
Safety, Tolerability, Pharmacokinetics, and Pharmacodynamic Effects of PF-06751979, a Potent and Selective Oral BACE1 Inhibitor: Results from Phase I Studies in Healthy Adults and Healthy Older Subjects
Abstract
PF-06751979 is a selective inhibitor of the beta-site amyloid precursor protein cleaving enzyme-1, which is a key aspartyl protease in the generation of amyloid-β (Aβ) peptides, thought to be critical for the cerebral degeneration observed in Alzheimer’s disease. Two Phase I studies (NCT02509117, NCT02793232) investigated the safety/tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of PF-06751979. Single-ascending doses up to 540 mg and multiple-ascending doses up to 275 mg once daily (QD) in healthy adults, and multiple doses of 50 mg or 125 mg QD in healthy older subjects were assessed. PF-06751979 was well tolerated at all doses given, and all treatment-related adverse events (AEs) were mild to moderate. PK parameters remained consistent across the PF-06751979 QD dosing regimens, and no notable food effects were observed. PD analysis showed that PF-06751979 reduced the cerebrospinal fluid (CSF) and plasma levels of Aβ peptides in a dose-dependent manner, with the greatest reductions observed in subjects treated with 275 mg QD (approximately 92% and 93% reduction in CSF Aβ1–40 and Aβ1–42 observed at 24 h after Day 14 dose, respectively). A drug interaction study (NCT03126721) using midazolam indicated that there was no clinically meaningful effect of multiple doses of PF-06751979 100 mg QD on the PK of single-dose midazolam in healthy adults. Overall, these data suggest that PF-06751979 with daily dosing is favorable for further clinical development.
INTRODUCTION
Alzheimer’s disease (AD) is a progressive, neurodegenerative disorder that is the most common form of dementia worldwide and one of the leading causes of disability and dependency in the elderly [1]. AD and other dementias are predicted to affect 115 million people by 2050 [2]. Current approved therapies, including cholinesterase inhibitors and an N-methyl-D-aspartate-receptor antagonist, provide symptomatic improvement but do not affect the progression of the disease [3, 4]. Therefore, there is an urgent need to develop new treatments that favorably alter the progression of AD by slowing, or even preventing, its associated neurodegeneration.
Cerebral degeneration in AD is characterized by the accumulation of intracellular neurofibrillary tangles and extracellular amyloid plaques [5, 6]. The primary components of amyloid plaques are 37 to 43 residue-long amyloid-β (Aβ) peptides [7]. The most abundant Aβ peptide identified in the post-mortem AD brain is 40 amino acids in length (Aβ40) [8]; however, the more pathological peptide is 42 amino acids long (Aβ42). This peptide has a higher aggregation tendency than other forms [9].
Aβ peptides have long been known to be derived from the sequential cleavage of the transmembrane amyloid-β protein precursor (AβPP) by β-site amyloid precursor protein cleaving enzyme (BACE) [10] followed by γ-secretase [11]. As processing of AβPP by BACE is necessary for the generation of Aβ peptides, BACE inhibition to reduce Aβ42 levels is a rational therapeutic strategy. Notably, soon after the original identification of BACE, a homolog, termed BACE2 was discovered [12]. While it is 75% homologous to BACE (BACE1), studies indicate that BACE2 does not play an active role in Aβ peptide production in the brain [13] and BACE2 inhibition can thus be considered off-target for the treatment of AD. Despite this, and although a number of BACE inhibitors have progressed to late-stage clinical trials, including AZD3293, MK-8931, JNJ-54861911, CNP520, and E2609 [14– 18], progress in designing a BACE inhibitor that has good selectivity for BACE1 over BACE2 has been limited. Indeed, aside from CNP520, which has shown some selectivity for BACE1 [16], current BACE inhibitors appear to inhibit BACE1 and BACE2 to a similar degree [19]. An understanding of the physiological impact of off-target BACE2 inhibition has emerged in recent years, with studies demonstrating that BACE2 knockout disrupts melanogenesis, resulting in a hypopigmentation phenotype [20, 21]. Several non-selective BACE inhibitors, such as AZD3293 and NB-360 have shown fur and skin pigment loss in mice, rats, or dogs following chronic dosing regimens [22, 23]. These data suggest that chronic BACE2 inhibition is undesirable in a therapeutic agent.
Table 1
Overview of the Phase I PF-06751979 clinical studies B8271001, B8271004, and B8271007
| Study | Study details |
| B8271001 SAD/MAD (NCT02509117) | SAD (Part A): single doses of placebo or PF-06751979 3, 12, 40, and 160 mg, administered in healthy adults (18–55 years of age) under fasted conditions |
| MAD (Part B): multiple doses of placebo or PF-06751979 5, 15, or 50 mg QD administered in healthy adults (18–55 years of age) for 14 days | |
| Multiple doses (Part C): multiple doses of placebo or PF-06751979 50 mg QD administered in healthy older subjects (60–85 years of age) for 14 days | |
| B8271004 SAD/MAD/food effect (NCT02793232) | SAD/food effect (Part A): single doses of placebo or PF-06751979 200, 400, or 540 mg administered in healthy adults (18–55 years of age) under fasted conditions, and PF-06751979 200 mg under fed conditions |
| MAD (Part B): multiple doses of placebo or PF-06751979 125 or 275 mg QD administered in healthy adults (18–55 years of age) for 14 days | |
| Multiple doses (Part C): multiple doses of placebo or PF-06751979 125 mg QD administered in healthy older subjects (60–85 years of age) for 14 days | |
| B8271007a (NCT03126721) | Midazolam DDI: multiple doses of PF-06751979 100 mg QD administered in healthy adults (18–55 years of age) for 11 days (steady state), with a single dose of midazolam 2 mg administered on Day 10 |
aOnly high-level results reported in this manuscript.
DDI, drug–drug interaction; SAD, single-ascending dose; MAD, multiple-ascending dose; QD, once daily.
PF-06751979 is a novel, brain-penetrable, small-molecule BACE inhibitor under investigation as a potential treatment for AD. The structure of PF-06751979 has previously been reported by O’Neill and colleagues (compound 64), and it has been shown to be a potent BACE inhibitor [19]. Using a radioligand binding or fluorescence polarization (FP) assay, PF-06751979 had 26.6-fold or 6.4-fold selectivity for BACE1 over BACE 2, respectively; broad selectivity over the related aspartyl protease Cathepsin D (Cat D) was also reported (∼2500-fold using an FP assay) [19]. Long-term toxicology studies in mice and dogs did not reveal pigmentation changes [19], thus supporting selectivity for BACE1 in vivo. Moreover, animal studies have demonstrated robust reductions of Aβ40 and Aβ42 in the brain and cerebrospinal fluid (CSF) following subcutaneous administration of PF-06751979 [19]. This observed change in the CSF Aβ profile represents a mechanism-relevant pharmacodynamic (PD) endpoint.
Here we report the findings of the first three Phase I studies investigating PF-06751979 in humans. Study B8271001 (NCT02509117) was a first-in-human trial and provided data to inform the use of higher doses in the subsequent study, B8271004 (NCT02793232), while B8271007 (NCT03126721) was a drug–drug interaction study with midazolam, a model substrate drug for CYP3A activity. Data from studies B8271001 and B8271004 were analyzed together to evaluate the safety, tolerability, and pharmacokinetics (PK) of single- and multiple-ascending doses of PF-06751979 in healthy adults and healthy older subjects, as well as the effect of multiple doses of PF-06751979 on plasma and CSF Aβ fragments in healthy adults. In addition, the effects of multiple-dose administration of PF-06751979 on the single-dose PK of oral midazolam in study B8271007 are briefly summarized.
MATERIALS AND METHODS
Study design
B8271001 and B8271004 were Phase I, randomized, double-blind, placebo-controlled studies in healthy adults (18–55 years of age) and healthy older subjects (60–85 years of age). B8271001 was conducted at Paraxel California Clinical Trials, Glendale, CA, USA, and B8271004 at Pfizer Clinical Research Unit (CRU), Brussels, Belgium. The studies were subject- and investigator-blind, and sponsor-open. Both studies were split into three parts (Parts A–C [Table 1]); this consistent design permitted data to be combined in this analysis.
Part A was a single-ascending dose (SAD) study of PF-06751979 (up to 160 mg in study B8271001 and up to 540 mg in B8271004) with a four-period, cross-over, placebo-substitution design in a single cohort of eight healthy adults. Subjects progressed through four treatment periods, with a single dose of placebo or PF-06751979 administered in each period. Subjects were randomized in a 1:1:1:1 ratio to one of four dosing sequences prior to receiving their first dose of study medication. In study B8271001, each subject could receive up to four study treatments (placebo and up to three PF-06751979 dose levels) administered orally following an overnight fast. In study B8271004, each subject could receive up to three treatments orally (placebo or up to three PF-06751979 dose levels following an overnight fast) in the first three periods. In period 4, placebo or PF-06751979 200 mg was adminstered following a high-fat breakfast to evaluate food effect. During each period, subjects were admitted to the CRU on Day –1 and confined for approximately 4 (B8271001) or 5 days (B8271004) following dosing. Each dose adminstration was seperated by a washout of at least 7 days in study B8271001 and 10 days in study B8271004.
Part B was a multiple-ascending dose (MAD) study, in which five cohorts of 10–16 healthy adults were randomized to receive placebo or multiple oral doses of PF-06751979 once daily (QD) for 14 days (5, 15, and 50 mg in study B8271001 and 125 and 275 mg in study B8271004). Randomization was performed in a 1:4 (5 and 15 mg cohort), 1:2 (50 mg cohort), or 1:3 (125 and 275 mg cohort) ratio. On Days 1, 7, and 14, treatment was administered following an overnight fast. On all other days, treatment was administered outside a window of±1 h of food. Subjects were admitted to the CRU on Day –1 (B8271001) or Day –4 (B8271004) and confined for approximately 21 days in study B8271001 and 22 days in study B8271004.
In Part C, two cohorts of 10–12 healthy older subjects were randomized in a 1:2 ratio in B8271001 or a 1:4 ratio in B8271004 to receive multiple oral doses of placebo or PF-06751979 (50 mg QD in study B8271001 and 125 mg QD in study B8271004) for 14 days. On Days 1, 7, and 14, treatment was administered following an overnight fast. On all other days, treatment was administered as in Part B. Subjects were admitted to the CRU on Day –1 (B8271001) or Day 0 (B8271004) and confined for approximately 21 days.
Study B8271007 was a Phase I, open-label, fixed-sequence study in a single cohort of 12 healthy adults to assess the effect of multiple doses of PF-06751979 100 mg QD for 11 days on the single-dose PK of oral midazolam (Table 1). The study comprised two periods. In the first, a single oral dose of midazolam 2 mg was administered. In the second, a single dose of midazolam 2 mg was administered on Day 10, with multiple doses of PF-06751979 100 mg QD administered from Days 1–11.
Safety assessments
The safety and tolerability of single- and multiple-oral doses of PF-06751979 in healthy adults and/or healthy older subjects were evaluated in studies B8271001 and B8271004. Safety and tolerability of multiple-oral doses of PF-06751979, with or without co-administration of midazolam, were evaluated in study B82710007. All subjects who received at least one dose of study medication were included in the safety analyses. Safety measures included an assessment of adverse events (AEs; spontaneous and solicited), blood and urine laboratory tests, vital signs, and electrocardiograms (ECGs).
Serious AEs (SAEs) were defined as any that resulted in death, were life-threatening, required inpatient hospitalization or prolongation of existing hospitalization, resulted in persistent or significant disability/incapacity or resulted in congenital anomaly/birth defect.
All treatment-emergent adverse events (TEAEs) and their severity and potential causality were recorded by investigators from the time the subject had taken at least one dose of study medication through their last follow-up.
PK assessment
In studies B8271001 and B8271004, plasma and/or urine samples were collected following single and multiple oral doses of PF-06751979. In both studies, during each period of Part A, blood samples were collected pre-dose on Day 1, and 0.5, 1, 2, 4, 8, 12, 16, 24, 36, 48, and 72 h post-dose. In study B8271001 an additional sample was collected 1.5 h post-dose; and in study B8271004 additional samples were collected on Day –1 (periods 2–4 only), and 6 and 96 h post-dose.
During Part B of both studies, on Days 1 and 7, blood samples were collected pre-dose and 0.5, 1, 2, 4, 8, 12, and 24 h post-dose. In study B8271001, an additional sample was collected 1.5 h post-dose, and in B8271004 an additional sample was collected 6 h post-dose. On Days 4 and 10, samples were taken pre-dose and 2 (B8271001) or 4 h (B8271004) post-dose. On Day 14, time points for blood sample collection were the same as those on Days 1 and 7 up to 24 h. Additional samples were then taken 48, 72, 96, and 120 h post-dose.
During Part C of both studies, blood samples were collected pre-dose on Day 1 and 0.5, 1, 2, 4, 8, 12, and 24 h post-dose. In study B8271001, an additional sample was collected 1.5 h post-dose, and in B8271004, an additional sample was collected 6 h post-dose. On Days 4, 7, and 10, samples were collected pre-dose and at 2 (B8271001) or 4 h (B8271004) post-dose. In addition, samples were also collected 12 and 24 h post-dose on Day 7 of study B8271001. On Day 14, time points for blood sample collection were the same as those on Day 1 up to 24 h. Additional samples were then taken 48, 72, 96, and 120 h post-dose.
In study B8271007, serial blood samples for midazolam PK analysis were taken over 24 h post-dose after midazolam was administered alone in period 1. In period 2, blood samples were taken up to 48 h post-dose after midazolam was co-administered with PF-06751979 on Day 10.
Samples were analyzed for PF-06751979 concentrations at Pfizer Inc (Groton, CT, USA) (B8271001) or York Bioanalytical Solutions (York, UK) (B8271004) using validated high-performance liquid chromatography tandem mass spectrometry. The lower limit of quantification (LLOQ) for PF-06751979 was 0.500 ng/mL (B8271001 only) or 5.00 ng/mL (B8271004 only) in plasma, 0.050 ng/mL in CSF, and 0.050 ng/mL in urine. Samples for midazolam concentrations were analyzed at York Bioanalytical Solutions (York, UK) using a validated, sensitive, and specific liquid chromatography tandem mass spectrometric method. The LLOQ for midazolam was 0.100 ng/mL.
The PK concentration population was defined as all enrolled, treated subjects who had at least one measurable PF-06751979 or midazolam (B8271007) concentration. The PK parameter population was defined as all enrolled, treated subjects who had at least one of the PK parameters of interest measured.
Plasma and urine PF-06751979 and midazolam PK parameters were calculated using non-compartmental analysis of concentration–time data.
PD assessments
In Part B of studies B8271001 and B8271004, the PD of multiple-ascending oral doses of PF-06751979 on CSF Aβ fragments in healthy adult subjects was evaluated in the PF-06751979 50, 125, and 275 mg QD cohorts. In addition, the PD of single- and multiple-ascending oral doses of PF-06751979 on plasma Aβ fragments in healthy adult subjects was evaluated in all cohorts in the SAD and MAD studies (Parts A and B).
Lumbar puncture to obtain CSF for PD analysis was conducted on Days 0 (∼24 h prior to the first dose on Day 1) and 15 (24 h after the last dose on Day 14) in 8271001 or on Days –3 (∼72 h prior to the first dose on Day 1) and 15 (24 h after the last dose on Day 14) in B8271004. During Part A of both studies, blood samples for plasma Aβ analyses were taken pre-dose on Day 1, and 8, 24, and 48 h post-dose, with additional samples collected 4 h post-dose (B8271001 only) and 6 h post-dose (B8271004 only). During Part B, blood samples were taken pre-dose on Days 1, 2, 4, 7, 10, and 14.
CSF samples were analyzed at a Pfizer CRU laboratory (NHCRU Biomarker Laboratory, New Haven, CT, USA) for concentrations of Aβx-38, Aβx-40, Aβx-42, Aβ1–38, Aβ1-40, Aβ1-42, total Aβ, soluble AβPPα (sAβPPα), and sAβPPβ. Samples for total Aβ were assayed using a validated dissociation-enhanced lanthanide fluorescent immunoassay (DELFIA®) method and the LLOQ was 391.0 pg/mL. Samples for all other peptides were assayed using a validated electrochemiluminescence method. The LLOQs were 47.0 pg/mL for Aβx-38, 14.0 pg/mL for Aβx-40, 2.06 pg/mL for Aβx-42, 27.7 pg/mL for Aβ1–38, 27.4 pg/mL for Aβ1–40, and 2.85 pg/mL for Aβ1–42. In study B8271001, the LLOQ for sAβPPα was 19.0 ng/mL, and in B8271004 it was 26.0 ng/mL. In both studies the LLOQ for sAβPPβ was 24.4 ng/mL.
Plasma samples were analyzed for Aβ1–40, Aβx-40, and total Aβ concentrations at the same laboratory and assayed using the DELFIA method. The LLOQ was 12.1 pg/mL for Aβ1–40, 28.4 pg/mL for Aβx-40, and 66.0 pg/mL for total Aβ.
Changes from baseline in CSF Aβ species after 14 days of dosing were natural log transformed [loge (Aβ post-dose) – loge (Aβ at baseline)] and analyzed using a linear model, analysis of covariance, with treatment as a fixed effect and loge baseline as a covariate. For these analyses, treatment mean was transformed to percent change from baseline, treatment versus pooled placebo difference was transformed to placebo-adjusted percent change from baseline, and mean estimates along with 2-sided 80% confidence intervals (CIs) were reported.
Changes from baseline in plasma Aβ species, at each of the time points indicated above, were log transformed and analyzed using a mixed model-repeated measures approach. In this analysis, treatment, time, and treatment by time were fixed effects, with subject as a random effect, and log baseline mean as a covariate.
Table 2
Demographics of subjects in studies B8271001 and B8271004
| Part A: PF-06751979 single-ascending dose in healthy adults | ||
| Study B8271001a | Study B8271004 | |
| Single cohort | Single cohort | |
| (N = 8) | (N = 8) | |
| Male, n (%) | 8 (100.0) | 8 (100.0) |
| Mean age (range), y | 32.8 (24.0–55.0) | 33.3 (23.0–51.0) |
| Race, n (%) | ||
| White | 4 (50.0) | 8 (100.0) |
| Black | 3 (37.5) | 0 (0.0) |
| Asian | 1 (12.5) | 0 (0.0) |
| Other | 0 (0.0) | 0 (0.0) |
| Mean BMI (range), kg/m2 | 25.7 (21.1–30.0) | 24.8 (20.6–29.6) |
| Part B: PF-06751979 multiple-ascending dose in healthy adults | |||||||
| Study B8271001 | Study B8271004 | ||||||
| Pooled PBO (N = 8) | 5 mg (N = 8) | 15 mg (N = 8) | 50 mg (N = 8) | Pooled PBO (N = 7) | 125 mg (N = 12) | 275 mg (N = 9) | |
| Male, n (%) | 8 (100.0) | 8 (100.0) | 8 (100.0) | 8 (100.0) | 7 (100.0) | 12 (100.0) | 9 (100.0) |
| Mean age (range), y | 38.0 (24.0–54.0) | 39.8 (26.0–50.0) | 35.1 (18.0–53.0) | 39.0 (32.0–53.0) | 37.9 (30.0–47.0) | 34.2 (24.0–46.0) | 35.4 (20.0–41.0) |
| Race, n (%) | |||||||
| White | 3 (37.5) | 5 (62.5) | 3 (37.5) | 3 (37.5) | 3 (42.9) | 7 (58.3) | 7 (77.8) |
| Black | 3 (37.5) | 3 (37.5) | 4 (50.0) | 4 (50.0) | 1 (14.3) | 0 (0.0) | 0 (0.0) |
| Asian | 1 (12.5) | 0 (0.0) | 0 (0.0) | 1 (12.5) | 3 (42.9)b | 5 (41.7)b | 2 (22.2)b |
| Other | 1 (12.5) | 0 (0.0) | 1 (12.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Mean BMI (range), kg/m2 | 25.1 (21.1–29.5) | 26.9 (23.1–29.5) | 24.3 (18.3–26.6) | 27.0 (19.4–30.4) | 25.0 (19.9–29.5) | 23.7 (18.7–29.6) | 26.0 (22.6–29.7) |
| Part C: PF-06751979 multiple doses in healthy older subjects | ||||
| Study B8271001 | Study B8271004 | |||
| PBO (N = 4) | 50 mg (N = 10) | PBO (N = 2) | 125 mg (N = 8) | |
| Male, n (%) | 1 (25.0) | 4 (40.0) | 1 (50.0) | 5 (62.5) |
| Mean age (range), y | 68.5 (64.0–75.0) | 69.0 (63.0–73.0) | 67.0 (67.0–67.0) | 66.4 (62.0–73.0) |
| Race, n (%) | ||||
| White | 3 (75.0) | 6 (60.0) | 2 (100.0) | 8 (100.0) |
| Black | 1 (25.0) | 3 (30.0) | 0 (0.0) | 0 (0.0) |
| Asian | 0 (0.0) | 1 (10.0) | 0 (0.0) | 0 (0.0) |
| Other | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Mean BMI (range), kg/m2 | 24.8 (21.8–26.4) | 26.1 (23.3–30.5) | 25.1 (24.1–26.0) | 25.7 (20.9–30.9) |
aData missing for one subject who discontinued after randomization prior to Period 1 dosing; bAll subjects in the Asian group were Japanese. BMI, body mass index; PBO, placebo.
PK/PD modeling for CSF Aβ1–40 and Aβ1-42
Using data from studies B8271001 and B8271004, a population PK/PD model of CSF Aβ1–40 and Aβ1–42 was developed to characterize the PF-06751979 plasma exposure and CSF Aβ-response relationship. All analyses were performed using NONMEM 7.3 (ICON Development Solutions, Gaithersburg, MD, USA).
Population PK was characterized using a two-compartment model with linear elimination and first-order absorption. Increases in relative bioavailability at higher doses (>100 mg), and slower absorption due to a high-fat meal were characterized also. To characterize PD effects, indirect response modeling was applied in which the rate of production of CSF Aβ was decreased as a function of PF-06751979 plasma concentration. PK data were included but population PK parameters were fixed (Population PK Parameters and Data Approach) [24].
Due to the limited amount of CSF Aβ data collected (only at baseline and a single trough measurement at steady state), the parameters for CSF Aβ turnover rates were fixed to 0.084/h for Aβ1–40 and 0.12/h for Aβ1–42, which were estimated in a separate, PK/PD study with serial CSF collections [25]. Due to the similarity in study populations, it was assumed that Aβ dynamics in the PK/PD model described here would be the same as those from the separate PK/PD study [25]. The estimated exposure-response relationship was specific to PF-06751979, based on the current multiple-dose data.
Aβ1–40 and Aβ1–42 data were simultaneously modeled. The same inhibitory maximum effect (Imax) and half maximum inhibitory concentration (IC50) were assumed for both species, based on the mechanism of action of BACE inhibitors, and the baseline correlation between both species was taken into account.
Ethical principles
All studies were conducted in compliance with the ethical principles of the Declaration of Helsinki, and International Conference on Harmonization Good Clinical Practice guidelines. The protocols were approved by the Independent Ethics Committee at the investigational centers. All subjects provided informed consent.
RESULTS
Subjects
A combined total of 101 subjects were randomized in studies B8271001 and B8271004; 100 subjects received treatment. Demographics for treated subjects are shown in Table 2.
In the SAD part (Part A), 17 adult males were randomized. In study B8271001, one subject withdrew between randomization and period one dosing. The remaining 16 subjects received treatment (mean age 33.1 years [range 23–55]; mean body mass index [BMI] 25.3 kg/m2; White n = 12, Black n = 3, Asian n = 1); all completed the study.
In the MAD part (Part B), 60 adult males were randomized (mean age 36.8 years [range 18–54]; mean BMI 25.3 kg/m2; White n = 31, Black n = 15, Asian n = 12, other n = 2). In Part B of B8271004, all Asian subjects (n = 10) were Japanese. Two subjects discontinued in B8271004 (placebo group, n = 1; PF-06751979 275 mg group, n = 1).
In the multiple-dose component (Part C), 24 older subjects were randomized (11 male, 13 female; mean age 67.9 years [range 62–75]; mean BMI 25.7 kg/m2; White n = 19, Black n = 4, Asian n = 1). Two were discontinued from the PF–06751979 50 mg group of study B8271001.
In study B8271007, a total of 12 adult males participated (mean age 32.1 years [range 21–54]; mean BMI 24.0 kg/m2; White n = 6, Black n = 6). All subjects completed the study.
Safety
The frequencies of TEAEs reported in studies B8271001 and B8271004 are summarized in Table 3. Overall, no SAEs or deaths were reported. All TEAEs reported in the two studies were mild to moderate in severity. Fatigue and headaches were the most frequently reported AEs (Table 3).
Table 3
Overview of all-causality treatment-emergent AEs in studies B8271001 and B8271004. The numbers of treatment-emergent AEs deemed to be treatment related are shown in parentheses
| Part A: PF-06751979 single-ascending dose in healthy adults | |||||||||
| Dose | PBO | 3 mg | 12 mg | 40 mg | 160 mg | 200 mg | 400 mg | 540 mg | 200 mg fed |
| Subjects evaluable | 16 | 5 | 6 | 6 | 6 | 6 | 6 | 6 | 6 |
| Number of AEs | 1 (1) | 1 (0) | 0 | 0 | 0 | 5 (3) | 5 (5) | 10 (6) | 7 (4) |
| Number of subjects with: | |||||||||
| AEs | 1 (1) | 1 (0) | 0 | 0 | 0 | 3 (3) | 2 (2) | 6 (3) | 4 (4) |
| SAEs | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Severe AEs | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Discontinuation due to AEs | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Temporary discontinuation or dose reduction due to AEs | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Most common AEs (MedDRA preferred term), occurring in≥2 subjects across all groups | |||||||||
| Fatigue | 0 | 0 | 0 | 0 | 0 | 2 (2) | 0 | 0 | 1 (1) |
| Headache | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1) | 2 (2) | 0 |
| Part B: PF-06751979 multiple-ascending dose in healthy adults | ||||||
| Dose | Pooled PBO | 5 mg | 15 mg | 50 mg | 125 mg | 275 mg |
| Subjects evaluable | 15 | 8 | 8 | 8 | 12 | 9 |
| Number of AEs | 15 (7) | 1 (1) | 2 (2) | 1 (1) | 22 (6) | 18 (13) |
| Number of subjects with: | ||||||
| AEs | 7 (3) | 1 (1) | 2 (2) | 1 (1) | 10 (4) | 8 (7) |
| SAEs | 0 | 0 | 0 | 0 | 0 | 0 |
| Severe AEs | 0 | 0 | 0 | 0 | 0 | 0 |
| Discontinuation due to AEs | 1 (1) | 0 | 0 | 0 | 0 | 1 (1) |
| Temporary discontinuation or dose reduction due to AEs | 0 | 0 | 0 | 0 | 0 | 0 |
| Most common AEs (MedDRA preferred term), occurring in≥2 subjects across all groups | ||||||
| Headache | 2 (2) | 0 | 1 (1) | 0 | 3 (1) | 1 (0) |
| Musculoskeletal discomfort | 1 (0) | 0 | 0 | 0 | 3 (0) | 2 (0) |
| Hepatic enzyme increased | 1 (1) | 0 | 1 (1) | 0 | 1 (0) | 1 (1) |
| Back pain | 1 (0) | 0 | 0 | 0 | 2 (0) | 0 |
| Fatigue | 1 (1) | 0 | 0 | 0 | 2 (2) | 0 |
| Insomnia | 1 (1) | 0 | 0 | 0 | 1 (1) | 0 |
| Paresthesia | 0 | 0 | 0 | 0 | 1 (0) | 1 (1) |
| Procedural pain | 0 | 0 | 0 | 0 | 2 (0) | 0 |
| Part C: PF-06751979 multiple doses in healthy older subjects | |||
| Dose | PBO | 50 mg | 125 mg |
| Subjects evaluable for AEs | 6 | 10 | 8 |
| Number of AEs | 9 (7) | 15 (9) | 11 (7) |
| Number of subjects with: | |||
| AEs | 5 (4) | 7 (6) | 6 (5) |
| SAEs | 0 | 0 | 0 |
| Severe AEs | 0 | 0 | 0 |
| Discontinuation due to AEs | 0 | 0 | 0 |
| Temporary discontinuation or dose reduction due to AEs | 0 | 0 | 0 |
| Most common AEs (MedDRA preferred term), occurring in≥2 subjects across all groups | |||
| Abnormal dreams | 1 (1) | 2 (2) | 0 |
| Headache | 1 (0) | 1 (0) | 1 (1) |
| Dizziness | 1 (1) | 1 (0) | 0 |
| Fatigue | 0 | 1 (1) | 1 (1) |
AE, adverse event; MedDRA, Medical Dictionary for Regulatory Activities; PBO, placebo; SAE, serious adverse event.
Following a single-ascending dose of PF-06751979 in healthy adults (Part A), 29 TEAEs were reported by 17 subjects, 19 of which were considered to be treatment related. In general, the frequency of TEAEs increased with dose. Following multiple doses of PF-06751979 in healthy adults (Part B), 59 TEAEs were reported by 29 subjects, 30 of which were considered to be treatment related. The frequency of treatment-related TEAEs was highest in the PF-06751979 275 mg treatment group; however, the TEAE incidence rate could not be deemed to be dose related due to the relatively small number of subjects in each treatment group. Following multiple doses of PF-06751979 in healthy older subjects (Part C), 35 TEAEs were reported by 18 subjects, 23 of which were deemed to be treatment related. The frequency of TEAEs was similar in the PF-06751979 50 mg and 125 mg treatment groups.
In Part B of B8271004, two subjects discontinued. One subject, randomized to the PF-06751979 275 mg cohort but treated with placebo, discontinued due to elevated liver enzymes. Alanine aminotransferase/aspartate aminotransferase (ALT/AST) elevation was identified in the subject on Day 4 (ALT, 6.2×upper limit of normal [ULN]; AST, 3.3 ×ULN), accompanied by elevated alkaline phosphatase (ALP, 1.2×ULN). The elevations were deemed to be treatment related and the subject was discontinued from the study on the same day. ALT/AST and ALP levels recovered quickly, without medical intervention, and were within normal range by Days 15 and 8, respectively. The second subject, treated with PF-06751979 275 mg QD, presented with a moderate maculopapular rash 6 h post-dose on Day 11, which was deemed to be treatment related and led to discontinuation from the study. Following discontinuation, the subject continued to undergo safety evaluations and a moderate increase in ALT/AST levels, deemed to be treatment related, was reported on Day 14 (ALT, 3.1 ×ULN; AST, 1.15 x ULN). ALT and AST levels recovered quickly, without medical intervention, and were within normal range by Days 24 and 14, respectively.
In Part B of B8271001, one subject treated with PF-06751979 15 mg QD presented with moderate ALT/AST elevation on Day 19, 5 days after the final dose of PF-06751979 and prior to discharge from the study. ALT and AST levels peaked on Day 21 (ALT, 7.7 ×ULN; AST, 5.6 ×ULN), and subsequently recovered quickly, without medical intervention, to fall within normal range on Days 32 and 26, respectively. Across both studies, transient ALT/AST elevations in PF-06751979-treated subjects were not accompanied by any other abnormality indicative of impaired liver function.
In Part C of B8271001, one subject was discontinued due to insomnia, prior to study medication being administered, and one subject treated with PF-06751979 50 mg QD was discontinued on Day 9, at the discretion of the principal investigator due to a medical history of memory problems. Neither discontinuation was determined to be treatment related by the investigator, and neither were recorded as AEs. There were no clinically meaningful findings in ECGs or vital signs, and no other clinically meaningful laboratory test findings in either study.
In study B8271007, following a single oral dose of 2 mg midazolam alone (period 1), three TEAEs were reported, none of which were deemed to be treatment related. Following multiple doses of PF-06751979 100 mg QD alone (period 2), nine TEAEs were reported, of which six were deemed to be treatment related. Following the co-administration of PF-06751979 100 mg QD and a single dose of midazolam 2 mg (Day 10, period 2), six TEAEs were reported, five of which were deemed to be treatment related. In period 2, one subject experienced moderate ALT/AST elevation on Day 7 (ALT, 2.12×ULN; AST, 1.23×ULN). ALT and AST levels peaked on Days 13 and 12, respectively (ALT, 3.33×ULN; AST, 1.75×ULN), and resolved without medical intervention, to reach normal levels on Days 22 and 15, respectively. The ALT/AST elevations were not accompanied by any other abnormality indicative of impaired liver function. All TEAEs reported in period 2 were mild to moderate in severity, with fatigue and headaches being the most commonly reported. No SAEs or deaths were reported and there were no dose reductions or discontinuations.
PK
Combined PK parameter data for studies B8271001 and B8271004 are shown in Table 4 and the mean plasma concentration–time curves are shown in Supplementary Figure 1.
Table 4
PK assessments in healthy adults and older subjects in studies B8271001 and B8271004
| Part A: PF-06751979 single-ascending dose in healthy adultsa | ||||||||
| Dose | 3 mg | 12 mg | 40 mg | 160 mg | 200 mg | 400 mg | 540 mg | 200 mg fed |
| N, n | 5, 3 | 6, 5 | 6, 5 | 6, 6 | 6, 6 | 6, 6 | 6, 6 | 6, 6 |
| Cmax, ng/mL | 8.4 (14) | 27.4 (36) | 78.7 (30) | 530.8 (41) | 643.2 (19) | 1436.0 (10) | 1829.0 (19) | 630.2 (9) |
| Tmax, h | 4.03 (4.00–8.00) | 4.11 (2.0–12.00) | 4.08 (2.00–8.05) | 3.03 (1.87–4.08) | 4.00 (2.00–6.07) | 3.02 (2.00–4.00) | 3.01 (2.00–6.02) | 6.00 (4.00–8.00) |
| AUCinf, ng.hr/mL | 289.7 (8) | 1142 (25) | 3121 (25) | 17050 (18) | 22850 (17) | 44980 (17) | 63420 (9) | 23260 (15) |
| t1/2, h | 29.30±0.60 | 32.94±4.92 | 32.12±3.64 | 39.33±8.20 | 35.20±8.97 | 34.70±8.97 | 35.85±8.10 | 33.38±6.25 |
| CL/F, mL/min | 172.3 (8) | 175.0 (25) | 213.4 (25) | 156.3 (18) | 145.8 (17) | 148.3 (17) | 142.0 (9) | 143.3 (15) |
| Vz/F, L | 436.8 (6) | 494.8 (20) | 591.4 (26) | 523.2 (25) | 433.7 (18) | 433.0 (19) | 431.1 (19) | 408.1 (16) |
| Part B: PF-06751979 multiple-ascending dose in healthy adultsa | |||||
| Dose | 5 mg | 15 mg | 50 mg | 125 mg | 275 mg |
| Day 1 | |||||
| N | 8 | 8 | 8 | 12 | 9 |
| Cmax, ng/mL | 10.12 (22) | 28.90 (22) | 120.6 (17) | 442.7 (20) | 983.0 (18) |
| Tmax, h | 3.98 (2.03–12.3) | 4.07 (4.00–7.83) | 2.99 (1.02–4.07) | 4.00 (2.00–6.03) | 4.00 (2.00–4.00) |
| AUCtau, ng.hr/mL | 167.7 (18) | 478.8 (20) | 1739 (14) | 6380 (15) | 13260 (18) |
| Day 7 | |||||
| N | 8 | 8 | 8 | 12 | 9 |
| Cmax, ng/mL | 20.68 (22) | 64.34 (12) | 241.1 (20) | 818.7 (19) | 2000 (14) |
| Tmax, h | 6.02 (3.95–8.05) | 4.02 (2.17–8.03) | 3.83 (1.43–4.07) | 3.00 (2.00–6.00) | 2.00 (2.00–4.00) |
| AUCtau, ng.h/mL | 395.2 (20) | 1149 (12) | 4181 (20) | 13810 (16) | 31210 (14) |
| Day 14 | |||||
| N, n | 8, 8 | 8, 8 | 8, 8 | 12, 12 | 8, 8 |
| Cmax, ng/mL | 22.41 (23) | 64.63 (14) | 245.7 (21) | 837.6 (16) | 1869 (14) |
| Tmax, h | 4.01 (3.98–8.02) | 4.05 (4.02–7.87) | 3.00 (0.88–8.08) | 4.00 (2.00–12.00) | 4.00 (2.00–4.00) |
| t1/2, h | 37.15±5.20 | 30.65±3.19 | 37.36±5.91 | 30.73±5.75 | 37.79±7.73 |
| CL/F, mL/min | 197.1 (20) | 216.9 (12) | 196.9 (24) | 149.0 (14) | 155.3 (13) |
| Vz/F, L | 628.7 (18) | 573.2 (12) | 629.0 (27) | 390.5 (23) | 498.3 (27) |
| AUCtau, ng.hr/mL | 422.2 (20) | 1152 (13) | 4236 (24) | 13990 (14) | 29470 (12) |
| Cmin, ng/mL | 12.55 (22) | 32.75 (14) | 120.4 (25) | 390.3 (18) | 868.5 (15) |
| Rac | 2.52 (12) | 2.41 (14) | 2.43 (15) | 2.19 (11) | 2.27 (11) |
| Part C: PF-06751979 multiple doses in healthy older subjectsa | ||
| Dose | 50 mg | 125 mg |
| Day 1 | ||
| N | 10 | 8 |
| Cmax, ng/mL | 102.9 (25) | 377.1 (30) |
| Tmax, h | 4.00 (2.02–7.98) | 4.00 (2.00–4.03) |
| AUCtau, ng.hr/mL | 1621 (22) | 5238 (24) |
| Day 14 | ||
| N, n | 8, 8 | 8, 8 |
| Cmax, ng/mL | 256.4 (23) | 879.4 (28) |
| Tmax, h | 4.00 (2.00–4.20) | 4.00 (2.00–4.03) |
| t1/2, h | 39.95±3.18 | 40.76±3.90 |
| CL/F, mL/min | 184.9 (20) | 141.4 (23) |
| Vz/F, L | 637.4 (23) | 496.1 (30) |
| AUCtau, ng.h/mL | 4505 (20) | 14750 (23) |
| Cmin, ng/mL | 135.8 (18) | 429.9 (23) |
aGeometric mean (% CV) for all PK parameters except: median (range) for Tmax; arithmetic mean±SD for t1/2. AUCinf, area under the plasma concentration–time curve from time 0 to infinity; AUCtau, area under the plasma concentration–time curve from over the dosing interval tau (τ) where τ equals 24 h for QD dosing; CL/F, apparent clearance; Cmin, minimum observed concentration during the dosing interval; Cmax,maximum observed concentration; CV, coefficient of variation; N, number of subjects in the treatment group and contributing to the summary statistics for the specified day; n, number of subjects for t1/2 and Vz/F; PK, pharmacokinetic; Rac, observed accumulation ratio; t1/2, terminal half-life; SD, standard deviation; Tmax, time to reach maximum concentration; Vz/F, apparent volume of distribution.
Following single-ascending doses of PF-06751979 ranging from 3 mg to 540 mg (Part A), and multiple doses up to 275 mg QD (Part B), median Tmax was 3–4 h in the fasted state. Plasma Cmax and AUCinf both increased with increasing dose. PK was linear up to 50 mg and from 125–275 mg after multiple dose administrations. Single- and multiple-dose PK were consistent, indicating no time-dependent PK change. Mean t1/2 ranged from 29–39 h across doses. When a single dose of PF-06751979 200 mg was administered in a fed state (Part A of study B8271004), Tmax increased to 6 h. However, there was no apparent food effect on plasma PF-06751979 exposure, with similar Cmax and AUCinf under both fed and fasted conditions.
Following multiple doses of PF-06751979 (Part B), steady state was achieved by Day 7 with 2.1- to 2.5-fold accumulation on Day 14. A mean of 7–10% of the PF-06751979 dose was recovered unchanged in the urine across the dose range studied.
Plasma PF-06751979 exposure in healthy older subjects receiving multiple doses of PF-06751979 50 mg or 125 mg QD (Part C) was similar to that in healthy adults, with a median Tmax of 4 h and steady state achieved by Day 7. The observed t1/2 was approximately 40 h, slightly longer than that seen in the healthy adult population. Plasma PF-06751979 accumulation observed at Day 14 was also slightly higher (2.8-fold) compared with the healthy adult population.
During study B8271007, following a single oral dose of midazolam 2 mg in the presence of multiple oral doses of PF-06751979 100 mg QD, there was a small decrease in both midazolam AUCinf (12%) and Cmax (11%). Neither reduction was considered to be clinically meaningful.
PD
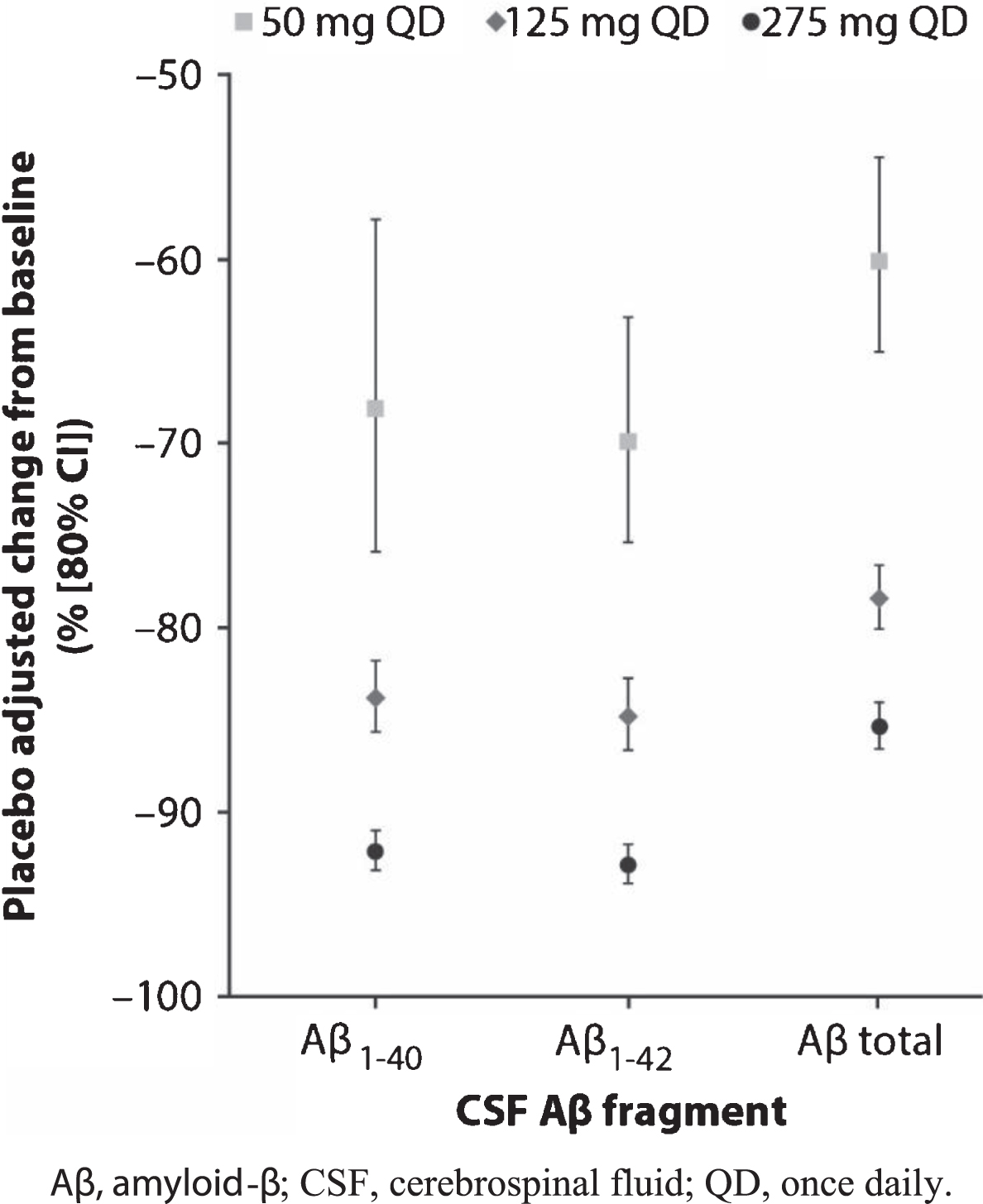
Robust and dose-dependent reductions from baseline in CSF Aβ fragments were observed following multiple doses of PF-06751979 ranging from 50–275 mg QD in Part B (Table 5; Fig. 1). The maximum observed placebo-adjusted reductions for Aβ1–40 and Aβ1–42 were 92.1% and 92.9%, respectively, at 275 mg QD. Reductions in the other CSF Aβ fragments analyzed (Aβ1–38, Aβx-38, Aβx-40, Aβx-42, and total Aβ) and sAβPPβ were similar. In all treatment groups, sAβPPα levels increased, with the greatest increase from baseline observed in the PF-06751979 275 mg QD treatment group (Table 5).
Table 5
Mean percent change from baseline to Day 15 (2-sided 80% CI) in CSF levels of Aβ fragments, sAβPPα, and sAβPPβ in healthy adults treated with multiple-ascending doses of PF-06751979 in studies B8271001 and B8271004
| CSF fragment | Percent change from baseline (2-sided 80% CI) | Placebo-adjusted percent change from baseline (2-sided 80% CI) | ||||||
| Study B8271001 | Study B8271004 | Study B8271001 | Study B8271004 | |||||
| Pooled PBO | PF-06751979 | Pooled | PF-06751979 | PF-06751979 | PF-06751979 | PF-06751979 | PF-06751979 | |
| (N = 4) | 50 mg (N = 8) | (N = 6) | 125 mg (N = 12) | 275 mg (N = 8) | 50 mg (N = 8) | 125 mg (N = 12) | 275 mg (N = 8) | |
| Aβ1–38 | –2.9 (–12.48, 7.63) | –58.4 (–61.34, –55.28) | –4.2 (–12.33, 4.63) | –80.3 (–81.51, –79.06) | –87.9 (–88.82, –86.98) | –57.2 (–62.28, –51.34) | –79.5 (–81.56, –77.11) | –87.4 (–88.80, –85.84) |
| Aβ1–40 | –6.5 (–24.69, 16.18) | –70.2 (–74.20, –65.52) | –10.0 (–18.48, –0.71) | –85.5 (–86.45, –84.38) | –92.9 (–93.54, –92.25) | –68.1 (–75.87, –57.86) | –83.8 (–85.65, –81.78) | –92.1 (–93.15, –90.98) |
| Aβ1–42 | –6.9 (–20.86, 9.61) | –71.9 (–74.97, –68.57) | –10.3 (–19.21, –0.48) | –86.4 (–87.40, –85.28) | –93.6 (–94.22, –92.93) | –69.9 (–75.38, –63.15) | –84.8 (–86.64, –82.74) | –92.9 (–93.85, –91.74) |
| Aβ total | –9.8 (–18.95, 0.30) | –64.0 (–66.62, –61.25) | –7.9 (–13.71, –1.73) | –80.1 (–81.01, –79.18) | –86.5 (–87.26, –85.74) | –60.1 (–65.03, –54.49) | –78.4 (–80.06, –76.62) | –85.4 (–86.57, –84.05) |
| Aβx-38 | –0.9 (–12.16, 11.87) | –60.2 (–63.42, –56.67) | –12.4 (–20.19, –3.77) | –82.0 (–83.18, –80.74) | –90.1 (–90.91, –89.23) | –59.8 (–65.42, –53.36) | –79.5 (–81.69, –76.96) | –88.7 (–90.06, –87.18) |
| Aβx-40 | 2.8 (–9.29, 16.49) | –60.4 (–63.71, –56.76) | –9.8 (–17.46, –1.40) | –81.0 (–82.16, –79.76) | –87.6 (–88.55, –86.62) | –61.5 (–66.98, –55.01) | –78.9 (–81.11, –76.52) | –86.3 (–87.82, –84.55) |
| Aβx-42 | –3.0 (–14.10, 9.64) | –64.3 (–67.21, –61.11) | –10.9 (–17.00, –4.29) | –81.9 (–82.76, –80.91) | –86.6 (–87.43, –85.76) | –63.2 (–68.37, –57.21) | –79.6 (–81.36, –77.78) | –85.0 (–86.34, –83.50) |
| sAβPPα | –9.9 (–18.34, –0.69) | 48.4 (38.56, 58.98) | –8.2 (–15.85, 0.06) | 58.7 (48.89, 69.05) | 81.7 (67.79, 96.85) | 64.8 (46.09, 85.94) | 72.9 (55.37, 92.40) | 98.0 (75.92, 122.96) |
| sAβPPβ | –13.8 (–20.39, –6.56) | –70.6 (–72.23, –68.93) | –5.6 (–12.11, 1.49) | –81.3 (–82.21, –80.27) | –84.3 (–85.28, –83.26) | –65.9 (–69.15, –62.40) | –80.2 (–81.84, –78.33) | –83.4 (–84.92, –81.69) |
Aβ, amyloid-β; CI, confidence interval; CSF, cerebrospinal fluid; sAβPP, soluble AβPP.
Fig.1
Mean placebo-adjusted changes from baseline to Day 15 in CSF levels of Aβ1–40, Aβ1–42, and total Aβ in healthy adults treated with multiple-ascending doses of PF-06751979 in studies B8271001 and B8271004. Aβ, amyloid-β; CSF, cerebrospinal fluid; QD, once daily.
Dose-dependent reductions from baseline in plasma Aβ fragments were observed following single-ascending and multiple doses of PF-06751979 in Parts A and B, respectively (Supplementary Table1). The maximum observed placebo-adjusted reductions for Aβ1–40 fragments following single-ascending doses was 90.3% with PF-06751979 540 mg (48 h post-dose), and following multiple-doses was 88.3% with PF-06751979 275 mg (on Day 1).
PK/PD modelling for CSF Aβ1–40 and Aβ1–42
The inhibitory effects of PF-06751979 on CSF Aβ40 and Aβ42 levels were estimated to be equal with an Imax of 0.984 (relative standard error [RSE], 1.71%) and IC50 of 63.7 ng/mL (RSE, 20.3%) characterized for both Aβ species simultaneously.
Goodness-of-fit plots of observed Aβ concentrations versus population- and individual-predicted Aβ concentrations showed that the PK/PD model described observed PD data well (data not shown).
Based on model parameter estimates and PK variability, the individual PD responses at steady state were simulated. It is predicted that PF-06751979 30, 75, and 110 mg QD, will result in respective average reductions of approximately 60%, 80%, and 85% of CSF Aβ1–40 and Aβ1–42 fragments, in approximately 50% of subjects at steady state (Supplementary Figure 2).
DISCUSSION
Three Phase I studies have provided insights into the safety, tolerability, PK, and PD effects of PF-06751979, a potent and selective oral BACE1 inhibitor that is being investigated as a potential treatment for AD.
Currently, investigational treatments targeting Aβ, such as anti-Aβ antibodies and BACE1 inhibitors, are the most advanced approaches in clinical development [16]. However, several trials in prodromal AD through moderate dementia have failed to show positive benefit:risk ratios or were stopped at interim analysis [26–28]. Recent evidence that Aβ deposition and measurable changes in brain biomarkers occur years before the onset of dementia suggests that, in these trials, treatment may have been given too late during the course of the disease to be effective [29, 30]. In addition, while treatment with MK-8931 reduced soluble Aβ levels in one study of mild-to-moderate dementia [26], data obtained using amyloid positron emission tomography in the same study indicated that amyloid plaque was reduced only slightly over 18 months, probably because of slow physiological clearance. This suggests that removal of soluble Aβ forms alone may not be sufficient to halt the Aβ-driven pathology in AD, and substantial amounts of deposited Aβ remain in the patient’s brain. The clinical development of drugs that target Aβ, particularly BACE inhibitors, is thus increasingly focused on treatment during the earlier stages of AD when disease-modifying therapy targeting Aβ will presumably be of greatest benefit (i.e., individuals with preclinical AD or minimal cognitive impairment) [30].
Mild cognitive worsening and behavioral disturbances have recently been reported with several BACE1 inhibitors [26, 31]. Whether this finding is the result of an off-target liability, such as BACE2 inhibition; an on-target, off-mechanism effect on a non-Aβ substrate such as neuregulin or seizure protein 6; or an on-target, on-mechanism effect such as interference with the physiologic role of Aβ is unknown. In addition to targeting patients who are earlier in the AD spectrum, future trials of BACE1 inhibitors may need to test doses that yield lower levels of inhibition in order to preserve normal synaptic functioning [32, 33].
Other than abnormal dreams, which were observed in the multiple-dose study of PF-06751979, no psychiatric or cognitive AEs were seen in the studies described in this paper. It should be noted, however, that given the low subject numbers and short duration of exposure in these studies, as well as the absence of dedicated cognitive testing, the three Phase I studies have limited sensitivity to detect such effects.
A major challenge in the development of BACE1 inhibitors for the treatment of AD is the selectivity over related aspartyl proteases such as CatD and BACE2. Although the small-molecule BACE inhibitors MK-8931 and AZD3293 have good selectivity for BACE1 over CatD [34, 35], they still exhibited undesirable characteristics, such as hair depigmentation in animal studies, presumably due to high BACE2 inhibition [15, 23]. Another BACE1 inhibitor currently in Phase III, CNP520, did not result in any hair depigmentation after long-term treatment in mice or dogs despite this compound displaying only a 3-fold selectivity for BACE1 over BACE2 in vitro. This in vitro–in vivo difference could potentially be due to low drug concentration in the skin tissues relative to IC50 for BACE2. Currently, CNP520 is being studied in two long-term pivotal clinical prevention trials in subjects at risk for AD (Generation studies 1 and 2) [36]. PF-06751979 displayed 6.4- and 26.6-fold selectivity for BACE1 over BACE2 in vitro in a fluorescence polarization assay and a binding assay, respectively [19]. In pigmented C57BL/6J mice, after 3 weeks of treatment with PF-06751979 or non-selective BACE inhibitors, hair color (pigmentation) change was not observed in the PF-06751979-treated mice while nonselective BACE inhibitors produced light color hair (hypo-pigmentation) in the mice [19]. In addition, chronic dosing of PF-06751979 for up to 9 months in dogs did not reveal any evidence of hair color change [19]. Together, the in vivo and in vitro data consistently suggested PF-06751979 is a very selective BACE1 inhibitor, a key differentiator over other BACE1 inhibitors that are nonselective against BACE2 [19]. The potential clinical importance of BACE1 inhibition selectivity remains to be fully elucidated, which will require an extended treatment duration in an appropriately designed study.
Overall, PF-06751979 was shown to be well tolerated with an acceptable safety profile in healthy adults and healthy older subjects following single- and multiple-dose administration, up to 2 weeks. TEAEs tended to become more frequent with increasing dose of PF-06751979, but there were no severe or serious AEs. Across the three Phase I studies, liver enzyme elevations, specifically of ALT/AST, were seen in three subjects treated with PF-06751979 and one subject treated with placebo. Liver toxicity has previously been seen with some BACE1 inhibitors, with concerns over abnormal liver enzymes leading to the termination of a Phase I study of RG7129 [37], a Phase II study of LY2886721 [38], and Phase II and Phase IIb/III studies of JNJ-54861911 [39]. However, for several other BACE1 inhibitors, including MK-8931, AZD3293, and CNP520, liver toxicity was not reported in clinical studies [14, 15, 26, 36], suggesting that the liver toxicity observed with some BACE1 inhibitors may not be mechanism-related. It is notable that the ALT/AST elevations seen with PF-06751979 were transient, asymptomatic, and were not accompanied by any additional abnormalities reflecting impaired liver function. We cannot exclude the possibility that longer dosing periods would lead to an increased risk of liver toxicity; therefore, liver function will continue to be monitored in studies of PF-06751979.
As BACE1 inhibition typically requires a basic amine for interaction with the catalytic aspartic acids at the active site of the BACE1 enzyme [40], BACE inhibitors in general are potential hERG ligands [36]. Most BACE1 inhibitors, as reported, inhibited hERG in in vitro studies [15, 17, 36], and displayed a dose-dependent effect on QT prolongation in early clinical studies [15, 17, 18]. However, these QT effects were small and not clinically significant at up to the highest clinically relevant (supratherapeutic) doses. In vitro and in vivo preclinical safety and toxicology studies of PF-06751979 have shown weak hERG inhibition [19]. In the SAD and MAD studies, a small but dose-dependent increase in QTc interval was observed in study B8271004 (higher doses). After a single dose of PF-06751979 at the highest dose of 540 mg, the maximum placebo-adjusted mean QTcF change from baseline was 14.9 ms (90% CI: 7.6, 22.2). In the MAD study, the maximum placebo-adjusted mean QTcF change from baseline was 12.8 ms (90% CI: 4.5, 21.1) after PF-06751979 275 mg QD for 14 days. Despite this small QT effect, no clinically meaningful findings (AEs) related to QTc and other ECG parameters were observed in healthy adults or older subjects in both studies. A preliminary concentration–QT analysis suggested that the maximum QTcF increase would be less than 10 ms at the upper bound of the 90% CI at the projected supratherapeutic dose of PF-06751979 (such as∼3-fold of top clinical dose anticipated; data not shown). A definitive QT study may be performed in the future to confirm the QT effect.
PF-06751979 had a PK profile well suited for QD dosing, with t1/2 ranging from 29–39 h in healthy adults, and approximately 40 h in healthy older subjects. Steady state was achieved by Day 7 in both patient populations. The Aβ profiles after multiple doses of PF-06751979 further support the hypothesis that QD dosing may be appropriate in future clinical studies. In addition, the food effect was minimal. Based on in vitro experiments, PF-06751979 was identified as a weak metabolism-dependent inhibitor of CYP3A4/5 (data on file); however, the B8271007 study, using the CYP3A4/5 probe midazolam, showed no drug interaction with midazolam PK in vivo.
Dose-dependent and robust Aβ (Aβ40 and Aβ42) reductions have been reported for other BACE1 inhibitors in clinical studies [14, 15, 41, 42]. These studies reported that Aβ reductions in CSF and plasma were observed after single- [14, 15, 41] and multiple-dose treatment [14, 15, 41, 42]. Furthermore, following multiple once-daily doses of JNJ-54861911, reductions were sustained over the 24-h interval at steady state [41]. These data also suggested that CSF Aβ reductions were very similar in magnitude in healthy adults [41], healthy older subjects [14, 41], and patients with mild-to-moderate AD [14, 15, 42]. Based on these data, we collected a single lumbar puncture at steady state to evaluate CSF PD effects after multiple-dose administration. CSF levels of Aβ1–40, Aβ1–42, and other Aβ fragments were robustly reduced after 14 days of dosing with PF-06751979 50 mg, 125 mg, or 275 mg QD. Reductions in CSF Aβ fragments were dose-dependent, with the greatest reductions from baseline observed in the 275 mg QD treatment group (92.1% and 92.9%, respectively). In addition, dose-dependent reductions of plasma levels of Aβ1–40, Aβx-40, and total Aβ occurred after a single dose of PF-06751979 and were sustained over the dosing interval (24 h) at steady state following multiple-dose administration.
The CSF Aβ fragment reductions observed with PF-06751979 were in line with those previously reported following the administration of other BACE1 inhibitors. Based on PK/PD modeling of the Phase I data reported here, about 60% and 80% reductions in CSF Aβ1–40 or Aβ1–42 fragments are expected following extended treatment with PF-06751979 30 and 75 mg QD, respectively. This is comparable to the reductions achieved by other BACE1 inhibitors at clinical doses studied in Phase III trials. For example, AZD3293 15 and 50 mg QD have been shown to produce approximately 51% and 76% reductions in CSF Aβ, respectively, in healthy older subjects, with similar effects seen in patients with mild-to-moderate AD [14]. MK-8931 reduced steady state CSF Aβ40 by an average of 57% and 79% in mild-to-moderate AD patients at 12 and 40 mg QD, respectively [15, 42]. Based on combined results observed in healthy adults and older subjects, JNJ-54861911 reduced CSF Aβ40 by approximately 50% and 80% at 5 and 25 mg QD, respectively, with similar reductions in CSF Aβ42 levels observed [41]. Taking this evidence together, a dose range of PF-06751979 25 to 75 mg QD, targeting various levels (modest to high) of Aβ lowering in the brain, may be appropriate to test in future studies.
While PF-06751979 demonstrated an acceptable safety profile, favorable PK characteristics and robust PD effect for future development during these Phase I studies, there are limitations associated with these results, including small sample size, short treatment duration (up to 2 weeks), and the inclusion of only healthy volunteers. As discussed above, the risk of PF-06751979 induced liver toxicity would need to be further addressed in larger study populations with treatment duration beyond 2 weeks.
Overall, the safety/tolerability, PK and PD profiles characterized in these Phase I studies suggest that PF-06751979 is suitable for further clinical development.
DATA SHARING STATEMENT
Upon request, and subject to certain criteria, conditions, and exceptions, see (https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual de-identified participant data from Pfizer-sponsored global interventional clinical studies conducted for medicines, vaccines and medical devices (1) for indications that have been approved in the US and/or EU or (2) in programs that have been terminated (i.e., development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de-identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
ACKNOWLEDGMENTS
The authors would like to thank the study subjects and investigators. The studies were sponsored by Pfizer Inc. Medical writing support under the guidance of the authors was provided by Kirsten Woollcott, MSc, on behalf of CMC Connect, a division of McCann Health Medical Communications Ltd, Glasgow, UK, and was funded by Pfizer Inc, New York, NY, USA in accordance with Good Publication Practice (GPP3) guidelines (Ann Intern Med 2015;163:461–464).
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/19-0228r1).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JAD-190228.
REFERENCES
[1] | World Health Organization (2017) Dementia: key facts. http://www.who.int/news-room/fact-sheets/detail/dementia. Accessed on: October 24, 2018. |
[2] | Wortmann M ((2012) ) Dementia: a global health priority – highlights from an ADI and World Health Organization report. Alzheimers Res Ther 4: , 40. |
[3] | Cummings JL , Morstorf T , Zhong K ((2014) ) Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res Ther 6: , 37. |
[4] | Anand R , Gill KD , Mahdi AA ((2014) ) Therapeutics of Alzheimer’s disease: Past, present and future. Neuropharmacology 76 Pt A: , 27–50. |
[5] | Selkoe DJ ((1991) ) The molecular pathology of Alzheimer’s disease. Neuron 6: , 487–498. |
[6] | Hardy J , Allsop D ((1991) ) Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci 12: , 383–388. |
[7] | Winkler E , Kamp F , Scheuring J , Ebke A , Fukumori A , Steiner H ((2012) ) Generation of Alzheimer disease-associated amyloid β42/43 peptide by γ-secretase can be inhibited directly by modulation of membrane thickness. J Biol Chem 287: 21326–21334. |
[8] | Murphy MP , LeVine H , 3rd ((2010) ) Alzheimer’s disease and the amyloid-beta peptide. J Alzheimers Dis 19: , 311–323. |
[9] | Burdick D , Soreghan B , Kwon M , Kosmoski J , Knauer M , Henschen A , Yates J , Cotman C , Glabe C ((1992) ) Assembly and aggregation properties of synthetic Alzheimer’s A4/beta amyloid peptide analogs. J Biol Chem 267: , 546–554. |
[10] | Yan R , Vassar R ((2014) ) Targeting the β secretase BACE1 for Alzheimer’s disease therapy. Lancet Neurol 13: , 319–329. |
[11] | Chow VW , Mattson MP , Wong PC , Gleichmann M ((2010) ) An overview of APP processing enzymes and products. Neuromolecular Med 12: , 1–12. |
[12] | Basi G , Frigon N , Barbour R , Doan T , Gordon G , McConlogue L , Sinha S , Zeller M ((2003) ) Antagonistic effects of beta-site amyloid precursor protein-cleaving enzymes 1 and 2 on beta-amyloid peptide production in cells. J Biol Chem 278: , 31512–31520. |
[13] | Ahmed RR , Holler CJ , Webb RL , Li F , Beckett TL , Murphy MP ((2010) ) BACE1 and BACE2 enzymatic activities in Alzheimer’s disease. J Neurochem 112: , 1045–1053. |
[14] | Cebers G , Alexander RC , Haeberlein SB , Han D , Goldwater R , Ereshefsky L , Olsson T , Ye N , Rosen L , Russell M , Maltby J , Eketjall S , Kugler AR ((2017) ) AZD3293: Pharmacokinetic and pharmacodynamic effects in healthy subjects and patients with Alzheimer’s disease. J Alzheimers Dis 55: , 1039–1053. |
[15] | Kennedy ME , Stamford AW , Chen X , Cox K , Cumming JN , Dockendorf MF , Egan M , Ereshefsky L , Hodgson RA , Hyde LA , Jhee S , Kleijn HJ , Kuvelkar R , Li W , Mattson BA , Mei H , Palcza J , Scott JD , Tanen M , Troyer MD , Tseng JL , Stone JA , Parker EM , Forman MS ((2016) ) The BACE1 inhibitor verubecestat (MK-8931) reduces CNS β-amyloid in animal models and in Alzheimer’s disease patients. Sci Transl Med 8: , 363ra150. |
[16] | Lopez Lopez C , Caputo A , Liu F , Riviere ME , Rouzade-Dominguez ML , Thomas RG , Langbaum JB , Lenz R , Reiman EM , Graf A , Tariot PN ((2017) ) The Alzheimer’s Prevention Initiative Generation Program: evaluating CNP520 efficacy in the prevention of Alzheimer’s disease. J Prev Alzheimers Dis 4: , 242–246. |
[17] | Timmers M , Sinha V , Darpo B , Smith B , Brown R , Xue H , Ferber G , Streffer J , Russu A , Tritsmans L , Solanki B , Bogert J , Van Nueten L , Salvadore G , Nandy P ((2018) ) Evaluating potential QT effects of JNJ-54861911, a BACE inhibitor in single- and multiple-ascending dose studies, and a thorough QT trial with additional retrospective confirmation, using concentration-QTc analysis. J Clin Pharmacol 58: , 952–964. |
[18] | Lai RYK , Darpo B , Dayal S , Hall N , Chang MK , Albala B , Ferry J , Bhaskar R ((2017) ) Elenbecestat, a novel oral BACE inhibitor, has no clinically meaningful effect on QTC interval up to a supratherapeutic dose of 200 mg. Alzheimers Dement 13: , 250–251. |
[19] | O’Neill BT , Beck EM , Butler CR , Nolan CE , Gonzales C , Zhang L , Doran SD , Lapham K , Buzon LM , Dutra JK , Barreiro G , Hou X , Martinez-Alsina LA , Rogers BN , Villalobos A , Murray JC , Ogilvie K , LaChapelle EA , Chang C , Lanyon LF , Steppan CM , Robshaw A , Hales K , Boucher GG , Pandher K , Houle C , Ambroise CW , Karanian D , Riddell D , Bales KR , Brodney MA ((2018) ) Design and synthesis of clinical candidate PF-06751979: a potent, brain penetrant, β-site amyloid precursor protein cleaving enzyme 1 (BACE1) inhibitor lacking hypopigmentation. J Med Chem 61: , 4476–4504. |
[20] | Rochin L , Hurbain I , Serneels L , Fort C , Watt B , Leblanc P , Marks MS , De Strooper B , Raposo G , van Niel G ((2013) ) BACE2 processes PMEL to form the melanosome amyloid matrix in pigment cells. Proc Natl Acad Sci U S A 110: , 10658–10663. |
[21] | Shimshek DR , Jacobson LH , Kolly C , Zamurovic N , Balavenkatraman KK , Morawiec L , Kreutzer R , Schelle J , Jucker M , Bertschi B , Theil D , Heier A , Bigot K , Beltz K , Machauer R , Brzak I , Perrot L , Neumann U ((2016) ) Pharmacological BACE1 and BACE2 inhibition induces hair depigmentation by inhibiting PMEL17 processing in mice. Sci Rep 6: , 21917. |
[22] | Neumann U , Rueeger H , Machauer R , Veenstra SJ , Lueoend RM , Tintelnot-Blomley M , Laue G , Beltz K , Vogg B , Schmid P , Frieauff W , Shimshek DR , Staufenbiel M , Jacobson LH ((2015) ) A novel BACE inhibitor NB-360 shows a superior pharmacological profile and robust reduction of amyloid-β and neuroinflammation in APP transgenic mice. Mol Neurodegener 10: , 44. |
[23] | Cebers G , Lejeune T , Attalla B , Soderberg M , Alexander RC , Budd Haeberlein S , Kugler AR , Ingersoll EW , Platz S , Scott CW ((2016) ) Reversible and species-specific depigmentation effects of AZD3293, a BACE inhibitor for the treatment of Alzheimer’s disease, are related to BACE2 inhibition and confined to epidermis and hair. J Prev Alzheimers Dis 3: , 202–218. |
[24] | Zhang L , Beal SL , Sheiner LB ((2003) ) Simultaneous vs. sequential analysis for population PK/PD data I: best-case performance. J Pharmacokinet Pharmacodyn 30: , 387–404. |
[25] | Ahn JE , Carrieri C , Dela Cruz F , Fullerton T , Hajos-Korcsok E , He P , Kantaridis C , Leurent C , Liu R , Mancuso J , Mendes da Costa L , Qiu R ((2019) ) Pharmacokinetic and pharmacodynamic effects of a γ-secretase modulator, PF-06648671, on CSF amyloid-β peptides in randomized Phase 1 studies. Clin Pharmacol Ther, doi: 10.1002/cpt.1570. |
[26] | Egan MF , Kost J , Tariot PN , Aisen PS , Cummings JL , Vellas B , Sur C , Mukai Y , Voss T , Furtek C , Mahoney E , Harper Mozley L , Vandenberghe R , Mo Y , Michelson D ((2018) ) Randomized trial of verubecestat for mild-to-moderate Alzheimer’s disease. N Engl J Med 378: , 1691–1703. |
[27] | Merck (2018) Merck announces discontinuation of APECS study evaluating verubecestat (MK-8931) for the treatment of people with prodromal Alzheimer’s disease. http://investors.merck.com/news/press-release-details/2018/Merck-Announces-Discontinuation-of-APECS-Study-Evaluating-Verubecestat-MK-8931-for-the-Treatment-of-People-with-Prodromal-Alzheimers-Disease/default.aspx. Accessed on: September 5, 2018. |
[28] | AstraZeneca (2018) Update on Phase III clinical trials of lanabecestat for Alzheimer’s disease. https://www.astrazeneca.com/media-centre/press-releases/2018/update-on-phase-iii-clinical-trials-of-lanabecestat-for-alzheimers-disease-12062018.html. Accessed on: September 5, 2018. |
[29] | Sperling R , Mormino E , Johnson K ((2014) ) The evolution of preclinical Alzheimer’s disease: implications for prevention trials. Neuron 84: , 608–622. |
[30] | Selkoe DJ , Hardy J ((2016) ) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8: , 595–608. |
[31] | Clinical Trials on Alzheimer’s Disease ((2018) ) Is there a role for BACE inhibition in Alzheimer’s treatment? http://www.ctad-alzheimer.com/files/files/BACE%20inhibitors%20press%20release%2025%20Oct%202018%20_0.pdf. Accessed on: October 24, 2018 |
[32] | Knopman DS ((2019) ) Lowering of amyloid-beta by β-secretase inhibitors – some informative failures. N Engl J Med 380: , 1476–1478. |
[33] | Ou-Yang MH , Kurz JE , Nomura T , Popovic J , Rajapaksha TW , Dong H , Contractor A , Chetkovich DM , Tourtellotte WG , Vassar R ((2018) ) Axonal organization defects in the hippocampus of adult conditional BACE1 knockout mice. Sci Transl Med 10: , pii: eaao5620. |
[34] | Scott JD , Li SW , Brunskill AP , Chen X , Cox K , Cumming JN , Forman M , Gilbert EJ , Hodgson RA , Hyde LA , Jiang Q , Iserloh U , Kazakevich I , Kuvelkar R , Mei H , Meredith J , Misiaszek J , Orth P , Rossiter LM , Slater M , Stone J , Strickland CO , Voigt JH , Wang G , Wang H , Wu Y , Greenlee WJ , Parker EM , Kennedy ME , Stamford AW ((2016) ) Discovery of the 3-imino-1,2,4-thiadiazinane 1,1-dioxide derivative verubecestat (MK-8931)-a β-site amyloid precursor protein cleaving enzyme 1 inhibitor for the treatment of Alzheimer’s disease. J Med Chem 59: , 10435–10450. |
[35] | Eketjall S , Janson J , Kaspersson K , Bogstedt A , Jeppsson F , Falting J , Haeberlein SB , Kugler AR , Alexander RC , Cebers G ((2016) ) AZD3293: a novel, orally active BACE1 inhibitor with high potency and permeability and markedly slow off-rate kinetics. J Alzheimers Dis 50: , 1109–1123. |
[36] | Neumann U , Ufer M , Jacobson LH , Rouzade-Dominguez ML , Huledal G , Kolly C , Luond RM , Machauer R , Veenstra SJ , Hurth K , Rueeger H , Tintelnot-Blomley M , Staufenbiel M , Shimshek DR , Perrot L , Frieauff W , Dubost V , Schiller H , Vogg B , Beltz K , Avrameas A , Kretz S , Pezous N , Rondeau JM , Beckmann N , Hartmann A , Vormfelde S , David OJ , Galli B , Ramos R , Graf A , Lopez Lopez C ((2018) ) The BACE-1 inhibitor CNP520 for prevention trials in Alzheimer’s disease. EMBO Mol Med 10: , e9316. |
[37] | Hung S-Y , Fu W-M ((2017) ) Drug candidates in clinical trials for Alzheimer’s disease. J Biomed Sci 24: , 47. |
[38] | May PC , Willis BA , Lowe SL , Dean RA , Monk SA , Cocke PJ , Audia JE , Boggs LN , Borders AR , Brier RA , Calligaro DO , Day TA , Ereshefsky L , Erickson JA , Gevorkyan H , Gonzales CR , James DE , Jhee SS , Komjathy SF , Li L , Lindstrom TD , Mathes BM , Martenyi F , Sheehan SM , Stout SL , Timm DE , Vaught GM , Watson BM , Winneroski LL , Yang Z , Mergott DJ ((2015) ) The potent BACE1 inhibitor LY2886721 elicits robust central Aβ pharmacodynamic responses in mice, dogs, and humans. J Neurosci 35: , 1199–1210. |
[39] | Janssen ((2018) ) Update on Janssen’s BACE Inhibitor Program. https://www.janssen.com/update-janssens-bace-inhibitor-program. Accessed on: September 10, 2018. |
[40] | Rajapakse HA , Nantermet PG , Selnick HG , Munshi S , McGaughey GB , Lindsley SR , Young MB , Lai MT , Espeseth AS , Shi XP , Colussi D , Pietrak B , Crouthamel MC , Tugusheva K , Huang Q , Xu M , Simon AJ , Kuo L , Hazuda DJ , Graham S , Vacca JP ((2006) ) Discovery of oxadiazoyl tertiary carbinamine inhibitors of beta-secretase (BACE-1). J Med Chem 49: , 7270–7273. |
[41] | Timmers M , van Broeck B , Ramael S , Slemmon J , Waepenaert KD , Russu A , Bogert J , Stieltjes H , Shaw LM , Engelborghs S , Moechars D , Marcken M , Liu E , Sinha V , Kemp J , Van Neuten L , Tritsmans L , Streffer JR ((2016) ) Profiling the dynamics of CSF and plasma Aβ reduction after treatment with JNJ-54861911, a potent oral BACE inhibitor. Alzheimers Dement 2: , 202–212. |
[42] | Forman M , Kleijn HJ , Dockendorf M , Palcza J , Tseng J , Canales C , Egan M , Kennedy M , Laterza O , Ma L , Scott J , Tanen M , Apter J , Backonja M , Ereshefsky L , Gevorkyan H , Jhee S , Rynders R , Zari A , Bryan E , Wagner J , Troyer MD , Stone JA ((2013) ) The novel BACE inhibitor MK-8931 dramatically lowers CSF beta-amyloid in patients with mild-to-moderate Alzheimer’s disease. Alzheimers Dement 9: , 139. |


