
Tetrahydrobiopterin Improves Recognition Memory in the Triple-Transgenic Mouse Model of Alzheimer’s Disease, Without Altering Amyloid-β and Tau Pathologies
Abstract
Background:
Alzheimer’s disease (AD) is a multifactorial disease, implying that multi-target treatments may be necessary to effectively cure AD. Tetrahydrobiopterin (BH4) is an enzymatic cofactor required for the synthesis of monoamines and nitric oxide that also exerts antioxidant and anti-inflammatory effects. Despite its crucial role in the CNS, the potential of BH4 as a treatment in AD has never been scrutinized.
Objective:
Here, we investigated whether BH4 peripheral administration improves cognitive symptoms and AD neuropathology in the triple-transgenic mouse model of AD (3xTg-AD), a model of age-related tau and amyloid-β (Aβ) neuropathologies associated with behavior impairment.
Methods:
Non-transgenic (NonTg) and 3xTg-AD mice were subjected to a control diet (5% fat – CD) or to a high-fat diet (35% fat - HFD) from 6 to 13 months to exacerbate metabolic disorders. Then, mice received either BH4 (15 mg/kg/day, i.p.) or vehicle for ten consecutive days.
Results:
This sub-chronic administration of BH4 rescued memory impairment in 13-month-old 3xTg-AD mice, as determined using the novel object recognition test. Moreover, the HFD-induced glucose intolerance was completely reversed by the BH4 treatment in 3xTg-AD mice. However, the HFD or BH4 treatment had no significant impact on Aβ and tau neuropathologies.
Conclusion:
Overall, our data suggest a potential benefit from BH4 administration against AD cognitive and metabolic deficits accentuated by HFD consumption in 3xTg-AD mice, without altering classical neuropathology. Therefore, BH4 should be considered as a candidate for drug repurposing, at least in subtypes of cognitively impaired patients experiencing metabolic disorders.
INTRODUCTION
Alzheimer’s disease (AD) is the most common form of dementia [1]. So far, there is no treatment proven efficient to prevent or stop the progression of the disease. AD is characterized by a cognitive decline, hyperphosphorylated tau aggregated into neurofibrillary tangles and the accumulation of amyloid-β (Aβ) into senile plaques [2]. Beside these canonical hallmarks, AD also involves defective monoamine neurotransmission [3, 4], neuroinflammation [5, 6], vascular impairment [7], and oxidative damage [8]. While the exact causes for the development of sporadic AD remain unknown, numerous risk factors have been identified such as age, apolipoprotein ɛ4 (ApoE4) carriage, and cardiovascular pathologies. More recently, obesity, diabetes, and metabolic syndrome have been added to the list of key risk factors of AD [9, 10]. Indeed, these metabolic disturbances lead to increased neuroinflammation and oxidative stress but also alterations in neuronal activity and vascular function associated with cognitive decline [11, 12]. Neuroinflammation and oxidative stress have long been associated with AD neuropathology and emerging evidence suggest these alterations may play a key role in the relationship between metabolic disorders and AD [9]. Impairments in dopaminergic and serotoninergic pathways have also been implicated in the constellation of cognitive or behavioral symptoms seen in AD [3, 13–15]. In light of these observations, the paradigm “one-drug-one-target” has been challenged and a more holistic approach has emerged, to include targets outside of the CNS [16, 17]. Overall, there is growing recognition that AD is a multifactorial disease and that multi-target treatments allowing the modulation of the multiple defects observed in AD are needed [9, 18].
Tetrahydrobiopterin (BH4) is mostly known as the main enzymatic cofactor required for the synthesis of serotonin (5-HT), dopamine (DA), and nitric oxide (NO) [19]. BH4 is synthesized by all cells from GTP and is readily oxidized to dihydrobiopterin (BH2), its inactive form [20]. Moreover, a fraction of BH2 can be re-oxidized in BH4 by the dihydrobiopterin reductase. BH4 plays crucial roles in the CNS, as a cellular antioxidant, regulating the redox function of endothelial NO synthase (eNOS) [21, 22], scavenging oxygen radicals [23] and monoamines production [24–27].
Interestingly, BH4 and its metabolites (BH2 and other oxidized biopterin) have been shown to be decreased in serum [28], cerebrospinal fluid [29], and temporal lobe from elderly or AD patients [27, 30]. Furthermore, neopterin, a BH4 derivative produced in inflammatory conditions, is increased in the plasma of AD patients and correlates with cognitive decline [31–33]. In vivo evidence obtained in Tg2576 mice, a model that overexpresses mutated human amyloid-β protein precursor (AβPP), suggests that Aβ peptides induce an increase in production of superoxide anion leading to BH4 oxidation in BH2 [34]. Conversely, it has been shown that a genetic increase in BH4 synthesis in endothelial cells of ApoE-knockout mice reduces vascular oxidative stress and restores NO signaling [35]. Interestingly, BH4 administration in mice and in humans was reported to improve glucose intolerance and insulin resistance, two markers of peripheral metabolic disorders, which are also risk factors for AD [36–38]. More recent evidence shows that BH4 crosses the blood-brain barrier, influencing dopamine release and improving performance in motivational tasks [39]. Although these data suggest that BH4 may act on several key aspects of AD pathogenesis, the potential of BH4 to improve symptoms and pathology has never been studied.
The triple-transgenic mouse model of AD (3xTg-AD) develops memory deficits and the two main neuropathological markers of the disease, namely amyloid plaques and neurofibrillary tangles, while aging [40–42]. Chronic consumption of high-fat diet (HFD) further increases neuropathologies and accelerates cognitive impairment [43–47] in these mice and other AD mouse models [48–54]. Several mechanisms have been proposed for the effect of HFD on AD pathology including changes in production/clearance ratio of Aβ [43, 46, 48, 55, 56], insulin-signaling dysregulations [54, 55, 57], and increased oxidative stress [44] and inflammation [58]. Finally, BH4 is already administered in patients suffering from genetic deficiency and its favorable safety profile has been shown in clinical trials [59]. Thus, given its ability to regulate metabolism, oxidative stress, and inflammation, BH4 appears to be promising to treat the HFD-induced increase in AD-like neuropathologies.
We hypothesized that BH4 administration could ameliorate memory impairment in 3xTg-AD mice owing to its action on different AD-relevant pathways. In order to exacerbate metabolic, inflammatory, and oxidative disturbances in 3xTg-AD mice, animals were fed either a HFD or a control diet from the age of 6 until 13 months. Then, mice were injected daily with either BH4 (15 mg/kg) or a control solution for ten consecutive days. The aim was to explore the effect of BH4 treatment on cognitive impairment, cerebral neuropathologies, and HFD-induced metabolic defects in the 3xTg-AD mouse model.
METHODS
Animals
All experiments were performed in accordance with the Canadian Council on Animal Care (CCAC) and received prior authorization from the Université Laval animal protection committee (CPAUL). Triple-transgenic 3xTg-AD mice (APPswe, PSIMI46V, tauP301L) (from Dr. LaFerla) [41] were maintained in our animal facilities and backcrossed every seven to ten generations to C57BL6/129SvJ mice. In this model, Aβ and tau pathologies become readily quantifiable at ∼12 months of age, co-occurring with cognitive deficits and glucose-related metabolic disturbances [42, 43, 55, 60–63]. Serum cytokines were previously studied in the 3xTg-AD model between 9 and 13 months of age [64].
Thus, the non-transgenic mice (NonTg), used as a control group, were on the same background as transgenic mice. Mice were housed one to five per cage with a 12:12 h light-dark cycle (light phase from 7 a.m. to 7 p.m). Animals had ad libitum access to water and diet. At the end of the experiment, mice were killed by intracardiac perfusion with phosphate saline buffer containing a cocktail of protease inhibitors (SIGMAFAST™ Protease Inhibitor Tablets, Sigma-Aldrich, St Louis, MO, USA) and phosphatase inhibitors (1 mM sodium pyrophosphate and 50 mM sodium fluoride) under deep anesthesia with ketamine/xylazine i.p. (100 mg/kg ketamine, 10 mg/kg xylazine). Brain was rapidly dissected on ice, frozen and kept at –80°C until processing. All groups were composed of males and females in equal proportion. All mice were randomly assigned to diet and BH4 treatment groups.
Diets and treatment
Mice received either a control diet (CD – 12 % kCal from fat) or a high-fat diet (HFD – 60 % kCal from fat) for 7 months, from the age of 6 months until the end of the experiment [43, 55]. The diet was manufactured by Research Diets Inc. (New Brunswick, NJ) and is described in detail in Supplementary Table 1. We used purified diet formulations standardized to ensure consistency and eliminate batch-to-batch variations, containing measured concentrations of macronutrients, vitamins, and minerals.
At the age of 13 months, mice were injected daily with either a BH4 solution (15 mg/kg, i.p.) or a control solution (0.1 M HCl diluted in saline, i.p.) for 10 consecutive days prior sacrifice. This treatment duration was selected from previous studies showing that IV or oral administration of BH4 for 7–10 days was sufficient to alter neurotransmitters such as DA, 5-HT and/or their metabolites [65–67]. Previous work shows that intraperitoneal administration of BH4 (50 mg/kg, ip) leads to increased BH4 concentrations in the brain as well as increased dopamine release and enhanced motivation in response to amphetamine [39]. Using in-situ brain perfusion, the brain uptake clearance (Clup) of BH4 was estimated at 0.08μl g–1.s–1, consistent with a modest rate of transport across the blood-brain barrier [39]. The dose of 15 mg/kg is in the range of doses used in previous clinical trials and animal studies [34, 68]. The BH4 stock solution (100 mg/mL) was prepared by dissolving 1 g of (6R)-5,6,7,8-Tetrahydro-L-biopterin dihydrochloride (Schricks laboratories) in 10 mL of 0.1 M HCl and stored at –80°C until use. Mice were injected 2 h before the end of the light phase (5 p.m.) and after metabolic and behavior experiments to avoid potential stress induction by the i.p. injection.
Behavioral testing
All behavioral and metabolic tests were performed from day 5 to day 10 of BH4 treatment, with a recovery time of at least 24 h between tests. All behavioral tests were conducted blindly, during the first 4 h of the light phase. To minimize stress, mice were acclimated to the testing room for 12 h before the test.
Spontaneous locomotion
Spontaneous locomotion was recorded at day 6 of BH4 treatment using an open-field apparatus consisting of clear boxes (40×40×40 cm) [42, 69]. Horizontal locomotor activity was recorded by the interruption of crossing photosensitive beams (San Diego Instrument). Mice were placed in the center of the open-field and their activity (total distance travelled in cm) was recorded for 1 h. The speed of mice was also calculated and expressed as cm/min.
Recognition memory
Recognition memory was assessed using the novel object recognition test (NOR) at days 7 and 8 of BH4 treatment, as previously described [55, 69]. The NOR test is based on the spontaneous tendency of rodents to explore a new object for longer than a familiar object. This task was selected based on the short period of time available for assessing both cognition and glucose tolerance in the same animals. In addition, compared with other frequently used behavior tests, it is less sensitive to sensorimotor dysfunction, anxiety or thermoregulatory deficits, which are commonly reported in the 3xTg-AD mouse [55, 61, 69–73].
During the acquisition phase, mice were placed in a clear box (29.2 x 19 x 12.7 cm) and allowed to explore two similar objects for 5 min. Mice were returned to their housing cage for 1 h and then presented with a familiar (F) and a novel (N) object (testing phase) in the same box for 5 min. The novel object was randomly placed at the exact location of one of the two familiar objects. The recognition index was defined as the time exploring the novel object divided by the total exploration time during the testing phase and expressed as a percentage. The NOR test assumes that a mouse not recognizing the object have a random chance of exploring each object (50%) [73]. A mouse which identifies one of the objects as new will spend more time exploring it. Therefore, its recognition index will be higher than 50%. Thus, a lack of discrimination between the novel and the familiar object, i.e., a recognition index of 50 % or below, was interpreted as impaired object recognition. Mice showing poor exploration activity (i.e., less than 7 s of exploration per object during the acquisition phase and less than 10 s of total exploration time during the testing phase) were excluded from the analysis of the experiment.
Glucose tolerance test
Glucose tolerance tests were performed at the days 9 and 10 of BH4 treatment, mice were individually housed in the testing room and fasted for 6 h before glucose tolerance test with free access to water. Mice received an injection of glucose (1 g/kg, i.p.) and glycemia was measured at 0, 15, 30, 45, 60, 90, and 120 min after glucose injection, in a drop of blood from the saphenous vein using a glucometer (OneTouch UltraMini, LifeScan, Milpitas, CA).
Protein extraction and western immunoblotting
The day following the last BH4 administration, parieto-temporal cortex and hippocampi were dissected, weighed and processed by successive centrifugation steps as previously described [55, 63], resulting in a soluble fraction (intra- and extracellular Tris buffer (TBS)-soluble proteins), a detergent-soluble fraction (membrane proteins) and a detergent-insoluble fraction (aggregated proteins extracted with formic acid). Briefly, tissues were homogenized in TBS (0.05 M Tris Base, 0.138 M NaCl, 2.7 mM KCl with 1 mM EDTA) containing a protease inhibitor cocktail (104 mM AEBSF, 80μM aprotinin, 5 mM bestatin, 1.5 mM E-64, 2 mM leupeptin and 1.5 mM pepstatin-A) and a phosphatase inhibitors cocktail (100 mM Sodium fluoride, 100 mM sodium orthovanadate, 400 mM sodium tartrate, 115 mM sodium molybdate, 200 mM imidazole, 2.5 mM (-)-p-bromotetramisole oxalate, 500μM cantharidin and 500 nM Microcystin LR, Microcystis aeruginosa) obtained from by Bimake (Houston, TX). Samples were sonicated (3×45 s) in a Sonic Dismembrator apparatus (Thermo Fisher Scientific, Waltham, MA) After a 20 min centrifugation at 100,000 g and collection of supernatant, the TBS-insoluble pellets were sonicated in lysis buffer (150 mM NaCl, 10 mM NaH2PO4, 1 mM EDTA, 1% Triton X-100, 0.5% SDS and 0.5% deoxycholate) containing the same protease and phosphatase inhibitors. After centrifugation, the remaining fraction containing insoluble proteins was resuspended in 99 % formic acid and divided in two parts, and dried to eliminate acid residue. One part was solubilized in 1X Laemmli’s buffer for western blotting and the other in guanidine hydrochloride (5 M in 0.05 M Tris-HCl) for subsequent ELISAs.
Protein concentrations were determined using bicinchoninic acid assay (Thermo-pierce). Twenty μL of each sample were added in Laemmli’s loading buffer to obtain a final concentration of 1.5μg/μL and heated to 70°C for 5 min. A total of 15–22.5μg of proteins per sample were separated by SDS-PAGE and electroblotted onto PVDF membranes (Immobilon, Millipore). Membranes were blocked in 0.5% BSA and 5% dry milk in PBS-0.1% tween-20 and then incubated with primary (listed in Supplementary Table 2) and secondary (HRP goat anti-rabbit or goat anti-mouse, 1/50000, Jackson ImmunoResearch) antibodies. Band intensities (optical density, OD) were revealed with Luminata Forte (Millipore) and detected using myECL imager (Thermo Fisher Scientific). Quantifications were performed blindly on ImageLab software (Millipore), using a background subtraction when necessary and results were expressed as relative OD.
Aβ40 and Aβ42 quantification
Human Aβ40 and Aβ42 contents were assessed in TBS-soluble and detergent-insoluble fractions of protein extracts from parietotemporal cortices of 3xTg-AD mice using High Sensitive human β-Amyloid ELISA kits for Aβ40 quantification (cat.no.292-62301, FUJIFILM Wako Pure Chemical Corporation) and for Aβ42 quantification (cat.no.296-6440, FUJIFILM Wako Pure Chemical Corporation) according to the manufacturer’s instructions [74]. Briefly, plates were coated with anti-human-Aβ[1–16] MoAb [Clone No. BAN50] which recognizes the N-terminal terminus of human Aβ. Then, HRP-conjugated Fab’ antibody BA27 (Aβ40) or BC05 (Aβ42) were used to specifically detects the C-terminal of each Aβ fragment. The cross reactivity of murine Aβ40 and Aβ42 are <0.1 and 0.5% for the Aβ40 and the Aβ42 ELISA kit, respectively.
High-performance liquid chromatography
Striatal monoamines and their main metabolites were quantified as previously described [75]. Briefly, striata were dissected on ice at the end of the 10 days of BH4-treatment, weighed and homogenized in perchloric acid (0.1 N), centrifuged (13 000 rpm, 50 min, 4°C) and supernatants were collected and injected on an HPLC apparatus. The mobile phase (8% methanol, 0.4 mM EDTA, 2 mM NaCl, 0.9 mM octanic sulfonic acid, 55 mM NaH2PO4, pH 2.9) was constantly delivered at 1.2 mL/min. Concentrations were calculated using daily injected standards and normalized to striatum weight.
Quantitative real-time PCR (qRT-PCR)
RNA from parietotemporal cortices was extracted using RNeasy lipid tissue mini kit (Qiagen). Two micrograms of RNA were reverse transcribed in cDNA (High capacity cDNA reverse transcription kit, Life technologies). Gene expression was measured using 80 nanograms of the resulting cDNA in duplicates and appropriate FAM-labeled Taqman primers (Taqman gene expression assays, IL1β (Mm00434228_m1), IL6 (Mm00446190_m1), TNF-α (Mm00443258_m1), GAPDH (Mm99999915_g1), Life technologies). Data were analyzed using the comparative threshold cycle method using GAPDH as a housekeeping gene. Results are expressed as relative fold change with untreated NonTg CD mice as the control group.
Nitrites/Nitrates brain levels
Nitrites and nitrates were measured as an indication of NO levels in the soluble fraction of parietotemporal cortices. Forty μL of each sample were assayed on a nitrate/nitrite colorimetric assay kit (Cayman) according to the manufacturers’ instructions. Results are expressed as the sum of nitrites and nitrates levels, in μmoles/mg of wet tissue.
Statistical analysis
The main effects and interaction of the independent variables (Sex, Diet, BH4-Treatment, and Genotype) were evaluated using multifactorial ANOVA. Unless a statistical effect of gender, males and females were grouped and a 3-way ANOVA was performed, with Time as a within-subjects repeated factor to compare recurrent measurements in the same animals. When variances were comparable between groups (Barlett’s test), one-way ANOVAs were performed, followed by Tukey’s tests. When variances were not comparable, Kruskal Wallis tests followed by Wilcoxon comparisons were used. All post-hoc comparisons were made using the Tukey test. For the test of recognition memory, a sample t-test was used to compare means to the theoretical value of 50% (i.e., random/no recognition); exploration times of the familiar and novel objects were compared using the Wilcoxon matched-pairs rank test.
Data are presented as mean±SEM. All statistical analyses were performed using GraphPad Prism 7 softwares. Statistical significance was set at p < 0.05.
RESULTS
The experimental design included male and female NonTg and 3xTg-AD mice, exposed to either a control or a HFD from the age of 6 months. After 7 months of diet, the mice received an injection of BH4 treatment for 10 days at 15 mg/kg/day. During the treatment period, behavioral and metabolic tests were performed. After the last day of treatment, the mice were sacrificed, and the brain tissue was removed for analysis (Fig. 1).
Fig.1
Experimental design. Non-transgenic mice (NonTg) and triple-transgenic mice (3xTg-AD) mice were fed from the age of 6 months a control (CD) or a high-fat diet (HFD) for 7 months. BH4 treatment (10 days, 15 mg/kg/day) was administered intra-peritoneally to mice at the age of 13 months. Behavioral (Open Field, Object Recognition) and metabolic (Glucose Tolerance Test) tests were performed between the 6th and 10th days of BH4 administration. On the day following the last injection, mice were sacrificed, and brain tissues were collected. All group (n = 11–17) were composed of males and females in equal proportion.
Mice were weighed at time of sacrifice, i.e., after 7 months of CD or HFD and 10 days after saline or BH4 treatment. The multifactorial ANOVA revealed a significant diet effect (p < 0.001), with a mean body weight of 36.6 g in CD versus 42.8 g in HFD mice (+17%), a genotype effect (p < 0.01), with a reduced body weight in 3xTg-AD mice versus NonTg (–10%). As expected, males had a significantly higher body weight than females (p < 0.001) (+34%). However, the administration of BH4 had no effect on the body weight of mice (Table 1).
Table 1
Animal weights and levels of monoamines and main metabolites in the striatum of NonTg and 3xTg-AD mice
| Genotype | Non Tg | 3xTg-AD | Multivariate analyses | ||||||
| Diet | CD | HFD | CD | HFD | |||||
| Treatment | Ctls | BH4 | Ctls | BH4 | Ctls | BH4 | Ctls | BH4 | |
| N | 11 | 12 | 12 | 14 | 17 | 15 | 13 | 15 | |
| Weight (g)(a) | 37.8±2.0 | 38.8±2.5 | 45.5±2.6 | 45.1±2.9 | 35.0±1.6 | 34.9±2.0 | 41.0±1.7 | 39.5±2.0 | Higher in males, p < 0.001 |
| Higher after HFD, p < 0.001 | |||||||||
| Lower in 3xTg-AD, p < 0.01 | |||||||||
| Weight Δ (g) (b) | –2.5±0.6 | –1.7±0.4 | –4.2±0.5 | –5.9±0.7 | –1.4±0.4 | –1.5±0.7 | –3.1±0.6 | –2.4±0.7 | More in HFD, p < 0.001 |
| Less in 3xTg-AD, p < 0.01 | |||||||||
| N | 11 | 12 | 10 | 10 | 16 | 14 | 13 | 13 | |
| DA | 21.85±2.12 | 17.19±1.69 | 19.75±1.49 | 22.38±2.33 | 21.79±1.36 | 20.91±1.75 | 24.66±3.35 | 24.59±1.37 | n.s |
| HVA | 2.94±0.30 | 2.66±0.23 | 2.81±0.31 | 3.56±0.33 | 3.28±0.18 | 3.37±0.29 | 3.62±0.38 | 3.61±0.24 | Higher in 3xTg-AD, p < 0.05 |
| DOPAC | 1.48±0.13 | 1.70±0.13 | 1.67±0.13 | 1.98±0.19 | 1.82±0.11 | 1.78±0.13 | 2.03±0.26 | 2.04±0.11 | Higher in HFD, p < 0.05 |
| 5-HT | 1.89±0.09 | 1.85±0.14 | 2.19±0.14 | 2.01±0.16 | 2.08±0.13 | 2.01±0.08 | 2.09±0.14 | 2.08±0.18 | n.s |
| 5-HIAA | 1.55±0.13 | 1.53±0.12 | 1.82±0.16 | 2.11±0.14† | 1.72±0.14 | 1.47±0.11 | 1.77±0.17 | 1.54±0.13 | Higher in HFD, p < 0.05 |
| 5-HIAA/5-HT | 0.82±0.06 | 0.84±0.06 | 0.82±0.04 | 1.06±0.06† | 0.83±0.04 | 0.73±0.03 | 0.84±0.05 | 0.75±0.03 | Genotype×Treatment, p < 0.001 |
| Higher in 3xTg-AD, p < 0.01 | |||||||||
| Higher in HFD, p < 0.05 | |||||||||
| [DOPAC + HVA]/DA | 0.21±0.01 | 0.25±0.01 | 0.23±0.01 | 0.26±0.1 | 0.24±0.01 | 0.25±0.01 | 0.24±0.01 | 0.23±0.01 | Higher with BH4, p < 0.01 |
| Genotype x Treatment, p < 0.05 | |||||||||
Dopamine (DA), serotonin (5-HT), and their metabolites, 3,4-Dihydroxyphenylacetic acid (DOPAC), homovanillic acid (HVA), and 5-hydroxyindolacetic acid (5-HIAA) respectively, were measured by HPLC-electrochemistry in striatum. Values are expressed as mean±SEM (ng/mg tissue). CD, control diet; HFD, high-fat diet; Ctls, control vehicle-treated mice; BH4, BH4-treated mice. a) Measured at sacrifice. b) Weight loss during the 10-day treatment, expressed as mean±SEM (g). †p < 0.05 versus all other groups (Kruskal Wallis followed by nonparametric Comparisons for each pair using Wilcoxon method).
BH4 administration improves recognition memory in 3xTg-AD mice without affecting spontaneous locomotion
We first evaluated object recognition memory after 6 days of BH4 administration in 3xTg-AD and NonTg mice using the NOR test (Fig. 2A). The NOR test assumes that a mouse not recognizing the new object have a random chance of exploring each object (50%). If a mouse explores more the novel object, the recognition index will be higher than 50%. All groups of NonTg mice exhibited a recognition index significantly different from 50%, indicating that they did recognize the new object, independently of the diet. Results showed that 3xTg-AD mice fed a CD or HFD had a recognition index that did not differ from the threshold of 50%, confirming memory impairments. Whereas BH4 administration had no effect on NOR in NonTg mice, the recognition index was higher in 3xTg-AD mice treated with BH4, independently of the diet, indicating that BH4 rescued memory deficits in these mice (Fig. 2B). Diet and BH4 treatment did not affect the exploration time during the testing phase. However, 3xTg-AD mice exhibited a slightly higher total exploration time (44±2 s) compared to NonTg mice (35±2 s) (Genotype effect, p = 0.0358). Memory deficits in 3xTg-AD mice were also illustrated by the time of exploration of the novel object during the testing phase, which did not differ from the time of exploration of the familiar object, in mice under CD and HFD diet, except for the mice treated by BH4 (p < 0.001) (Fig. 2C). Comparison of the two acquisition and testing phases showed that the exploration ratio was significantly higher in BH4-treated mice than in non-treated mice, only in the group fed the CD (p < 0.05) (Fig. 2D). BH4-treated animals were also compared directly to their respective untreated controls, confirming that the administration of BH4 increased the NOR index of 3xTg-AD mice compared to untreated controls, independently of the diet (p < 0.01). Multivariate analyses showed no effect of sex on the NOR index (p > 0.5).
Fig.2
BH4 administration improves recognition memory function in 3xTg-AD mice without affecting spontaneous locomotion. A) Recognition memory was evaluated using the novel object recognition (NOR) test. B) Recognition index: (time exploring the novel (N) object/total time exploring both N and familiar (F) objects during the testing phase)*100. C) Total exploration time of the N and the F objects during the testing phase. D) Ratio of total exploration time during the testing phase/acquisition phase. E) Spontaneous locomotion monitored for 1 h in an open-field (total distance travelled in cm). Values are represented as mean±SEM with N = 7–17/group. Statistical analyses: (B) ++p < 0.01; +++p < 0.001 compared to random selection of an object (one sample t-test versus 50%); # #p = 0.0013 (Effect of BH4 Treatment, two-way ANOVA within 3xTg-AD mice); (C) &p < 0.05, &&p < 0.01, &&&p < 0.001 between N and F objects (Wilcoxon matched paired rank test); (D) &p < 0.05 (Wilcoxon matched paired rank test); (E) **p < 0.01 Genotype effect, (Multimodal ANOVA).
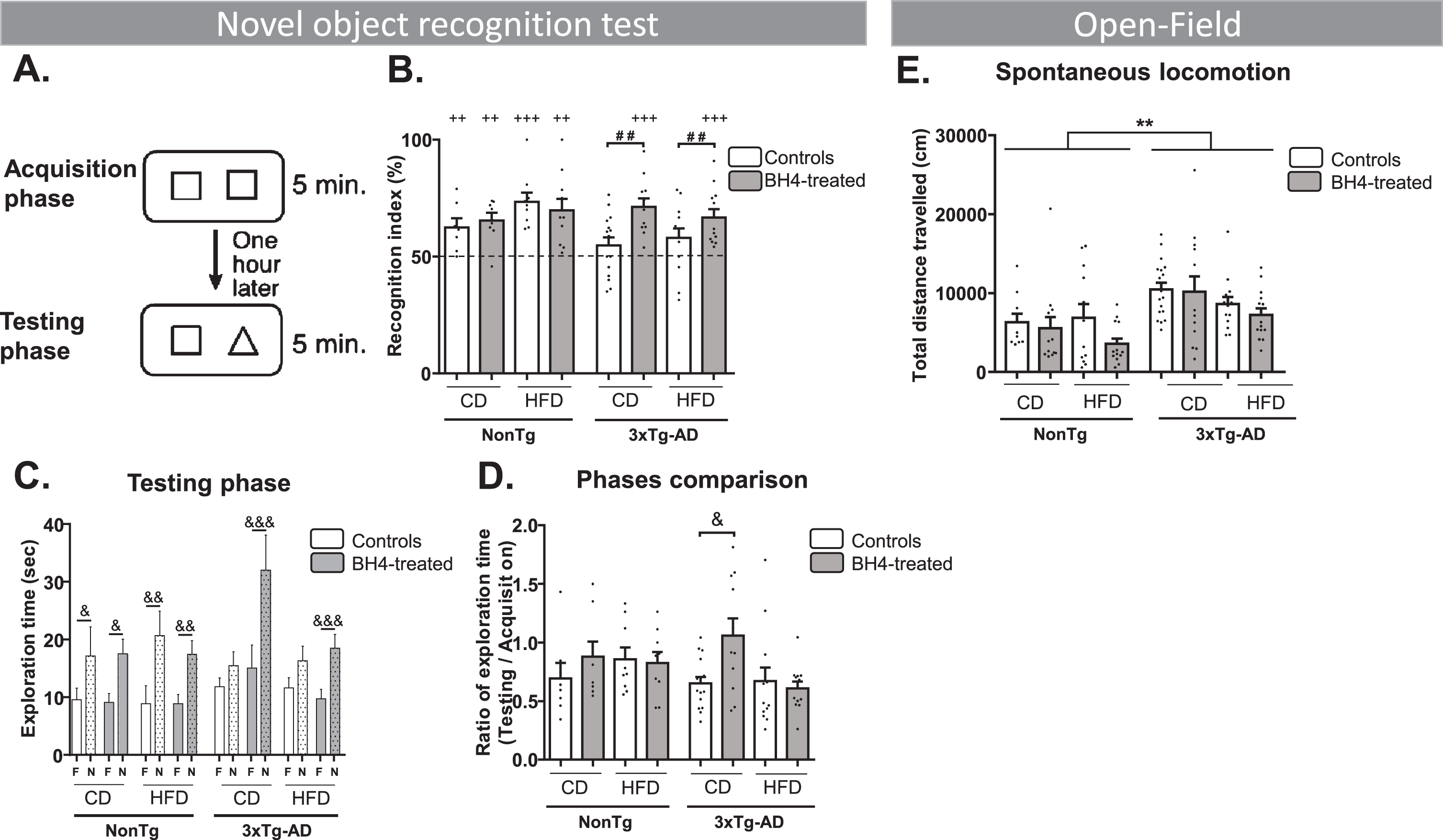
Spontaneous locomotion in the open-field assessed by the total distance travelled was significantly higher in 3xTg-AD compared to NonTg mice, without a BH4 treatment effect (Genotype effect, p < 0.001 and diet effect, p < 0.05). However, speed was not statistically different between genotypes (NonTg mice: 11.7±0.4 cm/min; 3xTg-AD: 13.2±0.2 cm/min), independently of diet or treatment (Fig. 2E).
BH4 administration had no effect on hippocampal tau and amyloid pathologies
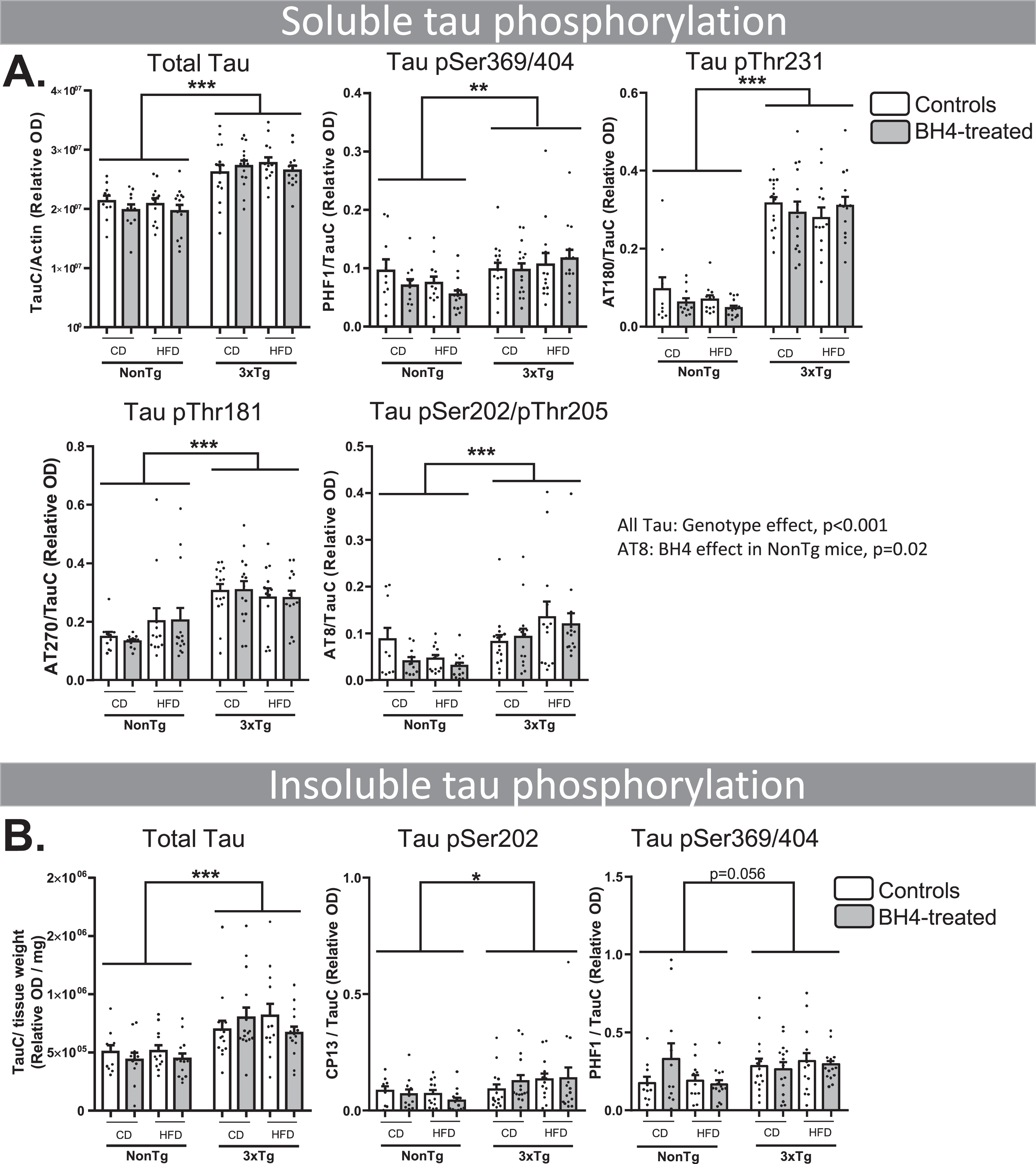
Since the 3xTg-AD mouse model displays an accretion of both tau and Aβ in the brain [41], we investigated whether the improvement in memory deficit we observed after BH4 administration was linked to changes in these two main AD neuropathological hallmarks. First, we quantified total tau protein, using an antibody specific for the C-terminal of both the murine endogenous form and the human transgenic form, in both cytosolic and insoluble fractions. As tau hyperphosphorylation is a key marker of AD, we also quantified tau phosphorylated levels at Ser369/Ser404 (PHF1), Thr231 (AT180), Thr181 (AT270), Ser202/Thr205 (AT8), and Ser202 (CP13) in soluble and insoluble fractions in the hippocampus As expected, 3xTg-AD mice displayed greater levels of total and phosphorylated tau in the soluble and insoluble fractions than NonTg mice (Fig. 3) (Genotype effects, all p < 0.001). Total soluble tau and phosphorylation status of each epitope assessed were higher in 3xTg-AD (p < 0.01 or p < 0.001) (Fig. 3A). Similarly, total insoluble tau and its phosphorylation status (PHF1 and CP13) were higher in 3xTg-AD compared to NonTg mice (Fig. 3B) (Genotype effect, p < 0.05, except p = 0.056 for CP13). BH4 administration and diet had no significant impact on the level of tau in 3xTg-AD mice, except for the levels of Ser202/Thr205 (AT8) phosphorylation significantly decreased by BH4 in NonTg animals (p < 0.05) (Fig. 3A). Western blots are shown in Supplementary Figure 1.
Fig.3
BH4 administration does not affect tau pathology. Hippocampal soluble (A) and insoluble (B) tau phosphorylation evaluated by western immunoblotting, confirming higher levels in 3xTg-AD compared to NonTg mice. Values are expressed as mean±SEM with N = 10–15/group. Statistical analyses: *p < 0.05, **p < 0.01, ***p < 0.001, Genotype effect (Multimodal ANOVA).
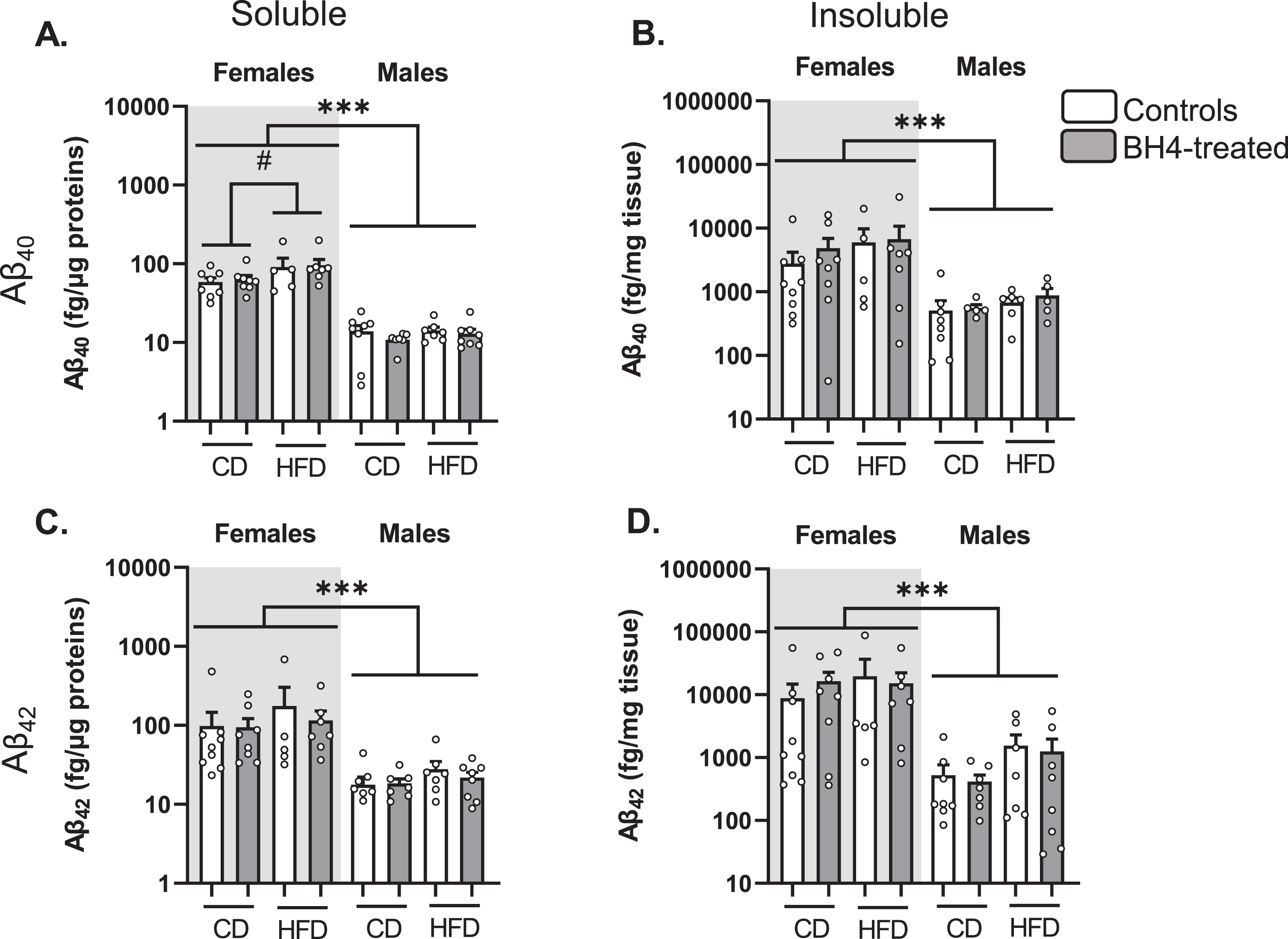
We next measured soluble and insoluble Aβ40 and Aβ42 levels in the parieto-temporal cortex of 3xTg-AD mice (Fig. 4). As previously described [55], we observed that females exhibited higher levels of Aβ40 (Fig. 4A, B) and Aβ42 (Fig. 4C, D) peptides than males (Sex effect, p < 0.001). HFD exposure significantly increased the levels of soluble Aβ40 in 3xTg-AD mice (p = 0.02). However, BH4 administration did not change levels of soluble or insoluble Aβ40 and Aβ42 peptides, nor insoluble/soluble ratios in the cortex of 3xTg-AD males and females (data not shown, see Supplementary Table 3 for statistical comparisons).
Fig.4
BH4 administration does not affect Aβ peptide levels in 3xTg-AD mice. Quantification of Aβ40 (A, B) and Aβ42 (C, D) peptides by ELISA in soluble and insoluble fractions of parieto-temporal cortex from 3xTg-AD mice. Females and males are represented separately on a logarithmic scale because of higher amyloid pathology in females. Values are expressed as mean±SEM with N = 5–8/group. Statistical analyses: ***p < 0.001, Sex effect (Multimodal ANOVA); #p < 0.05, HFD effect in females (Multimodal ANOVA).
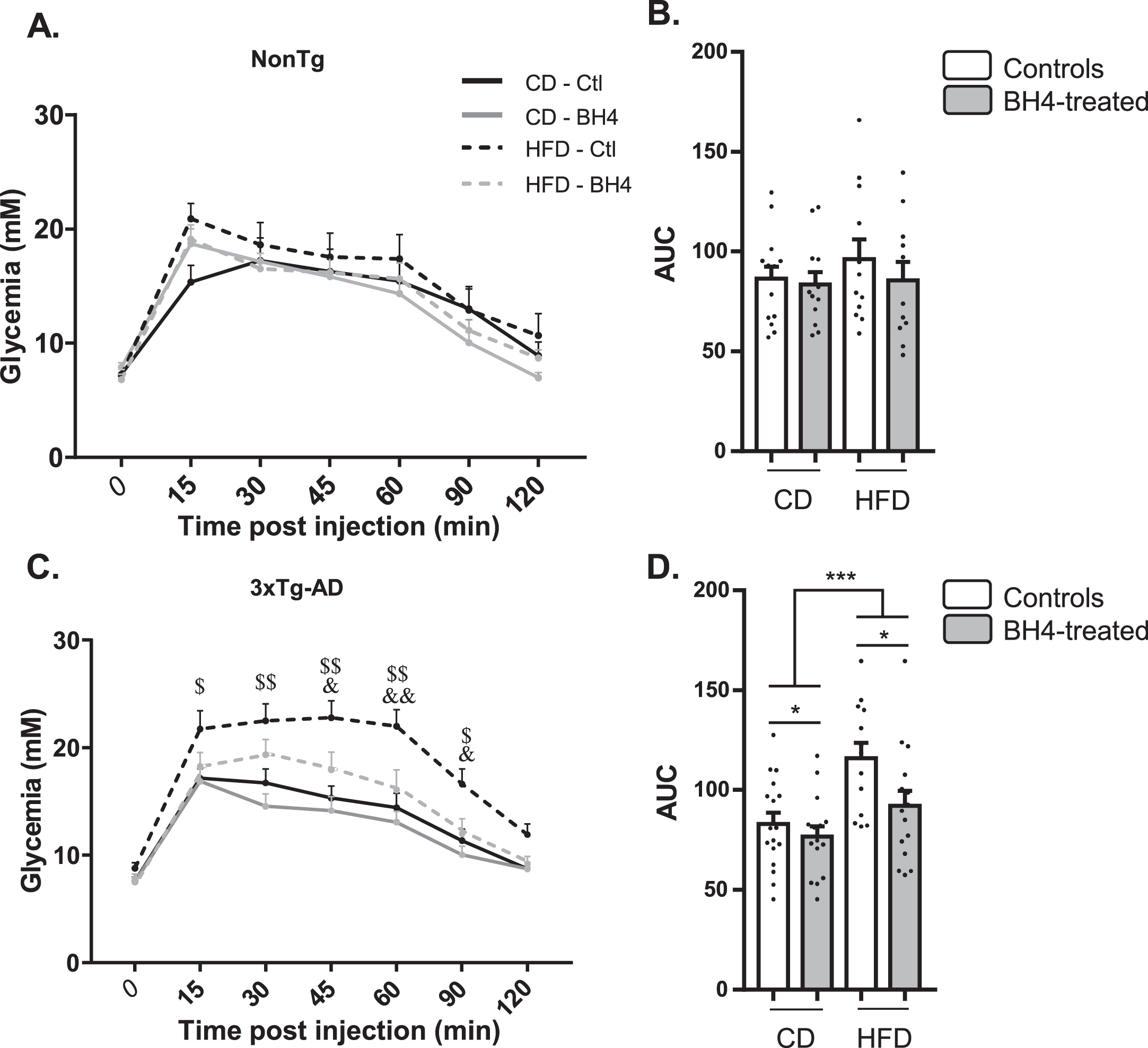
BH4 administration improves glucose tolerance in 3xTg-AD mice
We previously evidenced glucose intolerance in the 3xTg-AD mouse model, exacerbated by HFD [55]. To assess whether the beneficial effect of BH4 administration on memory could be associated to peripheral metabolic improvement, we performed glucose tolerance tests (Fig. 5). In NonTg mice, HFD and BH4 administration had no effect on the evolution of glycemia after glucose injection (Fig. 5A) nor on the AUC (Fig. 5B). However, results showed an HFD-induced failure in response to glucose injection in the 3xTg-AD mice (p < 0.001 RM ANOVA), which was totally restored 45 min after glucose injection in the group treated with BH4 (p < 0.05, RM ANOVA) (Fig. 5C, D).
Fig.5
BH4 administration improves glucose tolerance in 3xTg-AD mice. Glucose tolerance in response to the intraperitoneal administration of glucose (i.p. 1 g/kg) in fasted NonTg (A, B) and 3xTg-AD mice (C, D). B–D) Area under the curves (AUC) from t = 0 to t = 120 min is represented for all groups. Values shown are glycemia (mM) or AUC (mM/min), expressed as mean±SEM with N = 11–17/group. Statistical analyses: (C) $p < 0.05, $$p < 0.01 for each time point, significantly different between CD and HFD in Ctl mice; &p < 0.05, &&p < 0.01 for each time point, significantly different between BH4 treated and Ctl mice under HFD; (D) *p < 0.05 Main BH4 Treatment effect; ***p < 0.001 Main Diet effect (Multimodal ANOVA).
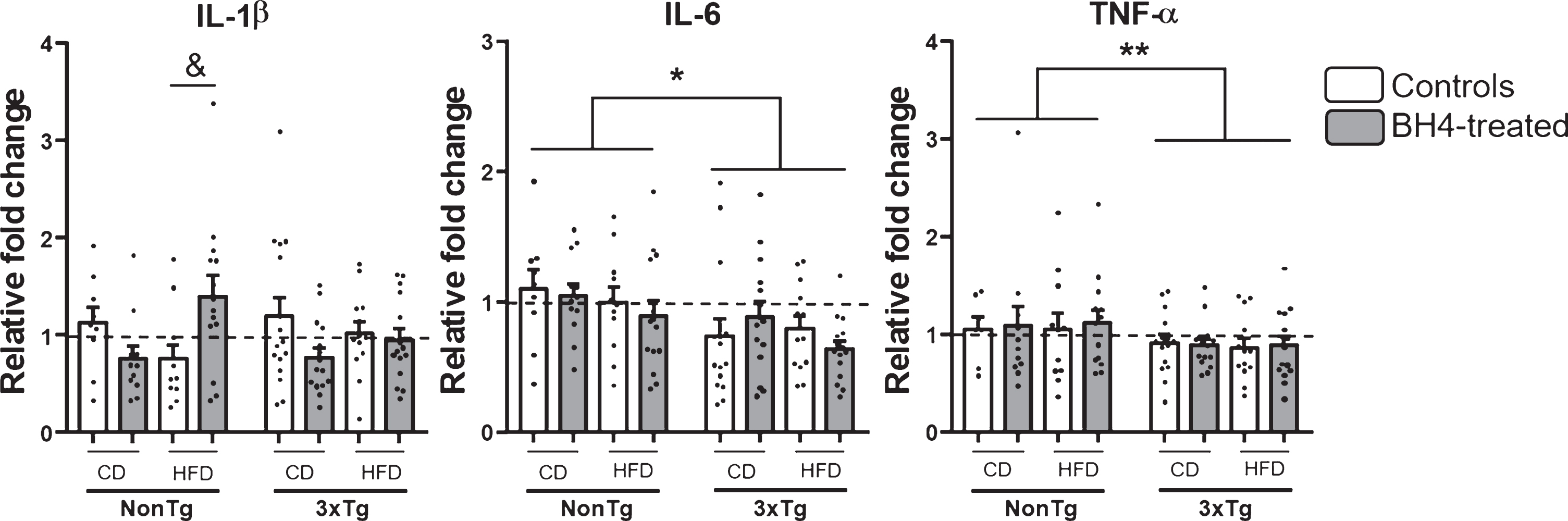
BH4 administration modulates cortical IL-1β cytokine expression according to the diet
Given the importance of neuroinflammation in AD etiopathology [5] and because BH4 was shown to exhibit anti-inflammatory properties [76], we next investigated the gene expression of major pro-inflammatory cytokines in the parieto-temporal cortex of mice (Fig. 6). Results showed a significant reduced expression of IL-6 (Genotype effect, p < 0.05) and TNF-α (Genotype effect, p < 0.01) mRNA in 3xTg-AD mice, compared to NonTg. The exposition to HFD had no effect on mRNA expression of the 3 proinflammatory cytokines studied. BH4 administration only induced a significant increase in mRNA expression of IL-1β in NonTg mice fed the HFD (Kruskal Wallis χ2 = 0.05, Wilcoxon, p = 0.02). No significant change of others pro-inflammatory markers such as the protein level measured by western blotting in the hippocampus of cyclooxygenase 2 (COX2), glial fibrillary acidic protein (GFAP), and superoxide dismutase (SOD1) were noted (data not shown).
Fig.6
Pro-inflammatory cytokines mRNA expression. Genic expression of IL1-β, IL-6, and TNF-α were measured by qRT-PCR in the parieto-temporal cortex of 3xTg-AD and NonTg mice. Values are expressed as relative fold changes using the comparative threshold method. Values are expressed as mean±SEM with N = 7–17/group. Statistical analyses: *p < 0.05, **p < 0.01 Main Genotype effect (Multimodal ANOVA); &p < 0.05 significantly different between controls and BH4-treated and HFD in NonTg HFD mice (Wilcoxon).
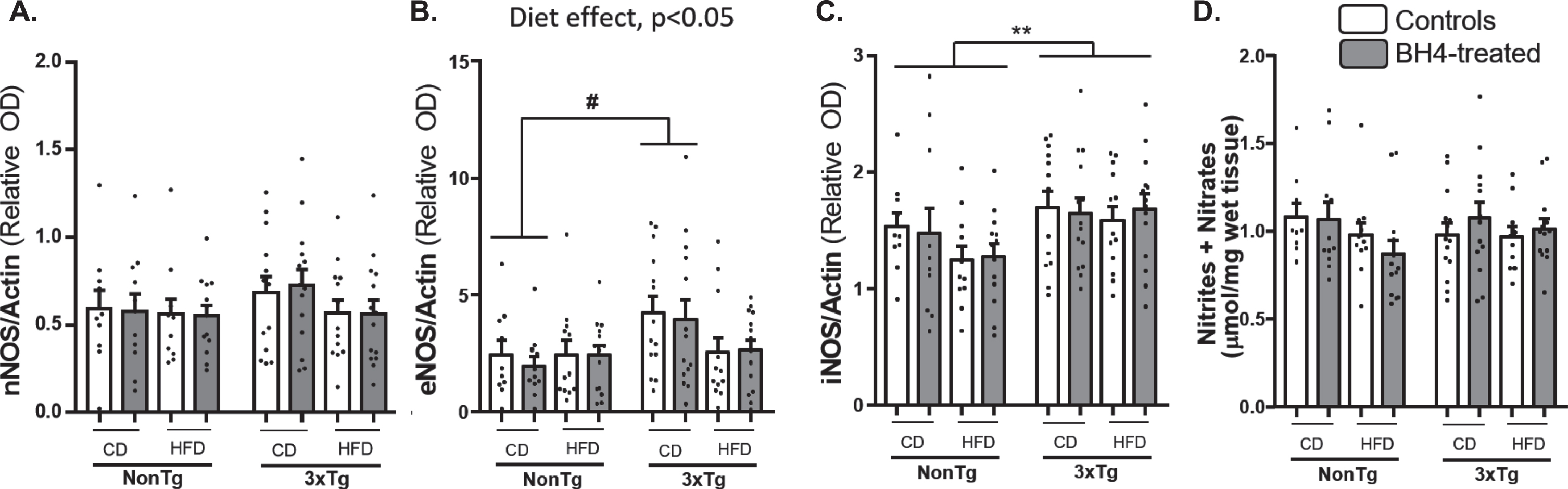
Effects of BH4 administration on NO and monoamines levels
BH4 is the mandatory cofactor of nitric oxide synthases (NOS) for the synthesis of NO which is a key modulator of vascularization but also involved in inflammatory responses. Thus, we evaluated the effect of BH4 administration on NOSs protein levels in hippocampus and on NO contents, representing as the total nitrite and nitrate level in parieto-temporal cortex (Fig. 7). Whereas neuronal NO synthase (nNOS) protein expression (Fig. 7A) and nitrite + nitrate levels (Fig. 7D) were equivalent in all experimental groups, the multivariate ANOVA revealed a significant Diet effect (p = 0.029) and a significant Diet x Genotype interaction (p = 0.041) on eNOS expression (Fig. 7B), with an increased expression in 3xTg-AD compared to NonTg mice, under CD. Moreover, results revealed a significant increase in inducible NO synthase (iNOS) protein (Fig. 7C) expression in the 3xTg-AD mice, as compared to NonTg mice (Genotype effect, p = 0.005). No significant effect of the BH4 administration was found on the different measures. Corresponding Western blots are shown in Supplementary Figure 2.
Fig.7
NOSs expression and total nitrites+nitrates. nNOS (A), eNOS (B), and iNOS (C) proteins content were measured by Western Blotting in hippocampus. Total nitrites and nitrates (D) were measured in parieto-temporal cortex of 3xTg-AD and NonTg mice and expressed as μmol/mg of wet tissue. Values are expressed as mean±SEM with N = 10–14/group. Statistical analyses: #p < 0.05 (Multimodal ANOVA, interaction Genotype x Diet); **p < 0.01, Genotype effect (Multimodal ANOVA).
BH4 is essential for monoamine synthesis, acting as a cofactor for phenylalanine hydroxylase (PheOH), tyrosine hydroxylase (TH), and tryptophane hydroxylase (TRP-OH) [20]. Thus, we measured the total amount of DA, 5-HT, and their main metabolites in the striatum of mice (Table 1). DA and 5-HT levels were not affected by genotype, diet or BH4-administration, whereas metabolites of DA (DOPAC) and 5-HT (5-HIAA), were significantly higher with HFD (Diet effect, p < 0.05). In addition, levels of the DA metabolite HVA were significantly higher in 3xTg-AD mice versus NonTg (Genotype effect, p < 0.05). Intriguingly, BH4 administration significantly increased the turnover of 5-HT, as illustrated by the increase in 5-HIAA levels and increased 5-HIAA/5-HT ratio (p < 0.05), specifically in NonTg mice fed the HFD, an effect not present in 3xTg-AD mice. Moreover, BH4 administration also significantly increased the [DOPAC+HVA]/DA ratio (p < 0.01), suggesting a higher turnover of DA.
DISCUSSION
BH4 is the essential cofactor for NO and monoamine synthesis. Given its potential involvement in key AD-associated defects, such as neuroinflammation, oxidative stress, and metabolic syndrome, we investigated the effects of BH4 administration in the 3xTg-AD mouse model of AD. We show that BH4 treatment reversed object recognition deficits in 3xTg-AD mice. This improvement was not accompanied with changes in the two classical neuropathological markers of AD, but with an improved glucose tolerance in HFD-fed 3xTg-AD mice.
BH4 administration leads to an improvement in memory deficits in 3xTg-AD mice
Recognition memory is known to be affected in patients in the earliest phase of AD [77]. The NOR test, based on the innate behavior of mice to explore novelty, is routinely used for the evaluation of memory deficits in AD mouse models [78] and has repeatedly been shown to distinguish 3xTg-AD mice from controls, from 12 months of age [42, 55, 69]. Here, we observed that a subchronic BH4 administration (15 mg/kg/day for 10 day) reversed the recognition memory deficits observed in the 3xTg-AD mouse model. This effect was present in all 3xTg-AD mice, either fed a control or HFD. NonTg mice performed well at the test, so these conditions do not allow exploring the potential beneficial effect of BH4 on memory in cognitively normal mice. Importantly, despite more locomotor activity recorded in 3xTg-AD mice, contrasting with previous reports [42, 79], locomotor behavior was not affected by BH4 administration indicating that the effect of BH4 on NOR test was specific to a memory improvement and not linked to a motor effect. It has been recently shown that BH4 improves memory acquisition, consolidation, and hippocampal plasticity in mice. More specifically, intra-cerebroventricular administration of BH4 enhances hippocampal long-term potentiation and glutamatergic transmission through an increase in NO activity [80], which participates to memory process and NOR performance [81]. Furthermore, BH4 supplementation in individuals with phenylketonuria (a genetic defect of phenylalanine hydroxylase) improves working memory and neuronal activity in the prefrontal cortex [82]. Then, consistent with previous data, the present work provides evidence to consider BH4 as a potential treatment for memory deficits observed in AD.
BH4 administration did not affect amyloid and tau pathologies
3xTg-AD mice develop the two main hallmarks of AD, amyloid plaques and tangles, as well as memory impairments [41, 42, 63, 79]. As previously described, accumulation of Aβ peptides was higher in females although cognitive impairment was similar between genders [42, 63, 79, 83, 84]. Although BH4 administration partly corrected memory deficits, it had no impact on cerebral Aβ burden or on total or phosphorylated tau. However, Aβ and tau modulation is not a prerequisite to cognitive and memory enhancement in mouse models. Indeed, numerous studies show cognitive improvement without changes in levels of amyloid and tau, particularly in 3xTg-AD mice [42, 85, 86] or other AD models [87, 88]. Reciprocally, other studies report cognitive deterioration independently of an aggravation of amyloid-β or tau pathologies [47, 62] and amyloid immunotherapies often fail to ameliorate cognition despite an amelioration of amyloid clearance and plaques [89]. In addition, interpretation of the present data should be limited to the study paradigm selected. Higher doses or a longer duration of BH4 administration could have exerted a more important impact on Aβ and tau pathologies. Similarly, using models with higher brain levels of Aβ deposits and tangles could also provide greater sensitivity to detect changes in AD markers. For example, several APP/PS1 transgenic models display much aggressive accumulation of Aβ plaques than the 3xTg-AD model and could be better suited to detect subtle change in Aβ pathologies. In any case, the present data confirms that cognitive improvement can occur irrespectively of changes in Aβ and tau pathologies in a genetic model of cognitive impairment.
Neuroinflammation status and brain monoamine levels in 3xTg-AD mice
BH4, as a cofactor, is required for the activity of NOSs, TH, and TRP-OH [20]. Thus, we assessed the status of these pathways throughout brain measures of protein expression for eNOS, iNOS, nNOS, NO, and monoamine levels in 3xTg-AD and NonTg mice under both diets. We also explored the effect of BH4 administration on these parameters. We showed that eNOS and iNOS protein expression was increased in the hippocampus of 3xTg-AD mice, whereas nNOS expression and NO levels were unchanged, independently of the diet. Higher expression of iNOS and eNOS have been described in AD mouse models and in the brains of AD patients, in particular in the vicinity of amyloid plaques [90, 91]. The increased nNOS levels and NO activity in AD brains have been postulated to initially serve a neuroprotective role as suggested by the selectively spared NOS-positive neurons in AD. However, sustained increase in NO may have deleterious effects, including oxidative stress, protein misfolding, mitochondrial stress and fragmentation, loss of synaptic function, and apoptosis [90].
The hypothesis of an increase in eNOS activity as a compensatory mechanism for NO which production is reduced in AD condition because of decrease in BH4 bioavailability is supported by a few studies in mouse transgenic models of AD [90, 92]. Santhanam et al. [34] observed a decreased BH4 level and NO production in isolated microvessels of the Tg2576 mouse model of AD, whereas these impairments are reversed by a ten-day BH4 treatment. BH4 has been shown in several studies to increase cerebral blood flow [93, 94]. In accordance with the impairment of cerebral blood flow and endothelial NO signaling dysfunctions previously described in 3xTg-AD mice [90], a beneficial effect of BH4 supplementation on brain vascularization could be involved in the memory improvement we observed in transgenic treated mice.
Neuroinflammation has been frequently observed to accompany AD neuropathology [5, 6, 89]. Our results showed that 3xTg-AD mice expressed lower TNF-α and IL-6 mRNA transcripts compared to NonTg mice. Previous studies reported an increase in pro-inflammatory IL-5 and IL-12 cerebral protein levels and an enhanced susceptibility to acute infection in 3xTg-AD mice [95]. Moreover, IL-1β has been shown to be involved in Aβ production and aggregation in mouse models of AD [96, 97], and genetic polymorphism of IL-1β is associated to a higher risk of developing AD [98]. Transient perturbations of pro-inflammatory cytokines expression are also observed in 6-month-old 3xTg-AD mice but not at 12 months [99]. The present observations are different from reports of increased cytokines in serum or plasma from AD patients [89, 100]. However, previous studies with transgenic mice modeling AD neuropathology (APP/PS1 or 3xTg-AD), at around the same age (13 and 14 months), showed equal or lower levels of IL-1β, IL-6, and TNF-α in serum or plasma compared to nontransgenic controls [45, 64, 101]. Altogether, these results suggest that 3xTg-AD mice do not express a classical neuroinflammation, with a global reduced expression of TNF-α and IL-6 but rather inflammatory dysfunctions that differ according to the age they are observed, but independent of the diet or BH4 administration.
Impairment of monoaminergic system in AD was shown in humans both in vivo and postmortem. Reductions of 5-HT or DA, along with their metabolites and receptors, have been reported in AD postmortem brain tissue [4]. In the 3xTg-AD model, abnormal 5-HT fiber sprouting in hippocampus has been described [102]. Here, we did not observe a significant difference in DA or 5-HT levels in striata of 3xTg-AD mice, although higher concentrations of DA metabolites HVA and DOPAC were detected in transgenic animals. BH4 administration induced a slight but significant increase of [DOPAC+HVA]/DA and 5-HIAA/5-HT ratios, common indexes of monoamine turnover, which could stem from a BH4-induced activation of monoamine oxidase. This effect of BH4 was mostly present in HFD-fed NonTg animals, suggesting a blunting effect of 3xTg-AD transgenes. The impact of peripheral administration of BH4 on brain DA and 5-HT contents is the subject of contrasting data in the literature. Some studies reported an increase in DA and 5-HT levels [103–106], while other detect no changes [107] following BH4 administration. Homma et al. [66] observed an increase in DA and 5-HT cerebral contents in hph-1 mice, a mouse model of peripheral and central BH4 and monoamine deficiency, after a 7-day treatment with BH4 at the dose of 50 mg/kg. Recently, we showed that acute administration of BH4 enhanced synaptic DA release in the ventral striatum, in association to a facilitated performance in a motivational task [39]. BH4 was also found to cross the blood-brain barrier with an uptake coefficient of 0.08μl.g–1.s–1 [19, 39]. The present data do not provide clear evidence of a sustained modulation of monoamine levels after chronic BH4 administration, beside a slight acceleration of DA and 5-HT metabolism.
Limited effect of the HFD
HFD had no significant impact on memory performance in 3xTg-AD mice, contrasting with previous work [44, 45, 55, 58, 108]. This may be due to a threshold or floor effect, as 3xTg-AD mice on the control diet are already cognitively impaired. Indeed, old NonTg and 3xTg-AD mice already failed to identify the new object.
Unexpectedly, the HFD did not have significant effect on markers of AD neuropathologies (expect on soluble Aβ40 in females), contrasting with previous reports in 3xTg-AD mice [55]. While HFD has been more rarely shown to affect tau levels [43, 47, 52, 53, 109, 110], many publications show an aggravating effect of a HFD on brain Aβ load in 3xTg-AD mice [43, 46, 55] and others AD model [52, 53, 58, 108, 111]. Here, the HFD increased soluble Aβ40 in females, while changes in Aβ42 remained non-significant. A simple explanation comes from the high variability of both markers within groups, precluding the detection of significant difference. Another is the loss of statistical power due to the separation of males versus females that was necessary before comparing Aβ concentrations.
When the metabolic status matters
Diabetes and obesity are recognized risk factors for AD [9, 112], and the interaction between the two comorbidities can be investigated in animal models using controlled diets [55]. The genetic induction of AD-like pathology in the brain has been shown to lead to peripheral glucose intolerance [63], aggravated by HFD [47, 55]. Here, we confirmed the vulnerability of 3xTg-AD mice to HFD-induced metabolic disturbances compared to NonTg mice, as they developed more prominent peripheral glucose intolerance [55]. However, quite interestingly, the glucose intolerance induced by this synergy of AD transgenes and HFD, was completely reversed by BH4 administration. This was not related to a reduction in calorie intake in BH4-treated animals of either diet. This beneficial effect of BH4 on metabolism is consistent with results from previous clinical studies and studies in mouse models of metabolic disturbances [36, 37, 113]. In particular, BH4 treatment has been reported to improve glucose tolerance and insulin resistance by suppressing hepatic gluconeogenesis [36].
The present data suggest that a peripheral metabolic regulation following BH4 administration could be involved in memory improvement observed in 3xTg-AD mice fed the HFD, with more limited central effects on classical AD pathology. This would indicate that BH4 treatment may be more beneficial in individuals with an impaired metabolic state, such as the elderly or obese people, in which alteration of BH4 pathway has been described [114, 115]. Thus, clinical studies with BH4 should consider the metabolic status of volunteer patients.
Other clinical utility of BH4
The present data could be relevant to other prevalent diseases [19]. Obviously, the observed effect of glucose tolerance support previous suggestion of an application for biopterins in metabolic diseases [36–38]. Because BH4 is strongly associated with the neurotransmission of dopamine and serotonin, neuropsychiatric diseases associated with alterations in monoamine function such as schizophrenia [116, 117] or depression [118–121] or autism [122, 123] could benefit from a better understanding on the impact of BH4 on behavior. In autism, some studies have shown an improvement of symptoms related to cognition, communication, or hyperactivity [122, 124–126] (review in [123]). Additional studies in animal models and in large patient cohorts are needed to fully understand BH4 mechanisms as well as it potential as a treatment in CNS diseases.
CONCLUSION
Altogether, the results of the present study indicate that the administration of BH4 for ten days attenuates memory dysfunction in the 3xTg-AD model of AD. The principal correlate observed was an improvement of metabolic determinants (i.e., glucose response), with no significant change in neuropathological endpoints. Given the positive effects of BH4 treatment on recognition memory and recent multiple failures of tau and amyloid-β targeted drugs in clinical trials, the present results suggest BH4 may represent a novel promising multi-target treatment with a favorable safety profile.
ACKNOWLEDGMENTS
This study was supported by INRAe, University of Bordeaux “Investissements d’avenir Initiative d’excellence” (Idex, ANR-10-IDEX-03-02), internal support to the International Associated Laboratory (LIA OptiNutriBrain), the Canadian Institutes of Health Research (MOP 102532), the Quebec Network for Research on Aging, the Alzheimer Society of Canada (#1502), and the Canadian Foundation for Innovation (#34480). M.T. was funded by a doctoral scholarship from the Alzheimer Society of Canada. F.C. is a Fonds de Recherche du Québec - Santé (FRQ-S) scholar (#253895). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/20-0637r1).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-200637.
REFERENCES
[1] | Tremblay C , Pilote M , Phivilay A , Emond V , Bennett DA , Calon F ((2007) ) Biochemical characterization of Aβ and tau pathologies in mild cognitive impairment and Alzheimer’s disease. J Alzheimers Dis 12: , 377–390. |
[2] | Tremblay C , Francois A , Delay C , Freland L , Vandal M , Bennett DA , Calon F ((2017) ) Association of neuropathological markers in the parietal cortex with antemortem cognitive function in persons with mild cognitive impairment and Alzheimer disease. J Neuropathol Exp Neurol 76: , 70–88. |
[3] | Krashia P , Nobili A , D’Amelio M ((2019) ) Unifying hypothesis of dopamine neuron loss in neurodegenerative diseases: Focusing on Alzheimer’s disease. Front Mol Neurosci 12: , 123. |
[4] | Šimić G , Babić Leko M , Wray S , Harrington CR , Delalle I , Jovanov-Milošević N , Bažadona D , Buée L , de Silva R , Di Giovanni G , Wischik CM , Hof PR ((2017) ) Monoaminergic neuropathology in Alzheimer’s disease. Prog Neurobiol 151: , 101–138. |
[5] | Heneka MT , Carson MJ , El Khoury J , Landreth GE , Brosseron F , Feinstein DL , Jacobs AH , Wyss-Coray T , Vitorica J , Ransohoff RM , Herrup K , Frautschy SA , Finsen B , Brown GC , Verkhratsky A , Yamanaka K , Koistinaho J , Latz E , Halle A , Petzold GC , Town T , Morgan D , Shinohara ML , Perry VH , Holmes C , Bazan NG , Brooks DJ , Hunot S , Joseph B , Deigendesch N , Garaschuk O , Boddeke E , Dinarello CA , Breitner JC , Cole GM , Golenbock DT , Kummer MP ((2015) ) Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14: , 388–405. |
[6] | Heppner FL , Ransohoff RM , Becher B ((2015) ) Immune attack: The role of inflammation in Alzheimer disease. Nat Rev Neurosci 16: , 358–372. |
[7] | Sweeney MD , Kisler K , Montagne A , Toga AW , Zlokovic BV ((2018) ) The role of brain vasculature in neurodegenerative disorders. Nat Neurosci 21: , 1318–1331. |
[8] | Ferreira ME , de Vasconcelos AS , da Costa Vilhena T , da Silva TL , da Silva Barbosa A , Gomes AR , Dolabela MF , Percário S ((2015) ) Oxidative stress in Alzheimer’s disease: Should we keep trying antioxidant therapies? Cell Mol Neurobiol 35: , 595–614. |
[9] | Kivipelto M , Mangialasche F , Ngandu T ((2018) ) Lifestyle interventions to prevent cognitive impairment, dementia and Alzheimer disease. Nat Rev Neurol 14: , 653–666. |
[10] | Barnes DE , Yaffe K ((2011) ) The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol 10: , 819–828. |
[11] | Verdile G , Keane KN , Cruzat VF , Medic S , Sabale M , Rowles J , Wijesekara N , Martins RN , Fraser PE , Newsholme P ((2015) ) Inflammation and oxidative stress: The molecular connectivity between insulin resistance, obesity, and Alzheimer’s disease. Mediators Inflamm 2015: , 105828. |
[12] | De Felice FG , Ferreira ST ((2014) ) Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer disease. Diabetes 63: , 2262–2272. |
[13] | Martorana A , Koch G ((2014) ) “Is dopamine involved in Alzheimer’s disease?” Front Aging Neurosci 6: , 252. |
[14] | Blin J , Baron JC , Dubois B , Crouzel C , Fiorelli M , Attar-Lévy D , Pillon B , Fournier D , Vidailhet M , Agid Y ((1993) ) Loss of brain 5-HT2 receptors in Alzheimer’s disease. In vivo assessment with positron emission tomography and [18F]setoperone. Brain 116: (Pt 3), 497–510. |
[15] | Chakraborty S , Lennon JC , Malkaram SA , Zeng Y , Fisher DW , Dong H ((2019) ) Serotonergic system, cognition, and BPSD in Alzheimer’s disease. Neurosci Lett 704: , 36–44. |
[16] | Bartus RT ((2000) ) On neurodegenerative diseases, models, and treatment strategies: Lessons learned and lessons forgotten a generation following the cholinergic hypothesis. Exp Neurol 163: , 495–529. |
[17] | Hughes RE , Nikolic K , Ramsay RR ((2016) ) One for all? Hitting multiple Alzheimer’s disease targets with one drug. Front Neurosci 10: , 177. |
[18] | Carreiras MC , Mendes E , Perry MJ , Francisco AP , Marco-Contelles J ((2013) ) The multifactorial nature of Alzheimer’s disease for developing potential therapeutics. Curr Top Med Chem 13: , 1745–1770. |
[19] | Fanet H , Capuron L , Castanon N , Calon F , Vancassel S ((2020) ) Tetrahydrobioterin (BH4) pathway: From metabolism to neuropsychiatry. Curr Neuropharmacol, doi: 10.2174/1570159X18666200729103529 |
[20] | Werner ER , Blau N , Thöny B ((2011) )Tetrahydrobiopterin: Biochemistry andathophysiology. Biochem J 438: , 397–414 . |
[21] | Channon KM ((2012) ) Tetrahydrobioptern: A vascular redox target to imrove endothelial function. Curr Vasc Pharmacol 10: , 705–708 . |
[22] | Chen DD , Chen LY , Xie JB , Shu C , Yang T , Zhou S , Yuan H , Chen AF ((2014) ) Tetrahydrobiopterin regulation of eNOS redox function. Curr Pharm Des 20: , 3554–3562. |
[23] | Heales SJ , Blair JA , Meinschad C , Ziegler I ((1988) ) Inhibition of monocyte luminol-dependent chemiluminescence by tetrahydrobiopterin, and the free radical oxidation of tetrahydrobiopterin, dihydrobiopterin and dihydroneopterin. Cell Biochem Funct 6: , 191–195. |
[24] | Ichinose H , Nomura T , Sumi-Ichinose C ((2008) ) Metabolism of tetrahydrobiopterin: Its relevance in monoaminergic neurons and neurological disorders. Chem Rec 8: , 378–385 . |
[25] | Miwa S , Watanabe Y , Hayaishi O ((1985) ) 6R-L-erythro-5,6,7,8-tetrahydrobiopterin as a regulator of dopamine and serotonin biosynthesis in the rat brain. Arch Biochem Biophys 239: , 234–241. |
[26] | Hyland K ((2007) ) Inherited disorders affecting dopamine and serotonin: Critical neurotransmitters derived from aromatic amino acids J Nutr 137: , 1568S–1572S; discussion 1573S-1575S. |
[27] | Foxton RH , Land JM , Heales SJ ((2007) ) Tetrahydrobiopterin availability in Parkinson’s and Alzheimer’s disease; potential pathogenic mechanisms. Neurochem Res 32: , 751–756. |
[28] | Aziz AA , Leeming RJ , Blair JA ((1983) ) Tetrahydrobiopterin metabolism in senile dementia of Alzheimer type. J Neurol Neurosurg Psychiatry 46: , 410–413. |
[29] | Williams AC , Levine RA , Chase TN , Lovenberg W , Calne DB ((1980) ) CFS hydroxylase cofactor levels in some neurological diseases. J Neurol Neurosurg Psychiatry 43: , 735–738. |
[30] | Barford PA , Blair JA , Eggar C , Hamon C , Morar C , Whitburn SB ((1984) ) Tetrahydrobiopterin metabolism in the temporal lobe of patients dying with senile dementia of Alzheimer type. J Neurol Neurosurg Psychiatry 47: , 736–738. |
[31] | Hull M , Pasinetti GM , Aisen PS ((2000) ) Elevated plasma neopterin levels in Alzheimer disease. Alzheimer Dis Assoc Disord 14: , 228–230. |
[32] | Blasko I , Knaus G , Weiss E , Kemmler G , Winkler C , Falkensammer G , Griesmacher A , Würzner R , Marksteiner J , Fuchs D ((2007) ) Cognitive deterioration in Alzheimer’s disease is accompanied by increase of plasma neopterin. J Psychiatr Res 41: , 694–701. |
[33] | Parker DC , Mielke MM , Yu Q , Rosenberg PB , Jain A , Lyketsos CG , Fedarko NS , Oh ES ((2013) ) Plasma neopterin level as a marker of peripheral immune activation in amnestic mild cognitive impairment and Alzheimer’s disease. Int J Geriatr Psychiatry 28: , 149–154. |
[34] | Santhanam AV , d’Uscio LV , He T , Das P , Younkin SG , Katusic ZS ((2015) ) Uncoupling of endothelial nitric oxide synthase in cerebral vasculature of Tg2576 mice. J Neurochem 134: , 1129–1138. |
[35] | Schmidt TS , McNeill E , Douglas G , Crabtree MJ , Hale AB , Khoo J , O’Neill CA , Cheng A , Channon KM , Alp NJ ((2010) ) Tetrahydrobiopterin supplementation reduces atherosclerosis and vascular inflammation in apolipoprotein E-knockout mice. Clin Sci (Lond) 119: , 131–142. |
[36] | Abudukadier A , Fujita Y , Obara A , Ohashi A , Fukushima T , Sato Y , Ogura M , Nakamura Y , Fujimoto S , Hosokawa M , Hasegawa H , Inagaki N ((2013) ) Tetrahydrobiopterin has a glucose-lowering effect by suppressing hepatic gluconeogenesis in an endothelial nitric oxide synthase-dependent manner in diabetic mice. Diabetes 62: , 3033–3043. |
[37] | Nyström T , Nygren A , Sjöholm A ((2004) ) Tetrahydrobiopterin increases insulin sensitivity in patients with type 2 diabetes and coronary heart disease. Am J Physiol Endocrinol Metab 287: , E919–925. |
[38] | Oguri Y , Fujita Y , Abudukadier A , Ohashi A , Goto T , Furuya F , Obara A , Fukushima T , Matsuo N , Kim M , Hosokawa M , Kawada T , Hasegawa H , Inagaki N ((2017) ) Tetrahydrobiopterin activates brown adipose tissue and regulates systemic energy metabolism. JCI Insight 2: , e91981. |
[39] | Fanet H , Ducrocq F , Tournissac M , Oummadi A , Lo A , Bourrassa P , De Smedt-Peyrusse V , Azzougen B , Capuron L , Layé S , Moussa F , Trifilieff P , Calon F , Vancassel S ((2020) ) Tetrahydrobiopterin administration facilitates amphetamine-induced dopamine release and motivation in mice. Behav Brain Res 379: , 112348. |
[40] | LaFerla FM , Oddo S ((2005) ) Alzheimer’s disease: Aβ, tau and synaptic dysfunction. Trends Mol Med 11: , 170–176. |
[41] | Oddo S , Caccamo A , Shepherd JD , Murphy MP , Golde TE , Kayed R , Metherate R , Mattson MP , Akbari Y , LaFerla FM ((2003) ) Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Aβ and synaptic dysfunction. Neuron 39: , 409–421. |
[42] | Dal-Pan A , Dudonne S , Bourassa P , Bourdoulous M , Tremblay C , Desjardins Y , Calon F , Neurophenols c ((2017) ) Cognitive-enhancing effects of a polyphenols-rich extract from fruits without changes in neuropathology in an animal model of Alzheimer’s disease. J Alzheimers Dis 55: , 115–135. |
[43] | Julien C , Tremblay C , Phivilay A , Berthiaume L , Emond V , Julien P , Calon F ((2010) ) High-fat diet aggravates amyloid-β and tau pathologies in the 3xTg-AD mouse model. Neurobiol Aging 31: , 1516–1531. |
[44] | Sah SK , Lee C , Jang JH , Park GH ((2017) ) Effect of high-fat diet on cognitive impairment in triple-transgenic mice model of Alzheimer’s disease. Biochem Biophys Res Commun 493: , 731–736. |
[45] | Sanguinetti E , Guzzardi MA , Panetta D , Tripodi M , De Sena V , Quaglierini M , Burchielli S , Salvadori PA , Iozzo P ((2019) ) Combined effect of fatty diet and cognitive decline on brain metabolism, food intake, body weight, and counteraction by intranasal insulin therapy in 3xTg mice. Front Cell Neurosci 13: , 188. |
[46] | Barron AM , Rosario ER , Elteriefi R , Pike CJ ((2013) ) Sex-specific effects of high fat diet on indices of metabolic syndrome in 3xTg-AD mice: Implications for Alzheimer’s disease. PLoS One 8: , e78554. |
[47] | Knight EM , Martins IV , Gümüsgöz S , Allan SM , Lawrence CB ((2014) ) High-fat diet-induced memory impairment in triple-transgenic Alzheimer’s disease (3xTgAD) mice is independent of changes in amyloid and tau pathology. Neurobiol Aging 35: , 1821–1832. |
[48] | Ho L , Qin W , Pompl PN , Xiang Z , Wang J , Zhao Z , Peng Y , Cambareri G , Rocher A , Mobbs CV , Hof PR , Pasinetti GM ((2004) ) Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. FASEB J 18: , 902–904. |
[49] | Maesako M , Uemura K , Kubota M , Kuzuya A , Sasaki K , Asada M , Watanabe K , Hayashida N , Ihara M , Ito H , Shimohama S , Kihara T , Kinoshita A ((2012) ) Environmental enrichment ameliorated high-fat diet-induced Aβ deposition and memory deficit in APP transgenic mice. Neurobiol Aging 33: , 1011.e1011–1023. |
[50] | Maesako M , Uemura K , Kubota M , Kuzuya A , Sasaki K , Hayashida N , Asada-Utsugi M , Watanabe K , Uemura M , Kihara T , Takahashi R , Shimohama S , Kinoshita A ((2012) ) Exercise is more effective than diet control in preventing high fat diet-induced β-amyloid deposition and memory deficit in amyloid precursor protein transgenic mice. J Biol Chem 287: , 23024–23033. |
[51] | Herculano B , Tamura M , Ohba A , Shimatani M , Kutsuna N , Hisatsune T ((2013) ) β-alanyl-L-histidine rescues cognitive deficits caused by feeding a high fat diet in a transgenic mouse model of Alzheimer’s disease. J Alzheimers Dis 33: , 983–997. |
[52] | Ramos-Rodriguez JJ , Ortiz-Barajas O , Gamero-Carrasco C , de la Rosa PR , Infante-Garcia C , Zopeque-Garcia N , Lechuga-Sancho AM , Garcia-Alloza M ((2014) ) Prediabetes-induced vascular alterations exacerbate central pathology in APPswe/PS1dE9 mice. Psychoneuroendocrinology 48: , 123–135. |
[53] | Ettcheto M , Petrov D , Pedros I , Alva N , Carbonell T , Beas-Zarate C , Pallas M , Auladell C , Folch J , Camins A ((2016) ) Evaluation of neuropathological effects of a high-fat diet in a presymptomatic Alzheimer’s disease stage in APP/PS1 mice. J Alzheimers Dis 54: , 233–251. |
[54] | Petrov D , Pedrós I , Artiach G , Sureda FX , Barroso E , Pallás M , Casadesús G , Beas-Zarate C , Carro E , Ferrer I , Vazquez-Carrera M , Folch J , Camins A ((2015) ) High-fat diet-induced deregulation of hippocampal insulin signaling and mitochondrial homeostasis deficiencies contribute to Alzheimer disease pathology in rodents. Biochim Biophys Acta 1852: , 1687–1699. |
[55] | Vandal M , White PJ , Tremblay C , St-Amour I , Chevrier G , Emond V , Lefrançois D , Virgili J , Planel E , Giguere Y , Marette A , Calon F ((2014) ) Insulin reverses the high-fat diet-induced increase in brain aβ and improves memory in an animal model of Alzheimer disease. Diabetes 63: , 4291–4301. |
[56] | Martino Adami PV , Galeano P , Wallinger ML , Quijano C , Rabossi A , Pagano ES , Olivar N , Reyes Toso C , Cardinali D , Brusco LI , Do Carmo S , Radi R , Gevorkian G , Castaño EM , Cuello AC , Morelli L ((2017) ) Worsening of memory deficit induced by energy-dense diet in a rat model of early-Alzheimer’s disease is associated to neurotoxic Aβ species and independent of neuroinflammation. Biochim Biophys Acta Mol Basis Dis 1863: , 731–743. |
[57] | King MR , Anderson NJ , Deciu M , Guernsey LS , Cundiff M , Hajizadeh S , Jolivalt CG ((2020) ) Insulin deficiency, but not resistance, exaggerates cognitive deficits in transgenic mice expressing human amyloid and tau proteins. Reversal by Exendin-4 treatment. J Neurosci Res 98: , 2357–2369. |
[58] | Walker JM , Dixit S , Saulsberry AC , May JM , Harrison FE ((2017) ) Reversal of high fat diet-induced obesity improves glucose tolerance, inflammatory response, β-amyloid accumulation and cognitive decline in the APP/PSEN1 mouse model of Alzheimer’s disease. Neurobiol Dis 100: , 87–98. |
[59] | Wasim M , Awan FR , Khan HN , Ayesha H ((2018) ) An overview of traditional and novel therapeutic options for the management of phenylketonuria. Crit Rev Eukaryot Gene Expr 28: , 177–185. |
[60] | Belfiore R , Rodin A , Ferreira E , Velazquez R , Branca C , Caccamo A , Oddo S ((2019) ) Temporal and regional progression of Alzheimer’s disease-like pathology in 3xTg-AD mice. Aging Cell 18: , e12873. |
[61] | Vandal M , White PJ , Tournissac M , Tremblay C , St-Amour I , Drouin-Ouellet J , Bousquet M , Traversy MT , Planel E , Marette A , Calon F ((2016) ) Impaired thermoregulation and beneficial effects of thermoneutrality in the 3xTg-AD model of Alzheimer’s disease. Neurobiol Aging 43: , 47–57. |
[62] | Arsenault D , Dal-Pan A , Tremblay C , Bennett DA , Guitton MJ , De Koninck Y , Tonegawa S , Calon F ((2013) ) PAK inactivation impairs social recognition in 3xTg-AD mice without increasing brain deposition of tau and Aβ. J Neurosci 33: , 10729–10740. |
[63] | Vandal M , White PJ , Chevrier G , Tremblay C , St-Amour I , Planel E , Marette A , Calon F ((2015) ) Age-dependent impairment of glucose tolerance in the 3xTg-AD mouse model of Alzheimer’s disease. FASEB J 29: , 4273–4284. |
[64] | St-Amour I , Bosoi CR , Pare I , Ignatius Arokia Doss PM , Rangachari M , Hebert SS , Bazin R , Calon F ((2019) ) Peripheral adaptive immunity of the triple transgenic mouse model of Alzheimer’s disease. J Neuroinflammation 16: , 3. |
[65] | Mizuma H , Mizutani M , Nozaki S , Iizuka H , Tohyama H , Nishimura N , Watanabe Y , Kohashi R ((2003) ) Improvement by repeated administration of 6R-tetrahydrobiopterin of 5,7-dihydroxytryptamine-induced abnormal behaviors in immature rats. Biochem Biophys Res Commun 302: , 156–161. |
[66] | Homma D , Katoh S , Tokuoka H , Ichinose H ((2013) ) The role of tetrahydrobiopterin and catecholamines in the developmental regulation of tyrosine hydroxylase level in the brain. J Neurochem 126: , 70–81. |
[67] | Scherer T , Allegri G , Sarkissian CN , Ying M , Grisch-Chan HM , Rassi A , Winn SR , Harding CO , Martinez A , Thöny B ((2018) ) Tetrahydrobiopterin treatment reduces brain L-Phe but only partially improves serotonin in hyperphenylalaninemic ENU1/2 mice. J Inherit Metab Dis 41: , 709–718. |
[68] | Canevari L , Land JM , Clark JB , Heales SJ ((1999) ) Stimulation of the brain NO/cyclic GMP pathway by peripheral administration of tetrahydrobiopterin in the hph-1 mouse. J Neurochem 73: , 2563–2568. |
[69] | Arsenault D , Julien C , Tremblay C , Calon F ((2011) ) DHA improves cognition and prevents dysfunction of entorhinal cortex neurons in 3xTg-AD mice. PLoS One 6: , e17397. |
[70] | St-Amour I , Pare I , Tremblay C , Coulombe K , Bazin R , Calon F ((2014) ) IVIg protects the 3xTg-AD mouse model of Alzheimer’s disease from memory deficit and Aβ pathology. J Neuroinflammation 11: , 54. |
[71] | Garvock-de Montbrun T , Fertan E , Stover K , Brown RE ((2019) ) Motor deficits in 16-month-old male and female 3xTg-AD mice. Behav Brain Res 356: , 305–313. |
[72] | España J , Giménez-Llort L , Valero J , Miñano A , Rábano A , Rodriguez-Alvarez J , LaFerla FM , Saura CA ((2010) ) Intraneuronal β-amyloid accumulation in the amygdala enhances fear and anxiety in Alzheimer’s disease transgenic mice. Biol Psychiatry 67: , 513–521. |
[73] | Antunes M , Biala G ((2012) ) The novel object recognition memory: Neurobiology, test procedure, and its modifications. Cogn Process 13: , 93–110. |
[74] | Tournissac M , Bourassa P , Martinez-Cano RD , Vu TM , Hebert SS , Planel E , Calon F ((2019) ) Repeated cold exposures protect a mouse model of Alzheimer’s disease against cold-induced tau phosphorylation. Mol Metab 22: , 110–120. |
[75] | Coulombe K , Kerdiles O , Tremblay C , Emond V , Lebel M , Boulianne AS , Plourde M , Cicchetti F , Calon F ((2018) ) Impact of DHA intake in a mouse model of synucleinopathy. Exp Neurol 301: , 39–49. |
[76] | Ghisoni K , Martins Rde P , Barbeito L , Latini A ((2015) ) Neopterin as a potential cytoprotective brain molecule. J Psychiatr Res 71: , 134–139. |
[77] | Ally BA ((2012) ) Using pictures and words to understand recognition memory deterioration in amnestic mild cognitive impairment and Alzheimer’s disease: A review. Curr Neurol Neurosci Rep 12: , 687–694. |
[78] | Bengoetxea X , Rodriguez-Perdigon M , Ramirez MJ ((2015) ) Object recognition test for studying cognitive impairments in animal models of Alzheimer’s disease. Front Biosci (Schol Ed) 7: , 10–29. |
[79] | Bories C , Guitton MJ , Julien C , Tremblay C , Vandal M , Msaid M , De Koninck Y , Calon F ((2012) ) Sex-dependent alterations in social behaviour and cortical synaptic activity coincide at different ages in a model of Alzheimer’s disease. PLoS One 7: , e46111. |
[80] | Latini A , de Bortoli da Silva L , da Luz Scheffer D , Pires ACS , de Matos FJ , Nesi RT , Ghisoni K , de Paula Martins R , de Oliveira PA , Prediger RD , Ghersi M , Gabach L , Pérez MF , Rubiales-Barioglio S , Raisman-Vozari R , Mongeau R , Lanfumey L , Aguiar AS ((2018) ) Tetrahydrobiopterin improves hippocampal nitric oxide-linked long-term memory. Mol Genet Metab 125: , 104–111. |
[81] | Pitsikas N ((2015) ) The role of nitric oxide in the object recognition memory. Behav Brain Res 285: , 200–207. |
[82] | Christ SE , Moffitt AJ , Peck D , White DA ((2013) ) The effects of tetrahydrobiopterin (BH4) treatment on brain function in individuals with phenylketonuria. Neuroimage Clin 3: , 539–547. |
[83] | Wolf AB , Braden BB , Bimonte-Nelson H , Kusne Y , Young N , Engler-Chiurazzi E , Garcia AN , Walker DG , Moses GS , Tran H , LaFerla F , Lue L , Emerson Lombardo N , Valla J ((2012) ) Broad-based nutritional supplementation in 3xTg mice corrects mitochondrial function and indicates sex-specificity in response to Alzheimer’s disease intervention. J Alzheimers Dis 32: , 217–232. |
[84] | Carroll JC , Rosario ER , Kreimer S , Villamagna A , Gentzschein E , Stanczyk FZ , Pike CJ ((2010) ) Sex differences in β-amyloid accumulation in 3xTg-AD mice: Role of neonatal sex steroid hormone exposure. Brain Res 1366: , 233–245. |
[85] | Xu J , Chatterjee M , Baguley TD , Brouillette J , Kurup P , Ghosh D , Kanyo J , Zhang Y , Seyb K , Ononenyi C , Foscue E , Anderson GM , Gresack J , Cuny GD , Glicksman MA , Greengard P , Lam TT , Tautz L , Nairn AC , Ellman JA , Lombroso PJ ((2014) ) Inhibitor of the tyrosine phosphatase STEP reverses cognitive deficits in a mouse model of Alzheimer’s disease. PLoS Biol 12: , e1001923. |
[86] | Medeiros R , Castello NA , Cheng D , Kitazawa M , Baglietto-Vargas D , Green KN , Esbenshade TA , Bitner RS , Decker MW , LaFerla FM ((2014) ) α7 Nicotinic receptor agonist enhances cognition in aged 3xTg-AD mice with robust plaques and tangles. Am J Pathol 184: , 520–529. |
[87] | Zhang W , Wang PJ , Sha HY , Ni J , Li MH , Gu GJ ((2014) ) Neural stem cell transplants improve cognitive function without altering amyloid pathology in an APP/PS1 double transgenic model of Alzheimer’s disease. Mol Neurobiol 50: , 423–437. |
[88] | Choi SH , Bylykbashi E , Chatila ZK , Lee SW , Pulli B , Clemenson GD , Kim E , Rompala A , Oram MK , Asselin C , Aronson J , Zhang C , Miller SJ , Lesinski A , Chen JW , Kim DY , van Praag H , Spiegelman BM , Gage FH , Tanzi RE ((2018) ) Combined adult neurogenesis and BDNF mimic exercise effects on cognition in an Alzheimer’s mouse model. Science 361: , eaan8821. |
[89] | St-Amour I , Cicchetti F , Calon F ((2016) ) Immunotherapies in Alzheimer’s disease: Too much, too little, too late or off-target? Acta Neuropathol 131: , 481–504. |
[90] | Lourenco CF , Ledo A , Barbosa RM , Laranjinha J ((2017) ) Neurovascular uncoupling in the triple transgenic model of Alzheimer’s disease: Impaired cerebral blood flow response to neuronal-derived nitric oxide signaling. Exp Neurol 291: , 36–43. |
[91] | Dubey H , Gulati K , Ray A ((2020) ) Alzheimer’s disease: A contextual link with nitric oxide synthase. Curr Mol Med 20: , 505–515. |
[92] | Tejero J , Stuehr D ((2013) ) Tetrahydrobiopterin in nitric oxide synthase. IUBMB Life 65: , 358–365. |
[93] | Mayhan WG , Arrick DM ((2017) ) Tetrahydrobiopterin rescues impaired responses of cerebral resistance arterioles during type 1 diabetes. Diab Vasc Dis Res 14: , 33–39. |
[94] | Cherian L , Hlatky R , Robertson CS ((2004) ) Comparison of tetrahydrobiopterin and L-arginine on cerebral blood flow after controlled cortical impact injury in rats. J Neurotrauma 21: , 1196–1203. |
[95] | Montacute R , Foley K , Forman R , Else KJ , Cruickshank SM , Allan SM ((2017) ) Enhanced susceptibility of triple transgenic Alzheimer’s disease (3xTg-AD) mice to acute infection. J Neuroinflammation 14: , 50. |
[96] | Heneka MT , Kummer MP , Stutz A , Delekate A , Schwartz S , Vieira-Saecker A , Griep A , Axt D , Remus A , Tzeng TC , Gelpi E , Halle A , Korte M , Latz E , Golenbock DT ((2013) ) NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493: , 674–678. |
[97] | Wang P , Guan PP , Wang T , Yu X , Guo JJ , Wang ZY ((2014) ) Aggravation of Alzheimer’s disease due to the COX-2-mediated reciprocal regulation of IL-1β and Aβ between glial and neuron cells. Aging Cell 13: , 605–615. |
[98] | Yuan H , Xia Q , Ge P , Wu S ((2013) ) Genetic polymorphism of interleukin 1β -511C/T and susceptibility to sporadic Alzheimer’s disease: A meta-analysis. Mol Biol Rep 40: , 1827–1834. |
[99] | Guedes JR , Custódia CM , Silva RJ , de Almeida LP , Pedroso de Lima MC , Cardoso AL ((2014) ) Early miR-155 upregulation contributes to neuroinflammation in Alzheimer’s disease triple transgenic mouse model. Hum Mol Genet 23: , 6286–6301. |
[100] | Lee KS , Chung JH , Choi TK , Suh SY , Oh BH , Hong CH ((2009) ) Peripheral cytokines and chemokines in Alzheimer’s disease. Dement Geriatr Cogn Disord 28: , 281–287. |
[101] | Yang SH , Kim J , Lee MJ , Kim Y ((2015) ) Abnormalities of plasma cytokines and spleen in senile APP/PS1/Tau transgenic mouse model. Sci Rep 5: , 15703. |
[102] | Noristani HN , Meadows RS , Olabarria M , Verkhratsky A , Rodríguez JJ ((2011) ) Increased hippocampal CA1 density of serotonergic terminals in a triple transgenic mouse model of Alzheimer’s disease: An ultrastructural study. Cell Death Dis 2: , e210. |
[103] | Tsukada H , Lindner KJ , Hartvig P , Långström B ((1994) ) Effect of 6R-L-erythro-5,6,7,8-tetrahydrobiopterin on the extracellular levels of dopamine and serotonin in the rat striatum: A microdialysis study with tyrosine or tryptophan infusion. Brain Res 635: , 59–67. |
[104] | Nagatsu T , Nakahara D , Kobayashi K , Morita S , Sawada H , Mizuguchi T , Kiuchi K ((1994) ) Peripherally administered (6R)-tetrahydrobiopterin increases in vivo tyrosine hydroxylase activity in the striatum measured by microdialysis both in normal mice and in transgenic mice carrying human tyrosine hydroxylase. Neurosci Lett 182: , 44–46. |
[105] | Sumi-Ichinose C , Urano F , Kuroda R , Ohye T , Kojima M , Tazawa M , Shiraishi H , Hagino Y , Nagatsu T , Nomura T , Ichinose H ((2001) ) Catecholamines and serotonin are differently regulated by tetrahydrobiopterin. A study from 6-pyruvoyltetrahydropterin synthase knockout mice. J Biol Chem 276: , 41150–41160. |
[106] | Winn SR , Scherer T , Thöny B , Harding CO ((2016) ) High dose sapropterin dihydrochloride therapy improves monoamine neurotransmitter turnover in murine phenylketonuria (PKU). Mol Genet Metab 117: , 5–11. |
[107] | Levine RA , Zoephel GP , Niederwieser A , Curtius HC ((1987) ) Entrance of tetrahydropterin derivatives in brain after peripheral administration: Effect on biogenic amine metabolism. J Pharmacol Exp Ther 242: , 514–522. |
[108] | Theriault P , ElAli A , Rivest S ((2016) ) High fat diet exacerbates Alzheimer’s disease-related pathology in APPswe/PS1 mice. Oncotarget 7: , 67808–67827. |
[109] | Gratuze M , Julien J , Morin F , Calon F , Hebert SS , Marette A , Planel E ((2016) ) High-fat, high-sugar, and high-cholesterol consumption does not impact tau pathogenesis in a mouse model of Alzheimer’s disease-like tau pathology. Neurobiol Aging 47: , 71–73. |
[110] | Leboucher A , Laurent C , Fernandez-Gomez F-J , Burnouf S , Troquier L , Eddarkaoui S , Demeyer D , Caillierez R , Zommer N , Vallez E , Bantubungi K , Breton C , Pigny P , Buée-Scherrer V , Staels B , Hamdane M , Tailleux A , Buée L , Blum D ((2013) ) Detrimental effects of diet-induced obesity on τ pathology are independent of insulin resistance in τ transgenic mice. Diabetes 62: , 1681–1688. |
[111] | Shie FS , Jin LW , Cook DG , Leverenz JB , LeBoeuf RC ((2002) ) Diet-induced hypercholesterolemia enhances brain Aβ accumulation in transgenic mice. Neuroreport 13: , 455–459. |
[112] | Norton S , Matthews FE , Barnes DE , Yaffe K , Brayne C ((2014) ) Potential for primary prevention of Alzheimer’s disease: An analysis of population-based data. Lancet Neurol 13: , 788–794. |
[113] | Akamine EH , Kawamoto EM , Scavone C , Nigro D , Carvalho MH , de Cássia ATR , Britto LR , Fortes ZB ((2006) ) Correction of endothelial dysfunction in diabetic female rats by tetrahydrobiopterin and chronic insulin. J Vasc Res 43: , 309–320. |
[114] | Capuron L , Geisler S , Kurz K , Leblhuber F , Sperner-Unterweger B , Fuchs D ((2014) ) Activated immune system and inflammation in healthy ageing: Relevance for tryptophan and neopterin metabolism. Curr Pharm Des 20: , 6048–6057. |
[115] | Capuron L , Schroecksnadel S , Féart C , Aubert A , Higueret D , Barberger-Gateau P , Layé S , Fuchs D ((2011) ) Chronic low-grade inflammation in elderly persons is associated with altered tryptophan and tyrosine metabolism: Role in neuropsychiatric symptoms. Biol Psychiatry 70: , 175–182. |
[116] | Richardson MA , Read LL , Reilly MA , Clelland JD , Clelland CL ((2007) ) Analysis of plasma biopterin levels in psychiatric disorders suggests a common BH4 deficit in schizophrenia and schizoaffective disorder. Neurochem Res 32: , 107–113. |
[117] | Richardson MA , Read LL , Taylor Clelland CL , Reilly MA , Chao HM , Guynn RW , Suckow RF , Clelland JD ((2005) ) Evidence for a tetrahydrobiopterin deficit in schizophrenia. Neuropsychobiology 52: , 190–201. |
[118] | Blair JA , Barford PA , Morar C , Pheasant AE , Hamon CG , Whitburn SB , Leeming RJ , Reynolds GP , Coppen A ((1984) ) Tetrahydrobiopterin metabolism in depression. Lancet 2: , 163. |
[119] | Curtius HC , Niederwieser A , Levine RA , Lovenberg W , Woggon B , Angst J ((1983) ) Successful treatment of depression with tetrahydrobiopterin. Lancet 1: , 657–658. |
[120] | Pan L , McKain BW , Madan-Khetarpal S , McGuire M , Diler RS , Perel JM , Vockley J , Brent DA ((2011) ) GTP-cyclohydrolase deficiency responsive to sapropterin and 5-HTP supplementation: Relief of treatment-refractory depression and suicidal behaviour. BMJ Case Rep 2011: , bcr0320113927. |
[121] | Knapp S , Irwin M ((1989) ) Plasma levels of tetrahydrobiopterin and folate in major depression. Biol Psychiatry 26: , 156–162. |
[122] | Danfors T , von Knorring AL , Hartvig P , Langstrom B , Moulder R , Stromberg B , Torstenson R , Wester U , Watanabe Y , Eeg-Olofsson O ((2005) ) Tetrahydrobiopterin in the treatment of children with autistic disorder: A double-blind placebo-controlled crossover study. J Clin Psychopharmacol 25: , 485–489. |
[123] | Frye RE , Huffman LC , Elliott GR ((2010) ) Tetrahydrobiopterin as a novel therapeutic intervention for autism. Neurotherapeutics 7: , 241–249. |
[124] | Frye RE , DeLatorre R , Taylor HB , Slattery J , Melnyk S , Chowdhury N , James SJ ((2013) ) Metabolic effects of sapropterin treatment in autism spectrum disorder: A preliminary study. Transl Psychiatry 3: , e237. |
[125] | Klaiman C , Huffman L , Masaki L , Elliott GR ((2013) ) Tetrahydrobiopterin as a treatment for autism spectrum disorders: A double-blind, placebo-controlled trial. J Child Adolesc Psychopharmacol 23: , 320–328. |
[126] | Fernell E , Watanabe Y , Adolfsson I , Tani Y , Bergström M , Hartvig P , Lilja A , von Knorring AL , Gillberg C , Långström B ((1997) ) Possible effects of tetrahydrobiopterin treatment in six children with autism–clinical and positron emission tomography data: A pilot study. Dev Med Child Neurol 39: , 313–318. |


