
APOE4 Copy Number-Dependent Proteomic Changes in the Cerebrospinal Fluid1
Abstract
Background:
APOE4 has been hypothesized to increase Alzheimer’s disease risk by increasing neuroinflammation, though the specific neuroinflammatory pathways involved are unclear.
Objective:
Characterize cerebrospinal fluid (CSF) proteomic changes related to APOE4 copy number.
Methods:
We analyzed targeted proteomic data from ADNI CSF samples using a linear regression model adjusting for age, sex, and APOE4 copy number, and additional linear models also adjusting for AD clinical status or for CSF Aβ, tau, or p-tau levels. False discovery rate was used to correct for multiple comparisons correction.
Results:
Increasing APOE4 copy number was associated with a significant decrease in a CRP peptide level across all five models (q < 0.05 for each), and with significant increases in ALDOA, CH3L1 (YKL-40), and FABPH peptide levels (q < 0.05 for each) except when controlling for AD clinical status or neurodegeneration biomarkers (i.e., CSF tau or p-tau). In all models except the one controlling for CSF Aβ levels, though not statistically significant, there was a consistent inverse direction of association between APOE4 copy number and the levels of all 24 peptides from all 8 different complement proteins measured. The odds of this happening by chance for 24 unrelated peptides would be less than 1 in 16 million.
Conclusion:
Increasing APOE4 copy number was associated with decreased CSF CRP levels across all models, and increased CSF ALDOA, CH3L1, and FABH levels when controlling for CSF Aβ levels. Increased APOE4 copy number may also be associated with decreased CSF complement pathway protein levels, a hypothesis for investigation in future studies.
INTRODUCTION
The best described genetic contributor to late onset Alzheimer’s disease (AD) is the ɛ4 polymorphism of the apolipoprotein E gene [1]. Individuals carrying a single APOE4 allele copy have a ∼3-fold increased risk of developing AD, and those who carry two APOE4 alleles have a greater than 10-fold risk of developing AD [2–5]. Additionally, the presence of an APOE4 allele is associated with worse neurologic outcomes including a higher index of disability in multiple sclerosis patients, worse cognitive outcomes following mild traumatic brain injury, and increased risk of death following subarachnoid hemorrhage [6–8], as well as increased atherosclerotic cardiovascular disease risk [9]. Likely due to these pleiotropic effects, APOE4 carriers live ∼4.2 years less than non-APOE4 carriers [10, 11]. Despite our knowledge of these multiple negative effects of APOE4, it remains unclear what the mechanisms are that explain how APOE4 contributes to AD risk and worse outcomes across these other disease states.
The ApoE protein has multiple biological roles, including cholesterol transport in the central nervous system (CNS), signaling through cell surface receptors, and modulating synaptic function by regulating the expression of syntaxin-1, PSD95, and NMDA and AMPA receptors [12]. Given this multitude of functions, it is unclear which mechanisms explain the increased AD risk in APOE4 carriers. Patients with an APOE4 allele are typically diagnosed with AD in their 7th or 8th decade of life, even though the ApoE protein is expressed within the CNS throughout life [13]. This suggests that APOE4 likely contributes to AD risk before cognitive deficits first appear [14, 15]. This idea is supported by fMRI studies demonstrating that young adult APOE4 carriers without AD have significant alterations in the default mode network when compared to non-carrier controls [16]. Additionally, young adult APOE4 carriers show increased activation of the bilateral medial temporal lobe during an encoding task [17], which may be a compensatory mechanism to achieve normal cognitive function in APOE4 carriers.
One mechanism hypothesized to underlie the link between APOE4 and AD is neuroinflammation. Indeed, neuroinflammation is a key contributor to AD pathogenesis in humans [18]. Recent evidence in murine models has shown that human APOE4 knock-in mice have increased glial activation in response to intracerebroventricular LPS injection and increased IL-1β, IL-6, and TNFα levels when compared to APOE2 or APOE3 allele knock-in mice [19]. Furthermore, microglia isolated from APOE4/4 targeted replacement mice, as compared to those from APOE3/3 mice, have increased pro-inflammatory cytokines such as IL-6, TNFα, and IL12p40 [20, 21]. Although these studies have linked APOE4 to increases in multiple inflammatory cytokines and pathways, it is unclear which of these inflammatory mechanisms are responsible for increased AD risk in APOE4 carriers.
Because neurodegeneration in AD itself is associated with inflammation, it is important to study the effect of APOE4 on the CNS of older adults who do not yet have dementia and neurodegeneration due to AD. Such studies provide an opportunity to discover how APOE4 affects the CNS and how it increases AD risk, before frank AD-related neurodegeneration begins. Thus, here we analyzed targeted cerebrospinal fluid (CSF) proteomic data from Alzheimer’s Disease Neuroimaging Institute (ADNI) research subjects, while controlling for AD clinical status, in order to find CSF protein level variation associated with APOE4 allele copy number.
METHODS
ADNI study and participants
The patient data and clinical annotations used in this study were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (http://adni.loni.usc.edu). ADNI is a longitudinal multicenter study that tracks and evaluates changes in cognition, brain structure and function, and biomarkers associated with the progression of mild cognitive impairment (MCI) and AD [22]. Further detail on ADNI is found in the Acknowledgments section. Each ADNI site received written informed consent from all participants and institutional review board approval.
Inclusion and exclusion criteria for the normal control (NC), MCI, and AD cohorts is available at http://adni.loni.usc.edu. Briefly, NC subjects were defined as having a Mini-Mental State Examination (MMSE) [23] score ≥24 and Clinical Dementia Rating (CDR) [24] score of 0 and having no confounding neurological or psychological disorders. MCI subjects had MMSE scores of 23–30, a CDR score of 0.5, objective memory loss as measured by Wechsler Memory Scale Revised—Logical Memory II [25], and preserved activities of daily living. AD patients met the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) [26] criteria for probable AD and had MMSE scores of 20–26 and CDR scores of 0.5–1.0. The current study includes CSF samples from 289 unique ADNI-1 subjects (85 normal control, 134 MCI, and 66 AD patients). Publicly available metadata such as age, gender, diagnosis at baseline, MMSE score, and APOE4 genotype were collected from the ADNI database. Cohort demographics are summarized in Table 1.
Table 1
Baseline characteristics of ADNI Patients, grouped by number of APOE4 alleles Values represent means (SD), or percentages in the case of gender (for females), or count per group (for AT classification)
| 0 APOE4 alleles (N = 148) | 1 APOE4 allele (N = 104) | 2 APOE4 alleles (N = 35) | p | |
| Age | 75.94 (6.88) | 75.43 (6.77) | 71.86 (6.88) | 0.0071 |
| Gender (Male) | 89 (60.1%) | 63 (60.6%) | 20 (57.1%) | 0.9352 |
| Race | 0.4733 | |||
| Asian | 3 (2.0%) | 0 (0.0%) | 0 (0.0%) | |
| Black/African American | 5 (3.4%) | 5 (4.8%) | 0 (0.0%) | |
| White | 140 (94.6%) | 99 (95.2%) | 35 (100.0%) | |
| Years of Education | 16 [14, 18] | 16 [14, 18] | 16 [14, 16] | 0.2274 |
| CSF Aβ* | 988.47 (397.74) | 643.16 (208.95) | 482.48 (160.98) | <0.0011 |
| CSF Tau** | 273.08 (115.89) | 335.57 (109.98) | 348.32 (120.79) | <0.0011 |
| CSF p-tau** | 25.72 (12.59) | 33.81 (12.73) | 35.64 (15.21) | <0.0011 |
| Clinical Status | <0.0012 | |||
| Normal | 65 (43.9%) | 19 (18.3%) | 2 (5.7%) | |
| MCI | 64 (43.2%) | 53 (51.0%) | 18 (51.4%) | |
| AD | 19 (12.8%) | 32 (30.8%) | 15 (42.9%) | |
| (ATN) Classification**, + | <0.0013 | |||
| A-T- | 59 (40.4%) | 8 (7.8%) | 0 (0.0%) | |
| A+T- | 37 (25.3%) | 26 (25.5%) | 9 (25.7%) | |
| A-T+ | 13 (8.9%) | 3 (2.9 %) | 0 (0.0%) | |
| A+T+ | 37 (25.3%) | 65 (63.7%) | 26 (74.3%) |
p-value key: 1ANOVA, 2Chi-square, 3Fisher’s Exact, 4Kruskal Wallis. *28 patients not included who returned values >1700; 4 patients with no BL CSF measures. **4 patients with no BL CSF measures. +A + defined as Aβ values below 1065 pg/ml, T + defined as p-tau values over 27 pg/ml. N classification was not given, because it cannot be determined based on CSF biomarker levels.
ADNI-1 CSF Collection and processing
CSF samples (0.5 mL) were obtained at ADNI visits, stored, transported, and processed according to published procedures [27, 28]. Technical details on the mass spectrometry platform data acquisition, quality control metrics, and validation protocols used in this study are described in the ADNI “Use of Targeted Multiplex Proteomic Strategies to Identify Novel CSF Biomarkers in AD” data primer and in [29]. Briefly, CSF samples were depleted of high abundance proteins using MARS-14 immunoaffinity resin, trypsin digested (1:10 protease:protein ratio), lyophilized, and desalted prior to LC/MRM-MS proteomic analysis on a QTRAP 5500 LC-MS/MS system. CSF multiplex multiple reaction monitoring (MRM) is a standardized peptide panel developed as a QC metric to verify the reproducibility of sample processing and mass spectrometry analysis [30]. A total of 320 peptides produced by tryptic digestion of 143 proteins were identified and met the QC criteria of the ADNI working group for inclusion in the original dataset [29]. These peptides were selected to measure the levels of proteins previously implicated in AD neuropathology and/or neuro-inflammation [29].
Patients in the ADNI proteomics study were classified by AT (i.e., amyloid and p-tau) status, using previously reported CSF Aβ and p-tau measurements made with the Roche Elecsys platform, and previously described Aβ, and p-tau thresholds [31, 32]. We used the AT schema rather than the full ATN classification, because CSF tau and p-tau levels are highly co-linear, such that every patient who would be T + would also be N + and vice versa (Shaw LM, personal communication, 6/16/2020).
Statistical analysis
Mass spectrometry data from the ADNI study was re-analyzed to compare peptide data by APOE4 allele count. Peptides with an expression value below zero were set to missing values. The intraclass correlation (ICC) across technical replicates was calculated for each peptide. Subsequent analysis included 294 peptides that had an ICC ≥ 0.6. The technical replicate for each individual with the smallest number of missing peptides was used in the analysis. We analyzed CSF targeted proteomic data from 289 research participants in the ADNI-1 study, 85 of whom were healthy controls, 134 of whom had MCI, and 66 of whom had dementia due to AD. Association between each of the variables of interest with each peptide was tested in a linear model framework with an empirical Bayes method for parameter estimation from the limma [33] Bioconductor [34] package. Age and gender were included as cofactors in all models; AD clinical status (i.e., normal, MCI, or dementia due to AD) was also included in model 2. Models 3, 4, and 5 also included either CSF tau or p-tau, or Aβ levels, respectively. False discovery rate was used to correct for multiple hypothesis testing within each statistical model.
RESULTS
ADNI patient cohort characteristics
Baseline characteristics of the ADNI-1 patients whose samples were used for targeted proteomics measurements [29] are presented in Table 1. Subjects with 0, 1, or 2 copies of the APOE4 allele were similar in terms of gender, race, and years of education. Consistent with prior work showing that the APOE4 allele is associated with reduced longevity [11], individuals with two APOE4 allele copies were ∼4 years younger those with zero or one copy of the APOE4 allele. As expected, the percentage of patients with MCI and AD increased among patients with either 1 or 2 APOE4 alleles. Consistent with prior work [35, 36], increasing APOE4 copy number was associated with lower CSF Aβ levels. Increasing APOE4 copy number was also associated with increases in CSF tau and p-tau levels (Table 1). Lastly, increasing APOE4 copy number was associated with decreases in the proportion of patients who were A-T- and increases in the proportion who were A + T+ (Table 1).
Model 1: CSF proteomic changes and APOE4 gene dosage
To identify protein-derived peptides whose level(s) differed as a function of APOE4 copy number, a linear model controlling for age and gender was used to test the relationship between APOE4 copy number and CSF peptide levels. Initial analysis evaluated 294 peptides with sufficient replicability (ICC ≥ 0.6) for measuring CSF expression variance by APOE4 copy number (Table 2). In this model, 12 of 294 peptides had significant expression changes (q ≤ 0.05) associated with increasing APOE4 copy number (Table 3 and Fig. 1A). CSF levels of an APOE4-specific peptide (APOE_LGADMEDVR) were substantially higher in APOE4 carriers versus non-carriers (q– = 6.53 x 10–81), consistent with prior studies [37, 38]. Two peptides found in all APOE isoforms had elevated CSF expression with increasing APOE4 copy number (APOE_LAVYQAGAR, q = 0.027; APOE_LGPLVEQGR, q = 0.027). CSF expression of an APOE2-specific peptide was found to decrease with higher APOE4 gene dosage (APOE_CLAVYQAGAR, q = 0.027).
Increasing APOE4 allele copy number was associated with reduced expression of a peptide from the acute inflammatory marker C-reactive protein (CRP) (q = 0.006). Increasing APOE4 copy number was also associated with increasing expression of peptides derived from the glycoprotein Chitinase 3-like protein 1 (CH3L1; also known as YKL-40) (q < 0.05), the cardiac injury biomarker heart-type fatty acid binding protein (FABPH) (q < 0.05), and the glycolytic enzyme fructose-bisphosphate aldolase A (ALDOA; q < 0.05).
Table 2
Proteins included in the ADNI Targeted CSF Proteomics Study
| 1433Z | CMGA | IFNB | NELL2 | SCG3 |
| A1AT | CNDP1 | IGSF8 | NEO1 | SDCB1 |
| A1AT | CNTF | IL10 | NEUS | SE6L1 |
| A1BG | CNTN1 | IL12B | NFH | SHSA7 |
| A2GL | CNTN2 | IL17 | NFL | SIAE |
| A2MG | CO2 | IL1A | NFM | SLIK1 |
| A4 | CO3 | IL27A | NGF | SMOC1 |
| AACT | CO4A | IL6 | NICA | SODC |
| AATM | CO5 | IL6RA | NLGN3 | SODE |
| AFAM | CO6 | ITIH1 | NPTX1 | SORC1 |
| ALDOA | CO8B | ITIH5 | NPTX2 | SORC2 |
| AMBP | COCH | ITM2B | NPTXR | SORC3 |
| AMD | CRP | JAK1 | NPY | SPON1 |
| APLP2 | CSTN1 | KAIN | NRCAM | SPRL1 |
| APOA | CSTN3 | KCC2B | NRX1A | STX12 |
| APOA1 | CUTA | KI67 | NRX2A | SV2A |
| APOB | CYTC | KLK10 | NRX3A | SYNJ1 |
| APOC1 | DAG1 | KLK11 | NSG1 | SYT11 |
| APOD | DIAC | KLK12 | OSTP | TADBP |
| APOE | ENOG | KLK3 | PCD17 | TAU |
| B2MG | ENPP2 | KLK6 | PCMD1 | TCRG1 |
| B3GN1 | EXTL2 | KLK9 | PCSK1 | TEN3 |
| BACE1 | FABP5 | KLKB1 | PDIA3 | TGFB1 |
| BASP1 | FABP6 | KNG1 | PDYN | TGFB2 |
| BDNF | FABP7 | KPCZ | PEDF | TGFB3 |
| BTD | FABPH | KPYM | PGRP2 | TGON2 |
| C1QA | FABPI | L1CAM | PIMT | THRB |
| C1QB | FAM3C | LAMB2 | PLDX1 | TIMP1 |
| C3AR | FBLN1 | LFTY2 | PLMN | TNF14 |
| CA2D1 | FBLN3 | LPHN1 | PPN | TNFA |
| CAD13 | FETUA | LRC4B | PRDX1 | TNR1B |
| CADM3 | FMOD | LTBP2 | PRDX2 | TNR21 |
| CAH1 | GFAP | MIME | PRDX3 | TNR6 |
| CATA | GLNA | MMP2 | PRDX4 | TRBM |
| CATD | GOGB1 | MMP9 | PRDX5 | TRFE |
| CATL1 | GOLM1 | MMRN2 | PRDX6 | TRFM |
| CCKN | GRIA4 | MOG | PTGDS | TTHY |
| CCL25 | HBA | MTHR | PTPRD | UBB |
| CD14 | HBB | MUC18 | PTPRN | UCHL1 |
| CD59 | HEMO | NBL1 | PVRL1 | VASN |
| CERU | HERC4 | NCAM1 | RIMS3 | VGF |
| CFAB | I18BP | NCAM2 | SAP | VTDB |
| CH3L1 | IBP2 | NCAN | SCG1 | X3CL1 |
| CLUS | IBP6 | NEGR1 | SCG2 |
Table 3
Expression of the Top 20 Peptides and for all complement peptides/proteins by APOE4 copy number in multivariate models accounting for age and gender (model 1). For full data set, see Supplementary File 1
| Top 20 proteins/peptides whose expression differed as a function of APOE4 copy number, controlling for age, gender, and clinical status (normal, MCI or AD) | |||||||
| Protein | UniProt ID | Peptide | Log Fold Change | Average Expression | t statistic | p | FDR Corrected p |
| APOE | P02649 | LGADMEDVR | 3.594 | 9.193 | 27.105 | 0.000 | 0.000 |
| CRP | P02741 | ESDTSYVSLK | –0.633 | 15.228 | –4.074 | 0.000 | 0.009 |
| APOE | P02649 | LGPLVEQGR | 0.287 | 22.419 | 3.588 | 0.000 | 0.038 |
| APOE | P02649 | LAVYQAGAR | 0.292 | 25.410 | 3.421 | 0.001 | 0.053 |
| ALDOA | P04075 | ALQASALK | 0.121 | 19.168 | 3.105 | 0.002 | 0.077 |
| CH3L1 | P36222 | ILGQQVPYATK | 0.112 | 23.114 | 3.133 | 0.002 | 0.077 |
| CH3L1 | P36222 | SFTLASSETGVGAPISGPGIPGR | 0.112 | 18.225 | 3.107 | 0.002 | 0.077 |
| FABPH | P05413 | SIVTLDGGK | 0.131 | 14.664 | 3.208 | 0.001 | 0.077 |
| ALDOA | P04075 | QLLLTADDR | 0.111 | 16.246 | 2.963 | 0.003 | 0.079 |
| AMBP | P02760 | FLYHK | –0.167 | 11.800 | –2.953 | 0.003 | 0.079 |
| APOE | P02649 | AATVGSLAGQPLQER | 0.247 | 20.145 | 3.039 | 0.003 | 0.079 |
| APOE | P02649 | CLAVYQAGAR | –0.630 | 8.715 | –2.919 | 0.004 | 0.079 |
| CH3L1 | P36222 | VTIDSSYDIAK | 0.109 | 21.303 | 2.915 | 0.004 | 0.079 |
| FABPH | P05413 | SLGVGFATR | 0.108 | 15.706 | 2.998 | 0.003 | 0.079 |
| KNG1 | P01042 | TVGSDTFYSFK | –0.185 | 15.118 | –2.898 | 0.004 | 0.079 |
| AMBP | P02760 | ETLLQDFR | –0.171 | 18.977 | –2.777 | 0.006 | 0.107 |
| A2GL | P02750 | DLLLPQPDLR | –0.148 | 25.898 | –2.701 | 0.007 | 0.127 |
| A2GL | P02750 | VAAGAFQGLR | –0.139 | 23.017 | –2.458 | 0.015 | 0.152 |
| AATC | P17174 | IVASTLSNPELFEEWTGNVK | 0.086 | 12.727 | 2.517 | 0.012 | 0.152 |
| AATM | P00505 | FVTVQTISGTGALR | 0.097 | 10.314 | 2.496 | 0.013 | 0.152 |
| CSF Complement proteins/peptides expression as a function of APOE4 copy number, controlling for age, gender, and clinical status (normal, MCI or AD) | |||||||
| Protein | UniProt ID | Peptide | Log Fold Change | Average Expression | t statistic | p | FDR Corrected p |
| C1QB | P02746 | LEQGENVFLQATDK | –0.042 | 17.128 | –1.241 | 0.216 | 0.622 |
| C1QB | P02746 | VPGLYYFTYHASSR | –0.053 | 17.604 | –1.135 | 0.257 | 0.669 |
| CFAB | P00751 | DAQYAPGYDK | –0.134 | 16.401 | –2.435 | 0.016 | 0.152 |
| CFAB | P00751 | VSEADSSNADWVTK | –0.111 | 16.220 | –2.471 | 0.014 | 0.152 |
| CFAB | P00751 | YGLVTYATYPK | –0.134 | 22.532 | –2.474 | 0.014 | 0.152 |
| CO2 | P06681 | DFHINLFR | –0.065 | 18.691 | –1.232 | 0.219 | 0.623 |
| CO2 | P06681 | HAIILLTDGK | –0.070 | 15.695 | –1.625 | 0.105 | 0.412 |
| CO2 | P06681 | SSGQWQTPGATR | –0.077 | 15.922 | –1.689 | 0.092 | 0.377 |
| CO3 | P01024 | IHWESASLLR | –0.267 | 14.006 | –2.136 | 0.034 | 0.235 |
| CO3 | P01024 | TELRPGETLNVNFLLR | –0.081 | 10.150 | –1.997 | 0.047 | 0.259 |
| CO4A | P0C0L4 | DHAVDLIQK | –0.038 | 22.142 | –0.745 | 0.457 | 0.858 |
| CO4A | P0C0L4 | GSFEFPVGDAVSK | –0.035 | 25.297 | –0.611 | 0.541 | 0.879 |
| CO4A | P0C0L4 | LGQYASPTAK | –0.041 | 21.701 | –0.760 | 0.448 | 0.857 |
| CO4A | P0C0L4 | NVNFQK | –0.019 | 18.387 | –0.392 | 0.695 | 0.978 |
| CO4A | P0C0L4 | VLSLAQEQVGGSPEK | –0.049 | 19.989 | –1.039 | 0.300 | 0.734 |
| CO4A | P0C0L4 | VTASDPLDTLGSEGALSPGGVASLLR | –0.029 | 18.008 | –0.712 | 0.477 | 0.871 |
| CO5 | P01031 | DINYVNPVIK | –0.045 | 16.181 | –0.630 | 0.529 | 0.876 |
| CO5 | P01031 | TLLPVSKPEIR | –0.050 | 17.209 | –0.757 | 0.449 | 0.857 |
| CO5 | P01031 | VFQFLEK | –0.043 | 18.210 | –0.644 | 0.520 | 0.873 |
| CO6 | P13671 | ALNHLPLEYNSALYSR | –0.113 | 16.297 | –1.772 | 0.077 | 0.345 |
| CO6 | P13671 | SEYGAALAWEK | –0.118 | 15.760 | –2.072 | 0.039 | 0.240 |
| CO8B | P07358 | IPGIFELGISSQSDR | –0.057 | 14.476 | –0.761 | 0.447 | 0.857 |
| CO8B | P07358 | SDLEVAHYK | –0.082 | 13.090 | –1.342 | 0.181 | 0.571 |
| CO8B | P07358 | YEFILK | –0.077 | 18.883 | –1.263 | 0.208 | 0.610 |
Log Fold Change - log2 fold change of peptide expression with each additional APOE4 allele copy in multivariate models for all analyzed CSF samples; Average expression –average peptide expression in all analyzed CSF samples; t statistic –hypothesis test statistic estimating the mean peptide expression (population mean) from the sampling distribution for each peptide in all analyzed CSF samples.
Fig. 1
Volcano Plot of CSF Protein/Peptide Expression by APOE genotype, for the top 20 proteins and the complement cascade proteins in model 1 (A), and for the top 20 proteins and the complement cascade proteins in model 2 (B).
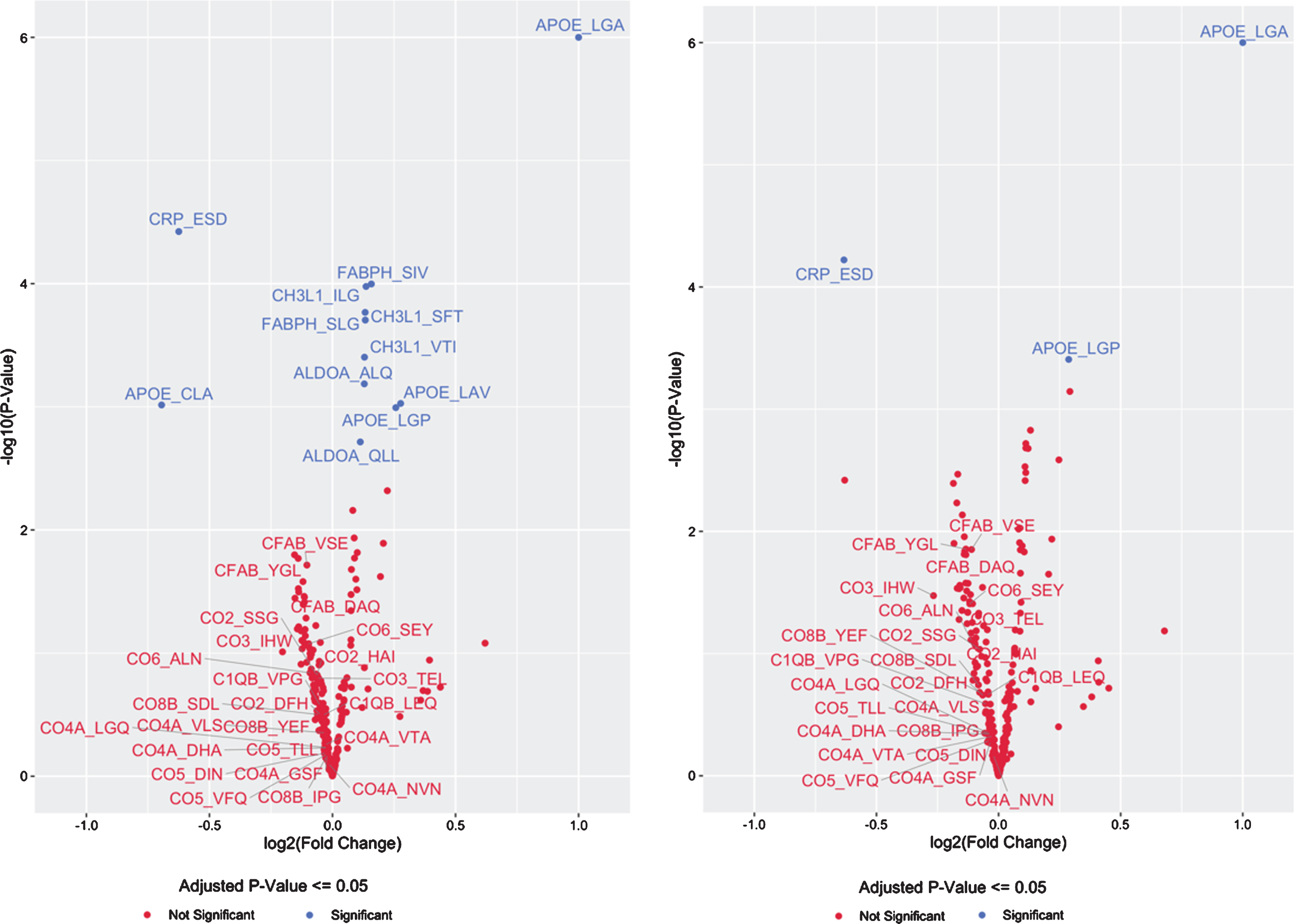
There was a consistent inverse direction of association between APOE4 copy number and CSF levels of all 24 peptides from all 8 complement pathway proteins measured (Table 3, Fig. 1A), although these effects were not significant for any individual complement protein-derived peptide (p > 0.05 for each, prior to multiple correction comparison). Nonetheless, the odds of this happening for 24 unrelated peptides by chance would be 1 over 224, or less than 1 in 16 million. Alternatively, since these 24 peptides were derived from 8 complement pathway proteins, the odds of 8 proteins at random all showing consistently lower expression as a function of APOE4 allele copy number, even if the change for each individual protein was not statistically significant, would be 1/28, or a 1 out of 256 chance.
Model 2: APOE4-dependent CSF peptide changes and clinical status
Because APOE4 is found in AD patients at disproportionately high frequencies compared to the general population, it is possible that the above findings reflect confounding by AD clinical status (and neurodegeneration) rather than changes directly related to increased APOE4 copy number itself. Therefore, a second linear model was used that corrected for clinical status (normal control, MCI, or dementia due to AD) in addition to the items in model 1, to test for associations between APOE4 copy number and CSF peptide expression levels. In this second model, only 3 of 294 peptides had statistically significant APOE4 copy number-related changes in CSF expression levels (Table 4 and Fig. 1B). Increasing APOE4-copy number was associated with increased expression of the APOE4-specific peptide (LGADMEDVR) (q < 0.01) and decreased expression the CRP-derived peptide (ESDTSYVSLK) (q < 0.01). A pan-APOE peptide (LGPLVEQGR) had increased expression associated with increased APOE4 copy number in this model (q = 0.038). CH3L1 (YKL-40)-, FABPH-, and ALDOA-derived peptides that showed significant APOE4- copy number-related changes in expression in model 1 (above) no longer remained statistically significant after correcting for disease status and multiple comparisons, although there was still a trend toward increased CSF protein expression for each (q = 0.077, q = 0.077, and q = 0.079, respectively). As in the first model (not controlling for AD clinical status), none of the 24 complement protein-derived peptides demonstrated statistically significant differences as a function of APOE4 copy number. Yet, as in the first model, in this model there was a consistent inverse direction of association between APOE4 copy number and expression of all 24 peptides from all 8 different complement proteins measured (Table 4). Although none of these inverse associations were statistically significant on their own (p > 0.05 for each, prior to multiple comparison) the odds of 24 unrelated peptides all showing this pattern of decreased expression by chance would be less than 1 over 224, or less than 1 in 16 million. Alternatively, all 24 of these peptides were derived from 8 complement pathway proteins. Thus the odds of 8 proteins all showing lower expression levels as a function of APOE4 allele copy number, even if the change in each individual protein was not statistically significant at p < 0.05, would be 1/28, or a 1 out of 256 chance.
Table 4
Expression of the Top 20 Peptides and for all complement peptides/proteins by APOE4 copy number in multivariate models accounting for age and gender and clinical status (Model 2). For full data set, see Supplementary File 1
| Top 20 proteins/peptides whose expression differed as a function of APOE4 copy number, controlling for age, gender, and clinical status (normal, MCI or AD) | |||||||
| Protein | UniProt ID | Peptide | Log Fold Change | Average Expression | t statistic | p | FDR Corrected p |
| APOE | P02649 | LGADMEDVR | 3.594 | 9.193 | 27.105 | 0.000 | 0.000 |
| CRP | P02741 | ESDTSYVSLK | –0.633 | 15.228 | –4.074 | 0.000 | 0.009 |
| APOE | P02649 | LGPLVEQGR | 0.287 | 22.419 | 3.588 | 0.000 | 0.038 |
| APOE | P02649 | LAVYQAGAR | 0.292 | 25.410 | 3.421 | 0.001 | 0.053 |
| ALDOA | P04075 | ALQASALK | 0.121 | 19.168 | 3.105 | 0.002 | 0.077 |
| CH3L1 | P36222 | ILGQQVPYATK | 0.112 | 23.114 | 3.133 | 0.002 | 0.077 |
| CH3L1 | P36222 | SFTLASSETGVGAPISGPGIPGR | 0.112 | 18.225 | 3.107 | 0.002 | 0.077 |
| FABPH | P05413 | SIVTLDGGK | 0.131 | 14.664 | 3.208 | 0.001 | 0.077 |
| ALDOA | P04075 | QLLLTADDR | 0.111 | 16.246 | 2.963 | 0.003 | 0.079 |
| AMBP | P02760 | FLYHK | –0.167 | 11.800 | –2.953 | 0.003 | 0.079 |
| APOE | P02649 | AATVGSLAGQPLQER | 0.247 | 20.145 | 3.039 | 0.003 | 0.079 |
| APOE | P02649 | CLAVYQAGAR | –0.630 | 8.715 | –2.919 | 0.004 | 0.079 |
| CH3L1 | P36222 | VTIDSSYDIAK | 0.109 | 21.303 | 2.915 | 0.004 | 0.079 |
| FABPH | P05413 | SLGVGFATR | 0.108 | 15.706 | 2.998 | 0.003 | 0.079 |
| KNG1 | P01042 | TVGSDTFYSFK | –0.185 | 15.118 | –2.898 | 0.004 | 0.079 |
| AMBP | P02760 | ETLLQDFR | –0.171 | 18.977 | –2.777 | 0.006 | 0.107 |
| A2GL | P02750 | DLLLPQPDLR | –0.148 | 25.898 | –2.701 | 0.007 | 0.127 |
| A2GL | P02750 | VAAGAFQGLR | –0.139 | 23.017 | –2.458 | 0.015 | 0.152 |
| AATC | P17174 | IVASTLSNPELFEEWTGNVK | 0.086 | 12.727 | 2.517 | 0.012 | 0.152 |
| AATM | P00505 | FVTVQTISGTGALR | 0.097 | 10.314 | 2.496 | 0.013 | 0.152 |
| CSF Complement proteins/peptides expression as a function of APOE4 copy number, controlling for age, gender, and clinical status (normal, MCI or AD) | |||||||
| Protein | UniProt ID | Peptide | Log Fold Change | Average Expression | t statistic | p | FDR Corrected p |
| C1QB | P02746 | LEQGENVFLQATDK | –0.042 | 17.128 | –1.241 | 0.216 | 0.622 |
| C1QB | P02746 | VPGLYYFTYHASSR | –0.053 | 17.604 | –1.135 | 0.257 | 0.669 |
| CFAB | P00751 | DAQYAPGYDK | –0.134 | 16.401 | –2.435 | 0.016 | 0.152 |
| CFAB | P00751 | VSEADSSNADWVTK | –0.111 | 16.220 | –2.471 | 0.014 | 0.152 |
| CFAB | P00751 | YGLVTYATYPK | –0.134 | 22.532 | –2.474 | 0.014 | 0.152 |
| CO2 | P06681 | DFHINLFR | –0.065 | 18.691 | –1.232 | 0.219 | 0.623 |
| CO2 | P06681 | HAIILLTDGK | –0.070 | 15.695 | –1.625 | 0.105 | 0.412 |
| CO2 | P06681 | SSGQWQTPGATR | –0.077 | 15.922 | –1.689 | 0.092 | 0.377 |
| CO3 | P01024 | IHWESASLLR | –0.267 | 14.006 | –2.136 | 0.034 | 0.235 |
| CO3 | P01024 | TELRPGETLNVNFLLR | –0.081 | 10.150 | –1.997 | 0.047 | 0.259 |
| CO4A | P0C0L4 | DHAVDLIQK | –0.038 | 22.142 | –0.745 | 0.457 | 0.858 |
| CO4A | P0C0L4 | GSFEFPVGDAVSK | –0.035 | 25.297 | –0.611 | 0.541 | 0.879 |
| CO4A | P0C0L4 | LGQYASPTAK | –0.041 | 21.701 | –0.760 | 0.448 | 0.857 |
| CO4A | P0C0L4 | NVNFQK | –0.019 | 18.387 | –0.392 | 0.695 | 0.978 |
| CO4A | P0C0L4 | VLSLAQEQVGGSPEK | –0.049 | 19.989 | –1.039 | 0.300 | 0.734 |
| CO4A | P0C0L4 | VTASDPLDTLGSEGALSPGGVASLLR | –0.029 | 18.008 | –0.712 | 0.477 | 0.871 |
| CO5 | P01031 | DINYVNPVIK | –0.045 | 16.181 | –0.630 | 0.529 | 0.876 |
| CO5 | P01031 | TLLPVSKPEIR | –0.050 | 17.209 | –0.757 | 0.449 | 0.857 |
| CO5 | P01031 | VFQFLEK | –0.043 | 18.210 | –0.644 | 0.520 | 0.873 |
| CO6 | P13671 | ALNHLPLEYNSALYSR | –0.113 | 16.297 | –1.772 | 0.077 | 0.345 |
| CO6 | P13671 | SEYGAALAWEK | –0.118 | 15.760 | –2.072 | 0.039 | 0.240 |
| CO8B | P07358 | IPGIFELGISSQSDR | –0.057 | 14.476 | –0.761 | 0.447 | 0.857 |
| CO8B | P07358 | SDLEVAHYK | –0.082 | 13.090 | –1.342 | 0.181 | 0.571 |
| CO8B | P07358 | YEFILK | –0.077 | 18.883 | –1.263 | 0.208 | 0.610 |
Log Fold Change - log2 fold change of peptide expression with each additional APOE4 allele copy in multivariate models for all analyzed CSF samples; Average expression –average peptide expression in all analyzed CSF samples; t statistic –hypothesis test statistic estimating the mean peptide expression (population mean) from the sampling distribution for each peptide in all analyzed CSF samples.
3.4Model 3: APOE4-dependent CSF peptide changes and CSF tau levels
To determine whether APOE4-dependent CSF proteomic changes were independent of changes in CSF tau levels (a non-specific indicator of neurodegeneration [39]), we constructed a third model for associations between APOE4 copy number and CSF peptide expression levels, correcting for CSF tau levels, age, and gender. In this third model, 183 of 294 peptides from 132 proteins had statistically significant APOE4 copy number-related changes in CSF expression levels (Table 5, Supplementary File 2). Similar to the findings in models 1 and 2, in model 3 increasing APOE4 copy number was associated with increased expression of the APOE4-specific peptide (LGADMEDVR) (q = 1.5×10–75) and decreased expression of the CRP-derived peptide (ESDTSYVSLK) (q = 0.0006; Supplementary File 2). CH3L1 (YKL-40)-, FABPH-, and ALDOA-derived peptides that showed significant APOE4 copy number-related changes in expression in model 1 (above) no longer remained statistically significant after correcting for CSF tau levels in model 3 (q > 0.05 for each; Supplementary File 2).
Table 5
Expression for the Top 20 Peptides and for all complement peptides/proteins by APOE4 copy number in multivariate models accounting for age, gender and CSF tau levels (Model 3). For full data set, see Supplementary File 2
| Proteins/peptides whose expression differed as a function of APOE4 copy number, controlling for age, gender, and CSF tau level (in order of significance). | |||||||
| Protein | UniProt ID | Peptide | Log Fold Change | Average Expression | t statistic | p | FDR Corrected p |
| APOE | P02649 | LGADMEDVR | 3.365 | 9.208 | 26.640 | 0.000 | 0.000 |
| A4 | P05067 | LVFFAEDVGSNK | –0.166 | 18.133 | –5.315 | 0.000 | 0.000 |
| NCAM2 | O15394 | IIELSQTTAK | –0.171 | 19.631 | –5.342 | 0.000 | 0.000 |
| VGF | O15240 | AYQGVAAPFPK | –0.254 | 17.733 | –5.299 | 0.000 | 0.000 |
| AMD | P19021 | IVQFSPSGK | –0.185 | 21.247 | –4.964 | 0.000 | 0.000 |
| CA2D1 | P54289 | FVVTDGGITR | –0.187 | 20.267 | –4.943 | 0.000 | 0.000 |
| CMGA | P10645 | YPGPQAEGDSEGLSQGLVDR | –0.237 | 15.851 | –5.008 | 0.000 | 0.000 |
| IGSF8 | Q969P0 | LQGDAVVLK | –0.133 | 18.301 | –4.885 | 0.000 | 0.000 |
| IGSF8 | Q969P0 | VVAGEVQVQR | –0.119 | 19.050 | –4.910 | 0.000 | 0.000 |
| NBL1 | P41271 | LALFPDK | –0.141 | 26.936 | –4.998 | 0.000 | 0.000 |
| SCG2 | P13521 | VLEYLNQEK | –0.202 | 20.916 | –4.897 | 0.000 | 0.000 |
| VGF | O15240 | NSEPQDEGELFQGVDPR | –0.252 | 19.459 | –4.923 | 0.000 | 0.000 |
| VGF | O15240 | THLGEALAPLSK | –0.244 | 16.748 | –4.919 | 0.000 | 0.000 |
| FAM3C | P84101 | SPFEQHIK | –0.159 | 19.027 | –4.815 | 0.000 | 0.000 |
| AMD | P19021 | IPVDEEAFVIDFKPR | –0.167 | 17.376 | –4.735 | 0.000 | 0.000 |
| AMD | P19021 | NGQWTLIGR | –0.180 | 16.249 | –4.725 | 0.000 | 0.000 |
| CA2D1 | P54289 | TASGVNQLVDIYEK | –0.171 | 13.242 | –4.712 | 0.000 | 0.000 |
| CYTC | P01034 | ALDFAVGEYNK | –0.114 | 34.102 | –4.717 | 0.000 | 0.000 |
| NPTX2 | P47972 | TESTLNALLQR | –0.282 | 10.581 | –4.723 | 0.000 | 0.000 |
| CAD13 | P55290 | YEVSSPYFK | –0.161 | 22.725 | –4.657 | 0.000 | 0.000 |
| CSF Complement proteins/peptides expression as a function of APOE4 copy number, controlling for age, gender, and tau | |||||||
| Protein | UniProt ID | Peptide | Log Fold Change | Average Expression | t statistic | p | FDR Corrected p |
| C1QB | P02746 | LEQGENVFLQATDK | –0.071 | 17.131 | –2.124 | 0.034 | 0.055 |
| C1QB | P02746 | VPGLYYFTYHASSR | –0.132 | 17.608 | –2.921 | 0.004 | 0.008 |
| CFAB | P00751 | DAQYAPGYDK | –0.111 | 16.394 | –2.015 | 0.045 | 0.068 |
| CFAB | P00751 | VSEADSSNADWVTK | –0.055 | 16.211 | –1.274 | 0.204 | 0.242 |
| CFAB | P00751 | YGLVTYATYPK | –0.124 | 22.526 | –2.270 | 0.024 | 0.039 |
| CO2 | P06681 | DFHINLFR | –0.088 | 18.688 | –1.670 | 0.096 | 0.129 |
| CO2 | P06681 | HAIILLTDGK | –0.101 | 15.692 | –2.360 | 0.019 | 0.032 |
| CO2 | P06681 | SSGQWQTPGATR | –0.102 | 15.920 | –2.228 | 0.027 | 0.043 |
| CO3 | P01024 | IHWESASLLR | –0.254 | 13.995 | –2.022 | 0.044 | 0.067 |
| CO3 | P01024 | TELRPGETLNVNFLLR | –0.083 | 10.147 | –2.028 | 0.044 | 0.067 |
| CO4A | P0C0L4 | DHAVDLIQK | –0.087 | 22.140 | –1.731 | 0.085 | 0.117 |
| CO4A | P0C0L4 | GSFEFPVGDAVSK | –0.098 | 25.295 | –1.774 | 0.077 | 0.109 |
| CO4A | P0C0L4 | LGQYASPTAK | –0.106 | 21.701 | –2.052 | 0.041 | 0.064 |
| CO4A | P0C0L4 | NVNFQK | –0.071 | 18.386 | –1.490 | 0.137 | 0.176 |
| CO4A | P0C0L4 | VLSLAQEQVGGSPEK | –0.062 | 19.985 | –1.317 | 0.189 | 0.229 |
| CO4A | P0C0L4 | VTASDPLDTLGSEGALSPGGVASLLR | –0.067 | 18.006 | –1.659 | 0.098 | 0.131 |
| CO5 | P01031 | DINYVNPVIK | –0.096 | 16.181 | –1.346 | 0.179 | 0.220 |
| CO5 | P01031 | TLLPVSKPEIR | –0.076 | 17.208 | –1.141 | 0.255 | 0.293 |
| CO5 | P01031 | VFQFLEK | –0.070 | 18.209 | –1.053 | 0.293 | 0.328 |
| CO6 | P13671 | ALNHLPLEYNSALYSR | –0.112 | 16.292 | –1.752 | 0.081 | 0.113 |
| CO6 | P13671 | SEYGAALAWEK | –0.104 | 15.755 | –1.817 | 0.070 | 0.102 |
| CO8B | P07358 | IPGIFELGISSQSDR | –0.045 | 14.471 | –0.595 | 0.552 | 0.584 |
| CO8B | P07358 | SDLEVAHYK | –0.055 | 13.083 | –0.883 | 0.378 | 0.413 |
| CO8B | P07358 | YEFILK | –0.045 | 18.876 | –0.727 | 0.468 | 0.499 |
Four peptides from the complement cascade proteins C1QB, CFAB and CO2 showed significantly reduced expression as a function of increasing APOE4 allele count in model 3 (q < 0.05 for each; Table 5). There was also a consistently inverse direction of association between APOE4 copy number and CSF levels for the 20 other peptides from all 8 complement proteins measured (Table 5), although these effects were not statistically significant after multiple comparison correction (q > 0.05 for each).
Model 4: APOE4-dependent CSF peptide changes and CSF p-tau levels
To determine whether APOE4-dependent CSF proteomic changes were independent of changes in CSF p-tau levels (a more specific indicator of AD-related neurodegeneration [39]), we constructed a fourth model for associations between APOE4 copy number and CSF peptide expression levels, correcting for CSF p-tau levels, age, and gender. In this fourth model, 184 of 294 peptides from 132 proteins had statistically significant APOE4 copy number-related changes in CSF expression levels (Table 6). Similar to the findings in models 1 and 2, in model 3 increasing APOE4-copy number was associated with increased expression of the APOE4-specific peptide (LGADMEDVR) (q = 4.1 x 10–74) and decreased expression of the CRP-derived peptide (ESDTSYVSLK) (q = 0.00094). As in model 3 (controlling for CSF tau levels), CH3L1 (YKL-40)-, FABPH-, and ALDOA-derived peptides that showed significant APOE4- copy number-related changes in expression observed in model 1 no longer remained statistically significant after correcting for CSF p-tau levels (q > 0.05 for each; Supplementary File 2).
Table 6
Expression of the Top 20 Indicated Peptides and for all complement peptides/proteins by APOE4 copy number in multivariate models accounting for age, gender and CSF p-tau levels (model 4). For full data set, see Supplementary File 2
| Proteins/peptides whose expression differed as a function of APOE4 copy number, controlling for age, gender, and ptau | |||||||
| Protein | UniProt ID | Peptide | Log Fold Change | Average Expression | t statistic | p | FDR Corrected p |
| APOE | P02649 | LGADMEDVR | 3.340 | 9.208 | 26.198 | 0.000 | 0.000 |
| A4 | P05067 | LVFFAEDVGSNK | –0.168 | 18.133 | –5.170 | 0.000 | 0.000 |
| NCAM2 | O15394 | IIELSQTTAK | –0.175 | 19.631 | –5.197 | 0.000 | 0.000 |
| VGF | O15240 | AYQGVAAPFPK | –0.258 | 17.733 | –5.151 | 0.000 | 0.000 |
| AMD | P19021 | IVQFSPSGK | –0.190 | 21.247 | –4.896 | 0.000 | 0.000 |
| CA2D1 | P54289 | FVVTDGGITR | –0.192 | 20.267 | –4.878 | 0.000 | 0.000 |
| CMGA | P10645 | YPGPQAEGDSEGLSQGLVDR | –0.245 | 15.851 | –4.931 | 0.000 | 0.000 |
| NBL1 | P41271 | LALFPDK | –0.143 | 26.936 | –4.855 | 0.000 | 0.000 |
| VGF | O15240 | NSEPQDEGELFQGVDPR | –0.258 | 19.459 | –4.857 | 0.000 | 0.000 |
| FAM3C | P84101 | SPFEQHIK | –0.165 | 19.027 | –4.768 | 0.000 | 0.000 |
| IGSF8 | Q969P0 | VVAGEVQVQR | –0.121 | 19.050 | –4.784 | 0.000 | 0.000 |
| SCG2 | P13521 | VLEYLNQEK | –0.207 | 20.916 | –4.822 | 0.000 | 0.000 |
| VGF | O15240 | THLGEALAPLSK | –0.249 | 16.748 | –4.799 | 0.000 | 0.000 |
| IGSF8 | Q969P0 | LQGDAVVLK | –0.135 | 18.301 | –4.735 | 0.000 | 0.000 |
| AMD | P19021 | IPVDEEAFVIDFKPR | –0.170 | 17.376 | –4.623 | 0.000 | 0.000 |
| AMD | P19021 | NGQWTLIGR | –0.183 | 16.249 | –4.615 | 0.000 | 0.000 |
| CA2D1 | P54289 | TASGVNQLVDIYEK | –0.175 | 13.242 | –4.641 | 0.000 | 0.000 |
| CAD13 | P55290 | YEVSSPYFK | –0.167 | 22.725 | –4.601 | 0.000 | 0.000 |
| CMGA | P10645 | SEALAVDGAGKPGAEEAQDPEGK | –0.216 | 17.989 | –4.597 | 0.000 | 0.000 |
| CMGA | P10645 | SGEATDGARPQALPEPMQESK | –0.206 | 19.085 | –4.642 | 0.000 | 0.000 |
| CSF Complement proteins/peptides expression as a function of APOE4 copy number, controlling for age, gender, and ptau | |||||||
| Protein | UniProt ID | Peptide | Log Fold Change | Average Expression | t statistic | p | FDR Corrected p |
| C1QB | P02746 | LEQGENVFLQATDK | –0.069 | 17.131 | –2.054 | 0.041 | 0.064 |
| C1QB | P02746 | VPGLYYFTYHASSR | –0.130 | 17.608 | –2.813 | 0.005 | 0.010 |
| CFAB | P00751 | DAQYAPGYDK | –0.107 | 16.394 | –1.925 | 0.055 | 0.083 |
| CFAB | P00751 | VSEADSSNADWVTK | –0.048 | 16.211 | –1.102 | 0.271 | 0.319 |
| CFAB | P00751 | YGLVTYATYPK | –0.119 | 22.526 | –2.169 | 0.031 | 0.049 |
| CO2 | P06681 | DFHINLFR | –0.086 | 18.688 | –1.610 | 0.109 | 0.146 |
| CO2 | P06681 | HAIILLTDGK | –0.100 | 15.692 | –2.293 | 0.023 | 0.038 |
| CO2 | P06681 | SSGQWQTPGATR | –0.099 | 15.920 | –2.137 | 0.033 | 0.053 |
| CO3 | P01024 | IHWESASLLR | –0.249 | 13.995 | –1.966 | 0.050 | 0.077 |
| CO3 | P01024 | TELRPGETLNVNFLLR | –0.082 | 10.147 | –1.982 | 0.048 | 0.074 |
| CO4A | P0C0L4 | DHAVDLIQK | –0.080 | 22.140 | –1.563 | 0.119 | 0.157 |
| CO4A | P0C0L4 | GSFEFPVGDAVSK | –0.091 | 25.295 | –1.596 | 0.112 | 0.149 |
| CO4A | P0C0L4 | LGQYASPTAK | –0.099 | 21.701 | –1.866 | 0.063 | 0.092 |
| CO4A | P0C0L4 | NVNFQK | –0.064 | 18.386 | –1.314 | 0.190 | 0.234 |
| CO4A | P0C0L4 | VLSLAQEQVGGSPEK | –0.053 | 19.985 | –1.114 | 0.266 | 0.314 |
| CO4A | P0C0L4 | VTASDPLDTLGSEGALSPGGVASLLR | –0.061 | 18.006 | –1.481 | 0.140 | 0.179 |
| CO5 | P01031 | DINYVNPVIK | –0.092 | 16.181 | –1.264 | 0.207 | 0.251 |
| CO5 | P01031 | TLLPVSKPEIR | –0.070 | 17.208 | –1.043 | 0.298 | 0.345 |
| CO5 | P01031 | VFQFLEK | –0.065 | 18.209 | –0.965 | 0.335 | 0.378 |
| CO6 | P13671 | ALNHLPLEYNSALYSR | –0.109 | 16.292 | –1.689 | 0.092 | 0.132 |
| CO6 | P13671 | SEYGAALAWEK | –0.101 | 15.755 | –1.744 | 0.082 | 0.119 |
| CO8B | P07358 | IPGIFELGISSQSDR | –0.036 | 14.471 | –0.478 | 0.633 | 0.660 |
| CO8B | P07358 | SDLEVAHYK | –0.049 | 13.083 | –0.781 | 0.435 | 0.472 |
| CO8B | P07358 | YEFILK | –0.038 | 18.876 | –0.613 | 0.541 | 0.570 |
Three peptides from the complement cascade proteins C1QB, CFAB, and CO2 showed significantly reduced expression as a function of increasing APOE4 allele count (q < 0.05 for each) in model 4. There was also a consistently inverse direction of association between APOE4 copy number and CSF levels for the 21 other peptides from all 8 complement proteins measured (Table 6), although these effects were not statistically significant after multiple comparison correction in model 4 (q > 0.05 for each).
Model 5: APOE4-dependent CSF peptide changes and CSF Aβ levels
To determine whether APOE4-dependent CSF proteomic changes were independent of changes in CSF Aβ levels, we constructed a fifth model for associations between APOE4 copy number and CSF peptide expression levels, correcting for CSF Aβ levels as well as age and gender. In this model, 176 of 294 peptides had statistically significant APOE4 copy number-related changes in CSF expression levels (Supplementary File 2). Increasing APOE4-copy number was associated with increased expression of the APOE4-specific peptide (LGADMEDVR) (q = 1.89 x 10–62) and decreased expression of the CRP-derived peptide (ESDTSYVSLK) (q < 0.045) after controlling for CSF Aβ levels. Increasing APOE4 copy number was associated with increased expression of a pan-APOE peptide (LGPLVEQGR) in this model (q = 1.69 x 10–7). As in models 1 and 2, all five CH3L1-, FABPH-, and ALDOA-derived peptides also showed statistically significant APOE4 copy number related increases in expression after correcting for CSF Aβ levels (q < 0.05 for each; Table 7).
Table 7
Expression of the Top 20 Indicated Peptides and for all complement peptides/proteins by APOE4 copy number in multivariate models accounting for age, gender and CSF Aβ levels (Model 5). For full data set, see Supplementary File 2
| Top 20 Proteins/Peptides whose Expression differed as function of APOE4 copy number, controlling for age, gender, and A β | |||||||
| Protein | UniProt ID | Peptide | Log Fold Change | Average Expression | t statistic | p | FDR Corrected p |
| APOE | P02649 | LGADMEDVR | 3.664 | 9.224 | 23.026 | 0.000 | 0.000 |
| APOE | P02649 | LGPLVEQGR | 0.565 | 22.385 | 6.309 | 0.000 | 0.000 |
| APOE | P02649 | LAVYQAGAR | 0.572 | 25.381 | 6.007 | 0.000 | 0.000 |
| APOE | P02649 | AATVGSLAGQPLQER | 0.515 | 20.108 | 5.631 | 0.000 | 0.000 |
| NRX1A | Q9ULB1 | ITTQITAGAR | 0.240 | 16.206 | 4.981 | 0.000 | 0.000 |
| AATM | P00505 | FVTVQTISGTGALR | 0.209 | 10.293 | 4.719 | 0.000 | 0.000 |
| NRX1A | Q9ULB1 | DLFIDGQSK | 0.209 | 16.186 | 4.688 | 0.000 | 0.000 |
| PCSK1 | P29120 | GEAAGAVQELAR | 0.245 | 21.388 | 4.676 | 0.000 | 0.000 |
| L1CAM | P32004 | LVLSDLHLLTQSQVR | 0.229 | 10.874 | 4.607 | 0.000 | 0.000 |
| CADM3 | Q8N126 | GNPVPQQYLWEK | 0.179 | 15.815 | 4.557 | 0.000 | 0.000 |
| AATC | P17174 | NLDYVATSIHEAVTK | 0.193 | 11.536 | 4.312 | 0.000 | 0.000 |
| ALDOA | P04075 | ALQASALK | 0.202 | 19.150 | 4.415 | 0.000 | 0.000 |
| ALDOA | P04075 | QLLLTADDR | 0.190 | 16.230 | 4.332 | 0.000 | 0.000 |
| CADM3 | Q8N126 | EGSVPPLK | 0.158 | 16.142 | 4.328 | 0.000 | 0.000 |
| CADM3 | Q8N126 | SLVTVLGIPQKPIITGYK | 0.191 | 16.167 | 4.424 | 0.000 | 0.000 |
| KPYM | P14618 | LDIDSPPITAR | 0.214 | 18.500 | 4.308 | 0.000 | 0.000 |
| NRX1A | Q9ULB1 | SDLYIGGVAK | 0.189 | 16.548 | 4.329 | 0.000 | 0.000 |
| PCSK1 | P29120 | ALAHLLEAER | 0.242 | 19.035 | 4.329 | 0.000 | 0.000 |
| PCSK1 | P29120 | NSDPALGLDDDPDAPAAQLAR | 0.227 | 13.959 | 4.351 | 0.000 | 0.000 |
| PRDX1 | Q06830 | DISLSDYK | 0.185 | 14.594 | 4.377 | 0.000 | 0.000 |
| CSF Complement Protein/Peptide Expression as a function of APOE4 copy number, controlling for age, gender, and A β | |||||||
| Protein | UniProt ID | Peptide | Log Fold Change | Average Expression | t statistic | p | FDR Corrected p |
| C1QB | P02746 | LEQGENVFLQATDK | 0.039 | 17.123 | 0.983 | 0.326 | 0.393 |
| C1QB | P02746 | VPGLYYFTYHASSR | 0.096 | 17.588 | 1.782 | 0.076 | 0.114 |
| CFAB | P00751 | DAQYAPGYDK | –0.009 | 16.383 | –0.132 | 0.895 | 0.904 |
| CFAB | P00751 | VSEADSSNADWVTK | –0.051 | 16.209 | –0.956 | 0.340 | 0.408 |
| CFAB | P00751 | YGLVTYATYPK | 0.007 | 22.512 | 0.119 | 0.905 | 0.911 |
| CO2 | P06681 | DFHINLFR | 0.085 | 18.670 | 1.381 | 0.168 | 0.232 |
| CO2 | P06681 | HAIILLTDGK | 0.050 | 15.673 | 1.008 | 0.315 | 0.384 |
| CO2 | P06681 | SSGQWQTPGATR | 0.054 | 15.901 | 1.026 | 0.306 | 0.378 |
| CO3 | P01024 | IHWESASLLR | –0.032 | 13.985 | –0.215 | 0.830 | 0.853 |
| CO3 | P01024 | TELRPGETLNVNFLLR | –0.011 | 10.138 | –0.227 | 0.821 | 0.847 |
| CO4A | P0C0L4 | DHAVDLIQK | 0.100 | 22.139 | 1.690 | 0.092 | 0.135 |
| CO4A | P0C0L4 | GSFEFPVGDAVSK | 0.140 | 25.290 | 2.132 | 0.034 | 0.056 |
| CO4A | P0C0L4 | LGQYASPTAK | 0.122 | 21.693 | 1.975 | 0.049 | 0.079 |
| CO4A | P0C0L4 | NVNFQK | 0.100 | 18.384 | 1.776 | 0.077 | 0.115 |
| CO4A | P0C0L4 | VLSLAQEQVGGSPEK | 0.055 | 19.994 | 1.009 | 0.314 | 0.384 |
| CO4A | P0C0L4 | VTASDPLDTLGSEGALSPGGVASLLR | 0.066 | 18.007 | 1.380 | 0.169 | 0.232 |
| CO5 | P01031 | DINYVNPVIK | 0.241 | 16.153 | 2.987 | 0.003 | 0.007 |
| CO5 | P01031 | TLLPVSKPEIR | 0.207 | 17.184 | 2.768 | 0.006 | 0.012 |
| CO5 | P01031 | VFQFLEK | 0.216 | 18.187 | 2.874 | 0.004 | 0.010 |
| CO6 | P13671 | ALNHLPLEYNSALYSR | 0.054 | 16.276 | 0.731 | 0.465 | 0.526 |
| CO6 | P13671 | SEYGAALAWEK | 0.029 | 15.745 | 0.434 | 0.665 | 0.706 |
| CO8B | P07358 | IPGIFELGISSQSDR | 0.107 | 14.458 | 1.208 | 0.228 | 0.298 |
| CO8B | P07358 | SDLEVAHYK | 0.049 | 13.073 | 0.679 | 0.498 | 0.560 |
| CO8B | P07358 | YEFILK | 0.061 | 18.865 | 0.844 | 0.399 | 0.468 |
In this model controlling for CSF Aβ levels (as well as age and sex), all three peptides from complement factor 5 (CO5) showed a statistically significant increase in expression as a function of increasing APOE4 copy number (q < 0.05 for each; Table 7). In contrast to the results from models 1–4, in this model (controlling for CSF Aβ levels) no consistent direction of association was seen between APOE4 copy number and the other 21 complement-derived peptides (Table 7).
DISCUSSION
Here we found that increasing APOE4 copy number was associated with decreased CSF CRP levels, which remained significant even after controlling for AD clinical status (Model 2), CSF tau levels (Model 3), CSF p-tau levels (Model 4), and/or CSF Aβ levels (Model 5; see Table 8 for summary of changes across models). In several models, we also found significant APOE4 copy number-related increases in pan-APOE peptides, and APOE4 copy number-related increases in peptides from CH3L1, FABPH, and ALDOA. Finally, except when controlling for CSF Aβ levels in model 5, in all other models a consistent inverse direction of association was observed between increasing APOE4 copy number and CSF levels of all 8 complement proteins measured int his targeted proteomics dataset.
Table 8
Summary of Change Direction for Top proteins and Complement Pathway Proteins in Statistical Models 1 –5. Up arrow indicates expression increase for the indicated protein as a function of increasing APOE4 copy number in the given statistical model; down arrow indicates expression decrease for the indicated protein as a function of increasing APOE4 copy number in the given statistical model
| Protein | UniProt ID | Peptide | Key functions | Statistical Model# | ||||
| 1 | 2 | 3 | 4 | 5 | ||||
| CRP | P02741 | ESDTSYVSLK | Acute phase reactant [40]. Mediator of inflammatory and apoptotic processes, including activation of classical complement pathway [41], opsonization of atherosclerotic plaques [42]. | ↓2 | ↓2 | ↓3 | ↓3 | ↓1 |
| CH3L1 | P36222 | ILGQQVPYATK | Glycoprotein thought to modulate tissue remodeling, and angiogenesis, highly expressed in reactive astrocytes after acute and chronic neuroinflammation [63–65]. Also expressed by activated microglia. | ↑ | ↑ | ↑ | ↑ | ↑ |
| VTIDSSYDIAK | ||||||||
| FABPH | P05413 | SIVTLDGGK | Lipid transporter regulates membrane composition and stability [71], may alter the activity of gamma-secretase or slow trans-membrane lipid transport to increase Aβ deposition in APOE4 carriers [72]. | ↑ | ↑ | ↑ | ↓ | ↑ |
| SLGVGFAT | ↓ | |||||||
| ALDOA | P04075 | ALQASALKQLLLTADDR | Glycolytic enzyme that breaks down fructose 1-6-diphosphate; decreased glucose metabolism has been observed in the brains of patients with AD [73], likely due to oxidative stress that impairs ATP production, induces synaptic dysfunction, and causes neuronal death [74]. | ↑ | ↑ | ↓ | ↓ | ↑3 |
| Complement pathway proteins | ||||||||
| C1QB | P02746 | LEQGENVFLQATDK | Complement pathway activation and complement factor-dependent synaptic phagocytosis thought to represent a neurodegeneration mechanism in AD [82, 83] | ↓ | ↓ | ↓ | ↓ | ↑ |
| VPGLYYFTYHASSR | ↓ | ↓ | ↓2 | ↓1 | ↑ | |||
| CFAB | P00751 | DAQYAPGYDK | ↓ | ↓ | ↓ | ↓ | ↓ | |
| VSEADSSNADWVTK | ↓ | ↓ | ↓ | ↓ | ↓ | |||
| YGLVTYATYPK | ↓ | ↓ | ↓1 | ↓1 | ↑ | |||
| CO2 | P06681 | DFHINLFR | ↓ | ↓ | ↓ | ↓ | ↑ | |
| HAIILLTDGK | ↓ | ↓ | ↓1 | ↓1 | ↑ | |||
| SSGQWQTPGATR | ↓ | ↓ | ↓1 | ↓1 | ↑ | |||
| CO3 | P01024 | IHWESASLLR | ↓ | ↓ | ↓ | ↓ | ↓ | |
| TELRPGETLNVNFLLR | ↓ | ↓ | ↓ | ↓ | ↓ | |||
| CO4A | P0C0L4 | DHAVDLIQK | ↓ | ↓ | ↓ | ↓ | ↑ | |
| GSFEFPVGDAVSK | ↓ | ↓ | ↓ | ↓ | ↑ | |||
| LGQYASPTAK | ↓ | ↓ | ↓ | ↓ | ↑ | |||
| NVNFQK | ↓ | ↓ | ↓ | ↓ | ↑ | |||
| VLSLAQEQVGGSPEK | ↓ | ↓ | ↓ | ↓ | ↑ | |||
| VTASDPLDTLGSEGALSPGGVASLLR | ↓ | ↓ | ↓ | ↓ | ↑ | |||
| CO5 | P01031 | DINYVNPVIK | ↓ | ↓ | ↓ | ↓ | ↑2 | |
| TLLPVSKPEIR | ↓ | ↓ | ↓ | ↓ | ↑1 | |||
| VFQFLEK | ↓ | ↓ | ↓ | ↓ | ↑2 | |||
| CO6 | P13671 | ALNHLPLEYNSALYSR | ↓ | ↓ | ↓ | ↓ | ↑ | |
| SEYGAALAWEK | ↓ | ↓ | ↓ | ↓ | ↑ | |||
| CO8B | P07358 | IPGIFELGISSQSDR | ↓ | ↓ | ↓ | ↓ | ↑ | |
| SDLEVAHYK | ↓ | ↓ | ↓ | ↓ | ↑ | |||
| YEFILK | ↓ | ↓ | ↓ | ↓ | ↑ | |||
#Statistical Models: Model 1: APOE4-dependent CSF peptide changes and age and gender. Model 2: APOE4-dependent CSF peptide changes and age, gender, and AD clinical status. Model 3: APOE4-dependent CSF peptide changes and age, gender, and CSF tau levels. Model 4: APOE4-dependent CSF peptide changes and age, gender, and CSF p-tau levels. Model 5: APOE4-dependent CSF peptide changes and age, gender, and CSF Aβ levels. 1FDR corrected p < 0.05. 2FDR corrected p < 0.01. 3FDR corrected p < 0.001.
One of the most consistent findings across all five statistical models examined here was that increasing APOE4 copy number was associated with reduced CSF levels of the CRP-derived peptide (ESDTSYVSLK, see Table 8). CRP is an acute phase reactant [40] and acts as a mediator of inflammatory and apoptotic processes, including activation of the classical complement pathway [41] and opsonization of atherosclerotic plaques [42]. Recent prospective studies have found that elevated serum CRP levels in midlife may predict increased AD risk, though there is paradoxical shift in which CRP levels decline with advancing age and before clinical AD symptoms appear [43–45]. Further, reduced CRP levels in peripheral blood [46–49] and in CSF [50, 51] correlate with increased cognitive dysfunction and further AD progression in an APOE4-dependent manner. These findings fit well with our observation of a significant decrease in CSF CRP levels as a function of increasing APOE4 copy number and suggests that reduced CRP levels may play a key biological role in APOE4-induced increased AD risk.
CRP is typically viewed as a marker of inflammation, so a straightforward interpretation of these results would be that increasing APOE4 copy number is associated with decreased CNS inflammation, which could play a role in increased AD risk if APOE4 leads to a reduction in inflammatory processes involved in clearing amyloid plaques or other neurotoxic pathology. Alternatively, although elevated CRP levels are typically viewed as indicative of active inflammation, low and low-normal CRP levels have been found in chronic inflammatory conditions with active disease such as lupus [52], rheumatoid arthritis [53], and inflammatory bowel disease [54, 55]. Thus, the reduced CSF CRP levels observed here may similarly reflect chronically increased inflammation within the CNS of APOE4 carriers, which could play a role in neurodegeneration and AD risk. CRP has been also implicated in the early development of amyloid plaque formation, neuronal damage, and AD risk [53–56]. The decreased CSF CRP levels observed here may reflect CRP deposition in Aβ plaques and neurofibrillary tangles, in which CRP might play a role in promoting the development of AD. Low CSF CRP levels could also reflect increased CRP consumption from opsonin-mediated glial phagocytosis in AD pathology, as previously suggested [62]. In this way, CSF CRP reductions might alternatively reflect a compensatory glial-dependent Aβ removal process in APOE4 carriers. Future studies should thus determine whether lower CSF CRP levels in APOE4 carriers represent a mechanism of neurodegeneration or a compensatory process against it, or even an APOE4-copy related process unrelated to the development of AD.
All five models studied here also demonstrated a strong positive correlation between APOE4 copy number and CSF expression of the APOE4 allele specific peptide LGADMEDVR [37, 38], which serves as a strong internal control for the validity of this dataset. We also found that increasing APOE4 copy number was associated with increased CSF pan-APOE peptide levels when controlling for AD clinical status or CSF Aβ levels (Models 2 and 5, respectively) but not when controlling for either CSF tau or CSF p-tau levels (Models 3 and 4, respectively). Taken together, these results are consistent with a paradigm in which APOE4 allele leads to increased CSF ApoE protein levels before and during the phase in which patients are developing brain Aβ pathology (as measured here by CSF Aβ levels). Then, as frank neurodegeneration begins (as evidenced by rising CSF tau and p-tau levels), there is no longer a significant increase in CSF ApoE protein levels as a function of increasing APOE4 allele count. This could help explain conflicting prior studies on CSF ApoE protein levels in APOE4 carriers [57–59], since it suggests that the relationship between APOE4 copy number and CSF ApoE protein levels would change as neurodegeneration begins (i.e., as defined by increasing CSF tau and p-tau levels). Yet, it is unclear whether the increased AD risk in APOE4 carriers is due to APOE4 copy number-related increases in ApoE protein levels versus functional or structural changes in the APOE4 allele-encoded ApoE protein. Indeed, it remains debated in the field to what extent APOE4–related increased AD risk represents a toxic gain of function(s) or a loss of protective function(s) (reviewed in [60]).
In models 1 and 5, but not in models 2–4 (Table 8), increasing APOE4 copy number was also associated with increased expression of peptides derived from CH3L1 (also known as YKL-40) (q < 0.05), FABPH (q < 0.05), and ALDOA (q < 0.05), as seen in another recent paper [61]. These findings suggest that APOE4 copy number is associated with increased CSF CH3L1, FABPH, and CH3L1 levels independent of CSF Aβ level changes, but these increases dissipate after controlling for neurodegeneration (i.e., via rising CSF tau or p-tau levels) or by controlling for neurodegeneration-related clinical status (i.e., MCI or dementia).
Our finding of APOE4-related increases in CSF CH3L1 levels is corroborated by another recent study using ELISA assays that found increased CSF CH3L1 levels in APOE4 carriers [62], CH3L1 is a glycoprotein hypothesized to modulate tissue remodeling, and angiogenesis and is highly expressed in reactive astrocytes after acute and chronic neuroinflammation [63–65]. After traumatic brain injuries, CH3L1 knockout mice demonstrated greater astrogliosis, immune cell infiltration, and neurologic impairment compared to wild-type mice, suggesting CH3L1 plays a role in regulating neuro-inflammation [66]. Astrocytes are known regulators of neuroinflammation and oxidative stress in the nervous system, and CH3L1-expressing astrocytes have also been shown to cluster around Aβ plaques and vessels with β-amyloid angiopathy in AD [67]. Taken together, these findings and the data presented here suggest that CH3L1 may contributes to pathologic astrocyte activation in APOE4 carriers while Aβ pathology is developing, prior to actual neurodegeneration.
Our finding of a positive correlation between APOE4 copy number and CSF FABPH levels when controlling for Aβ levels, but not when controlling for CSF neurodegeneration markers (i.e., CSF tau and p-tau; Table 8), is consistent with prior studies suggesting a role for FABPH in AD [29, 37, 68–70]. FABPH is a lipid transporter that regulates membrane composition and stability [71], and may alter the activity of gamma-secretase or slow trans-membrane lipid transport to increase Aβ deposition in APOE4 carriers [72]. Indeed, prior studies have found that CSF FAPBH levels in APOE4 carriers are associated with low CSF Aβ levels and atrophy of the entorhinal cortex and other AD-vulnerable brain regions [69]. These findings indicate a potential role for FABPH in APOE4-mediated Aβ accumulation and later neurodegeneration. The findings presented here further support this idea, and further suggest that altered FABPH levels likely play a role in the development of early Aβ pathology in APOE4 carriers, before CSF tau and p-tau levels begin to decline.
We also found that increasing APOE4 copy number was associated with increased CSF ALDOA levels in models one and five (Table 8). ALDOA is a glycolytic enzyme that catalyzes the breakdown of fructose 1-6-diphosphate, which is interesting because decreased glucose metabolism has been observed in the brains of patients with AD [73], likely due to oxidative stress that impairs ATP production, induces synaptic dysfunction, and causes neuronal death [74]. A recent study has also proposed that CSF ALDOA levels are a sensitive and specific biomarker of cognitive impairment due to AD [75]. Thus, our finding of an APOE4 copy number-dependent increase in CSF ALDOA levels may reflect altered glucose metabolism within the CNS of APOE4 carriers, which has also been observed in AD. It is unclear whether increased ALDOA levels are part of the mechanism for impaired glucose metabolism in APOE4 carriers and in AD, or whether these increased ALDOA levels reflects a secondary biologic compensation as the result of dysregulated glucose metabolism in APOE4 carriers. As in the case of CH3L1 and ALDOA, the data presented here suggest that APOE4 copy number is likely associated with increased CSF ALDOA levels while individuals are clinically normal, and either completely free of AD neuropathology or when they show CSF evidence of Aβ, but not tau or p-tau, pathology.
Further, while not statistically significant, there was a consistent inverse direction of association between APOE4 copy number and expression of all 24 peptides from all 8 different complement proteins measured (Fig 1A, B) in model 1. These findings were present even after controlling for AD clinical status in model 2, suggesting that the lower complement levels seen as a function of APOE4 copy number do not simply reflect confounding due increased AD dementia frequency in APOE4 carriers. Further, this consistent inverse direction of association between APOE4 copy number and expression of all 24 peptides from all 8 different complement proteins measured here was also observed after controlling for CSF tau and p-tau levels in models 3 and 4, respectively. These findings suggest that that these potential APOE4-related reductions in CSF complement levels are not due to confounding from neurodegeneration in APOE4 carriers.
Nonetheless, this consistent inverse direction of association between APOE4 copy number and CSF complement protein expression was not observed in model 5, which controlled for CSF Aβ levels. These findings would fit with a scenario in which APOE4 leads to reductions in both CSF Aβ levels and CSF complement protein levels, and these APOE4-dependent reductions in CSF Aβ and complement levels are closely associated with one another (or co-linear). In this case, increasing APOE4 copy number would be expected to be associated with reduced complement levels in all situations except when controlling for falling CSF Aβ levels. This framework is consistent with the finding that complement proteins deposit into Aβ plaques [76, 77], which would offer a potential biochemical explanation for a co-linear relationship between reductions in CSF Aβ and complement levels as a function of increasing APOE4 copy number.
The finding that APOE4 copy number may be associated with decreased CSF complement protein levels is supported by two other recent studies that also found lower CSF complement protein levels in APOE4 carriers [61, 78]. While the absolute magnitude of the reduction in each complement-protein derived peptide was modest, the chance that the level of 24 peptides (or 8 proteins) would all decrease due to chance alone is extremely low, suggesting that this likely represents a true biological finding. These lower complement protein levels could represent either decreased complement factor transcription/translation or increased degradation in APOE4 carriers. The former possibility is unlikely, though, because prior work has shown that APOE4 is not associated with alterations in the transcription or translation of complement pathway proteins [79]. These data therefore suggest that increasing APOE4 copy number may be associated with increased complement pathway protein degradation in the CNS.
Complement protein degradation can be caused by complement cascade activation, which involves cleavage and degradation of complement proteins [80]. Thus, the relationship between increasing APOE4-copy number and potential reductions in CSF complement protein levels may reflect increased complement pathway activation. Recent work suggests that the ApoE protein is a negative regulator of complement pathway activation [81]. Taken together with our results, this raises the possibility that the APOE4 allele results in reduced inhibition (i.e., disinhibition) of the complement pathway, thus resulting in APOE4 copy number-dependent reductions in CSF levels of complement pathway proteins (due to complement degradation).
Complement pathway activation and complement factor-dependent synaptic phagocytosis is thought to represent a neurodegeneration mechanism in AD [78, 82]; thus, our results raise the possibility that APOE4 may contribute to AD risk by increasing complement pathway activation and resultant synaptic phagocytosis and neurodegeneration. Overall, even though the APOE4-related reductions in CSF complement protein levels seen here were generally not statistically significant after multiple comparison correction, the convergence of our human findings with data from cellular [79] and mouse models [83] suggest that further studies are warranted on the relationship between APOE4 copy number and CSF complement protein levels.
This work has several limitations. First, the data set studied here included very few African-American APOE4 allele carriers, raising the question of how well these data may generalize to African-American or other non-Caucasian populations. Future work should study the effects of the APOE4 allele on the CSF proteome in other racial and ethnic populations to develop a broader understanding of how APOE4 may increase AD risk. Second, the data analyzed here were originally obtained to study CSF proteomic correlates of dementia due to AD or MCI [29], rather than to study the effects of APOE4 copy number on the CSF proteome. Although we controlled for clinical status in model 2, the relatively smaller number of study patients within each clinical status cohort (i.e., normal, MCI, or dementia due to AD) likely limited statistical power to detect effects of APOE4 on the CSF proteome itself. Thus, future studies on this topic should focus on larger clinically homogenous study populations (i.e., all cognitively normal individuals, or all individuals with dementia due to AD) to reduce variance within each genotype group, and to improve statistical power. Third, the data reported here were from a targeted proteomic platform that only measured 222 proteins total, including just 8 of the over 30 proteins in the full complement pathway [84]. Future studies should focus on quantitating the CSF levels of a broader array of proteins including all complement pathway protein to develop a more complete understanding of the relationship between APOE4 allele copy number and the classical, lectin and alternative complement cascades and other protein pathways.
Nonetheless, the data presented here provide strong support for the hypothesis that the increased AD risk in APOE4 carriers is related to changes within CRP-related biological processes, and to changes in biological mechanisms involving CH3L1, FABPH, ALDOA, and the complement pathway. The results from our five different statistical models also demonstrate key differences across the neuropathologic phases of preclinical AD in the CSF proteomic correlates of APOE4 copy number. Thus, future CSF proteomic studies designed to identify the earliest mechanisms by which APOE4 contributes to AD risk may need to focus on patients without significant Aβ or neurodegenerative pathology.
ACKNOWLEDGMENTS
Dr. Berger acknowledges support from K76-AG057022, and additional support from NIH grants P30AG028716 and UH2AG056925; a program to advance clinical trials (PACT) grant from the Alzheimer’s Drug Discovery Foundation, the inaugural Ann Bussel award from the Ruth K. Broad Foundation at Duke University, and the Duke Anesthesiology Department. Dr. Devinney acknowledges support from a research fellowship grant from the Foundation for Anesthesia Education and Research.
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (http://www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/20-0747r2).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-200747.
REFERENCES
[1] | Corder E , Saunders A , Strittmatter W , Schmechel D , Gaskell P , Small G , Roses A , Haines J , Pericak-Vance M ((1993) ) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261: , 921–923. |
[2] | Neu SC , Pa J , Kukull W , Beekly D , Kuzma A , Gangadharan P , Wang LS , Romero K , Arneric SP , Redolfi A , Orlandi D , Frisoni GB , Au R , Devine S , Auerbach S , Espinosa A , Boada M , Ruiz A , Johnson SC , Koscik R , Wang JJ , Hsu WC , Chen YL , Toga AW ((2017) ) Apolipoprotein E Genotype and Sex Risk Factors for Alzheimer Disease: A Meta-analysis. JAMA Neurol 74: , 1178–1189. |
[3] | Loy CT , Schofield PR , Turner AM , Kwok JBJ ((2014) ) Genetics of dementia. Lancet 383: , 828–840. |
[4] | Holtzman DM , Herz J , Bu G ((2012) ) Apolipoprotein E and apolipoprotein E receptors: Normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med 2: , a006312. |
[5] | Michaelson DM ((2014) ) APOE ɛ4: The most prevalent yet understudied risk factor for Alzheimer’s disease. Alzheimers Dement 10: , 861–868. |
[6] | Fazekas F , Strasser–Fuchs S , Kollegger H , Berger T , Kristoferitsch W , Schmidt H , Enzinger C , Schiefermeier M , Schwarz C , Kornek B , Reindl M , Huber K , Grass R , Wimmer G , Vass K , Pfeiffer KH , Hartung HP , Schmidt R ((2001) ) Apolipoprotein E ɛ4 is associated with rapid progression of multiple sclerosis. Neurology 57: , 853–857. |
[7] | Yue JK , Robinson CK , Burke JF , Winkler EA , Deng H , Cnossen MC , Lingsma HF , Ferguson AR , McAllister TW , Rosand J , Burchard EG , Sorani MD , Sharma S , Nielson JL , Satris GG , Talbott JF , Tarapore PE , Korley FK , Wang KKW , Yuh EL , Mukherjee P , Diaz-Arrastia R , Valadka AB , Okonkwo DO , Manley GT , TRACK-TBI Investigators ((2017) ) Apolipoprotein E epsilon 4 (APOE-epsilon4) genotype is associated with decreased 6-month verbal memory performance after mild traumatic brain injury. Brain Behav 7: , e00791. |
[8] | Martinez-Gonzalez NA , Sudlow CL ((2006) ) Effects of apolipoprotein E genotype on outcome after ischaemic stroke, intracerebral haemorrhage and subarachnoid haemorrhage. J Neurol Neurosurg Psychiatry 77: , 1329–1335. |
[9] | Mahley RW ((2016) ) Apolipoprotein E: From cardiovascular disease to neurodegenerative disorders. J Mol Med (Berl) 94: , 739–746. |
[10] | Kulminski AM , Arbeev KG , Culminskaya I , Arbeeva L , Ukraintseva SV , Stallard E , Christensen K , Schupf N , Province MA , Yashin AI ((2014) ) Age, gender, and cancer but not neurodegenerative and cardiovascular diseases strongly modulate systemic effect of the Apolipoprotein E4 allele on lifespan. PLoS Genet 10: , e1004141. |
[11] | Deelen J , Evans DS , Arking DE , Tesi N , Nygaard M , Liu X , Wojczynski MK , Biggs ML , van der Spek A , Atzmon G , Ware EB , Sarnowski C , Smith AV , Seppala I , Cordell HJ , Dose J , Amin N , Arnold AM , Ayers KL , Barzilai N , Becker EJ , Beekman M , Blanche H , Christensen K , Christiansen L , Collerton JC , Cubaynes S , Cummings SR , Davies K , Debrabant B , Deleuze JF , Duncan R , Faul JD , Franceschi C , Galan P , Gudnason V , Harris TB , Huisman M , Hurme MA , Jagger C , Jansen I , Jylha M , Kahonen M , Karasik D , Kardia SLR , Kingston A , Kirkwood TBL , Launer LJ , Lehtimaki T , Lieb W , Lyytikainen LP , Martin-Ruiz C , Min J , Nebel A , Newman AB , Nie C , Nohr EA , Orwoll ES , Perls TT , Province MA , Psaty BM , Raitakari OT , Reinders MJT , Robine JM , Rotter JI , Sebastiani P , Smith J , Sorensen TIA , Taylor KD , Uitterlinden AG , van der Flier W , van der Lee SJ , van Duijn CM , van Heemst D , Vaupel JW , Weir D , Ye K , Zeng Y , Zheng W , Holstege H , Kiel DP , Lunetta KL , Slagboom PE , Murabito JM ((2019) ) A meta-analysis of genome-wide association studies identifies multiple longevity genes. Nat Commun 10: , 3669. |
[12] | Yamazaki Y , Zhao N , Caulfield TR , Liu CC , Bu G ((2019) ) Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat Rev Neurol 15: , 501–518. |
[13] | Iacono D , Feltis G . Impact of Apolipoprotein E gene polymorphism during normal and pathological conditions of the brain across the lifespan. Aging 11: , 787–816. |
[14] | Ghisays V , Goradia DD , Protas H , Bauer 3rd RJ , Devadas V , Tariot PN , Lowe VJ , Knopman DS , Petersen RC , Jack CR Jr. , Caselli RJ , Su Y , Chen K , Reiman EM ((2020) ) Brain imaging measurements of fibrillar amyloid-beta burden, paired helical filament tau burden, and atrophy in cognitively unimpaired persons with two, one, and no copies of the APOE epsilon4 allele. Alzheimers Dement 16: , 598–609. |
[15] | Jansen WJ , Ossenkoppele R , Knol DL , Tijms BM , Scheltens P , Verhey FR , Visser PJ , Amyloid Biomarker Study G, Aalten P , Aarsland D , Alcolea D , Alexander M , Almdahl IS , Arnold SE , Baldeiras I , Barthel H , van Berckel BN , Bibeau K , Blennow K , Brooks DJ , van Buchem MA , Camus V , Cavedo E , Chen K , Chetelat G , Cohen AD , Drzezga A , Engelborghs S , Fagan AM , Fladby T , Fleisher AS , van der Flier WM , Ford L , Forster S , Fortea J , Foskett N , Frederiksen KS , Freund-Levi Y , Frisoni GB , Froelich L , Gabryelewicz T , Gill KD , Gkatzima O , Gomez-Tortosa E , Gordon MF , Grimmer T , Hampel H , Hausner L , Hellwig S , Herukka SK , Hildebrandt H , Ishihara L , Ivanoiu A , Jagust WJ , Johannsen P , Kandimalla R , Kapaki E , Klimkowicz-Mrowiec A , Klunk WE , Kohler S , Koglin N , Kornhuber J , Kramberger MG , Van Laere K , Landau SM , Lee DY , de Leon M , Lisetti V , Lleo A , Madsen K , Maier W , Marcusson J , Mattsson N , de Mendonca A , Meulenbroek O , Meyer PT , Mintun MA , Mok V , Molinuevo JL , Mollergard HM , Morris JC , Mroczko B , Van der Mussele S , Na DL , Newberg A , Nordberg A , Nordlund A , Novak GP , Paraskevas GP , Parnetti L , Perera G , Peters O , Popp J , Prabhakar S , Rabinovici GD , Ramakers IH , Rami L , Resende de Oliveira C , Rinne JO , Rodrigue KM , Rodriguez-Rodriguez E , Roe CM , Rot U , Rowe CC , Ruther E , Sabri O , Sanchez-Juan P , Santana I , Sarazin M , Schroder J , Schutte C , Seo SW , Soetewey F , Soininen H , Spiru L , Struyfs H , Teunissen CE , Tsolaki M , Vandenberghe R , Verbeek MM , Villemagne VL , Vos SJ , van Waalwijk van Doorn LJ , Waldemar G , Wallin A , Wallin AK , Wiltfang J , Wolk DA , Zboch M , Zetterberg H ((2015) ) Prevalence of cerebral amyloid pathology in persons without dementia: A meta-analysis. JAMA 313: , 1924–1938. |
[16] | Hodgetts CJ , Shine JP , Williams H , Postans M , Sims R , Williams J , Lawrence AD , Graham KS ((2019) ) Increased posterior default mode network activity and structural connectivity in young adult APOE-epsilon4 carriers: A multimodal imaging investigation. Neurobiol Aging 73: , 82–91. |
[17] | Dennis NA , Browndyke JN , Stokes J , Need A , Burke JR , Welsh-Bohmer KA , Cabeza R ((2010) ) Temporal lobe functional activity and connectivity in young adult APOE ɛ4 carriers. Alzheimers Dement 6: , 303–311. |
[18] | Molinuevo JL , Ayton S , Batrla R , Bednar MM , Bittner T , Cummings J , Fagan AM , Hampel H , Mielke MM , Mikulskis A , O’Bryant S , Scheltens P , Sevigny J , Shaw LM , Soares HD , Tong G , Trojanowski JQ , Zetterberg H , Blennow K ((2018) ) Current state of Alzheimer’s fluid biomarkers. Acta Neuropathol 136: , 821–853. |
[19] | Zhu Y , Nwabuisi-Heath E , Dumanis SB , Tai LM , Yu C , Rebeck GW , LaDu MJ ((2012) ) APOE genotype alters glial activation and loss of synaptic markers in mice. Glia 60: , 559–569. |
[20] | Vitek MP , Brown CM , Colton CA ((2009) ) APOE genotype-specific differences in the innate immune response. Neurobiol Aging 30: , 1350–1360. |
[21] | Tsoi L-M , Wong K-Y , Liu Y-M , Ho Y-Y ((2007) ) Apoprotein E isoform-dependent expression and secretion of pro-inflammatory cytokines TNF-α and IL-6 in macrophages. Arch Biochem Biophys 460: , 33–40. |
[22] | Weiner MW , Aisen PS , Jack CR Jr. , Jagust WJ , Trojanowski JQ , Shaw L , Saykin AJ , Morris JC , Cairns N , Beckett LA , Toga A , Green R , Walter S , Soares H , Snyder P , Siemers E , Potter W , Cole PE , Schmidt M , Alzheimer’s Disease Neuroimaging Initiative ((2010) ) The Alzheimer’s Disease Neuroimaging Initiative: Progress report and future plans. Alzheimers Dement 6: , 202–211 e207. |
[23] | Arevalo-Rodriguez I , Smailagic N , Roque IFM , Ciapponi A , Sanchez-Perez E , Giannakou A , Pedraza OL , Bonfill Cosp X , Cullum S ((2015) ) Mini-Mental State Examination (MMSE) for the detection of Alzheimer’s disease and other dementias in people with mild cognitive impairment (MCI). Cochrane Database Syst Rev 2015: , CD010783. |
[24] | Morris JC ((1997) ) Clinical Dementia Rating: A reliable and valid diagnostic and staging measure for dementia of the Alzheimer type. Int Psychogeriatr 9: , 173. |
[25] | Wechsler D ((1987) ) WMS-R: Wechsler Memory Scale-Revised: Manual, Psychological Corporation. |
[26] | McKhann GM , Knopman DS , Chertkow H , Hyman BT , Jack CR Jr. , Kawas CH , Klunk WE , Koroshetz WJ , Manly JJ , Mayeux R , Mohs RC , Morris JC , Rossor MN , Scheltens P , Carrillo MC , Thies B , Weintraub S , Phelps CH ((2011) ) The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 263–269. |
[27] | Shaw LM , Vanderstichele H , Knapik-Czajka M , Figurski M , Coart E , Blennow K , Soares H , Simon AJ , Lewczuk P , Dean RA , Siemers E , Potter W , Lee VM , Trojanowski JQ , Alzheimer’s Disease Neuroimaging Initiative ((2011) ) Qualification of the analytical and clinical performance of CSF biomarker analyses in ADNI. Acta Neuropathol 121: , 597–609. |
[28] | Shaw LM , Vanderstichele H , Knapik-Czajka M , Clark CM , Aisen PS , Petersen RC , Blennow K , Soares H , Simon A , Lewczuk P , Dean R , Siemers E , Potter W , Lee VM , Trojanowski JQ , Alzheimer’s Disease Neuroimaging Initiative ((2009) ) Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol 65: , 403–413. |
[29] | Spellman DS , Wildsmith KR , Honigberg LA , Tuefferd M , Baker D , Raghavan N , Nairn AC , Croteau P , Schirm M , Allard R , Lamontagne J , Chelsky D , Hoffmann S , Potter WZ , Alzheimer’s Disease Neuroimaging Initiative, Foundation for NIH (FNIH) Biomarkers Consortium CSF Proteomics Project Team ((2015) ) Development and evaluation of a multiplexed mass spectrometry based assay for measuring candidate peptide biomarkers in Alzheimer’s Disease Neuroimaging Initiative (ADNI) CSF. Proteomics Clin Appl 9: , 715–731. |
[30] | Choi YS , Lee KH ((2016) ) Multiple reaction monitoring assay based on conventional liquid chromatography and electrospray ionization for simultaneous monitoring of multiple cerebrospinal fluid biomarker candidates for Alzheimer’s disease. Arch Pharm Res 39: , 390–397. |
[31] | Shaw LM , Arias J , Blennow K , Galasko D , Molinuevo JL , Salloway S , Schindler S , Carrillo MC , Hendrix JA , Ross A , Illes J , Ramus C , Fifer S ((2018) ) Appropriate use criteria for lumbar puncture and cerebrospinal fluid testing in the diagnosis of Alzheimer’s disease. Alzheimers Dement 14: , 1505–1521. |
[32] | Blennow K , Shaw LM , Stomrud E , Mattsson N , Toledo JB , Buck K , Wahl S , Eichenlaub U , Lifke V , Simon M , Trojanowski JQ , Hansson O ((2019) ) Predicting clinical decline and conversion to Alzheimer’s disease or dementia using novel Elecsys Aβ(1-42), pTau and tTau CSF immunoassays. Sci Rep 9: , 19024. |
[33] | Ritchie ME , Phipson B , Wu D , Hu Y , Law CW , Shi W , Smyth GK ((2015) ) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43: , e47. |
[34] | Gentleman R , Carey V , Bates D , Bolstad B , Dettling M , Dudoit S , Ellis B , Gautier L , Ge Y , Gentry J , Hornik K , Hothorn T , Huber W , Iacus S , Irizarry R , Leisch F , Li C , Maechler M , Rossini A , Sawitzki G , Smith C , Smyth G , Tierney L , Yang J , Zhang J Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol 5: , R80. |
[35] | Millard SP , Lutz F , Li G , Galasko DR , Farlow MR , Quinn JF , Kaye JA , Leverenz JB , Tsuang D , Yu CE , Peskind ER , Bekris LM ((2014) ) Association of cerebrospinal fluid Aβ42 with A2M gene in cognitively normal subjects. Neurobiol Aging 35: , 357–364. |
[36] | Kok E , Haikonen S , Luoto T , Huhtala H , Goebeler S , Haapasalo H , Karhunen PJ ((2009) ) Apolipoprotein E-dependent accumulation of Alzheimer disease-related lesions begins in middle age. Ann Neurol 65: , 650–657. |
[37] | Llano DA , Bundela S , Mudar RA , Devanarayan V , Alzheimer’s Disease Neuroimaging I ((2017) ) A multivariate predictive modeling approach reveals a novel CSF peptide signature for both Alzheimer’s Disease state classification and for predicting future disease progression. PLoS One 12: , e0182098. |
[38] | Simon R , Girod M , Fonbonne C , Salvador A , Clement Y , Lanteri P , Amouyel P , Lambert JC , Lemoine J ((2012) ) Total ApoE and ApoE4 isoform assays in an Alzheimer’s disease case-control study by targeted mass spectrometry (n=669): A pilot assay for methionine-containing proteotypic peptides. Mol Cell Proteomics 11: , 1389–1403. |
[39] | Jack CR Jr. , Bennett DA , Blennow K , Carrillo MC , Dunn B , Haeberlein SB , Holtzman DM , Jagust W , Jessen F , Karlawish J , Liu E , Molinuevo JL , Montine T , Phelps C , Rankin KP , Rowe CC , Scheltens P , Siemers E , Snyder HM , Sperling R ((2018) ) NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement 14: , 535–562. |
[40] | Sproston NR , Ashworth JJ ((2018) ) Role of C-reactive protein at sites of inflammation and infection. Front Immunol 9: , 754. |
[41] | Mihlan M , Blom AM , Kupreishvili K , Lauer N , Stelzner K , Bergstrom F , Niessen HW , Zipfel PF ((2011) ) Monomeric C-reactive protein modulates classic complement activation on necrotic cells. FASEB J 25: , 4198–4210. |
[42] | Torzewski J , Torzewski M , Bowyer D , Frohlich M , Koenig W , Waltenberger J , Fitzsimmons C , Hombach V ((1998) ) C-reactive protein frequently colocalizes with the terminal complement complex in the intima of early atherosclerotic lesions of human coronary arteries. Arterioscler Thromb Vasc Biol 18: , 1386–1392. |
[43] | Nilsson K , Gustafson L , Hultberg B ((2011) ) C-reactive protein level is decreased in patients with Alzheimer’s disease and related to cognitive function and survival time. Clin Biochem 44: , 1205–1208. |
[44] | O’Bryant SE , Waring SC , Hobson V , Hall JR , Moore CB , Bottiglieri T , Massman P , Diaz-Arrastia R ((2010) ) Decreased C-reactive protein levels in Alzheimer disease. J Geriatr Psychiatry Neurol 23: , 49–53. |
[45] | Gabin JM , Saltvedt I , Tambs K , Holmen J ((2018) ) The association of high sensitivity C-reactive protein and incident Alzheimer disease in patients 60 years and older: The HUNT study, Norway. Immun Ageing 15: , 4. |
[46] | Royall DR , Al-Rubaye S , Bishnoi R , Palmer RF ((2017) ) Few serum proteins mediate APOE’s association with dementia. PLoS One 12: , e0172268. |
[47] | Haan MN , Aiello AE , West NA , Jagust WJ ((2008) ) C-reactive protein and rate of dementia in carriers and non carriers of Apolipoprotein APOE4 genotype. Neurobiol Aging 29: , 1774–1782. |
[48] | Hubacek JA , Peasey A , Pikhart H , Stavek P , Kubinova R , Marmot M , Bobak M ((2010) ) APOE polymorphism and its effect on plasma C-reactive protein levels in a large general population sample. Hum Immunol 71: , 304–308. |
[49] | Locascio JJ , Fukumoto H , Yap L , Bottiglieri T , Growdon JH , Hyman BT , Irizarry MC ((2008) ) Plasma amyloid beta-protein and C-reactive protein in relation to the rate of progression of Alzheimer disease. Arch Neurol 65: , 776–785. |
[50] | Brosseron F , Traschutz A , Widmann CN , Kummer MP , Tacik P , Santarelli F , Jessen F , Heneka MT ((2018) ) Characterization and clinical use of inflammatory cerebrospinal fluid protein markers in Alzheimer’s disease. Alzheimers Res Ther 10: , 25. |
[51] | Schuitemaker A , Dik MG , Veerhuis R , Scheltens P , Schoonenboom NS , Hack CE , Blankenstein MA , Jonker C ((2009) ) Inflammatory markers in AD and MCI patients with different biomarker profiles. Neurobiol Aging 30: , 1885–1889. |
[52] | Suh CH , Chun HY , Ye YM , Park HS ((2006) ) Unresponsiveness of C-reactive protein in the non-infectious inflammation of systemic lupus erythematosus is associated with interleukin 6. Clin Immunol 119: , 291–296. |
[53] | Kay J , Morgacheva O , Messing SP , Kremer JM , Greenberg JD , Reed GW , Gravallese EM , Furst DE ((2014) ) Clinical disease activity and acute phase reactant levels are discordant among patients with active rheumatoid arthritis: Acute phase reactant levels contribute separately to predicting outcome at one year. Arthritis Res Ther 16: , R40. |
[54] | Yang DH , Yang SK , Park SH , Lee HS , Boo SJ , Park JH , Na SY , Jung KW , Kim KJ , Ye BD , Byeon JS , Myung SJ ((2015) ) Usefulness of C-reactive protein as a disease activity marker in Crohn’s disease according to the location of disease. Gut Liver 9: , 80–86. |
[55] | Florin TH , Paterson EW , Fowler EV , Radford-Smith GL ((2006) ) Clinically active Crohn’s disease in the presence of a low C-reactive protein. Scand J Gastroenterol 41: , 306–311. |
[56] | Rea IM , Gibson DS , McGilligan V , McNerlan SE , Alexander HD , Ross OA ((2018) ) Age and age-related diseases: Role of inflammation triggers and cytokines. Front Immunol 9: , 586. |
[57] | van Harten AC , Jongbloed W , Teunissen CE , Scheltens P , Veerhuis R , van der Flier WM ((2017) ) CSF ApoE predicts clinical progression in nondemented APOEɛ4 carriers. Neurobiol Aging 57: , 186–194. |
[58] | Martínez-Morillo E , Hansson O , Atagi Y , Bu G , Minthon L , Diamandis EP , Nielsen HM ((2014) ) Total apolipoprotein E levels and specific isoform composition in cerebrospinal fluid and plasma from Alzheimer’s disease patients and controls. Acta Neuropathol 127: , 633–643. |
[59] | Minta K , Brinkmalm G , Janelidze S , Sjödin S , Portelius E , Stomrud E , Zetterberg H , Blennow K , Hansson O , Andreasson U ((2020) ) Quantification of total apolipoprotein E and its isoforms in cerebrospinal fluid from patients with neurodegenerative diseases. Alzheimers Res Ther 12: , 19. |
[60] | Safieh M , Korczyn AD , Michaelson DM ((2019) ) ApoE4: An emerging therapeutic target for Alzheimer’s disease. BMC Med 17: , 64. |
[61] | Reus LM , Stringer S , Posthuma D , Teunissen CE , Scheltens P , Pijnenburg YAL , Visser PJ , Tijms BM ((2020) ) Degree of genetic liability for Alzheimer’s disease associated with specific proteomic profiles in cerebrospinal fluid. Neurobiol Aging 93: , 144.e141-144.e115. |
[62] | Wang L , Gao T , Cai T , Li K , Zheng P , Liu J ((2020) ) Cerebrospinal fluid levels of YKL-40 in prodromal Alzheimer’s disease. Neurosci Lett 715: , 134658. |
[63] | Johansen JS , Baslund B , Garbarsch C , Hansen M , Stoltenberg M , Lorenzen I , Price PA ((1999) ) YKL-40 in giant cells and macrophages from patients with giant cell arteritis. Arthritis Rheum 42: , 2624–2630. |
[64] | Junker N , Johansen JS , Andersen CB , Kristjansen PE ((2005) ) Expression of YKL-40 by peritumoral macrophages in human small cell lung cancer.. Lung Cancer 48: , 223–231. |
[65] | Létuvé S , Kozhich A , Arouche N , Grandsaigne M , Reed J , Dombret MC , Kiener PA , Aubier M , Coyle AJ , Pretolani M ((2008) ) YKL-40 is elevated in patients with chronic obstructive pulmonary disease and activates alveolar macrophages. J Immunol 181: , 5167–5173. |
[66] | Wiley CA , Bonneh-Barkay D , Dixon CE , Lesniak A , Wang G , Bissel SJ , Kochanek PM ((2015) ) Role for mammalian chitinase 3-like protein 1 in traumatic brain injury. Neuropathology 35: , 95–106. |
[67] | Llorens F , Thune K , Tahir W , Kanata E , Diaz-Lucena D , Xanthopoulos K , Kovatsi E , Pleschka C , Garcia-Esparcia P , Schmitz M , Ozbay D , Correia S , Correia A , Milosevic I , Andreoletti O , Fernandez-Borges N , Vorberg IM , Glatzel M , Sklaviadis T , Torres JM , Krasemann S , Sanchez-Valle R , Ferrer I , Zerr I ((2017) ) YKL-40 in the brain and cerebrospinal fluid of neurodegenerative dementias. Mol Neurodegener 12: , 83. |
[68] | Sepe FN , Chiasserini D , Parnetti L ((2018) ) Role of FABP3 as biomarker in Alzheimer’s disease and synucleinopathies. Future Neurol 13: , 199–207. |
[69] | Desikan RS , Thompson WK , Holland D , Hess CP , Brewer JB , Zetterberg H , Blennow K , Andreassen OA , McEvoy LK , Hyman BT , Dale AM ((2013) ) Heart fatty acid binding protein and Aβ-associated Alzheimer’s neurodegeneration. Mol Neurodegener 8: , 39. |
[70] | Bjerke M , Kern S , Blennow K , Zetterberg H , Waern M , Borjesson-Hanson A , Ostling S , Kern J , Skoog I ((2016) ) Cerebrospinal fluid fatty acid-binding protein 3 is related to dementia development in a population-based sample of older adult women followed for 8 years. J Alzheimers Dis 49: , 733–741. |
[71] | Dyszy F , Pinto AP , Araújo AP , Costa-Filho AJ ((2013) ) Probing the interaction of brain fatty acid binding protein (B-FABP) with model membranes. PLoS One 8: , e60198. |
[72] | Osenkowski P , Ye W , Wang R , Wolfe MS , Selkoe DJ ((2008) ) Direct and potent regulation of gamma-secretase by its lipid microenvironment. J Biol Chem 283: , 22529–22540. |
[73] | An Y , Varma VR , Varma S , Casanova R , Dammer E , Pletnikova O , Chia CW , Egan JM , Ferrucci L , Troncoso J , Levey AI , Lah J , Seyfried NT , Legido-Quigley C , O’Brien R , Thambisetty M ((2018) ) Evidence for brain glucose dysregulation in Alzheimer’s disease. Alzheimers Dement 14: , 318–329. |
[74] | Butterfield DA , Halliwell B ((2019) ) Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat Rev Neurosci 20: , 148–160. |
[75] | Zhou M , Haque RU , Dammer EB , Duong DM , Ping L , Johnson ECB , Lah JJ , Levey AI , Seyfried NT ((2020) ) Targeted mass spectrometry to quantify brain-derived cerebrospinal fluid biomarkers in Alzheimer’s disease. Clin Proteomics 17: , 19. |
[76] | Fonseca MI , Kawas CH , Troncoso JC , Tenner AJ ((2004) ) Neuronal localization of C1q in preclinical Alzheimer’s disease. Neurobiol Dis 15: , 40–46. |
[77] | Stoltzner SE , Grenfell TJ , Mori C , Wisniewski KE , Wisniewski TM , Selkoe DJ , Lemere CA ((2000) ) Temporal accrual of complement proteins in amyloid plaques in Down’s syndrome with Alzheimer’s disease. Am J Pathol 156: , 489–499. |
[78] | Konijnenberg E , Tijms BM , Gobom J , Dobricic V , Bos I , Vos S , Tsolaki M , Verhey F , Popp J , Martinez-Lage P , Vandenberghe R , Lleo A , Frolich L , Lovestone S , Streffer J , Bertram L , Blennow K , Teunissen CE , Veerhuis R , Smit AB , Scheltens P , Zetterberg H , Visser PJ ((2020) ) APOE epsilon4 genotype-dependent cerebrospinal fluid proteomic signatures in Alzheimer’s disease.. Alzheimers Res Ther 12: , 65. |
[79] | Lin YT , Seo J , Gao F , Feldman HM , Wen HL , Penney J , Cam HP , Gjoneska E , Raja WK , Cheng J , Rueda R , Kritskiy O , Abdurrob F , Peng Z , Milo B , Yu CJ , Elmsaouri S , Dey D , Ko T , Yankner BA , Tsai LH ((2018) ) APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron 98: , 1141–1154 e1147. |
[80] | Vignesh P , Rawat A , Sharma M , Singh S ((2017) ) Complement in autoimmune diseases. Clin Chim Acta 465: , 123–130. |
[81] | Yin C , Ackermann S , Ma Z , Mohanta SK , Zhang C , Li Y , Nietzsche S , Westermann M , Peng L , Hu D , Bontha SV , Srikakulapu P , Beer M , Megens RTA , Steffens S , Hildner M , Halder LD , Eckstein HH , Pelisek J , Herms J , Roeber S , Arzberger T , Borodovsky A , Habenicht L , Binder CJ , Weber C , Zipfel PF , Skerka C , Habenicht AJR ((2019) ) ApoE attenuates unresolvable inflammation by complex formation with activated C1q. Nat Med 25: , 496–506. |
[82] | Hong S , Beja-Glasser VF , Nfonoyim BM , Frouin A , Li S , Ramakrishnan S , Merry KM , Shi Q , Rosenthal A , Barres BA , Lemere CA , Selkoe DJ , Stevens B ((2016) ) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352: , 712–716. |
[83] | Chung WS , Verghese PB , Chakraborty C , Joung J , Hyman BT , Ulrich JD , Holtzman DM , Barres BA ((2016) ) Novel allele-dependent role for APOE in controlling the rate of synapse pruning by astrocytes. Proc Natl Acad Sci U S A 113: , 10186–10191. |
[84] | Mayilyan KR ((2012) ) Complement genetics, deficiencies, and disease associations. Protein Cell 3: , 487–496. |


