
Cognition at Each Stage of Lewy Body Disease with Co-occurring Alzheimer’s Disease Pathology1
Abstract
Background:
Alzheimer’s disease neuropathologic change (ADNC) may contribute to dementia in patients with Lewy body disease (LBD) pathology.
Objective:
To examine how co-occurring ADNC impacts domain specific cognitive impairments at each pathologic stage (brainstem, limbic, cerebral cortical) of LBD.
Methods:
2,433 participants with antemortem longitudinal neuropsychological assessment and postmortem neuropathological assessment from the National Alzheimer’s Coordinating Center’s Uniform Data Set were characterized based on the evaluation of ADNC and LBD. Longitudinal mixed-models were used to derive measures of cumulative cognitive deficit for each cognitive domain at each pathologic stage of LBD (brainstem, limbic, and cerebral cortical).
Results:
111 participants with a pathologic diagnosis of LBD, 741 participants with combined LBD and ADNC, 1,357 participants with ADNC only, and 224 with no pathology (healthy controls) were included in the analyses. In the executive/visuospatial domain, combined LBD and ADNC showed worse deficits than LBD only when Lewy bodies were confined to the brainstem, but no difference when Lewy bodies extended to the limbic or cerebral cortical regions. The cerebral cortical LBD only group exhibited greater executive/visuospatial deficits than the ADNC only group. By contrast, the ADNC only group and the combined pathology group both demonstrated significantly greater cumulative memory deficits relative to Lewy body disease only, regardless of stage.
Conclusion:
The impact of co-occurring ADNC on antemortem cumulative cognitive deficits varies not only by domain but also on the pathological stage of Lewy bodies. Our findings stress the cognitive impact of different patterns of neuropathological progression in Lewy body diseases.
INTRODUCTION
Historically, the strongest predictor of cognitive impairment and dementia in patients with clinical Parkinson’s disease and dementia with Lewy Bodies is the progression of Lewy body disease (LBD) pathology to the cortex [1, 2]. However, co-occu-rring Alzheimer’s disease neuropathologic change (ADNC) is also a common pathologic finding in patients with LBD [3]. Indeed, clinical-pathological series indicate that 10–59%of patients with dementia due to LBD exhibit co-occurring ADNC [4, 5], which may act synergistically with LBD on domain specific cognitive impairments [6–8].
In both LBD and ADNC, the progression of the underlying pathology generally corresponds to a dec-line in cognitive functioning [9, 10], accompanied by unique patterns of decline depending on the differences in pathological spread. For instance, the initial episodic memory impairment in early Alzheimer’s di-sease is associated with tau tangles in the medial temporal lobes, which is followed by development of language and semantic memory deficits as tau tangle pathology progresses to affect semantic language net-works in the broader temporal lobe [11, 12]. Interestingly, some patients with LBD exhibit a similar pattern of cognitive decline, which has been sugge-sted to occur, or is more pronounced, in the presence of co-morbid ADNC [13]. In contrast, most patients with LBD experience primary executive and visuospatial deficits, suggesting that these deficits are closely related to LBD neurodegenerative processes irrespective of ADNC [14]. However, it is unknown if the impact of ADNC on cognition is different at each pathologic stage of LBD (brainstem, limbic, and cerebral cortical).
We compared cognitive impairment in patients with LBD, ADNC, and combined ADNC/LBD at three pathologic stages of LBD (brainstem, limbic, and cerebral cortical). We examined a novel measurement of cumulative cognitive deficit that quantifies the extent of impairment statistically modeled acr-oss the available data six years prior to the patient’s death using longitudinal pre-mortem cognitive domain-specific assessments. We hypothesized the domain-specific cumulative cognitive deficits will be different in participants with ADNC co-morbidity across increasing severity of LBD pathologic lesions.
MATERIALS AND METHODS
Participants
The current study used data from the U.S. National Alzheimer’s Coordinating Center’s (NACC) Uniform Data Set (UDS) collected between November 2005 and June 2019 from 35 past and present NIA-funded Alzheimer’s Disease Centers [15]. Participants were evaluated approximately annually at each Center using a standardized protocol beginning in 2005. Par-ticipants were enrolled with any level of cognition, ranging from normal to demented. Written informed consent was obtained from all participants and their study coparticipants; institutional review board approval was obtained from all individual Centers.
Participants selected for the current study met the following criteria (Fig. 1): 1) had both neuropath-ologic [16] and neuropsychological data, version C1 [17] available; and 2) time between the most recent UDS in-person visit and death was not greater than 2 years to ensure that the neuropsychological test results reflected cognitive status proximate to death, consistent with previous reports [18, 19]. Exclusion criteria included the presence of any of the follo-wing at autopsy: Down’s syndrome, prion disease, early-onset autosomal dominant genetic diseases, frontotemporal lobar degeneration, progressive sup-ranuclear palsy, cortical basal degeneration, multiple system atrophy, CADASIL, other 3R and/or 4R tauopathies, argyrophilic grains, chronic traumatic encephalopathy, TDP-43, tangle dominance disease, pigment-spheroid degeneration, malformation of co-rtical development, metabolic/storage disorders, leukodystrophy, multiple sclerosis or other demyelinating disease, neoplasm, cerebral microbleeds and microinfarcts (as the latter have the most support in the literature regarding impact on cognition) [20, 21].
Fig. 1
Flow Diagram of Exclusion Criteria. “Pure” Lewy body disease pathology (LBD) groups exhibited low Alzheimer’s disease neuropathologic change (ADNC). Low ADNC includes Neuritic plaque score (CERAD[26]) equivalent to C0 or C1, and Braak Neurofibrillary Tangle Score (Braak Stage; equivalent to B0 or B1 [27]). LBD groups were further delineated into brainstem (LBD-b; most commonly observed in the substantia nigra or locus coeruleus), limbic (LBD-l; cingulate, entorhinal, amygdala), and cerebral cortical (LBD-c) groups. Combined pathology groups (ADNC/LBD) exhibited intermediate or high ADNC and the presence of Lewy body neuropathology and were further delineated into brainstem (ADNC/LBD-b), limbic (ADNC/LBD-l), and cerebral cortical (ADNC/LBD-c) groups. “Pure” ADNC exhibited intermediate or high ADNC and no LBD. Healthy controls (HC) exhibited low ADNC and no LBD and no diagnosis of dementia at any evaluation. NACC, National Alzheimer’s Coordinating Center; UDS, Uniform Data Set; FTD, frontotemporal dementia; CERAD, Consortium to Establish a Registry for Alzheimer’s disease criteria.
![Flow Diagram of Exclusion Criteria. “Pure” Lewy body disease pathology (LBD) groups exhibited low Alzheimer’s disease neuropathologic change (ADNC). Low ADNC includes Neuritic plaque score (CERAD[26]) equivalent to C0 or C1, and Braak Neurofibrillary Tangle Score (Braak Stage; equivalent to B0 or B1 [27]). LBD groups were further delineated into brainstem (LBD-b; most commonly observed in the substantia nigra or locus coeruleus), limbic (LBD-l; cingulate, entorhinal, amygdala), and cerebral cortical (LBD-c) groups. Combined pathology groups (ADNC/LBD) exhibited intermediate or high ADNC and the presence of Lewy body neuropathology and were further delineated into brainstem (ADNC/LBD-b), limbic (ADNC/LBD-l), and cerebral cortical (ADNC/LBD-c) groups. “Pure” ADNC exhibited intermediate or high ADNC and no LBD. Healthy controls (HC) exhibited low ADNC and no LBD and no diagnosis of dementia at any evaluation. NACC, National Alzheimer’s Coordinating Center; UDS, Uniform Data Set; FTD, frontotemporal dementia; CERAD, Consortium to Establish a Registry for Alzheimer’s disease criteria.](https://ip.ios.semcs.net:443/media/jad/2021/80-3/jad-80-3-jad201187/jad-80-jad201187-g001.jpg)
Neuropathological characterization
Each Center conducted neuropathologic assessments following consensus guidelines and uploaded the data to the NACC using neuropathology forms 9 and 10 [22]. The presence and stage of ADNC [23, 24] and LBD [25] were used to characterize the extent of neuropathology and assign participants to one of the following eight groups (detailed in Fig. 1): Healthy controls (HC), LBD only [differentiated into brainstem (LBD-b), limbic (LBD-l), and cerebral cor-tical (LBD-c)], and combined pathology ADNC/LBD [differentiated by stage of LBD: brainstem (ADNC/LBD-b), limbic (ADNC/LBD-l), and cerebral cortical (ADNC/LBD-c)]. Specifically, consensus guidelines for LBD were used to determine the presence and stage of LBD with an NIA-AA Lewy body score of either 1, 2, or 3, corresponding to bra-instem, limbic/transitional, or cerebral cortical [25]. ADNC only and combined pathology groups (ADNC/LBD) exhibited intermediate or high ADNC. Low ADNC was defined as no/sparse neuritic plaques [Consortium to Establish a Registry for Alzheimer’s disease criteria (CERAD) score equivalent to C0 or C1] [26] and Braak stage 0-II for neurofibrillary degeneration [27] (equivalent to B0 or B1). Intermediate ADNC was defined as moderate or frequent neuritic plaques (CERAD C2 or C3) and Braak B2 (stage III-IV). High ADNC was defined as moderate or frequent neuritic plaques (CERAD C2 or C3) and Braak B3 (stage V-VI). A dichotomous variable quantifying the presence of vascular brain injury (other than those excluded for cerebral microbleeds and microinfarcts) was created to use as a covariate in statistical models.
Demographic and clinical characteristics
We report demographic data, including age, sex, education, and race/ethnicity, health histories, and clinical diagnoses as of the last study visit in the UDS (within 2 years of death). Diagnoses of normal cognition, mild cognitive impairment (MCI), or dementia were made at each Center either by a single clinician or consensus group of clinicians after a review of all evaluation information available.
Neuropsychological testing
As the majority of patients with neuropathological data available were administered the UDS C1 version of the neuropsychological assessment, only those participants were included in the current analyses. The Mini-Mental State Examination (MMSE) and neuropsychological tests administered as part of the UDS C1 are described in detail elsewhere [17]. We calculated normative z-scores using age- and sex-corrected demographic adjustments based on previously published regression norms derived using the UDS [28]. Tests were grouped into cognitive domains based on previous investigations [29]. In addition, we ensured that the within-domain subtest correlations were greater than the between-cognitive tests correlations within this sample (Fig. 2A). Domain scores were calculated by averaging the sum of the z-scores for the test scores within each of the following domains: executive/visuospatial, memory, language, and attention (Fig. 2A). A separate visuospatial domain score could not be generated since the UDS C1 neuropsychological test battery includes no stand-alone visuospatial test; however, the UDS C1 tests of executive function rely on visuospatial information processing and therefore this domain is referred to as the executive/visuospatial domain.
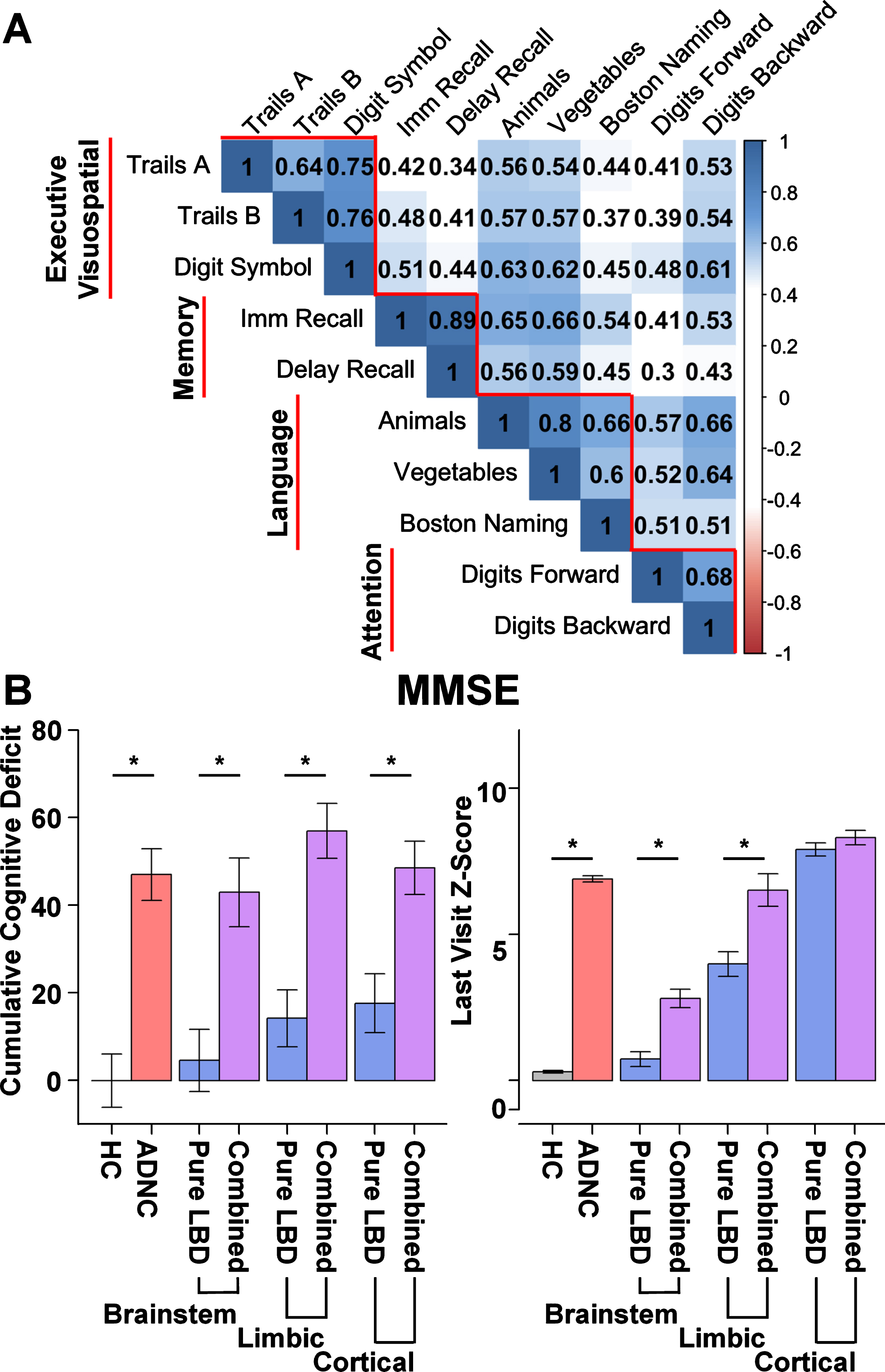
Fig. 2
Neuropsychological correlations and cumulative global deficit by group. A) Correlation plot depicting greater within domain correlations relative to between domain correlations among neuropsychological tests. Subtests (horizontal) contributing to each domain score (vertical). Executive Visuospatial domain includes: Trails A, Trail Making Test A time; Trails B, Trail Making Test B time; Digit Symbol, Digit Symbol Coding from the Wechsler Adult Intelligence Scale –Revised. Memory domain includes: Imm Recall, Logical memory Immediate Recall; Delay Recall, Logical Memory Delayed Recall. Language domain includes: number of correct words on Animal and Vegetable List generation; score on the modified Boston Naming Test (30 odd item version); Attention Domain includes: Digits Forward, Digit Span Forward total score Wechsler Memory Scale –Revised (WMS-R); Digits Backward, Digit Span Backwards total score from the WMS-R. B) Cumulative global deficit on Mini-Mental Status Exam (MMSE) and average MMSE Z-score at the participant’s last visit (within 2 years of death) by group. Cumulative global deficit is significantly greater for Alzheimer’s disease neuropathologic change (ADNC, red) and the combined ADNC and Lewy body disease groups (ADNC/LBD, purple) relative to Healthy Controls (HC, gray) and LBD only (blue), irrespective of stage of LBD (Brainstem, Limbic, or Cortical).
Statistical analyses
Participants were recruited at various stages of cognitive impairment with different lengths of follow up prior to their death, raising challenges to using traditional measures of longitudinal change (e.g., annualized rate of change). To overcome this, we cal-culated a measure of cumulative cognitive deficit derived using the estimated summary score trajectory based on the fitted mixed effects regression model. Specifically, we performed mixed effects regression analyses with longitudinal global cognitive ability (MMSE) or domain-specific summary scores (i.e., executive/visuospatial, memory, language, attention) as the outcome and time as the independent varia-ble of primary interest. Within-person correlations between the longitudinally measured summary sco-res are accounted for by subject-specific random intercept and slope, performed within each of the eight neuropathological subgroups. Models were adj-usted for age, gender, education, and the presence of vascular factors. The domain-specific models included the MMSE score as a covariate. Cumulative cognitive deficits are the average deviation for each cognitive domain between normative performance (a z-score of 0) and estimated summary score trajectory based on the fitted mixed effects regression model that was derived using available timepoints within a six-year time window prior to death. This allows us to summarize the cumulative impairment for patients who had less than six-year follow-up prior to their death. Additionally, the statistical model was used to estimate the underlying cumulative exposure after adjusting for a set of potential confounders.
We evaluated selected contrasts of interest with a focus on interactions between ADNC and various levels of LBD (e.g., the effect of ADNC on participants with LBD-b versus the effect of ADNC on participants with LBD-c) and applied appropriate follow-up simple effects contrasts. Statistical inferences were made via a nonparametric bootstrap method in which we repeatedly calculated the estimated cumulative deficit based on bootstrapped data. Planned sensitivity analyses were conducted to determine if results were consistent when the lowest possible score on a given test was substituted for data missing due to cognitive reasons. Two-sided probabilities of p < 0.0.0125 (0.05/4) were considered significant, after applying Bonferroni correction for the four comparisons within each model. All statistical analyses were conducted using R (R 3.6.1 The R Foundation for Statistical Computing).
RESULTS
Participants
Demographic, clinical, and pathological characte-ristics of the sample are presented in Table 1. Among the 2,433 autopsied UDS participants included in the current study, the mean age at death was 82.18 years (SD = 10.19 years) and the average interval between last clinical visit and death was 8.78 months (SD = 5.94 months). Across the entire sample, 111 (4.6) had LBD only [33 (1.4%) LBD-b, 43 (1.8%) LBD-l, and 35 (1.5%) LBD-c], 741 (30.5%) had combined ADNC/LBD [56 (2.3%) ADNC/LBD-b, 337 (13.9%) ADNC/LBD-l and 348 (14.3%) ADNC/LBD-c], 1,357 (55.8%) had ADNC only, and 224 (9.2%) had neither ADNC nor LBD and did not have dementia at any visit (HC) including their last visit within 2 years of death. Across all participants with LBD (N = 852), 111 (13%) had LBD only and the remainder had combined ADNC/LBD (N = 741). Clinical diagnoses for each of the neuropathology defined groups are presented in Table 1. The number and frequency of clinical diagnoses were as follows: (1) within LBD, 34 (30.6%) Parkinson’s disease, 50 (45.0%) dementia with Lewy bodies, and 20 (18.0%) Alzheimer’s disease; (2) within the ADNC/LBD, 70 (9.4%) Parkinson’s disease, 127 (17.1%) dementia with Lewy bodies, and 531 (71.7%) Alzheimer’s disease; (3) within the ADNC, 26 (1.9%) Parkinson’s disease, 36 (2.7%) dementia with Lewy bodies, and 1,089 (80.3%) Alzheimer’s disease.
Table 1
Demographic, Clinical, and Pathologic Characteristics
| LBD (n = 111) | ADNC/LBD (n = 741) | |||||||
| Pathologic Diagnostic Group (n) | HC (224) | ADNC (1357) | Brainstem (33) | Limbic (43) | Cortical (35) | Brainstem (56) | Limbic (337) | Cortical (348) |
| Age at death, mean (SD) | 84.1 (9.0) | 80.7 (10.7) | 82.2 (8.7) | 79.4 (8.9) | 78.9 (9.5) | 81.3 (8.7) | 78.3 (11.0) | 77.2 (9.4) |
| Female, n (%) | 111 (49.6) | 644 (47.5) | 16 (48.5) | 4 (9.3) | 8 (22.9) | 29 (51.8) | 152 (45.1) | 122 (35.1) |
| Years Education, mean (SD) | 15.3 (2.9) | 15.2 (3.3) | 15.9 (3.0) | 16.8 (3.1) | 15.6 (2.9) | 14.3 (3.6) | 15.7 (3.0) | 15.5 (3.2) |
| Non-Hispanic, n (%) | 211 (94.2) | 1,287 (94.8) | 32 (97.0) | 43 (100.0) | 35 (100.0) | 49 (87.5) | 319 (94.7) | 332 (95.4) |
| White, n (%) | 210 (93.8) | 1,279 (94.3) | 33 (100.0) | 41 (95.3) | 33 (94.3) | 52 (92.9) | 316 (93.8) | 322 (92.5) |
| Number of visits, mean (SD) | 4.8 (2.6) | 4.3 (2.6) | 4.6 (2.8) | 4.2 (2.7) | 4.3 (2.5) | 3.9 (2.1) | 4.9 (2.8) | 3.8 (2.4) |
| Baseline MMSE, mean (SD) | 28.4 (2.0) | 20.0 (8.5) | 26.6 (5.9) | 25.2 (4.6) | 23.9 (5.8) | 20.8 (7.9) | 18.3 (8.5) | 18.8 (8.5) |
| Last visit MMSE, mean (SD) | 28.1 (1.9) | 15.5 (10.2) | 24.5 (7.3) | 26.7 (13.2) | 20.5 (7.1) | 16.4 (9.5) | 13.7 (12.0) | 12.5 (8.4) |
| CDR, mean (SD) | 0.62 (1.44) | 12.54 (6.02) | 3.77 (5.41) | 7.48 (6.21) | 8.64 (5.65) | 10.74 (6.27) | 13.71 (5.38) | 13.80 (5.02) |
| Clinical Diagnosis, n (%) | ||||||||
| Parkinson’s disease | 3 (1.3) | 26 (1.9) | 5 (15.2) | 17 (39.5) | 12 (34.3) | 4 (7.1) | 24 (7.1) | 42 (12.1) |
| Mild cognitive impairment | 42 (18.8) | 85 (6.3) | 6 (18.2) | 7 (16.3) | 3 (8.6) | 3 (5.4) | 13 (3.9) | 2 (0.5) |
| Dementia at any visit | 0 (0) | 1200 (88.4) | 11 (33.3) | 29 (67.4) | 28 (80) | 47 (83.9) | 315 (93.4) | 342 (98.2) |
| DLB primary | 2 (0.01) | 36 (0.03) | 7 (21.2) | 25 (58.1) | 18 (51.4) | 5 (8.9) | 21 (6.2) | 101 (29.0) |
| AD primary | 0 (0) | 1,089 (80.3) | 6 (18.18) | 8 (18.6) | 6 (17.1) | 38 (67.9) | 273 (81.0) | 220 (63.2) |
| ADNC Pathologic | ||||||||
| Braak Stage, n (%) | ||||||||
| III/IV | 0 (0) | 284 (20.9) | 0 (0) | 0 (0) | 0 (0) | 22 (39.3) | 49 (14.5) | 107 (30.7) |
| V/VI | 0 (0) | 1073 (78.9) | 0 (0) | 0 (0) | 0 (0) | 34 (60.7) | 288 (85.5) | 241 (69.2) |
| CERAD (C score), n (%) | ||||||||
| C2 | 0 (0) | 371 (27.3) | 0 (0) | 0 (0) | 0 (0) | 21(37.5) | 69 (20.5) | 116 (33.3) |
| C3 | 0 (0) | 982 (72.4) | 0 (0) | 0 (0) | 0 (0) | 35 (62.5) | 268 (79.5) | 231 (66.4) |
| Vascular Pathology, n (%) | ||||||||
| Infarcts and lacunes | 56 (25.0) | 260 (19.2) | 7 (21.1) | 2 (4.7) | 7 (20.0) | 14 (25.0) | 46 (13.6) | 40 (11.5) |
HC, healthy control; LBD, Lewy body disease; ADNC/LBD, Alzheimer’s disease neuropathologic change and Lewy body disease pathology; SD, standard deviation; CDR, Clinical Dementia Rating Standard sum of boxes; DLB, dementia with Lewy bodies; AD, Alzheimer’s disease; CERAD, Consortium to Establish a Registry for Alzheimer’s Disease. Number of participants for each group specified by the column label. In addition, percentage of the group or standard deviation are presented in parenthesis.
Global cognitive ability
The ADNC only and all three combined pathology groups (ADNC/LBD-b, ADNC/LBD-l, and ADNC/LBD-c) all exhibited significantly greater cumulative MMSE deficit than the HC and LBD only groups (LBD-b, LBD-l, and LBD-c). Of the LBD only groups, only those with limbic and cerebral cortical stages of LBD (LBD-l and LBD-c) had significantly greater cumulative MMSE deficit than the HC group (Table 2; Fig. 2B).
Table 2
Results
| Cognitive Domain | Main/Simple Effects | Interaction [(ADNC/LBD –LB)–(ADNC –HC)] | ||
| difference (CI) | p | difference (CI) | p | |
| MMSE | ||||
| Brainstem LBD × ADNC | –8.65 (–21.49 4.20) | 0.187 | ||
| LBD-b –HC | 4.63 (–3.50 12.75) | 0.264 | ||
| ADNC –LBD-b | 42.38 (34.10 50.65) | < 0.001 | ||
| ADNC/LBD-b –LBD-b | 38.36 (26.76 50.95) | < 0.001 | ||
| ADNC/LBD-b –ADNC | –4.02 (14.22 6.18) | 0.440 | ||
| Limbic LBD × ADNC | –4.26 (–11.74 3.21) | 0.264 | ||
| LBD-l –HC | 14.23 (8.32 20.20) | < 0.001 | ||
| ADNC –LBD-l | 32.78 (26.85 38.69) | < 0.001 | ||
| ADNC/LBD-l –LBD-l | 42.74 (35.52 49.96) | < 0.001 | ||
| ADNC/LBD-l –AD | 9.97 (5.64 14.30) | < 0.001 | ||
| Cortical LBD × ADNC | –16.11 (–24.43 –7.79) | <0.001 | ||
| LBD-c –HC | 17.66 (11.18 24.14) | < 0.001 | ||
| ADNC –LBD-c | 29.35 (22.44 36.25) | < 0.001 | ||
| ADNC/LBD-c –LBD-c | 30.89 (23.32 38.47) | < 0.001 | ||
| ADNC/LBD-c –ADNC | 1.55 (–2.80 5.90) | 0.485 | ||
| Executive/Visuospatial | ||||
| Brainstem LBD × ADNC | 0.80 (–3.20 4.81) | 0.694 | ||
| LBD-b –HC | –0.49 (–2.39 1.41) | 0.614 | ||
| ADNC –LBD-b | 6.40 (4.38 8.42) | < 0.001 | ||
| ADNC/LBD-b –LBD-b | 6.71 (2.96 10.46) | < 0.001 | ||
| ADNC/LBD-b –ADNC | 0.32 (–3.09 3.72) | 0.860 | ||
| Limbic LBD × ADNC | –6.55 (–10.71 –2.94) | <0.001 | ||
| LBD-l –HC | 8.65 (5.84 11.46) | < 0.001 | ||
| ADNC –LBD-l | –2.74 (–5.60 0.12) | 0.060 | ||
| ADNC/LBD-l –LBD-l | –0.65 (–3.99 2.70) | 0.705 | ||
| ADNC/LBD-l –ADNC | 2.10 (0.18 4.02) | 0.032 | ||
| Cortical LBD × ADNC | –9.17 (–15.40 –2.95) | 0.004 | ||
| LBD-c –HC | 13.70 (7.53 19.87) | < 0.001 | ||
| ADNC –LBD-c | –7.79 (–13.86 –1.73) | 0.012 | ||
| ADNC/LBD-c –LBD-c | –3.26 (–9.46 2.93) | 0.302 | ||
| ADNC/LBD-c –ADNC | 4.53 (3.13 5.92) | < 0.001 | ||
| Memory | ||||
| Brainstem LBD × ADNC | –3.51 (–5.90 –1.12) | 0.004 | ||
| LBD-b –HC | 2.25 (0.42 4.08) | 0.016 | ||
| ADNC –LBD-b | 6.73 (4.96 8.51) | < 0.001 | ||
| ADNC/LBD-b –LBD-b | 5.47 (3.21 7.74) | < 0.001 | ||
| ADNC/LBD-b –ADNC | –1.26 (–2.70 0.18) | 0.087 | ||
| Limbic LBD × ADNC | –3.49 (–5.31 –1.67) | <0.001 | ||
| LBD-l –HC | 4.04 (2.54 5.53) | < 0.001 | ||
| ADNC –LBD-l | 4.94 (3.49 6.41) | < 0.001 | ||
| ADNC/LBD-l –LBD-l | 5.50 (3.90 7.10) | < 0.001 | ||
| ADNC/LBD-l –ADNC | 0.55 (–0.11 1.21) | 0.102 | ||
| Cortical LBD × ADNC | –5.02 (–7.10 –2.94) | <0.001 | ||
| LBD-c –HC | 4.13 (2.17 6.09) | < 0.001 | ||
| ADNC –LBD-c | 5.21 (3.29 7.14) | < 0.001 | ||
| ADNC/LBD-c –LBD-c | 3.97 (2.05 5.88) | < 0.001 | ||
| ADNC/LBD-c –ADNC | –0.89 (–1.53 –0.26) | 0.006 | ||
| Language | ||||
| Brainstem LBD × ADNC | –0.83 (–2.89 1.24) | 0.432 | ||
| LBD-b –HC | 0.95 (–0.64 2.55) | 0.242 | ||
| ADNC –LBD-b | 3.15 (1.60 4.70) | < 0.001 | ||
| ADNC/LBD-b –LBD-b | 3.28 (1.28 5.27) | 0.001 | ||
| ADNC/LBD-b –ADNC | 0.13 (–1.18 1.43) | 0.850 | ||
| Limbic LBD × ADNC | –1.13 (–2.65 0.40) | 0.148 | ||
| LBD-l –HC | 1.15 (0.14 2.15) | 0.025 | ||
| ADNC –LBD-l | 2.96 (2.03 3.88) | < 0.001 | ||
| ADNC/LBD-l –LBD-l | 2.98 (1.53 4.43) | < 0.001 | ||
| ADNC/LBD-l –ADNC | 0.02 (–1.24 1.28) | 0.974 | ||
| Cortical LBD × ADNC | –1.65 (–3.20 –0.09) | 0.038 | ||
| LBD-c –HC | 1.66 (0.29 3.03) | 0.018 | ||
| ADNC –LBD-c | 2.45 (1.12 3.78) | < 0.001 | ||
| ADNC/LBD-c –LBD-c | 2.50 (1.00 3.92) | 0.001 | ||
| ADNC/LBD-c –ADNC | 0.01 (–0.66 0.69) | 0.968 | ||
| Attention | ||||
| Brainstem LBD × ADNC | –0.36 (–1.28 1.46) | 0.699 | ||
| LBD-b –HC | 0.11 (–1.23 1.44) | 0.874 | ||
| ADNC –LBD-b | 0.96 (–0.31 2.23) | 0.139 | ||
| ADNC/LBD-b –LBD-b | 0.71 (–1.05 2.47) | 0.430 | ||
| ADNC/LBD-b –ADNC | –0.25 (–1.47 0.97) | 0.687 | ||
| Limbic LBD × ADNC | 0.10 (–1.36 1.56) | 0.893 | ||
| LBD-l –HC | –0.00 (–1.20 1.20) | 0.996 | ||
| ADNC –LBD-l | 1.07 (–0.09 2.23) | 0.070 | ||
| ADNC/LBD-l –LBD-l | 1.17 (–0.15 2.49) | 0.082 | ||
| ADNC/LBD-l –ADNC | 0.10 (–0.51 0.71) | 0.755 | ||
| Cortical LBD × ADNC | –0.50 (–1.98 0.97) | 0.503 | ||
| LBD-c –HC | 0.93 (–0.35 2.20) | 0.156 | ||
| ADNC –LBD-c | 0.14 (–1.12 1.41) | 0.824 | ||
| ADNC/LBD-c –LBD-c | 0.56 (–0.77 1.90) | 0.406 | ||
| ADNC/LBD-c –ADNC | 0.42 (–0.10 0.94) | 0.114 | ||
HC, healthy control; ADNC, Alzheimer’s disease neuropathologic change; LBD-b, brainstem Lewy body disease; ADNC/LBD-b, Alzheimer’s disease neuropathologic change and brainstem Lewy body disease; LBD-l, limbic Lewy body disease; ADNC/LBD-l, Alzheimer’s disease neuropathologic change and limbic Lewy body disease; LBD-c, cerebral cortical Lewy body disease; ADNC/LBD-c, Alzheimer’s disease neuropathologic change and cerebral cortical Lewy body disease. Bold indicates the groups within each model that demonstrated a significantly greater deficit within each contrast, with significance set at p≤0.0125 (0.05/4) after correction for multiple comparisons within each model.
Executive/visuospatial
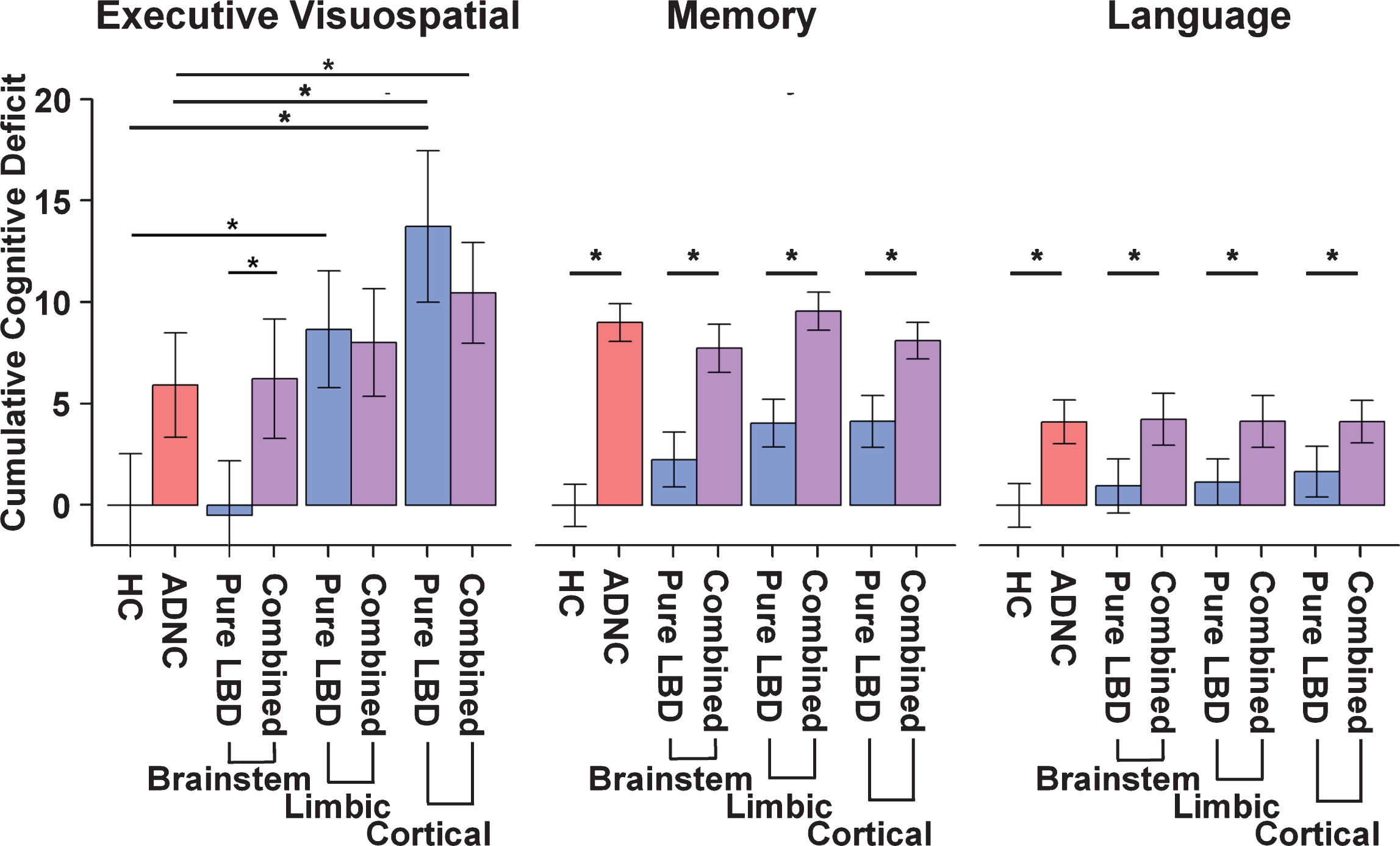
There was a significant interaction between ADNC and LBD in the limbic and cerebral cortical stages of LBD (Table 2; Fig. 3). The limbic LBD and cerebral cortical LBD groups exhibited a significantly greater cumulative executive/visuospatial deficit than the HC group; however, only the cerebral cortical LBD group showed greater cumulative deficit than the ADNC only group. The reverse was seen for the LBD-b group, which showed significantly less cumulative executive/visuospatial deficit than the ADNC only group. Interestingly, limbic and cerebral cortical LBD groups (LBD-l and LBD-c) had sim-ilar cumulative executive/visuospatial deficit as com-bined pathology groups (ADNC/LBD-l and ADNC/LBD-c), but the LBD-b group exhibited less cum-ulative executive/visuospatial deficit as the combined ADNC/LBD-b group. Finally, the combined pathologies groups (ADNC/LBD-l and ADNC/LBD-c) also exhibited a significantly greater cumulative deficit than the ADNC only group, but only the ADNC/LBD-c group remained significant after Bonferroni correction.
Fig. 3
Barplots depicting the cumulative deficit for cognitive domains by group. In the Memory and Language domains combined LBD and ADNC (purple) showed a significantly greater cumulative deficit than pure LBD (blue) at each stage of Lewy body pathology. By contrast, in the Executive/Visuospatial domain combined LBD and ADNC showed greater cumulative deficit than pure LBD only when the Lewy body pathology was confined to the brainstem. Limbic and Cortical Lewy body pathology showed no differences in cumulative deficit with or without combined ADNC. HC, healthy control; ADNC, Alzheimer’s disease neuropathologic change; LBD, Lewy body disease; ADNC/LBD, Alzheimer’s disease neuropathologic change and Lewy body disease.
Memory
There was a significant interaction between ADNC and LBD in all stages of LBD (brainstem, limbic, and cerebral cortical). Unlike the executive/visuospatial domain, the ADNC only and combined pathology groups (ADNC/LBD-b, ADNC/LBD-l, and ADNC/LBD-c) all exhibited significantly greater cumulative memory deficit than the LBD only groups, regardless of pathologic stage (LBD-b, LBD-l, and LBD-c). The LBD-l and LBD-c exhibited significantly greater cumulative memory deficit than the HC group, but not the LBD-b group.
Language
There was a significant interaction between ADNC and LBD only in the cerebral cortical LBD stage. Similar to the memory domain, the ADNC only and combined pathology groups (ADNC/LBD-b, ADNC/LBD-l, ADNC/LBD-c) all exhibited greater cumulative language deficit than the LBD only groups, regardless of pathologic stage (LBD-b, LBD-l, and LBD-c).
Attention
No significant interaction was observed for ADNC and LBD (brainstem, limbic, and cerebral cortical examined separately). Simple effects indicated no significant differences.
Missing data and imputation
Findings were similar in models with neuropsychological test scores imputed as the worst score when data was missing due to cognitive reasons. Ra-tes of missing data due to cognitive reasons varied based on the cognitive test. For instance, 15%of ADNC, 13%of ADNC/LBD-l, 14%of ADNC/LBD-c, 10%of ADNC/LBD-b and 0%of the remaining groups were missing delayed memory test scores. In contrast, 21%of ADNC, 20%of ADNC/LBD-l, 24%of ADNC/LBD-c, 18%of ADNC/LBD-b, 12%LBD-c, 6%LBD-l and 0%of the remaining groups were missing scores on the Trail Making Test Part B.
Findings were also similar when limited to a subset of participants that were demented at their last visit (within two years of death). These analyses did not include the brain stem LBD groups since this subsample was too small. Findings were also similar when analyses included the time between last clinical visit and site as additional covariates.
Data availability
All data used in the current analyses is available for download from the National Alzheimer’s Coordinating Center (https://www.alz.washington.edu/).
DISCUSSION
Increasing evidence suggests ADNC may contri-bute to or exacerbate the impact of LBD on cognitive dysfunction [8, 30, 31]. Our study is the first to show the cognitive sequelae of ADNC co-morbidity is domain-specific and depends on the pathologic stage of LBD.
The interaction between ADNC and LBD stage is domain-specific
Individuals with limbic and cerebral cortical LBD (LBD-l and LBD-c) exhibited executive/visuospatial deficits that are the same or worse than those of individuals with ADNC, suggesting that LBD uniquely impacts this cognitive domain to a greater degree, but only after it has progressed past the brainstem. Additionally, executive/visuospatial deficits did not significantly differ between brainstem LBD and HC, further suggesting impairment in executive/visuospatial function reflects a progression of LBD beyond the brainstem. In contrast, memory impairment is significantly greater when there is co-occurring ADNC across all stages of LBD. Interestingly, the deficit with combined pathology is not greater than when there is ADNC alone. This suggests that more severe memory impairment is driven by ADNC, regardless of the severity of the LBD pathology.
Executive and visuospatial impairments in LBD
As participants were recruited in the study with any stage of cognitive deficits (e.g., no cognitive impairment, mild cognitive impairment, dementia), examination of annualized rate of change in this cohort is not appropriate as many individuals may have significant impairment upon enrollment into the study. To overcome this, we used a measure of cumulative deficit that quantifies the extent of impairment modeled using the available data six years prior to the participant’s death. Patients with Lewy body dementias exhibit visuospatial and executive function impairment that is relatively more pronounced than in patients with Alzheimer’s disease dementia [13, 29, 32–38]. The presence of early visuospatial deficits may be the most salient difference in cognitive profiles of dementia with Lewy bodies and Alzheimer’s disease dementia, and predicts the rate of decline in clinical and neuropathological cohorts [34, 35, 39, 40]. The executive/visuospatial domain score in the current study includes primarily executive functioning measures that rely on visuospatial information. Therefore, this measure is not specific to each of these domains and changes may reflect deficits in either of these functions. In contrast to memory, the executive/visuospatial deficit in the cerebral cortical LBD group was more pronounced than in the ADNC group and was equally severe in the limbic LBD and ADNC groups. These findings suggest that LBD is a determinant of executive/visuospatial dysfunction independent of ADNC. Further, the progression of Lewy Bodies from brainstem to limbic to cortical regions corresponds to a greater decrement in executive/visuospatial deficits. Additionally, combined pathology did not have a greater impact on executive/visuospatial deficits than LBD only. This is consistent with prior notions that the phenotypic expression of LBD may be less pronounced in the presence of co-pathology [35] and different LBD clinical subtypes may be associated with less ADNC [41].
Pathology underlying memory and language impairments
While memory impairment is a well-established predictor of dementia in Parkinson’s disease and dementia with Lewy bodies [42, 43], it is unclear to what extent memory impairment is due to LBD versus ADNC. In Parkinson’s disease patients, early memory changes may primarily be due to retrieval difficulties secondary to executive impairments (a non-amnestic memory impairment), although storage deficits related to medial temporal changes may also contribute (amnestic memory impairment) [42]. When characterizing patients based on amnestic MCI or non-amnestic MCI, prior work highlights that amnestic MCI have a propensity for increased neurofibrillary tangles in select anatomic regions, where whereas non-amnestic MCI exhibited increased Lewy body pathology [44]. The Logical Memory test used in the current study is not particularly sensitive to early memory changes [45] and may be less impacted by executive deficits, and therefore less sensitive to LBD, than other types of memory tests (e.g., word-list learning tests) since the to-be-remembered information is inherently organized and easier to retrieve [46, 47]. In the current study, we found significantly greater memory impairment in individuals with ADNC or combined ADNC/LBD than in those with LBD only, consistent with previous reports [14, 48]. While the memory deficit increased as LBD progressed across brainstem to limbic to cerebral cortical LBD groups, it never reached the same level as that of the ADNC only group. Combined ADNC/LBD did not have a significantly greater effect on memory than ADNC alone, perhaps because memory performance in the ADNC only group is near floor and any impact of LBD co-pathology cannot be discerned. With regard to language, the ADNC group had substantially greater language deficits than all LBD groups, consistent with reports of relatively preserved language in Lewy body dementias [49, 50].
Combined versus “pure” pathology in dementia syndromes
The prevalence of a dementia diagnosis at the final UDS visit varied across groups with the highest prevalence for the combined ADNC/LBD-l (93.5%) and ADNC/LBD-c (98.3%) groups. These values were higher than for those of the ADNC only (88.4%), ADNC/LBD-b (83.9%) or LBD only (LBD-b = 33.3%, LBD-l = 67.4%, LBD-c = 80.0%) groups. These results highlight that individuals with combined pathology are more likely than those with LBD only to be diagnosed with a dementia syndrome during life.
Only 13%of individuals with LBD exhibited “pure” LBD and the remainder exhibited combined ADNC/LBD with moderate or high levels of ADNC. Those with combined ADNC/LBD typically had more severe stages of α-synuclein than “pure” LBD (cerebral cortical and limbic). These results suggest a relatively lower prevalence of pure LBD relative to other studies [4, 51], likely due to the referral bias inherent in recruiting patients from memory clinics relative to movement disorders clinics. The more severe stages of LBD corresponding to greater rates of ADNC, however, is consistent with the existing literature [30]. An emerging framework proposes that the frequent co-occurrence of ADNC and LBD suggests a converging disease mechanism that promotes protein aggregation [2, 6, 52]. Our findings of a greater global cumulative cognitive deficit (i.e., MMSE) across all combined pathology groups (ADNC/LBD-b, ADNC/LBD-l and ADNC/LBD-c) than in LBD alone and HC (Fig. 2) are consistent with this framework, however, this is given the limitation that the MMSE may have fewer items sensitive to the changes that is observed in LBD pathology. Examination of the individual cognitive domains suggests that the impact of individual and combined pathologies differs depending on the domain examined. This is likely due to the regional impact of neuropathology, and warrants further investigation.
Methodological considerations
The current study is the first to evaluate a novel measurement of domain-specific cumulative cognitive deficits in a large sample of individuals with neuropathologically confirmed LBD and ADNC; however, there are several methodological considerations. One major limitation is that the UDS test battery used for the current study cannot differentiate between visuospatial and executive function deficits. Despite this, we were able to examine the relative contributions of LBD and ADNC to deficits on executive function tasks that have a strong visuospatial component. Second, there is a referral bias due to the nature of participants recruited to Alzheimer’s Disease Research Centers, such that mixed ADNC and LBD is likely more common relative to what would be observed in a movement disorders or general neurology clinic. Therefore, the current cohort likely does not reflect the relative distribution of these pathologies in the community. Third, we cannot assess the impact of cognitive or neural reserve on the differential effects of the two pathologies and their interaction on deficits in various cognitive domains. Fourth, the available NACC forms only include limited vascular information, therefore, we cannot systematically examine vascular effects on our findings. Fifth, the cumulative impairment is estimated via random effects model to allow adjusting for selected baseline covariates and including patients whose follow-up time prior to death is less than six years in the analysis. The validity and precision of the estimated cumulative impairment depends on the model assumptions, which warrants further examination in a larger cohort. Finally, the lack of detailed information regarding the distribution of LBD (e.g., cortical pathology differentially impacting parietal relative to temporal lobe) precludes the ability to fully examine domain-specific cognitive-neuropathology correlates. Future studies examining stages and regional LBD and ADNC distributions would be helpful to further understand the impact of pathology location on domain specific cognitive impairment [53, 54].
CONCLUSIONS AND IMPLICATIONS
In summary, in the latest stages of LBD pathology, there is a greater level of executive/visuospatial impairment relative to cognitive impairments from ADNC. By contrast, the extent of global and memory impairment is significantly greater in individuals with ADNC, regardless of stage of LBD pathology. An important implication of these findings is that detailed characterization of cognitive phenotypes of LBD is crucial for the development of effective disease-modifying therapeutic trials. For instance, treatments that target α-synuclein may have a greater impact on executive/visuospatial abilities than on memory. Conversely, treatments targeting tau (or perhaps ADNC more broadly) may affect memory and global cognition to a greater degree than executive/visuospatial measures. While recent work suggests that memory and language are most sensitive outcome measures of cognitive change in dementia with Lewy bodies relative to visuospatial and executive function in Parkinson’s disease dementia [55], the study did not aim to differentiate the impact of each pathology on cognition, but rather the nature of cognitive changes associated with each clinical diagnosis. Notably, dementia with Lewy bodies typically exhibit concomitant ADNC pathology, which may have driven the decline in memory and language in this particular study [55]. As disease modifying therapies for specific neuropathology emerge, it will be increasingly important to utilize outcome measures directly impacted by that specific pathology rather than co-occurring pathologies, which is often the case in dementia with Lewy bodies.
ACKNOWLEDGMENTS
This study was supported by the National Institutes of Health (NIH) –National Institute on Aging (NIA; P30 AG047366, P30 AGAG066515, and P50 AG062429) and the NIH –National Institute of Neurological Disorders and Stroke (NINDS; P50 NS062684, R01 NS115114). The NACC database is funded by NIA/NIH Grant U01 AG016976. NACC data are contributed by the NIA-funded ADCs: P30 AG019610 (PI Eric Reiman, MD), P30 AG013846 (PI Neil Kowall, MD), P30 AG062428-01 (PI James Leverenz, MD) P50 AG008702 (PI Scott Small, MD), P50 AG025688 (PI Allan Levey, MD, PhD), P50 AG047266 (PI Todd Golde, MD, PhD), P30 AG010133 (PI Andrew Saykin, PsyD), P50 AG005146 (PI Marilyn Albert, PhD), P30 AG062421-01 (PI Bradley Hyman, MD, PhD), P30 AG062422-01 (PI Ronald Petersen, MD, PhD), P50 AG005138 (PI Mary Sano, PhD), P30 AG008051 (PI Thomas Wisniewski, MD), P30 AG013854 (PI Robert Vassar, PhD), P30 AG008017 (PI Jeffrey Kaye, MD), P30 AG010161 (PI David Bennett, MD), P30 AG010129 (PI Charles DeCarli, MD), P50 AG016573 (PI Frank LaFerla, PhD), P30 AG062429-01(PI James Brewer, MD, PhD), P50 AG023501 (PI Bruce Miller, MD), P30 AG035982 (PI Russell Swerdlow, MD), P30 AG028383 (PI Linda Van Eldik, PhD), P30 AG053760 (PI Henry Paulson, MD, PhD), P30 AG010124 (PI John Trojanowski, MD, PhD), P50 AG005133 (PI Oscar Lopez, MD), P50 AG005142 (PI Helena Chui, MD), P30 AG012300 (PI Roger Rosenberg, MD), P30 AG049638 (PI Suzanne Craft, PhD), P50 AG005136 (PI Thomas Grabowski, MD), P30 AG062715-01 (PI Sanjay Asthana, MD, FRCP), P50 AG005681 (PI John Morris, MD), P50 AG047270 (PI Stephen Strittmatter, MD, PhD).
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/20-1187r2).
REFERENCES
[1] | Schneider J , Arvanitakis Z , Yu L , Boyle P , Leurgans S , Bennett D ((2012) ) Cognitive impairment, decline and fluctuations in older community-dwelling subjects with Lewy bodies. Brain 135: , 3005–3014. |
[2] | Graff-Radford J , Aakre J , Savica R , Boeve B , Kremers WK , Ferman TJ , Jones DT , Kantarci K , Knopman DS , Dickson DW ((2017) ) Duration and pathologic correlates of Lewy body disease. JAMA Neurol 74: , 310–315. |
[3] | Walker L , McAleese KE , Thomas AJ , Johnson M , Martin-Ruiz C , Parker C , Colloby SJ , Jellinger K , Attems J ((2015) ) Neuropathologically mixed Alzheimer’s and Lewy body disease: Burden of pathological protein aggregates differs between clinical phenotypes. Acta Neuropathol 129: , 729–748. |
[4] | Irwin DJ , White MT , Toledo JB , Xie SX , Robinson JL , Van Deerlin V , Lee VM , Leverenz JB , Montine TJ , Duda JE ((2012) ) Neuropathologic substrates of Parkinson disease dementia. Ann Neurol 72: , 587–598. |
[5] | Kotzbauer PT , Cairns NJ , Campbell MC , Willis AW , Racette BA , Tabbal SD , Perlmutter JS ((2012) ) Pathologic accumulation of α-synuclein and Aβ in Parkinson disease patients with dementia. Arch Neurol 69: , 1326–1331. |
[6] | Coughlin D , Xie SX , Liang M , Williams A , Peterson C , Weintraub D , McMillan CT , Wolk DA , Akhtar RS , Hurtig HI ((2019) ) Cognitive and pathological influences of tau pathology in Lewy body disorders. Ann Neurol 85: , 259–271. |
[7] | Irwin DJ , Grossman M , Weintraub D , Hurtig HI , Duda JE , Xie SX , Lee EB , Van Deerlin VM , Lopez OL , Kofler JK ((2017) ) Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: A retrospective analysis. Lancet Neurol 16: , 55–65. |
[8] | Irwin DJ , Lee VM-Y , Trojanowski JQ ((2013) ) Parkinson’s disease dementia: Convergence of α-synuclein, tau and amyloid-β pathologies. Nat Rev Neurosci 14: , 626. |
[9] | Brenowitz WD , Hubbard RA , Keene CD , Hawes SE , Longstreth WT , Woltjer RL , Kukull WA ((2017) ) Mixed neuropathologies and associations with domain-specific cognitive decline. Neurology 89: , 1773–1781. |
[10] | Brenowitz WD , Keene CD , Hawes SE , Hubbard RA , Longstreth WT Jr , Woltjer RL , Crane PK , Larson EB , Kukull WA ((2017) ) Alzheimer’s disease neuropathologic change, Lewy body disease, and vascular brain injury in clinic-and community-based samples. Neurobiol Aging 53: , 83–92. |
[11] | Bejanin A , Schonhaut DR , La Joie R , Kramer JH , Baker SL , Sosa N , Ayakta N , Cantwell A , Janabi M , Lauriola M ((2017) ) Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer’s disease. Brain 140: , 3286–3300. |
[12] | Bondi MW , Jak AJ , Delano-Wood L , Jacobson MW , Delis DC , Salmon DP ((2008) ) Neuropsychological contributions to the early identification of Alzheimer’s disease. Neuropsychol Rev 18: , 73–90. |
[13] | Kraybill ML , Larson EB , Tsuang DW , Teri L , McCormick WC , Bowen JD , Kukull WA , Leverenz JB , Cherrier MM ((2005) ) Cognitive differences in dementia patients with autopsy-verified AD, Lewy body pathology, or both. Neurology 64: , 2069–2073. |
[14] | Gurnani AS , Gavett BE ((2017) ) The differential effects of alzheimer’s disease and lewy body pathology on cognitive performance: A meta-analysis. Neuropsychol Rev 27: , 1–17. |
[15] | Beekly DL , Ramos EM , Lee WW , Deitrich WD , Jacka ME , Wu J , Hubbard JL , Koepsell TD , Morris JC , Kukull WA ((2007) ) The National Alzheimer’s Coordinating Center (NACC) database: The uniform data set. Alzheimer Dis Assoc Disord 21: , 249–258. |
[16] | Morris JC , Weintraub S , Chui HC , Cummings J , DeCarli C , Ferris S , Foster NL , Galasko D , Graff-Radford N , Peskind ER ((2006) ) The Uniform Data Set (UDS): Clinical and cognitive variables and descriptive data from Alzheimer Disease Centers. Alzheimer Dis Assoc Disord 20: , 210–216. |
[17] | Weintraub S , Salmon D , Mercaldo N , Ferris S , Graff-Radford NR , Chui H , Cummings J , DeCarli C , Foster NL , Galasko D ((2009) ) The Alzheimer’s disease centers’ uniform data set (UDS): The neuropsychological test battery. Alzheimer Dis Assoc Disord 23: , 91. |
[18] | Cholerton B , Larson E , Baker LD , Craft S , Crane PK , Millard SP , Sonnen JA , Montine TJ ((2013) ) Neuropathologic correlates of cognition in a population-based sample. J Alzheimers Dis 36: , 699–709. |
[19] | Flanagan M , Larson EB , Latimer CS , Cholerton B , Crane PK , Montine KS , White LR , Keene CD , Montine TJ ((2016) ) Clinical-pathologic correlations in vascular cognitive impairment and dementia. Biochim Biophys Acta 1862: , 945–951. |
[20] | Smith EE , Schneider JA , Wardlaw JM , Greenberg SM ((2012) ) Cerebral microinfarcts: The invisible lesions. Lancet Neurol 11: , 272–282. |
[21] | Sonnen JA , Leverenz JB , Crane PK , Larson EB , Montine TJ ((2007) ) Cognitive impairment and dementia due to Alzheimer disease and cerebrovascular pathology in autopsied ACT study patients. FASEB J 21: , A72. |
[22] | Besser LM , Kukull WA , Teylan MA , Bigio EH , Cairns NJ , Kofler JK , Montine TJ , Schneider JA , Nelson PT ((2018) ) The revised National Alzheimer’s Coordinating Center’s Neuropathology Form—available data and new analyses. J Neuropathol Exp Neurol 77: , 717–726. |
[23] | Hyman BT , Phelps CH , Beach TG , Bigio EH , Cairns NJ , Carrillo MC , Dickson DW , Duyckaerts C , Frosch MP , Masliah E ((2012) ) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 8: , 1–13. |
[24] | Montine TJ , Phelps CH , Beach TG , Bigio EH , Cairns NJ , Dickson DW , Duyckaerts C , Frosch MP , Masliah E , Mirra SS ((2012) ) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: A practical approach. Acta Neuropathol (Berl) 123: , 1–11. |
[25] | McKeith IG , Boeve BF , Dickson DW , Halliday G , Taylor J-P , Weintraub D , Aarsland D , Galvin J , Attems J , Ballard CG ((2017) ) Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology 89: , 88–100. |
[26] | Mirra SS , Heyman A , McKeel D , Sumi SM , Crain BJ , Brownlee LM , Vogel FS , Hughes JP , van Belle G , Berg L ((1991) ) The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41: , 479–486. |
[27] | Braak H , Braak E ((1991) ) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 82: , 239–259. |
[28] | Shirk SD , Mitchell MB , Shaughnessy LW , Sherman JC , Locascio JJ , Weintraub S , Atri A ((2011) ) A web-based normative calculator for the uniform data set (UDS) neuropsychological test battery. Alzheimers Res Ther 3: , 32. |
[29] | Johnson DK , Morris JC , Galvin JE ((2005) ) Verbal and visuospatial deficits in dementia with Lewy bodies. Neurology 65: , 1232–1238. |
[30] | Irwin DJ , Xie SX , Coughlin D , Nevler N , Akhtar RS , McMillan CT , Lee EB , Wolk DA , Weintraub D , Chen-Plotkin A ((2018) ) CSF tau and β-amyloid predict cerebral synucleinopathy in autopsied Lewy body disorders. Neurology 90: , e1038–e1046. |
[31] | Van Steenoven I , Aarsland D , Weintraub D , Londos E , Blanc F , Van der Flier WM , Teunissen CE , Mollenhauer B , Fladby T , Kramberger MG ((2016) ) Cerebrospinal fluid Alzheimer’s disease biomarkers across the spectrum of Lewy body diseases: Results from a large multicenter cohort. J Alzheimers Dis 54: , 287–295. |
[32] | Ferman TJ , Smith GE , Boeve BF , Graff-Radford NR , Lucas JA , Knopman DS , Petersen RC , Ivnik RJ , Wszolek Z , Uitti R ((2006) ) Neuropsychological differentiation of dementia with Lewy bodies from normal aging and Alzheimer’s disease. Clin Neuropsychol 20: , 623–636. |
[33] | Guidi M , Paciaroni L , Paolini S , De Padova S , Scarpino O ((2006) ) Differences and similarities in the neuropsychological profile of dementia with Lewy bodies and Alzheimer’s disease in the early stage. J Neurol Sci 248: , 120–123. |
[34] | Hamilton JM , Landy KM , Salmon DP , Hansen LA , Masliah E , Galasko D ((2012) ) Early visuospatial deficits predict the occurrence of visual hallucinations in autopsy-confirmed dementia with Lewy bodies. Am J Geriatr Psychiatry 20: , 773–781. |
[35] | Hamilton JM , Salmon DP , Galasko D , Raman R , Emond J , Hansen LA , Masliah E , Thal LJ ((2008) ) Visuospatial deficits predict rate of cognitive decline in autopsy-verified dementia with Lewy bodies. Neuropsychology 22: , 729. |
[36] | Hansen L , Salmon D , Galasko D , Masliah E , Katzman R , DeTeresa R , Thal L , Pay MM , Hofstetter R , Klauber M ((1990) ) The Lewy body variant of Alzheimer’s disease: A clinical and pathologic entity. Neurology 40: , 1. |
[37] | McGuinness B , Barrett SL , Craig D , Lawson J , Passmore AP ((2010) ) Executive functioning in Alzheimer’s disease and vascular dementia. Int J Geriatr Psychiatry 25: , 562–568. |
[38] | Metzler-Baddeley C ((2007) ) A review of cognitive impairments in dementia with Lewy bodies relative to Alzheimer’s disease and Parkinson’s disease with dementia. Cortex 43: , 583–600. |
[39] | Landy KM , Salmon DP , Galasko D , Filoteo JV , Festa EK , Heindel WC , Hansen LA , Hamilton JM ((2015) ) Motion discrimination in dementia with Lewy bodies and Alzheimer disease. Neurology 85: , 1376–1382. |
[40] | Mitolo M , Hamilton JM , Landy KM , Hansen LA , Galasko D , Pazzaglia F , Salmon DP ((2016) ) Visual perceptual organization ability in autopsy-verified dementia with Lewy bodies and Alzheimer’s disease. J Int Neuropsychol Soc 22: , 609–619. |
[41] | Dugger BN , Boeve BF , Murray ME , Parisi JE , Fujishiro H , Dickson DW , Ferman TJ ((2012) ) Rapid eye movement sleep behavior disorder and subtypes in autopsy-confirmed dementia with Lewy bodies. Mov Disord 27: , 72–78. |
[42] | Aarsland D ((2016) ) Cognitive impairment in Parkinson’s disease and dementia with Lewy bodies. Parkinsonism Relat Disord 22: , S144–S148. |
[43] | Levy G , Jacobs DM , Tang M , Côté LJ , Louis ED , Alfaro B , Mejia H , Stern Y , Marder K ((2002) ) Memory and executive function impairment predict dementia in Parkinson’s disease. Mov Disord 17: , 1221–1226. |
[44] | Dugger BN , Davis K , Malek-Ahmadi M , Hentz JG , Sandhu S , Beach TG , Adler CH , Caselli RJ , Johnson TA , Serrano GE ((2015) ) Neuropathological comparisons of amnestic and nonamnestic mild cognitive impairment. BMC Neurol 15: , 146. |
[45] | Rabin LA , Paré N , Saykin AJ , Brown MJ , Wishart HA , Flashman LA , Santulli RB ((2009) ) Differential memory test sensitivity for diagnosing amnestic mild cognitive impairment and predicting conversion to Alzheimer’s disease. Aging Neuropsychol Cogn 16: , 357–376. |
[46] | Lamar M , Charlton R , Zhang A , Kumar A ((2012) ) Differential associations between types of verbal memory and prefrontal brain structure in healthy aging and late life depression. Neuropsychologia 50: , 1823–1829. |
[47] | Tremont G , Halpert S , Javorsky DJ , Stern RA ((2000) ) Differential impact of executive dysfunction on verbal list learning and story recall. Clin Neuropsychol 14: , 295–302. |
[48] | Adamowicz DH , Roy S , Salmon DP , Galasko DR , Hansen LA , Masliah E , Gage FH ((2017) ) Hippocampal α-synuclein in dementia with Lewy bodies contributes to memory impairment and is consistent with spread of pathology. J Neurosci 37: , 1675–1684. |
[49] | Galasko D , Katzman R , Salmon DP , Hansen L ((1996) ) Clinical and neuropathological findings in Lewy body dementias. Brain Cogn 31: , 166–175. |
[50] | Peavy GM , Edland SD , Toole BM , Hansen LA , Galasko DR , Mayo AM ((2016) ) Phenotypic differences based on staging of Alzheimer’s neuropathology in autopsy-confirmed dementia with Lewy bodies. Parkinsonism Relat Disord 31: , 72–78. |
[51] | Pletnikova O , West N , Lee MK , Rudow GL , Skolasky RL , Dawson TM , Marsh L , Troncoso JC ((2005) ) Aβ deposition is associated with enhanced cortical α-synuclein lesions in Lewy body diseases. Neurobiol Aging 26: , 1183–1192. |
[52] | Clinton LK , Blurton-Jones M , Myczek K , Trojanowski JQ , LaFerla FM ((2010) ) Synergistic interactions between Aβ, tau, and α-synuclein: Acceleration of neuropathology and cognitive decline. J Neurosci 30: , 7281–7289. |
[53] | Beach TG , Adler CH , Lue L , Sue LI , Bachalakuri J , Henry-Watson J , Sasse J , Boyer S , Shirohi S , Brooks R ((2009) ) Unified staging system for Lewy body disorders: Correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol (Berl) 117: , 613–634. |
[54] | Toledo JB , Gopal P , Raible K , Irwin DJ , Brettschneider J , Sedor S , Waits K , Boluda S , Grossman M , Van Deerlin VM ((2016) ) Pathological α-synuclein distribution in subjects with coincident Alzheimer’s and Lewy body pathology. Acta Neuropathol (Berl) 131: , 393–409. |
[55] | Smirnov DS , Galasko D , Edland SD , Filoteo JV , Hansen LA , Salmon DP ((2020) ) Cognitive decline profiles differ in Parkinson disease dementia and dementia with Lewy bodies. Neurology 94: , e2076–e2087. |


