA results in less substrate for CS and ultimately less citrate and further less acetyl-CoA (serve as direct energy source to neuronal cell and also lipogenesis for brain tissue) decreases ACh synthesis in neuronal cytoplasm [245, 246], which starts the silent phase pathogenesis of AD.
The shift in energy utilization from the oxidative phosphorylation pathway to glycolysis causes a decrease in ROS production by decreasing mitochondrial activity as the reduction in oxidative phosphorylation and reduction in the inducing effects on oxidative phosphorylation by calcium in the matrix of mitochondria [247], represses the transcriptional mitochondrial calcium uniporter via calcium-Npas4induction [248], and reduces the harmful conversion of mitochondrial energy at the cost of low ATP yield by glucose metabolism or glycolysis [247]. A recent study also shows that the soluble form of Aβ aggregates is found in the silent phase of AD [249].
Oxidative stress-mediated damage if found in an early phase of AD and it decreases as the disease progresses, and increased deposition of Aβ is associated with reduced oxidative damage [250]. In humans, studies find enlargement of the resting synapse as a result of compensatory response that allows the system to perform well and counter balance this initial damage [251, 252]. So that in humans, the progression of AD from an early phase to symptomatic phase takes a long time [253]. The upregulation of GLUT3 and PDK3 can be biomarkers for the silent phase of AD. While GLUT3 transporter is reduced in the brain of AD patients, results show decreased glucose metabolism [81], which again hamper overall brain functions, cellular bioenergetics, ACh synthesis in AD, and cause loss of memory and cognitive functions. It also reduces acetyl-CoA mediated lipogenesis and shrinks the brain tissue over the time (Fig. 3).
Fig. 3
Pathophysiology of silent phase of Alzheimer’s disease.

In recent reports, these above-mentioned factors, like changes in energy metabolism, etc., result in normal aging and are responsible for the pathogenesis of LOAD. The cells of LOAD also show impaired mitochondrial metabolic and redox potential, decreased metabolism of NAD+, altered activity of the TCA cycle, and shifting of the energy production towards glycolysis [254].
CONCLUSION
According to this review, mitochondrial CS activity and citrate are important factors in AD. It suggests that AD is a metabolic disorder more than a neurodegenerative disease. In clinical studies of AD patients, mitochondrial enzyme CS activity is found to be lower in different regions of brain tissue, platelet samples, and cybrid cells. Decreased CS reduces citrate, a substrate for cellular bioenergetics in TCA cycle, and acetyl-CoA, ACh synthesis in cytosol. Decreased citrate, cellular bioenergetics, and neurocytoplasmic acetyl-CoA reduce ACh synthesis and results in memory and cognition impairments. Furthermore, reduced acetyl-CoA also decreases lipogenesis that leads to shrinking of the brain tissues. A lower energy environment in the cell favors Aβ aggregation. Therefore, citrate can be a more effective treatment for AD because it maintains cellular energy, improves ACh synthesis physiologically, lipogenesis, and inhibition Aβ aggregation. Citrate can also be utilized in the treatment of type 2 diabetes.
In AD’s silent phase, higher oxidative stress, free radicals, and ROS upregulate GLUT3 and PDK3. GLUT3 promotes more glucose uptake, whereas PDK3 inhibits PDH and inhibit acetyl-CoA, which is the substrate of CS, and same pathway of CS related to citrate, cellular bioenergetics, and ACh synthesis is inhibited. So GLUT3 and PDK3 can be biomarkers for the silent phase of AD. For further development, inhibition of PDK3 can play an important role in the silent phase of AD; behavioral therapy of breathing exercises and intermittent fasting can also prevent this oxidative stress and improve the mitochondrial quality control system in initial stages. Oxidative stress can be overcome by decreasing the exposure of environmental pollutants having oxidizing properties, by increasing the levels of endogenous and exogenous antioxidants, and by stabilizing the production and efficiency of mitochondrial energy. Importantly, citrate maintains mitochondrial energy production as well as having antioxidant properties.
In the future, an animal model can be develop by reducing the CS and citrate for the silent phase of AD. Current treatments like acetylcholinesterase inhibitors and NMDA antagonists only slow down the disease progression. However citrate can be a new and more effective therapeutic option by improving mitochondrial health, cellular bioenergetics, acetyl-CoA, and ACh synthesis, than the current treatments of AD.
ACKNOWLEDGMENTS
We thank the whole Department of Pharmacology, PGIMER, Chandigarh, Dr. Prem Prakash Khosla, Abhishek Chhimpa, Dr, Harmanjit Singh, Dr. Parvinder, Dr. Meenakshi Raju, Dr. Shikha Raheja, Dr Sudeep Bhardwaj, Dr. Ashutosh Agarwal and Dr. Muskan Malhotra.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | DeTure MA , Dickson DW ((2019) ) The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener 14: , 32. |
[2] | Burns A , Jacoby R , Levy R ((1990) ) Psychiatric phenomena in Alzheimer’s disease. I: Disorders of thought content. Br J Psychiatry 157: , 72–76. |
[3] | ((2021) ) 2021 Alzheimer’s disease facts and figures. Alzheimers Dement 17: , 327–406. |
[4] | Holtzman DM , John CM , Goate A ((2011) ) Alzheimer’s disease: The challenge of the second century. Sci Transl Med 3: , 77sr1. |
[5] | Dumas JA , Newhouse PA ((2011) ) The cholinergic hypothesis of cognitive aging revisited again: Cholinergic functional compensation. Pharmacol Biochem Behav 99: , 254–261. |
[6] | Weinstock M ((1999) ) Selectivity of cholinesterase inhibition: Clinical implications for the treatment of Alzheimer’s disease. CNS Drugs 12: , 307–323. |
[7] | Hardy JA , Higgins GA ((1992) ) Alzheimer’s disease: The amyloid cascade hypothesis. Science 256: , 184–185. |
[8] | Chhimpa N , Saifuddin PK , Kaur H , Sidhu SK , Puri N , Shree R ((2020) ) Pharmacotherapy of Alzheimer’s disease. Drug Bulletin 45: , 52–63. |
[9] | Wiegand G , Remington SJ ((1986) ) Citrate synthase: Structure, control, and mechanism. Annu Rev Biophys Biophys Chem 15: , 97–117. |
[10] | Wlassics ID , Stille C , Anderson VE ((1988) ) Coenzyme A dithioesters: Synthesis, characterization and reaction with citrate synthase and acetyl-CoA: Choline O-acetyltransferase. Biochim Biophys Acta 952: , 269–276. |
[11] | Suematsu N , Isohashi F ((2006) ) Molecular cloning and functional expression of human cytosolic acetyl-CoA hydrolase. Acta Biochim Pol 53: , 553–561. |
[12] | Lysiak W , Szutowicz A , Angielski S ((1976) ) Pyruvate metabolism in rat brain mitochondria. Acta Biochim Pol 23: , 325–333. |
[13] | Anderson NM , Mucka P , Kern JG , Feng H ((2018) ) The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell 9: , 216–237. |
[14] | Gabriel BM , Al-Tarrah M , Alhindi Y , Kilikevicius A , Venckunas T , Gray SR , Lionikas A , Ratkevicius A ((2017) ) H55N polymorphism is associated with low citrate synthase activity which regulates lipid metabolism in mouse muscle cells. PLoS One 12: , e0185789. |
[15] | Pal S , Paul S ((2020) ) ATP controls the aggregation of Aβ16-22 peptides. J Phys Chem B 124: , 210–223. |
[16] | Ronowska A , Szutowicz A , Bielarczyk H , Gul-Hinc S , Klimaszewska-Łata J , Dyś A , Zyśk M , Jankowska-Kulawy A ((2018) ) The regulatory effects of Acetyl-CoA distribution in the healthy and diseased brain. Front Cell Neurosci 12: , 169. |
[17] | Rimessi A , Giorgi C , Pinton P , Rizzuto R ((2008) ) The versatility of mitochondrial calcium signals: From stimulation of cell metabolism to induction of cell death. Biochim Biophys Acta 1777: , 808–816. |
[18] | Calvo-Rodriguez M , Bacskai BJ ((2021) ) Mitochondria and calcium in Alzheimer’s disease: From cell signaling to neuronal cell death. Trends Neurosci 44: , 136–151. |
[19] | Pérez MJ , Ponce DP , Aranguiz A , Behrens MI , Quintanilla RA ((2018) ) Mitochondrial permeability transition pore contributes to mitochondrial dysfunction in fibroblasts of patients with sporadic Alzheimer’s disease. Redox Biol 19: , 290–300. |
[20] | Pérez MJ , Ponce DP , Osorio-Fuentealba C , Behrens MI , Quintanilla RA ((2017) ) Mitochondrial bioenergetics is altered in fibroblasts from patients with sporadic Alzheimer’s disease. Front Neurosci 11: , 553. |
[21] | Manczak M , Reddy PH ((2012) ) Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: Implications for mitochondrial dysfunction and neuronal damage. Hum Mol Genet 21: , 2538–2547. |
[22] | Kim DI , Lee KH , Gabr AA , Choi GE , Kim JS , Ko SH , Han HJ ((2016) ) Aβ-Induced Drp1 phosphorylation through Akt activation promotes excessive mitochondrial fission leading to neuronal apoptosis. Biochim Biophys Acta 1863: , 2820–2834. |
[23] | Fang EF , Hou Y , Palikaras K , Adriaanse BA , Kerr JS , Yang B , Lautrup S , Hasan-Olive MM , Caponio D , Dan X , Rocktäschel P , Croteau DL , Akbari M , Greig NH , Fladby T , Nilsen H , Cader MZ , Mattson MP , Tavernarakis N , Bohr VA ((2019) ) Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci 22: , 401–412. |
[24] | Nixon RA , Yang DS ((2011) ) Autophagy failure in Alzheimer’s disease—locating the primary defect. Neurobiol Dis 43: , 38–45. |
[25] | PDB101:Molecule of the Month: Citrate Synthase RCSB: PDB-101. |
[26] | Liu X , Cooper DE , Cluntun AA , Warmoes MO , Zhao S , Reid MA , Liu J , Lund PJ , Lopes M , Garcia BA , Wellen KE , Kirsch DG , Locasale JW ((2018) ) Acetate production from glucose and coupling to mitochondrial metabolism in mammals. Cell 175: , 502–513.e13. |
[27] | Gillen JB , Martin BJ , MacInnis MJ , Skelly LE , Tarnopolsky MA , Gibala MJ ((2016) ) Twelve weeks of sprint interval training improves indices of cardiometabolic health similar to traditional endurance training despite a five-fold lower exercise volume and time commitment. PLoS One 11: , e0154075. |
[28] | Jacobs RA , Díaz V , Meinild AK , Gassmann M , Lundby C ((2013) ) The C57Bl/6 mouse serves as a suitable model of human skeletal muscle mitochondrial function. Exp Physiol 98: , 908–921. |
[29] | Alhindi Y , Vaanholt LM , Al-Tarrah M , Gray SR , Speakman JR , Hambly C , Alanazi BS , Gabriel BM , Lionikas A , Ratkevicius A ((2019) ) Low citrate synthase activity is associated with glucose intolerance and lipotoxicity. J Nutr Metab 2019: , 8594825. |
[30] | Yubero D , Adin A , Montero R , Jou C , Jiménez-Mallebrera C , García-Cazorla A , Nascimento A , O’Callaghan MM , Montoya J , Gort L , Navas P , Ribes A , Ugarte MD , Artuch R ((2016) ) A statistical algorithm showing coenzyme Q10 and citrate synthase as biomarkers for mitochondrial respiratory chain enzyme activities. Sci Rep 6: , 15. |
[31] | Szutowicz A , Jankowska A , Blusztajn JK , Tomaszewicz M ((1999) ) Acetylcholine and acetyl-CoA metabolism in differentiating SN56 septal cell line. J Neurosci Res 57: , 131–136. |
[32] | McCommis KS , Finck BN ((2015) ) Mitochondrial pyruvate transport: A historical perspective and future research directions. Biochem J 466: , 443–454. |
[33] | Srere PA ((1965) ) The molecular physiology of citrate. Nature 205: , 766–770. |
[34] | Angielski S , Szutowicz A ((1967) ) Tissue content of citrate and citrate-cleavage enzyme activity during starvation and refeeding. Nature 213: , 1252–1253. |
[35] | Szutowicz A , Bielarczyk H , Lysiak W ((1981) ) The role of citrate derived from glucose in the acetylcholine synthesis in rat brain synaptosomes. . Int J Biochem 13: , 887–892. |
[36] | Szutowicz A , Tomaszewicz M , Bielarczyk H ((1996) ) Disturbances of acetyl-CoA, energy and acetylcholine metabolism in some encephalopathies. Acta Neurobiol Exp (Wars) 56: , 323–339. |
[37] | Gnoni GV , Priore P , Geelen MJH , Siculella L ((2009) ) The mitochondrial citrate carrier: Metabolic role and regulation of its activity and expression. IUBMB Life 61: , 987–994. |
[38] | Sterling GH , O’Neill JJ ((1978) ) Citrate as the precursor of the acetyl moiety of acetylcholine. JNeurochem 31: , 525–530. |
[39] | Nachmansohn D , Machado AL ((1943) ) The formation of acetylcholine. a new enzyme: “Choline acetylase.”. J Neurophysiol 6: , 397–403. |
[40] | Jope RS , Jenden DJ ((1980) ) The utilization of choline and acetyl coenzyme A for the synthesis of acetylcholine. J Neurochem 35: , 318–325. |
[41] | Blokland A ((1995) ) Acetylcholine: A neurotransmitter for learning and memory? Brain Res Brain Res Rev 21: , 285–300. |
[42] | Patel MS , Owen OE ((1976) ) Lipogenesis from ketone bodies in rat brain. Evidence for conversion of acetoacetate into acetyl-coenzyme A in the cytosol. Biochem J 56: , 603–607. |
[43] | Bhaduri A , Srere PA ((1963) ) The incorporation of citrate carbon into fatty acids. Biochim Biophys Acta 70: , 221–230. |
[44] | Sorbi S , Bird ED , Blass JP ((1983) ) Decreased pyruvate dehydrogenase complex activity in Huntington and Alzheimer brain. Ann Neurol 13: , 72–78. |
[45] | Bowen DM , White P , Spillane JA , Goodhardt MJ , Curzon G , Iwangoff P , Meier-Ruge W , Davison AN ((1979) ) Accelerated ageing or selective neuronal loss as an important cause of dementia? Lancet 1: , 11–14. |
[46] | Perry EK , Perry RH , Tomlinson BE , Blessed G , Gibson PH ((1980) ) Coenzyme a-acetylating enzymes in Alzheimer’s disease: Possible cholinergic ‘compartment’ of pyruvate dehydrogenase. Neurosci Lett 18: , 105–110. |
[47] | Bird ED , Gale JS , Spokes EG ((1977) ) Huntington’s chorea: Post mortem activity of enzymes involved in cerebral glucose metabolism. J Neurochem 29: , 539–545. |
[48] | Szutowicz A , Bielarczyk H , Jankowska-Kulawy A , Pawełczyk T , Ronowska A ((2013) ) Acetyl-CoA the key factor for survival or death of cholinergic neurons in course of neurodegenerative diseases. . Neurochem Res 38: , 1523–1542. |
[49] | Szutowicz A , Stcepień M , Bielarczyk H , Kabata J , Lysiak W ((1982) ) ATP citrate lyase in cholinergic nerve endings. Neurochem Res 7: , 799–810. |
[50] | Gibson GE , Shimada M ((1980) ) Studies on the metabolic pathway of the acetyl group for acetylcholine synthesis. Biochem Pharmacol 29: , 167–174. |
[51] | Patel TB , Clark JB ((1980) ) Lipogenesis in the brain of suckling rats. Studies on the mechanism of mitochondrial-cytosolic carbon transfer. Biochem J 188: , 163–168. |
[52] | Ito T , Quastel JH ((1970) ) Acetoacetate metabolism in infant and adult rat brain. Biochem J 116: , 641–655. |
[53] | Wellen KE , Hatzivassiliou G , Sachdeva UM , Bui TV , Cross JR , Thompson CB ((2009) ) ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324: , 1076–1080. |
[54] | Swerdlow RH , Kish SJ ((2002) ) Mitochondria in Alzheimer’s disease. Int Rev Neurobiol 53: , 341–385. |
[55] | Mark RJ , Blanc EM , Mattson MP ((1996) ) Amyloid beta-peptide and oxidative cellular injury in Alzheimer’s disease. Mol Neurobiol 12: , 211–224. |
[56] | Cardoso SM , Santos S , Swerdlow RH , Oliveira CR ((2001) ) Functional mitochondria are required for amyloid beta-mediated neurotoxicity. FASEB J 15: , 1439–1441. |
[57] | Arriagada PV , Growdon JH , Hedley-Whyte ET , Hyman BT ((1992) ) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42: , 631–639. |
[58] | Ihara Y , Nukina N , Miura R , Ogawara M ((1986) ) Phosphorylated tau protein is integrated into paired helical filaments in Alzheimer’s disease. J Biochem 99: , 1807–1810. |
[59] | Ishiguro K , Takamatsu M , Tomizawa K , Omori A , Takahashi M , Arioka M , Uchida T , Imahori K ((1992) ) Tau protein kinase I converts normal tau protein into A68-like component of paired helical filaments. J Biol Chem 267: , 10897–10901. |
[60] | Hoshi M , Takashima A , Noguchi K , Murayama M , Sato M , Kondo S , Saitoh Y , Ishiguro K , Hoshino T , Imahori K ((1996) ) Regulation of mitochondrial pyruvate dehydrogenase activity by tau protein kinase I/glycogen synthase kinase 3beta in brain. Proc Natl Acad Sci U S A 93: , 2719–23. |
[61] | Readnower RD , Sauerbeck AD , Sullivan PG ((2011) ) Mitochondria, amyloid β, and Alzheimer’s disease. Int J Alzheimers Dis 2011: , 104545. |
[62] | Wertkin AM , Turner RS , Pleasure SJ , Golde TE , Younkin SG , Trojanowski JQ , Lee VM ((1993) ) Human neurons derived from a teratocarcinoma cell line express solely the 695-amino acid amyloid precursor protein and produce intracellular beta-amyloid or A4 peptides. Proc Natl Acad Sci U S A 90: , 9513–9517. |
[63] | Munir M , Lu L , McGonigle P ((1995) ) Excitotoxic cell death and delayed rescue in human neurons derived from NT2 cells. J Neurosci 15: , 7847–7860. |
[64] | Behl C , Davis J , Cole GM , Schubert D ((1992) ) Vitamin E protects nerve cells from amyloid beta protein toxicity. Biochem Biophys Res Commun 186: , 944–950. |
[65] | Gibson GE , Huang HM ((2002) ) Oxidative processes in the brain and non-neuronal tissues as biomarkers of Alzheimer’s disease. Front Biosci 7: , 1007–1015. |
[66] | Grundke-Iqbal I , Iqbal K , Tung YC , Quinlan M , Wisniewski HM , Binder LI ((1986) ) Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A 83: , 4913–4917. |
[67] | Mori H , Kondo J , Ihara Y ((1987) ) Ubiquitin is a component of paired helical filaments in Alzheimer’s disease. Science 235: , 1641–1644. |
[68] | Weingarten MD , Lockwood AH , Hwo SY , Kirschner MW ((1975) ) A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A 72: , 1858–1862. |
[69] | Yamaguchi H , Ishiguro K , Uchida T , Takashima A , Lemere CA , Imahori K ((1996) ) Preferential labeling of Alzheimer neurofibrillary tangles with antisera for tau protein kinase (TPK) I/glycogen synthase kinase-3 beta and cyclin-dependent kinase 5, a component of TPK II. Acta Neuropathol 92: , 232–241. |
[70] | Hoshi M , Sato M , Kondo S , Takashima A , Noguchi K , Takahashi M , Ishiguro K , Imahori K ((1995) ) Different localization of tau protein kinase I/glycogen synthase kinase-3 beta from glycogen synthase kinase-3 alpha in cerebellum mitochondria. J Biochem 118: , 683–685. |
[71] | Imahori K , Uchida T ((1997) ) Physiology and pathology of tau protein kinases in relation to Alzheimer’s disease. J Biochem 121: , 179–188. |
[72] | Pettegrew JW , Panchalingam K , Klunk WE , McClure RJ , Muenz LR ((1994) ) Alterations of cerebral metabolism in probable Alzheimer’s disease: A preliminary study. Neurobiol Aging 15: , 117–132. |
[73] | Kennedy AM , Frackowiak RS , Newman SK , Bloomfield PM , Seaward J , Roques P , Lewington G , Cunningham VJ , Rossor MN ((1995) ) Deficits in cerebral glucose metabolism demonstrated by positron emission tomography in individuals at risk of familial Alzheimer’s disease. Neurosci Lett 186: , 17–20. |
[74] | Reiman EM , Caselli RJ , Yun LS , Chen K , Bandy D , Minoshima S , Thibodeau SN , Osborne D ((1996) ) Preclinical evidence of Alzheimer’s disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. N Engl J Med 334: , 752–758. |
[75] | Hoyer S , Oesterreich K , Wagner O ((1988) ) Glucose metabolism as the site of the primary abnormality in early-onset dementia of Alzheimer type? J Neurol 235: , 143–148. |
[76] | Kalaria RN , Harik SI ((1989) ) Reduced glucose transporter at the blood-brain barrier and in cerebral cortex in Alzheimer disease. J Neurochem 53: , 1083–1088. |
[77] | Jagust WJ , Seab JP , Huesman RH , Valk PE , Mathis CA , Reed BR , Coxson PG , Budinger TF ((1991) ) Diminished glucose transport in Alzheimer’s disease: Dynamic PET studies. J Cereb Blood Flow Metab 11: , 323–330. |
[78] | Blass JP ((1993) ) Metabolic alterations common to neural and non-neural cells in Alzheimer’s disease. Hippocampus 3: , 45–53. |
[79] | Novelli A , Reilly JA , Lysko PG , Henneberry RC ((1988) ) Glutamate becomes neurotoxic via the N-methyl-D-aspartate receptor when intracellular energy levels are reduced. Brain Res 451: , 205–212. |
[80] | Cheng B , Mattson MP ((1992) ) Glucose deprivation elicits neurofibrillary tangle-like antigenic changes in hippocampal neurons: Prevention by NGF and bFGF. Exp Neurol 117: , 14–23. |
[81] | Simpson IA , Chundu KR , Davies-Hill T , Honer WG , Davies P ((1994) ) Decreased concentrations of GLUT1 and GLUT3 glucose transporters in the brains of patients with Alzheimer’s disease. Ann Neurol 35: , 546–551. |
[82] | Selkoe DJ ((1991) ) The molecular pathology of Alzheimer’s disease. Neuron 6: , 487–498. |
[83] | Behl C , Davis JB , Lesley R , Schubert D ((1994) ) Hydrogen peroxide mediates amyloid beta protein toxicity. Cell 77: , 817–827. |
[84] | Mark RJ , Hensley K , Butterfield DA , Mattson MP ((1995) ) Amyloid beta-peptide impairs ion-motive ATPase activities: Evidence for a role in loss of neuronal Ca2+ homeostasis and cell death. J Neurosci 15: , 6239–6249. |
[85] | Mattson MP , Tomaselli KJ , Rydel RE ((1993) ) Calcium-destabilizing and neurodegenerative effects of aggregated beta-amyloid peptide are attenuated by basic FGF. Brain Res 621: , 35–49. |
[86] | Loo DT , Copani A , Pike CJ , Whittemore ER , Walencewicz AJ , Cotman CW ((1993) ) Apoptosis is induced by beta-amyloid in cultured central nervous system neurons. Proc Natl Acad Sci U SA 90: , 7951–7955. |
[87] | Harris ME , Wang Y , Pedigo NW , Hensley K , Butterfield DA , Carney JM ((1996) ) Amyloid beta peptide (25-35) inhibits Na+-dependent glutamate uptake in rat hippocampal astrocyte cultures. J Neurochem 67: , 277–286. |
[88] | Kelly JF , Furukawa K , Barger SW , Rengen MR , Mark RJ , Blanc EM , Roth GS , Mattson MP ((1996) ) Amyloid beta-peptide disrupts carbachol-induced muscarinic cholinergic signal transduction in cortical neurons. Proc Natl Acad Sci U S A 93: , 6753–6758. |
[89] | Lovell MA , Ehmann WD , Butler SM , Markesbery WR ((1996) ) Elevated thiobarbituric acid-reactive substances and antioxidant enzyme activity in the brain in Alzheimer’s disease. Neurology 45: , 1594–1601. |
[90] | Smith CD , Carney JM , Starke-Reed PE , Oliver CN , Stadtman ER , Floyd RA , Markesbery WR ((1991) ) Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proc Natl Acad Sci U S A 88: , 10540–10543. |
[91] | Smith MA , Sayre LM , Monnier VM , Perry G ((1995) ) Radical AGEing in Alzheimer’s disease. Trends Neurosci 18: , 172–176. |
[92] | Mark RJ , Lovell MA , Markesbery WR , Uchida K , Mattson MP ((1997) ) A role for 4-hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid beta-peptide. J Neurochem 68: , 255–264. |
[93] | Mark RJ , Pang Z , Geddes JW , Uchida K , Mattson MP ((1997) ) Amyloid beta-peptide impairs glucose transport in hippocampal and cortical neurons: Involvement of membrane lipid peroxidation. J Neurosci 17: , 1046–1054. |
[94] | Small GW , Mazziotta JC , Collins MT , Baxter LR , Phelps ME , Mandelkern MA , Kaplan A , La Rue A , Adamson CF , Chang L ((1995) ) Apolipoprotein E type 4 allele and cerebral glucose metabolism in relatives at risk for familial Alzheimer disease. JAMA 273: , 942–947. |
[95] | Chang S , ran Ma T , Miranda RD , Balestra ME , Mahley RW , Huang Y ((2005) ) Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc Natl Acad Sci U S A 102: , 18694–18699. |
[96] | Chen HK , Ji ZS , Dodson SE , Miranda RD , Rosenblum CI , Reynolds IJ , Freedman SB , Weisgraber KH , Huang Y , Mahley RW ((2011) ) Apolipoprotein E4 domain interaction mediates detrimental effects on mitochondria and is a potential therapeutic target for Alzheimer disease. J Biol Chem 286: , 5215–5221. |
[97] | Mahley RW , Huang Y ((2012) ) Apolipoprotein E sets the stage: Response to injury triggers neuropathology. Neuron 76: , 871–885. |
[98] | Wilkins HM , Koppel SJ , Bothwell R , Mahnken J , Burns JM , Swerdlow RH ((2017) ) Platelet cytochrome oxidase and citrate synthase activities in APOE ɛ4 carrier and non-carrier Alzheimer’s disease patients. Redox Biol 12: , 828–832. |
[99] | Martire S , Fuso A , Mosca L , Forte E , Correani V , Fontana M , Scarpa S , Maras B , d’Erme M ((2016) ) Bioenergetic impairment in animal and cellular models of Alzheimer’s disease: PARP-1 inhibition rescues metabolic dysfunctions. J Alzheimers Dis 54: , 307–324. |
[100] | Wilkins HM , Swerdlow RH ((2017) ) Amyloid precursor protein processing and bioenergetics. Brain Res Bull 133: , 71–79. |
[101] | Schaefer PM , Einem B von , Walther P , Calzia E , Arnim CAF von ((2016) ) Metabolic characterization of intact cells reveals intracellular amyloid beta but not its precursor protein to reduce mitochondrial respiration. PLoS One 11: , e0168157. |
[102] | Pera M , Larrea D , Guardia-Laguarta C , Montesinos J , Velasco KR , Agrawal RR , Xu Y , Chan RB , Di Paolo G , Mehler MF , Perumal GS , Macaluso FP , Freyberg ZZ , Acin-Perez R , Enriquez JA , Schon EA , Area-Gomez E ((2017) ) Increased localization of APP-C99 in mitochondria-associated ER membranes causes mitochondrial dysfunction in Alzheimer disease. EMBO J 36: , 3356–3371. |
[103] | Sweeney G , Song J ((2016) ) The association between PGC-1α and Alzheimer’s disease. Anat Cell Biol 49: , 1–6. |
[104] | Qin W , Haroutunian V , Katsel P , Cardozo CP , Ho L , Buxbaum JD , Pasinetti GM ((2009) ) PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch Neurol 66: , 352–361. |
[105] | Pedrós I , Petrov D , Allgaier M , Sureda F , Barroso E , Beas-Zarate C , Auladell C , Pallàs M , Vázquez-Carrera M , Casadesús G , Folch J , Camins A ((2014) ) Early alterations in energy metabolism in the hippocampus of APPswe/PS1dE9 mouse model of Alzheimer’s disease. Biochim Biophys Acta 1842: , 1556–1566. |
[106] | Handschin C , Spiegelman BM ((2006) ) Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev 27: , 728–735. |
[107] | Hauptmann S , Scherping I , Dröse S , Brandt U , Schulz KL , Jendrach M , Leuner K , Eckert A , Müller WE ((2009) ) Mitochondrial dysfunction: An early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol Aging 30: , 1574–1586. |
[108] | Calkins MJ , Manczak M , Mao P , Shirendeb U , Reddy PH ((2011) ) Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum Mol Genet 20: , 4515–4529. |
[109] | Zhang C , Rissman RA , Feng J ((2015) ) Characterization of ATP alternations in an Alzheimer’s disease transgenic mouse model. J Alzheimers Dis 44: , 375–378. |
[110] | Rönnbäck A , Pavlov PF , Mansory M , Gonze P , Marlière N , Winblad B , Graff C , Behbahani H ((2016) ) Mitochondrial dysfunction in a transgenic mouse model expressing human amyloid precursor protein (APP) with the Arctic mutation. J Neurochem 136: , 497–502. |
[111] | Monteiro-Cardoso VF , Oliveira MM , Melo T , Domingues MR , Moreira PI , Ferreiro E , Peixoto F , Videira RA ((2015) ) Cardiolipin profile changes are associated to the early synaptic mitochondrial dysfunction in Alzheimer’s disease. J Alzheimers Dis 43: , 1375–1392. |
[112] | Pohland M , Pellowska M , Asseburg H , Hagl S , Reutzel M , Joppe A , Berressem D , Eckert SH , Wurglics M , Schubert-Zsilavecz M , Eckert GP ((2018) ) MH84 improves mitochondrial dysfunction in a mouse model of early Alzheimer’s disease. Alzheimers Res Ther 10: , 18. |
[113] | Gabuzda D , Busciglio J , Chen LB , Matsudaira P , Yankner BA ((1994) ) Inhibition of energy metabolism alters the processing of amyloid precursor protein and induces a potentially amyloidogenic derivative. J Biol Chem 269: , 13623–13628. |
[114] | Leuner K , Schütt T , Kurz C , Eckert SH , Schiller C , Occhipinti A , Mai S , Jendrach M , Eckert GP , Kruse SE , Palmiter RD , Brandt U , Dröse S , Wittig I , Willem M , Haass C , Reichert AS , Müller WE ((2012) ) Mitochondrion-derived reactive oxygen species lead to enhanced amyloid beta formation. Antioxid Redox Signal 16: , 1421–1433. |
[115] | Webster MT , Pearce BR , Bowen DM , Francis PT ((1998) ) The effects of perturbed energy metabolism on the processing of amyloid precursor protein in PC12 cells. J Neural Transm (Vienna) 105: , 839–853. |
[116] | Gasparini L , Racchi M , Benussi L , Curti D , Binetti G , Bianchetti A , Trabucchi M , Govoni S ((1997) ) Effect of energy shortage and oxidative stress on amyloid precursor protein metabolism in COS cells. Neurosci Lett 231: , 113–117. |
[117] | Yao J , Irwin RW , Zhao L , Nilsen J , Hamilton RT , Brinton RD ((2009) ) Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 106: , 14670–14675. |
[118] | Lin CC , Cheng TL , Tsai WH , Tsai HJ , Hu KH , Chang HC , Yeh CW , Chen YC , Liao CC , Chang WT ((2012) ) Loss of the respiratory enzyme citrate synthase directly links the Warburg effect to tumor malignancy. Sci Rep 2: , 785. |
[119] | Kish SJ , Bergeron C , Rajput A , Dozic S , Mastrogiacomo F , Chang LJ , Wilson JM , DiStefano LM , Nobrega JN ((1992) ) Brain cytochrome oxidase in Alzheimer’s disease. J Neurochem 59: , 776–779. |
[120] | Maurer I , Zierz S , Möller HJ ((2000) ) A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol Aging 21: , 455–462. |
[121] | Cardoso SM , Proença MT , Santos S , Santana I , Oliveira CR ((2004) ) Cytochrome c oxidase is decreased in Alzheimer’s disease platelets. . Neurobiol Aging 25: , 105–110. |
[122] | Fišar Z , Hansíková H , Křížová J , Jirák R , Kitzlerová E , Zvěřová M , Hroudová J , Wenchich L , Zeman J , Raboch J ((2019) ) Activities of mitochondrial respiratory chain complexes in platelets of patients with Alzheimer’s disease and depressive disorder. Mitochondrion 48: , 67–77. |
[123] | Bubber P , Haroutunian V , Fisch G , Blass JP , Gibson GE ((2005) ) Mitochondrial abnormalities in Alzheimer brain: Mechanistic implications. Ann Neurol 57: , 695–703. |
[124] | Harish G , Venkateshappa C , Mahadevan A , Pruthi N , Bharath MMS , Shankar SK ((2013) ) Mitochondrial function in human brains is affected by pre- and post mortem factors. Neuropathol Appl Neurobiol 39: , 298–315. |
[125] | Cooper JM , Wischik C , Schapira AHV ((1993) ) Mitochondrial function in Alzheimer’s disease. Lancet 341: , 969–970. |
[126] | Kish SJ , Mastrogiacomo F , Guttman M , Furukawa Y , Taanman JW , Dozić S , Pandolfo M , Lamarche J , DiStefano L , Chang LJ ((1999) ) Decreased brain protein levels of cytochrome oxidase subunits in Alzheimer’s disease and in hereditary spinocerebellar ataxia disorders: A nonspecific change? J Neurochem 72: , 700–707. |
[127] | Mastrogiacoma F , Lindsay JG , Bettendorff L , Rice J , Kish SJ ((1996) ) Brain protein and alpha-ketoglutarate dehydrogenase complex activity in Alzheimer’s disease. Ann Neurol 39: , 592–598. |
[128] | Cardoso SM , Santana I , Swerdlow RH , Oliveira CR ((2004) ) Mitochondria dysfunction of Alzheimer’s disease cybrids enhances Aβ toxicity. . J Neurochem 89: , 1417–1426. |
[129] | Dong XX , Wang Y , Qin ZH ((2009) ) Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol Sin 30: , 379–387. |
[130] | Brus L ((2008) ) Noble metal nanocrystals: Plasmon electron transfer photochemistry and single-molecule Raman spectroscopy. Acc Chem Res 41: , 1742–1749. |
[131] | Yang HD , Son I , Lee S , Park Y ((2008) ) Protective effect of citrate against A beta-induced neurotoxicity in PC12 cells. Mol Cell Toxicol 4: , 157–163. |
[132] | Pike CJ , Walencewicz-Wasserman AJ , Kosmoski J , Cribbs DH , Glabe CG , Cotman CW ((1995) ) Structure-activity analyses of beta-amyloid peptides: Contributions of the beta 25-35 region to aggregation and neurotoxicity. J Neurochem 64: , 253–265. |
[133] | Park YH , Kim YJ , Son IH , Yang HD ((2009) ) Inhibition of β-amyloid 1-40 peptide aggregation and neurotoxicity by citrate. Korean J Physiol Pharmacol 13: , 273–279. |
[134] | Koh JY , Yang LL , Cotman CW ((1990) ) Beta-amyloid protein increases the vulnerability of cultured cortical neurons to excitotoxic damage. Brain Res 533: , 315–320. |
[135] | Pike CJ , Burdick D , Walencewicz AJ , Glabe CG , Cotman CW ((1993) ) Neurodegeneration induced by beta-amyloid peptides: The role of peptide assembly state. J Neurosci 13: , 1676–1687. |
[136] | Howlett DR , Jennings KH , Lee DC , Clark MS , Brown F , Wetzel R , Wood SJ , Camilleri P , Roberts GW ((1995) ) Aggregation state and neurotoxic properties of Alzheimer beta-amyloid peptide. Neurodegeneration 4: , 23–32. |
[137] | Seilheimer B , Bohrmann B , Bondolfi L , Müller F , Stüber D , Döbeli H ((1997) ) The toxicity of the Alzheimer’s beta-amyloid peptide correlates with a distinct fiber morphology. J Struct Biol 119: , 59–71. |
[138] | Watanabe K , Segawa T , Nakamura K , Kodaka M , Okuno H , Konakahara T ((2001) ) Identification of the molecular interaction site of amyloid β peptide by using a fluorescence assay. J Pept Res 58: , 342–346. |
[139] | Jarrett JT , Berger EP , Lansbury PT ((1993) ) The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: Implications for the pathogenesis of Alzheimer’s disease. Biochemistry 32: , 4693–4697. |
[140] | Wood SJ , Maleeff B , Hart T , Wetzel R ((1996) ) Physical, morphological and functional differences between pH 5.8 and 7.4 aggregates of the Alzheimer’s amyloid peptide Abeta. J Mol Biol 256: , 870–877. |
[141] | Lazo ND , Grant MA , Condron MC , Rigby AC , Teplow DB ((2005) ) On the nucleation of amyloid beta-protein monomer folding. Protein Sci 14: , 1581–1596. |
[142] | Lührs T , Ritter C , Adrian M , Riek-Loher D , Bohrmann B , Döbeli H , Schubert D , Riek R ((2005) ) 3D structure of Alzheimer’s amyloid-beta(1-42) fibrils. Proc Natl Acad Sci U S A 102: , 17342–17347. |
[143] | Khandogin J , Brooks CL 3rd ((2007) ) Linking folding with aggregation in Alzheimer’s β-amyloid peptides. Proc Natl Acad Sci U S A 104: , 16880–16885. |
[144] | Rochet JC , Lansbury PT Jr ((2000) ) Amyloid fibrillogenesis: Themes and variations. Curr Opin Struct Biol 10: , 60–68. |
[145] | Lomakin A , Chung DS , Benedek GB , Kirschner DA , Teplow DB ((1996) ) On the nucleation and growth of amyloid beta-protein fibrils: Detection of nuclei and quantitation of rate constants. Proc Natl Acad Sci U S A 93: , 1125–1129. |
[146] | Marcinowski KJ , Shao H , Clancy EL , Zagorski MG ((1998) ) Solution structure model of residues 1–28 of the Amyloid β-peptide when bound to micelles. J Am Chem Soc 120: , 11082–11091. |
[147] | Wang SS , Chen YT , Chou SW ((2005) ) Inhibition of amyloid fibril formation of β-amyloid peptides via the amphiphilic surfactants. Biochim Biophys Acta 1741: , 307–313. |
[148] | Cao M , Han Y , Wang J , Wang Y ((2007) ) Modulation of fibrillogenesis of amyloid β(1–40) peptide with cationic gemini surfactant. J Phys Chem B 111: , 13436–13443. |
[149] | Haerer AF ((1972) ) Pyruvate, citrate, alpha ketoglutarate and glucose in the CSF and blood of neurologic patients. Acta Neurol Scand 48: , 306–312. |
[150] | Kumar S , Sharma P , Arora K , Raje M , Guptasarma P ((2014) ) Calcium binding to Beta-2-microglobulin at physiological pH drives the occurrence of conformational changes which cause the protein to precipitate into amorphous forms that subsequently transform into amyloid aggregates. PLoS One 9: , e95725. |
[151] | Li Z , Okamoto KI , Hayashi Y , Sheng M ((2004) ) The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119: , 873–887. |
[152] | Verstreken P , Ly CV , Venken KJT , Koh TW , Zhou Y , Bellen HJ ((2005) ) Synaptic mitochondria are critical for mobilization of reserve pool vesicles at drosophila neuromuscular junctions. Neuron 47: , 365–378. |
[153] | Gazit N , Vertkin I , Shapira I , Helm M , Slomowitz E , Sheiba M , Mor Y , Rizzoli S , Slutsky I ((2016) ) IGF-1 receptor differentially regulates spontaneous and evoked transmission via mitochondria at hippocampal synapses. Neuron 89: , 583–597. |
[154] | Yi M , Weaver D , Hajnóczky G ((2004) ) Control of mitochondrial motility and distribution by the calcium signal: A homeostatic circuit. J Cell Biol 167: , 661–672. |
[155] | Glater EE , Megeath LJ , Stowers RS , Schwarz TL ((2006) ) Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J Cell Biol 173: , 545–557. |
[156] | Russo GJ , Louie K , Wellington A , Macleod GT , Hu F , Panchumarthi S , Zinsmaier KE ((2009) ) Drosophila Miro is required for both anterograde and retrograde axonal mitochondrial transport. J Neurosci 29: , 5443–5455. |
[157] | Sheng ZH , Cai Q ((2012) ) Mitochondrial transport in neurons: Impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci 13: , 77–93. |
[158] | Quinlan CL , Perevoshchikova IV , Hey-Mogensen M , Orr AL , Brand MD ((2013) ) Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol 1: , 304–312. |
[159] | CipakGasparovic A , Zarkovic N , Zarkovic K , Semen K , Kaminskyy D , Yelisyeyeva O , Bottari SP ((2017) ) Biomarkers of oxidative and nitro-oxidative stress: Conventional and novel approaches. Br J Pharmacol 174: , 1771–1783. |
[160] | Cobley JN , Fiorello ML , Bailey DM ((2018) ) 13 reasons why the brain is susceptible to oxidative stress. Redox Biol 15: , 490–503. |
[161] | Mishra P , Chan DC ((2016) ) Metabolic regulation of mitochondrial dynamics. J Cell Biol 212: , 379–387. |
[162] | Sanchis-Gomar F , García-Giménez JL , Gómez-Cabrera MC , Pallardó FV ((2014) ) Mitochondrial biogenesis in health and disease. Molecular and therapeutic approaches. Curr Pharm Des 20: , 5619–5633. |
[163] | Scarpulla RC ((2011) ) Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. . Biochim Biophys Acta 1813: , 1269–1278. |
[164] | Kang I , Chu CT , Kaufman BA ((2018) ) The mitochondrial transcription factor TFAM in neurodegeneration: Emerging evidence and mechanisms. FEBS Lett 592: , 793–811. |
[165] | Krols M , Bultynck G , Janssens S ((2016) ) ER-Mitochondria contact sites: A new regulator of cellular calcium flux comes into play. J Cell Biol 214: , 367–370. |
[166] | Rizzuto R , Pozzan T ((2006) ) Microdomains of intracellular Ca2+: Molecular determinants and functional consequences. Physiol Rev 86: , 369–408. |
[167] | McCormack JG , Halestrap AP , Denton RM ((1990) ) Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev 70: , 391–425. |
[168] | De Stefani D , Raffaello A , Teardo E , Szabò I , Rizzuto R ((2011) ) A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476: , 336–340. |
[169] | Bernardi P ((1999) ) Mitochondrial transport of cations: Channels, exchangers, and permeability transition. Physiol Rev 79: , 1127–1155. |
[170] | Green DR , Kroemer G ((2004) ) The pathophysiology of mitochondrial cell death. Science 305: , 626–629. |
[171] | Werth JL , Thayer SA ((1994) ) Mitochondria buffer physiological calcium loads in cultured rat dorsal root ganglion neurons. J Neurosci 14: , 348–356. |
[172] | Billups B , Forsythe ID ((2002) ) Presynaptic mitochondrial calcium sequestration influences transmission at mammalian central synapses. J Neurosci 22: , 5840–5847. |
[173] | Voos W ((2013) ) Chaperone-protease networks in mitochondrial protein homeostasis. Biochim Biophys Acta 1833: , 388–399. |
[174] | Youle RJ , Narendra DP ((2011) ) Mechanisms of mitophagy. Nat Rev Mol Cell Biol 12: , 9–14. |
[175] | Rüb C , Wilkening A , Voos W ((2017) ) Mitochondrial quality control by the Pink1/Parkin system. Cell Tissue Res 367: , 111–123. |
[176] | Cenini G , Voos W ((2019) ) Mitochondria as potential targets in Alzheimer disease therapy: An update. Front Pharmacol 10: , 902. |
[177] | Haass C , Selkoe DJ ((2007) ) Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol 8: , 101–112. |
[178] | Tillement L , Lecanu L , Papadopoulos V ((2011) ) Alzheimer’s disease: Effects of β-Amyloid on mitochondria. Mitochondrion 11: , 13–21. |
[179] | Jiao Y , Zhang Y , Wei Y , Liu Z , An W , Guo M ((2012) ) Direct observation of internalization and ROS generation of amyloid β-peptide in neuronal cells at subcellular resolution. Chembiochem 13: , 2335–2338. |
[180] | Bigl M , Brückner MK , Arendt T , Bigl V , Eschrich K ((1999) ) Activities of key glycolytic enzymes in the brains of patients with Alzheimer’s disease. J Neural Transm (Vienna) 106: , 499–511. |
[181] | Blass JP , Sheu RK , Gibson GE ((2000) ) Inherent abnormalities in energy metabolism in Alzheimer disease. Interaction with cerebrovascular compromise. Ann N Y Acad Sci 903: , 204–221. |
[182] | Eckert A , Schulz KL , Rhein V , Götz J ((2010) ) Convergence of amyloid-beta and tau pathologies on mitochondria. Mol Neurobiol 41: , 107–114. |
[183] | Jurica MS , Mesecar A , Heath PJ , Shi W , Nowak T , Stoddard BL ((1998) ) The allosteric regulation of pyruvate kinase by fructose-1,6-bisphosphate. Structure 6: , 195–210. |
[184] | Chaneton B , Gottlieb E ((2012) ) Rocking cell metabolism: Revised functions of the key glycolytic regulator PKM2 in cancer. Trends Biochem Sci 37: , 309–316. |
[185] | Gibson GE , Shi Q ((2010) ) A mitocentric view of Alzheimer’s disease suggests multi-faceted treatments. J Alzheimers Dis 20: , S591–S607. |
[186] | Kapogiannis D , Mattson MP ((2011) ) Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and Alzheimer’s disease. Lancet Neurol 10: , 187–198. |
[187] | Johnson AB , Blum NR ((1970) ) Nucleoside phosphatase activities associated with the tangles and plaques of Alzheimer’s disease: A histochemical study of natural and experimental neurofibrillary tangles. J Neuropathol Exp Neurol 29: , 463–478. |
[188] | Hirai K , Aliev G , Nunomura A , Fujioka H , Russell RL , Atwood CS , Johnson AB , Kress Y , Vinters HV , Tabaton M , Shimohama S , Cash AD , Siedlak SL , Harris PL , Jones PK , Petersen RB , Perry G , Smith MA ((2001) ) Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci 21: , 3017–3023. |
[189] | Butterfield DA , Halliwell B ((2019) ) Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat Rev Neurosci 20: , 148–160. |
[190] | Wang J , Markesbery WR , Lovell MA ((2006) ) Increased oxidative damage in nuclear and mitochondrial DNA in mild cognitive impairment. J Neurochem 96: , 825–832. |
[191] | Area-Gomez E , de Groof A , Bonilla E , Montesinos J , Tanji K , Boldogh I , Pon L , Schon EA ((2018) ) A key role for MAM in mediating mitochondrial dysfunction in Alzheimer disease. Cell Death Dis 9: , 335. |
[192] | Calkins MJ , Reddy PH ((2011) ) Amyloid beta impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer’s disease neurons. Biochim Biophys Acta 1812: , 507–513. |
[193] | Beck SJ , Guo L , Phensy A , Tian J , Wang L , Tandon N , Gauba E , Lu L , Pascual JM , Kroener S , Du H ((2016) ) Deregulation of mitochondrial F1FO-ATP synthase via OSCP in Alzheimer’s disease. Nat Commun 7: , 11483. |
[194] | García-Blanco A , Baquero M , Vento M , Gil E , Bataller L , Cháfer-Pericás C ((2017) ) Potential oxidative stress biomarkers of mild cognitive impairment due to Alzheimer disease. J Neurol Sci 373: , 295–302. |
[195] | Mark RJ , Keller JN , Kruman I , Mattson MP ((1997) ) Basic FGF attenuates amyloid beta-peptide-induced oxidative stress, mitochondrial dysfunction, and impairment of Na+/K+-ATPase activity in hippocampal neurons. Brain Res 756: , 205–214. |
[196] | Butterfield DA , Poon HF , St Clair D , Keller JN , Pierce WM , Klein JB , Markesbery WR ((2006) ) Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: Insights into the development of Alzheimer’s disease. Neurobiol Dis 22: , 223–232. |
[197] | Du H , Guo L , Yan S , Sosunov AA , McKhann GM , Yan S ((2010) ) Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc Natl Acad Sci U S A 107: , 18670–18675. |
[198] | Tramutola A , Abate G , Lanzillotta C , Triani F , Barone E , Iavarone F , Vincenzoni F , Castagnola M , Marziano M , Memo M , Garrafa E , Butterfield DA , Perluigi M , Di Domenico F , Uberti D ((2018) ) Protein nitration profile of CD3+ lymphocytes from Alzheimer disease patients: Novel hints on immunosenescence and biomarker detection. Free Radic Biol Med 129: , 430–439. |
[199] | Mangialasche F , Baglioni M , Cecchetti R , Kivipelto M , Ruggiero C , Piobbico D , Kussmaul L , Monastero R , Brancorsini S , Mecocci P ((2015) ) Lymphocytic mitochondrial aconitase activity is reduced in Alzheimer’s disease and mild cognitive impairment. J Alzheimers Dis 44: , 649–660. |
[200] | Birnbaum JH , Wanner D , Gietl AF , Saake A , Kündig TM , Hock C , Nitsch RM , Tackenberg C ((2018) ) Oxidative stress and altered mitochondrial protein expression in the absence of amyloid-β and tau pathology in iPSC-derived neurons from sporadic Alzheimer’s disease patients. Stem Cell Res 27: , 121–130. |
[201] | Wang X , Su B , Fujioka H , Zhu X ((2008) ) Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer’s disease patients. Am J Pathol 173: , 470–482. |
[202] | Wang X , Su B , Lee HG , Li X , Perry G , Smith MA , Zhu X ((2009) ) Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci 29: , 9090–9103. |
[203] | Gan X , Huang S , Wu L , Wang Y , Hu G , Li G , Zang H , Yu H , Swerdlow RH , Chen ZX , Yan SS ((2014) ) Inhibition of ERK-DLP1 signaling and mitochondrial division alleviates mitochondrial dysfunction in Alzheimer’s disease cybrid cell. Biochim Biophys Acta 1842: , 220–231. |
[204] | Cho DH , Nakamura T , Fang J , Cieplak P , Godzik A , Gu Z , Lipton SA ((2009) ) S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 324: , 102–105. |
[205] | Manczak M , Reddy PH ((2012) ) Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer’s disease. Hum Mol Genet 21: , 5131–5146. |
[206] | Kim YJ , Park JK , Kang WS , Kim SK , Han C , Na HR , Park HJ , Kim JW , Kim YY , Park MH , Paik JW ((2017) ) Association between Mitofusin 2 gene polymorphisms and late-onset Alzheimer’s disease in the Korean population. Psychiatry Investig 14: , 81–85. |
[207] | de Brito OM , Scorrano L ((2008) ) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456: , 605–610. |
[208] | Filadi R , Greotti E , Turacchio G , Luini A , Pozzan T , Pizzo P ((2016) ) Presenilin 2 modulates endoplasmic reticulum-mitochondria coupling by tuning the antagonistic effect of Mitofusin 2. Cell Rep 15: , 2226–2238. |
[209] | Wang X , Su B , Siedlak SL , Moreira PI , Fujioka H , Wang Y , Casadesus G , Zhu X ((2008) ) Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci U S A 105: , 19318–19323. |
[210] | Zhao XL , Wang WA , Tan JX , Huang JK , Zhang X , Zhang BZ , Wang YH , Cheng HY , Zhu HL , Sun XJ , Huang FD ((2010) ) Expression of beta-amyloid induced age-dependent presynaptic and axonal changes in Drosophila. J Neurosci 30: , 1512–1522. |
[211] | Wang W , Yin J , Ma X , Zhao F , Siedlak SL , Wang Z , Torres S , Fujioka H , Xu Y , Perry G , Zhu X ((2017) ) Inhibition of mitochondrial fragmentation protects against Alzheimer’s disease in rodent model. Hum Mol Genet 26: , 4118–4131. |
[212] | Sheng B , Wang X , Su B , Lee HG , Casadesus G , Perry G , Zhu X ((2012) ) Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J Neurochem 120: , 419–429. |
[213] | Song C , Li M , Xu L , Shen Y , Yang H , Ding M , Liu X , Xie Z ((2018) ) Mitochondrial biogenesis mediated by melatonin in an APPswe/PS1dE9 transgenic mice model. Neuroreport 29: , 1517–1524. |
[214] | Cagin U , Duncan OF , Gatt AP , Dionne MS , Sweeney ST , Bateman JM ((2015) ) Mitochondrial retrograde signaling regulates neuronal function. Proc Natl Acad Sci U S A 112: , E6000–E6009. |
[215] | Lee JH , Yu WH , Kumar A , Lee S , Mohan PS , Peterhoff CM , Wolfe DM , Martinez-Vicente M , Massey AC , Sovak G , Uchiyama Y , Westaway D , Cuervo AM , Nixon RA ((2010) ) Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 141: , 1146–1158. |
[216] | Lin MT , Simon DK , Ahn CH , Kim LM , Beal MF ((2002) ) High aggregate burden of somatic mtDNA point mutations in aging and Alzheimer’s disease brain. Hum Mol Genet 11: , 133–145. |
[217] | Rui Y , Tiwari P , Xie Z , Zheng JQ ((2006) ) Acute impairment of mitochondrial trafficking by beta-amyloid peptides in hippocampal neurons. J Neurosci 26: , 10480–10487. |
[218] | Hansson Petersen CA , Alikhani N , Behbahani H , Wiehager B , Pavlov PF , Alafuzoff I , Leinonen V , Ito A , Winblad B , Glaser E , Ankarcrona M ((2008) ) The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc Natl Acad Sci U S A 105: , 13145–13150. |
[219] | Anandatheerthavarada HK , Biswas G , Robin MA , Avadhani NG ((2003) ) Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J Cell Biol 161: , 41–54. |
[220] | Cenini G , Rüb C , Bruderek M , Voos W ((2016) ) Amyloid β-peptides interfere with mitochondrial preprotein import competence by a coaggregation process. Mol Biol Cell 27: , 3257–3272. |
[221] | Dubey M , Chaudhury P , Kabiru H , Shea TB ((2008) ) Tau inhibits anterograde al transport and perturbs stability in growing axonal neurites in part by displacing kinesin cargo: Neurofilaments attenuate tau-mediated neurite instability. Cell Motil Cytoskeleton 65: , 89–99. |
[222] | Manczak M , Calkins MJ , Reddy PH ((2011) ) Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum Mol Genet 20: , 2495–2509. |
[223] | Eckert A , Nisbet R , Grimm A , Götz J ((2014) ) March separate, strike together–role of phosphorylated TAU in mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta 1842: , 1258–1266. |
[224] | Melov S , Adlard PA , Morten K , Johnson F , Golden TR , Hinerfeld D , Schilling B , Mavros C , Masters CL , Volitakis I , Li QX , Laughton K , Hubbard A , Cherny RA , Gibson B , Bush AI ((2007) ) Mitochondrial oxidative stress causes hyperphosphorylation of tau. PLoS One 2: , e536. |
[225] | Chacinska A , Koehler CM , Milenkovic D , Lithgow T , Pfanner N ((2009) ) Importing mitochondrial proteins: Machineries and mechanisms. Cell 138: , 628–644. |
[226] | Devi L , Prabhu BM , Galati DF , Avadhani NG , Anandatheerthavarada HK ((2006) ) Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J Neurosci 26: , 9057–9068. |
[227] | Subramanian S , Gottschalk WK , Kim SY , Roses AD , Chiba-Falek O ((2017) ) The effects of PPARγ on the regulation of the TOMM40-APOE-C1 genes cluster. Biochim Biophys Acta Mol Basis Dis 1863: , 810–816. |
[228] | Saunders AM , Strittmatter WJ , Schmechel D , George-Hyslop PH , Pericak-Vance MA , Joo SH , Rosi BL , Gusella JF , Crapper-MacLachlan DR , Alberts MJ ((1993) ) Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 43: , 1467–1472. |
[229] | Davies G , Harris SE , Reynolds CA , Payton A , Knight HM , Liewald DC , Lopez LM , Luciano M , Gow AJ , Corley J , Henderson R , Murray C , Pattie A , Fox HC , Redmond P , Lutz MW , Chiba-Falek O , Linnertz C , Saith S , Haggarty P , McNeill G , Ke X , Ollier W , Horan M , Roses AD , Ponting CP , Porteous DJ , Tenesa A , Pickles A , Starr JM , Whalley LJ , Pedersen NL , Pendleton N , Visscher PM , Deary IJ ((2014) ) A genome-wide association study implicates the APOE locus in nonpathological cognitive ageing. Mol Psychiatry 19: , 76–87. |
[230] | Roses AD , Lutz MW , Amrine-Madsen H , Saunders AM , Crenshaw DG , Sundseth SS , Huentelman MJ , Welsh-Bohmer KA , Reiman EM ((2010) ) A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer’s disease. Pharmacogenomics J 10: , 375–384. |
[231] | Johnson SC , La Rue A , Hermann BP , Xu G , Koscik RL , Jonaitis EM , Bendlin BB , Hogan KJ , Roses AD , Saunders AM , Lutz MW , Asthana S , Green RC , Sager MA ((2011) ) The effect of TOMM40 poly-T length on gray matter volume and cognition in middle-aged persons with APOE ɛ3/ɛ3 genotype. Alzheimers Dement 7: , 456–465. |
[232] | Payton A , Sindrewicz P , Pessoa V , Platt H , Horan M , Ollier W , Bubb VJ , Pendleton N , Quinn JP ((2016) ) A TOMM40 poly-T variant modulates gene expression and is associated with vocabulary ability and decline in nonpathologic aging. Neurobiol Aging 39: , 217.e1–7. |
[233] | Amtul Z ((2016) ) Why therapies for Alzheimer’s disease do not work: Do we have consensus over the path to follow? Ageing Res Rev 25: , 70–84. |
[234] | Krstic D , Knuesel I ((2013) ) Deciphering the mechanism underlying late-onset Alzheimer disease. Nat Rev Neurol 9: , 25–34. |
[235] | Sun N , Youle RJ , Finkel T ((2016) ) The mitochondrial basis of aging. Mol Cell 61: , 654–666. |
[236] | de la Monte SM ((2017) ) Insulin resistance and neurodegeneration: Progress towards the development of new therapeutics for Alzheimer’s disease. Drugs 77: , 47–65. |
[237] | Hall CN , Klein-Flügge MC , Howarth C , Attwell D ((2012) ) Oxidative phosphorylation, not glycolysis, powers presynaptic and postsynaptic mechanisms underlying brain information processing. J Neurosci 32: , 8940–8951. |
[238] | Warburg O ((1956) ) On the origin of cancer cells. Science 123: , 309–314. |
[239] | Bading H ((2013) ) Nuclear calcium signalling in the regulation of brain function. Nat Rev Neurosci 14: , 593–608. |
[240] | Sun J , Ren X , Qi W , Yuan D , Simpkins JW ((2016) ) Geissoschizine methyl ether protects oxidative stress-mediated cytotoxicity in neurons through the ‘Neuronal Warburg Effect.’. J Ethnopharmacol 187: , 249–258. |
[241] | Bas-Orth C , Tan YW , Lau D , Bading H ((2017) ) Synaptic activity drives a genomic program that promotes a neuronal Warburg effect. J Biol Chem 292: , 5183–5194. |
[242] | Zhang SJ , Zou M , Lu L , Lau D , Ditzel DA , Delucinge-Vivier C , Aso Y , Descombes P , Bading H ((2009) ) Nuclear calcium signaling controls expression of a large gene pool: Identification of a gene program for acquired neuroprotection induced by synaptic activity. PLoS Gene 5: , e1000604. |
[243] | Zhang SJ , Steijaert MN , Lau D , Schütz G , Delucinge-Vivier C , Descombes P , Bading H ((2007) ) Decoding NMDA receptor signaling: Identification of genomic programs specifying neuronal survival and death. Neuron 53: , 549–562. |
[244] | Mobasheri A , Richardson S , Mobasheri R , Shakibaei M , Hoyland JA ((2005) ) Hypoxia inducible factor-1 and facilitative glucose transporters GLUT1 and GLUT3: Putative molecular components of the oxygen and glucose sensing apparatus in articular chondrocytes. Histol Histopathol 20: , 1327–1338. |
[245] | Hitosugi T , Fan J , Chung TW , Lythgoe K , Wang X , Xie J , Ge Q , Gu TL , Polakiewicz RD , Roesel JL , Chen GZ , Boggon TJ , Lonial S , Fu H , Khuri FR , Kang S , Chen J ((2011) ) Tyrosine phosphorylation of mitochondrial pyruvate dehydrogenase kinase 1 is important for cancer metabolism. Mol Cell 44: , 864–877. |
[246] | Sutendra G , Michelakis ED ((2013) ) Pyruvate dehydrogenase kinase as a novel therapeutic target in oncology. Front Oncol 3: , 38. |
[247] | Glancy B , Balaban RS ((2012) ) Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 51: , 2959–2973. |
[248] | Qiu J , Tan YW , Hagenston AM , Martel MA , Kneisel N , Skehel PA , Wyllie DJ , Bading H , Hardingham GE ((2013) ) Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat Commun 4: , 2034. |
[249] | Sideris DI , Danial JSH , Emin D , Ruggeri FS , Xia Z , Zhang YP , Lobanova E , Dakin H , De S , Miller A , Sang JC , Knowles TPJ , Vendruscolo M , Fraser G , Crowther D , Klenerman D ((2021) ) Soluble amyloid beta-containing aggregates are present throughout the brain at early stages of Alzheimer’s disease. Brain Commun 3: , fcab147. |
[250] | Nunomura A , Perry G , Aliev G , Hirai K , Takeda A , Balraj EK , Jones PK , Ghanbari H , Wataya T , Shimohama S , Chiba S , Atwood CS , Petersen RB , Smith MA ((2001) ) Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol 60: , 759–767. |
[251] | DeKosky ST , Scheff SW ((1990) ) Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann Neurol 27: , 457–464. |
[252] | Scheff SW , Price DA ((1993) ) Synapse loss in the temporal lobe in Alzheimer’s disease. Ann Neurol 33: , 190–199. |
[253] | Selkoe DJ ((2012) ) Preventing Alzheimer’s disease. Science 337: , 1488–1492. |
[254] | Sonntag KC , Ryu WI , Amirault KM , Healy RA , Siegel AJ , McPhie DL , Forester B , Cohen BM ((2017) ) Late-onset Alzheimer’s disease is associated with inherent changes in bioenergetics profiles. Sci Rep 7: , 14038. |


