
Epitomic Characterization of the Specificity of the Anti-Amyloid Aβ Monoclonal Antibodies 6E10 and 4G8
Abstract
The monoclonal antibodies 6E10 and 4G8 are among the first anti-amyloid monoclonal antibodies against Aβ and the most widely used antibodies in Alzheimer’s disease research. Although the epitopes for 6E10 and 4G8 have been reported to correspond to residues 1–16 and 17–24, a more recent high-resolution mapping approach indicates that 6E10 maps to residues 4–10 while 4G8 maps to residues 18–23. To characterize the binding specificity of both antibodies in greater detail, we used immunoselection of random sequences from phage display library followed by deep sequencing and analysis of resulting patterns from thousands of immunoselected sequences. We found that the minimum sequence required for 6E10 binding is R-x-D with over half (53%) of the immunoselected sequences conforming to this pattern. The vast majority of these sequences contain an H at position x (R-H-D), corresponding to residues 5–7 of the Aβ target sequences, but Y is also permitted at this position in a minority of sequences. For 4G8 we found that the most frequent pattern is F-x-A contained in approximately 30% of the sequences, followed by F-A, L-x(3)-A, L-x-F, and F-F each accounting for approximately 18% of the sequences. The F-x-A motif also occurs in islet amyloid poly peptide which may explain why 4G8 also recognizes amyloid fibrils of this peptide. Immunoselection of random sequences and deep sequencing may also be a facile and efficient means of determining residues critical for antibody binding and validating the specificity of monoclonal antibodies and polyclonal antisera.
INTRODUCTION
Anti-amyloid antibodies have long been an important focus of research for neurodegenerative diseases such as Alzheimer’s disease (AD). They are important research tools and potential therapeutic agents [1]. Immunotherapy targeting amyloid-β (Aβ) is one of the leading approaches for therapeutic development including both active vaccination by using Aβ antigens and passive vaccination using the anti-Aβ antibodies [2]. Since active vaccination against Aβ was found to be associated with the occurrence of unacceptable levels of meningoencephalitis [3], more work has been focused on monoclonal antibodies [4]. The recent report that the monoclonal antibody Aducanumab slows cognitive decline in human clinical trials has heightened interest in anti-Aβ antibodies [5]. Although over 100 different monoclonal antibodies have been developed against amyloid Aβ, two of the most commonly used monoclonals are 6E10 and 4G8, which were among the first mouse monoclonals to be cloned and commercially available [6, 7]. Both 4G8 and 6E10 were raised against a 24 residue synthetic peptide identified in cerebrovascular amyloid corresponding to residues 1–24 of Aβ [6]. Although the epitopes of 4G8 and 6E10 have been described as corresponding to residues 17–24 and 1–16, respectively, the epitopes for these commonly used antibodies have not been characterized in detail until recently when a high resolution mapping approach using successive 10 residue segments of Aβ that overlap by one residue indicated that 6E10 maps to residues 4–10 while 4G8 maps to residues 18–23 [1].
Conformation dependent antibodies that are specific for epitopes associated with different aggregation states of Aβ are also very useful antibodies because they can distinguish among several different oligomeric and fibrillar structures and they do not react with monomeric Aβ or AβPP [1, 8]. Conformation dependent antisera such as A11, OC, and aAPF also recognize generic aggregation specific epitopes that are common to the same aggregation state of different amyloid forming proteins and peptides and independent of the underlying amino acid sequence [1, 9, 10]. The ability of these antibodies to recognize sequence independent epitopes is also displayed by many of the monoclonal antibodies derived from these antisera and it is also displayed by 4G8 even though all of the antibodies appear to recognize linear segments of the Aβ sequence [1, 9, 10]. The epitopes recognized by these antisera are unknown as is the molecular basis for why they recognize different aggregate structures in a sequence-independent fashion. The goal of this investigation is to characterize the specificity of 6E10 and 4G8 in detail. Here we used immunoselection of random sequences from a phage display library and deep sequencing to determine the epitome of peptide sequences that specifically bind to the antibodies and gain insight into how they recognize the specific aggregation state independently of the underlying amino acid sequence.
MATERIALS AND METHODS
Antibodies used
The monoclonal antibodies 4G8 and 6E10 were purchased from BioLegend, San Diego, CA, USA.
Phage immunoselection
Immunoselection of phage was carried out according to the manufacturer’s detailed directions according to the protocol labeled “Solution phase panning with affinity bead capture” using protein A conjugated magnetic beads (Dynabeads Protein A, 10002D, Novex Life Technologies). Briefly, 50μl of a 50% aqueous suspension of beads was mixed with 1 ml of TBS+0.1% Tween (TBST) and suspended by gentle vortexing. The beads were immobilized by magnetic capture and the supernatant pipetted away and discarded. A 100-fold representation of the library (2×1011 plaque forming units) and 1μg of antibody were mixed in a final volume of 200μl with TBST, added to the beads, mixed and incubated for 15 min at room temperature. The resin was then magnetically immobilized, and the supernatant removed and discarded, and the beads washed 10 times with 1 ml of TBST. The bound phages were eluted by suspending the beads in 1 ml of 0.2 M Glycine-HCl, pH 2.2, 1 mg/ml BSA, incubated for 10 min at room temperature and then immobilizing the beads and removing the supernatant and transferring it to a tube containing 150μl of 1 M Tris-HCl, pH 9.1. The eluted phages were amplified as described in the manufacturer’s protocol and the panning repeated two times.
Phage DNA isolation and library preparation
Phage DNA was isolated using a standard phenol:chloroform method [11]. Quality was assessed by visualization in a 1% agarose gel, and its concentration measured by spectrophotometry. The random sequences obtained by immunoselection were amplified by PCR in two steps. The first included the addition of an identifier sequence at the 5’ end which used to identify all of the sequences belonging to a particular sample in the NGS process. The first PCR product size after adding the identifiers is 253 bp, and the primers used were:
Reverse overhang primer sequence of – 96 gIII (5’GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGCCCTCATAGTTAGCGTAACG3’) and forward overhang primer sequence of – 28 gIII (5’TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGTTATTCGCAATTCCTTTAGTG3’.
The second PCR was to add indexes and adapters specific for the Illumina Mi-Seq NGS technology used which are necessary to distinguish and link the amplicons to the chip during the analysis. The final size of the amplicon after both PCR reactions is 340 bp. The barcoded phage DNA amplicons were pooled, and a 10 nM library was sequenced commercially on an Illumina MiSeq platform (Laragen Inc, Culver City, CA, USA).
Data analysis
In order to analyze the Illumina sequencing data, we wrote a program to extract the DNA sequences coding for the dodecapeptides and translate them to protein. Duplicates were removed and counted, and the peptide sequences were sorted by number of times it was observed and written to text file in FASTA format. The sequences were analyzed using PRATT 2.1 program (downloaded from http://ftp.ebi.ac.uk) [12], a tool to discover patterns conserved in a set of protein or peptide sequences. The program was edited and recompiled to accommodate 50,000 input sequences by editing the max_nr parameter definition from “max_nr = 10000;” to “max_nr = 50000;” in the “sequence.c” file and then recompiling the code. A number of parameters used by PRATT were adjusted as described in the text to optimize the epitope patterns found. Copies of the data set can be obtained from the corresponding author.
RESULTS
Immunoselection of random sequences from a phage display library has long been used to characterize the binding specificity of proteins and monoclonal antibodies [13, 14], before next generation sequencing has been available. The traditional approach recommended by the manufacturer of the library is to use the antibody to pan for phage that bind and then elute the phage and amplify them by infection of E. coli. This process is repeated two more times to enrich for the phage that specifically bind to the antibody and then individual phage plaques are selected and sequenced to reveal the common pattern of amino acids in the cognate phage. While this process should select for phage that specifically bind to the antibody, it may introduce bias in the observed sequences by selecting for phage sequences that amplify more rapidly than others. Traditionally, individual plaques were purified, sequenced and the sequences aligned to identify amino acid residues that are important for binding. Next generation sequencing eliminates the need for purifying individual plaques because all of the phage that bind can be sequenced simultaneously. Sequences that bind non-specifically to the protein A magnetic beads and all antibodies can be easily identified in appropriate controls and subtracted from the sequences specific to the antibodies of interest, potentially eliminating the need for subsequent pannings. We sequenced all three sequential pannings after amplification and also sequenced the 3rd panning after elution of the phage and before amplification to test the effect of phage replication on observed sequence abundance and diversity.
For 4G8 antibody, we obtained 8,068 unique sequences for the first amplified panning round, 826 for the second amplified round, 5,923 for the third unamplified panning, and 13,555 for the third amplified round. It is not clear why there were significantly fewer unique sequences in the second amplified round, but it may be due to an inefficient sequencing run due to a poor PCR amplification product rather than a failure of the phage to replicate because the number of unique sequences increased greatly in the next panning and these sequences must have been present in the second round. We ranked the sequences according to the number of times a particular sequence was encountered in a sequencing run and the top 32 sequences are shown in Fig 1. The top ten sequences for the first amplified panning were color coded to make their identification more readily apparent. Of the top 10 sequences in the first panning, all of them are found in the top 32 sequences in the second panning and 9 out of 10 sequences are found in the top 32 sequences are found in the third amplified and unamplified panning. Sequence #9 (TLSQVFHADEWL) fell out of the top 32 and is ranked 72 and 77 in the third amplified and unamplified, respectively. These results indicate that while most of the top sequences remain in the top 32 in subsequent pannings, some sequences are relatively less abundant while some sequences increase in ranking. The total number of unique sequences actually increases from the first to the third amplified round (8,068 versus 13,555), indicating that few unique sequences are lost during panning. However, the total number of times a unique sequence was observed increases greatly, possibly due to replication of the phage. Many of the top 10 sequences in the first panning increase 10 to 100-fold in the third panning. In addition, we compared the unamplified and amplified (replicated) sequences in the third round and found that while there were more than twice as many unique sequences identified in the amplified sample, the top sequences were very similar, indicating that phage replication may not be necessary if enough phage are eluted from the antibody bound to the magnetic beads. These results suggest that it is possible to just sequence the ensemble of initially immunoselected phage and that the absence of a need to replicate the phage and conduct multiple pannings would be important for high throughput applications of this approach.
Fig. 1.
Top ranked 32 unique sequences for 4G8. The top 10 sequences in the first panning are color coded to make their recognition more readily apparent. Of the top 10 sequences in the first panning, all ten are found in the top 32 of the second panning and 9 out of 10 are found in the third round of panning, indicating that their relative abundance does not change much with subsequent pannings.
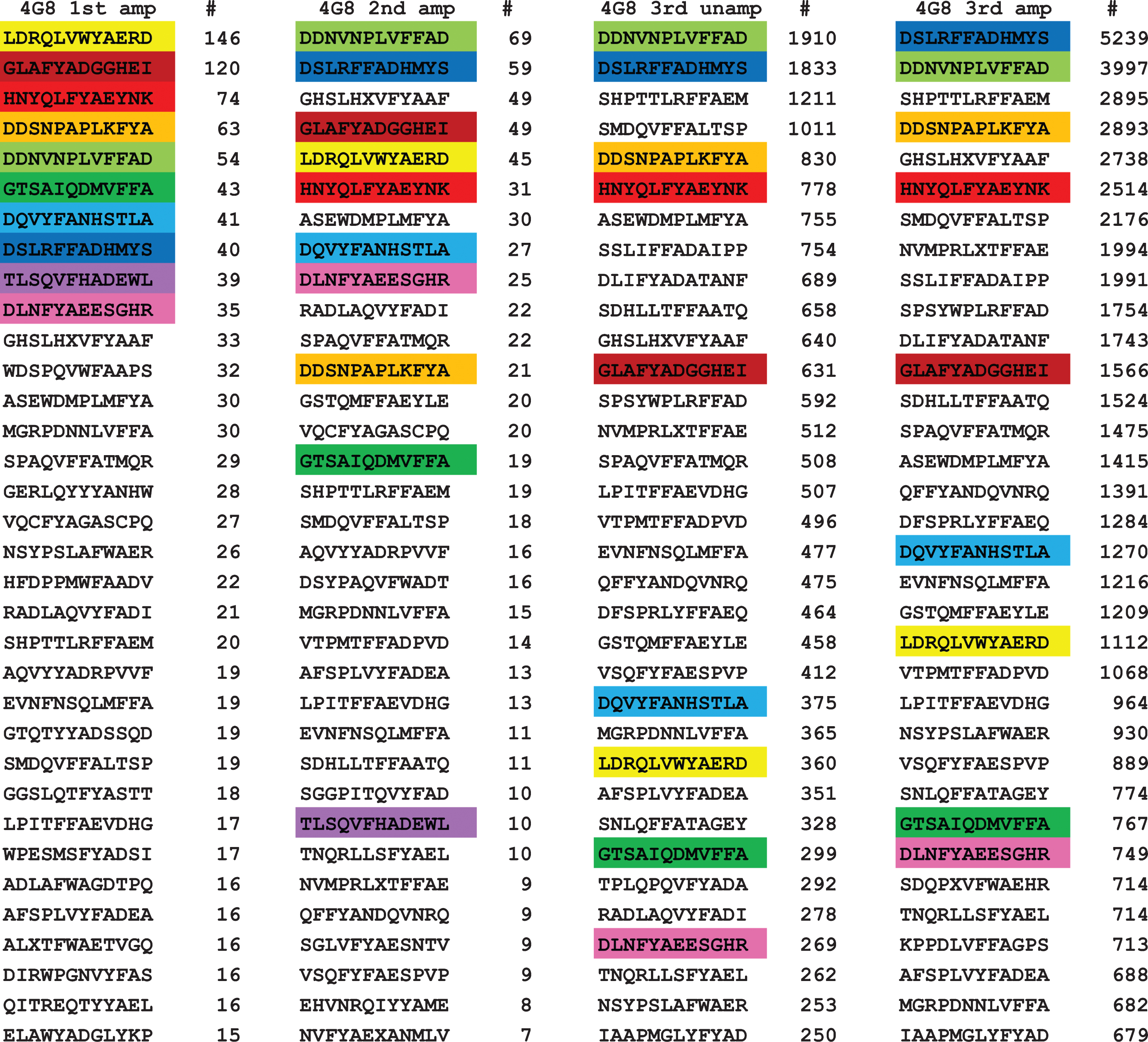
It is readily apparent from visual inspection of the data that most of the top ranked sequences contain a sequence segment related to the putative epitope of 4G8 (residues 17–23 of Aβ, LVFFAED) [1]. For example, sequence 5 from the first panning, DDNVNPLVFFAD, contains the subsequence LVFFA, which is a close match to the putative epitope. In order to obtain a more detailed understanding of the critical sequence elements necessary for 4G8 binding, we analyzed the sequences from the first amplified panning using the protein and peptide sequence pattern recognition program Pratt 2.1 [15]. We edited the program and recompiled it to accommodate 50,000 sequences rather than 10,000 sequences in the program available for downloading. We also evaluated the effect of a number of Pratt parameters that are important for finding the most significant and abundant patterns. CM is the minimum number of the total input sequences to match in order to be scored as a pattern. If CM is set too high, Pratt will find no patterns and if set too low, Pratt will find thousands of patterns where most of the patterns are subpatterns of the dominant pattern. We evaluated CM values corresponding to approximately 50%, 10% 5%, and 1% of the total unique sequences. Although Pratt will find the same patterns at low CM values as high values, it is more difficult to discern the dominant patterns at low CM values. Other parameters that have an influence on the number of patterns include PL (pattern length), FN (maximum number of flexible length spacers), and E (search “greediness”). We chose values PL = 6, FN = 0, and E = 0 as optimal values. ON, the maximum number of patterns output to a text file, must be set high enough to accommodate the number of patterns found. Pratt outputs the pattern in Prosite notation with a fitness score that is a non-statistical indication of how unique or significant the pattern is, the number of “hits”, which is the number of times the pattern was observed in the sequence set and the number of sequences containing the indicated pattern. The number of hits can be greater than the number of sequences if the pattern occurs more than once within a sequence. The higher the fitness score, the more unique or non-random the pattern is. After analyzing the sequence set for patterns, Pratt refines the pattern by determining the set of preferred amino acid residues at each flexible X position and displays them in square brackets, e.g., [FHWY]. If no restricted subset is discernable, an X is displayed.
For a monoclonal antibody like 4G8, you would expect that most of the sequences would match a pattern defining the epitope that binds to the antibody. However, when you analyze the 13,555 sequences from the third amplified panning with a CM of 5,000, Pratt fails to find any patterns. The patterns obtained for the third amplified panning which contains the largest number of unique sequences was analyzed with CM values of 2,000 and 100 and the resulting patterns are shown in Table 1. The most abundant pattern found was F-x-A, with 3,841 sequences out of 13,555 conforming to this pattern. Other abundant patterns contained two of the residues from the sequence LVFFA, such as FF (2,430 sequences), FA (2,626 sequences), L-x(3)-A (2,563 sequences), and L-x-F (2,467 sequences). Dropping the minimum to 100 yields several patterns of higher fitness that match more closely the Aβ sequence from residues 17–23 that are much less common than the patterns found at the CM value of 2,000. For example, pattern 3 is LVFFA, which corresponds to residues 17–21. The number of sequences observed in the third panning containing LVFFA in Fig. 1 (3397 light green) is less than the number of sequences containing only FFA (5239, blue) even though the number of sequences observed in the first panning was roughly the same (40 versus 54) indicating that a higher number of residues matching the target sequence does not confer a detectable binding advantage. There are several other examples where FFA and FYA containing sequences seem to bind as efficiently as LVFFA, including 5 of the top 10 and 3 of the top 10 sequences in the 3rd amplified round that contain FYA rather than FFA. Using a low CM value of 100 also returns hundreds of lower fitness patterns, most of which are subpatterns of the higher fitness patterns (data not shown). The fact that many of the patterns contain critical binding residues every other amino acid suggests that 4G8 may prefer an extended β conformation for a binding site and the fact that the most common patterns contain only two critical residues out of the total of 5 residues from the target Aβ sequence suggests that 4G8 may have a relatively loose fit binding site.
Table 1
Top sequences patterns for 4G8 using CM 2000 and 100
| Top sequence patterns for 4G8 using CM = 2000 | |||
| Best Patterns before refinement: | |||
| fitness | hits (seqs) | Pattern | |
| 1: | 8.3401 | 2489 (2430) | F-F |
| 2: | 8.3401 | 2643 (2626) | F-A |
| 3: | 8.3401 | 2591 (2563) | L-x(3)-A |
| 4: | 8.3401 | 2534 (2467) | L-x-F |
| 5: | 8.3401 | 4049 (3841) | F-x-A |
| Best Patterns (after refinement phase): | |||
| fitness | hits (seqs) | Pattern | |
| 1: | 12.7631 | 2070 (2070 | F-[F-A-[ADEGNQST] |
| 2: | 11.7711 | 2054 (2051) | L-x-[FW]-x-A |
| 3: | 11.5192 | 2010 (2006) | L-x-F-x-[AS] |
| 4: | 10.9372 | 2037 (2035) | F-F-[ASV] |
| 5: | 10.6366 | 2007 (2007) | L-x(2)-[FHWY]-A |
| 6: | 10.5573 | 2139 (2084) | F-x-A-[ADES] |
| 7: | 8.3401 | 2643 (2626) | F-A |
| 8: | 8.3401 | 2591 (2563) | L-x(3)-A |
| Top sequence patterns for 4G8 using CM = 100 | |||
| Best Patterns before refinement: | |||
| fitness | hits (seqs) | Pattern | |
| 1: | 20.8503 | 217 (217) | L-x-F-F-A-D |
| 2: | 20.8503 | 155 (155) | S-L-x-F-F-A |
| 3: | 20.8503 | 134 (134) | L-V-F-F-A |
| 4: | 20.8503 | 168 (168) | P-L-x-F-F-A |
| 5: | 20.8503 | 139 (139) | L-T-F-F-A |
| 6: | 20.8503 | 129 (129) | L-x-F-Y-A-D |
| Best Patterns (after refinement phase): | |||
| fitness | hits (seqs) | Pattern | |
| 1: | 23.4972 | 109 (109) | L-R-F-F-A-[ADE] |
| 2: | 22.6198 | 112 (112) | L-[AV]-F-[FWY]-A-D |
| 3: | 22.0887 | 101 (101) | L-T-F-F-A-[ADEGNQST] |
| 4: | 22.0819 | 100 (100) | L-A-F-[FY]-A-[ADES] |
| 5: | 22.0548 | 102 (102) | L-[ERS]-F-[FWY]-A-D |
| 6: | 22.0510 | 103 (103) | P-L-[ASTV]-[FY]-F-A |
For 6E10 antibody, we obtained 7,214 unique sequences for the first amplified panning round, 5,858 for the second amplified, 7,722 for the third unamplified, and 9,791 for the third amplified round. Similar to what we observed for 4G8, the second amplified round of 6E10 was lower than the first. Figure 2 shows the top 32 sequences of 6E10 in the four rounds arranged also according to their frequency number among the sequences obtained. Once again, the top 10 sequences for the first amplified panning are color coded to facilitate visualization. Unlike 4G8, of the top 10 sequences from the 1st amplified panning, only 7 remained in the top 32 in the 2nd amplified panning, and only 4 remained in the 3rd unamplified and amplified panning. Three of these sequences fell out of the top 32 sequences, while only four sequences remained in the top 32 in the successive three rounds. Sequences number 2 and 6 (SVRHDAGFAPMQ, NGLSHMANRHDH respectively) of top 10 in the first amplified panning alternated between first and second place in the successive rounds. Sequence 3 of the first amplified panning (GGSGPHDHRHDA), while keeping its ranking in the second amplified panning at position 4, dropped to 10 and 21 in the following rounds of the third unamplified and amplified panning, respectively. Another sequence which ranked at 4 (TSVNNPNAYRHD) lost its ranking to 78, 340, and 289 in the successive rounds respectively. Unlike 4G8, only two of the top 10 sequences from the first amplified panning remained in the top 10 until the last the round. The remaining sequences kept decreasing in ranking in the successive rounds of panning and amplification. The number of unique sequences in the unamplified and amplified third panning were close with slight increase in the amplified (7,722 and 9,791, respectively), which reinforces the conclusion that phage replication may not be necessary.
Fig. 2.
Top ranked 32 unique sequences for 6E10. The top 10 sequences in the first panning were color coded to make their recognition more readily apparent. Of the top 10 sequences in the first panning, 7 were found in the top 32 of the second panning and 4 out of 10 were found in the third round of panning, indicating that their relative abundance does not change much with subsequent pannings.

Since the epitope of 6E10 has been variously described as residues 1–16 to residues 4–8, it is readily apparent that almost all of the top 32 contain residues 5–7 (RHD). Peptide 7 has the sequence IRYDTGSYHIH, which has a RYD sequence instead of RHD. Fewer sequences had the sequence RHDS (residues 5–8) such as sequence 10 (VPNSLKHVRHDS) from first amplified panning, and with much less frequency segment (FRYD) such as sequence 3 from the third unamplified panning (FRYDSGYELSSK).
Pratt analysis for 6E10 was carried out on the third amplified panning which had the highest number of unique sequences with different CM values as described above for 4G8. With the CM value set to 5,000 corresponding to approximately 50% of the Table 2. The highest fitness pattern observed is R-H-D-x-G, which corresponds to residues 5–9 of Aβ. The most abundant patterns are H-D, R-H and R-H-D. Setting the CM value to 100 or approximately 1% of the total sequences returns several patterns with a higher fitness score than R-H-D-x-G, but they all contain non-Aβ amino acid residues as part of the pattern. The results indicate that any two residues within residues 5–7 (R-H, H-D, D-S, R-x-D) and D-x-G is sufficient for 6E10 binding, while the highest binding exact match of the Aβ sequence RHD displayed by 4,045 of the 9,791 sequences.
Table 2
Top sequence patterns for 6E10 using CM 1000 and 100
| Top sequence patterns for 6E10 using CM = 1000 | |||
| Best Patterns (after refinement phase): | |||
| fitness | hits (seqs) | Pattern | |
| 1: | 16.6802 | 1096 (1095) | R-H-D-x-G |
| 2: | 15.1605 | 1137 (1137) | R-H-D-[ANS] |
| 3: | 12.5102 | 1680 (1679) | R-x-D-x-G |
| 4: | 12.5102 | 1266 (1265) | H-D-x-G |
| 5: | 12.5102 | 1160 (1159) | R-H-x(2)-G |
| 6: | 11.5192 | 1092 (1087) | H-D-[AS] |
| 7: | 11.5192 | 1018 (1015) | R-H-x-[AS] |
| 8: | 11.5192 | 1253 (1253) | R-x-D-[AS] |
| 9: | 10.2298 | 1034 (1029) | D-[ANSTV]-G |
| 10: | 9.7532 | 1036 (1035) | R-x(2)-[ADGNSTV]-G |
| 11: | 8.3401 | 1169 (1130) | D-S |
| 12: | 8.3401 | 1016 (1004) | S-x-R |
| 13: | 8.3401 | 2148 (2114) | D-x-G |
| 14: | 8.3401 | 2148 (2114) | H-x(2)-G |
| 15: | 8.3401 | 1905 (1903) | R-x(3)-G |
| Top sequence patterns for 6E10 using CM = 100 | |||
| Best Patterns (after refinement phase): | |||
| fitness | hits (seqs) | Pattern | |
| 1: | 20.8503 | 111 (111) | L-R-H-D-x-G |
| 2: | 20.8503 | 129 (129) | R-H-D-L-G |
| 3: | 20.8503 | 142 (142) | R-H-D-x-G-L |
| 4: | 20.8503 | 149 (149) | R-H-D-A-G |
| 5: | 20.8503 | 186 (186) | R-H-D-S-G |
| 6: | 20.8503 | 120 (120) | V-R-H-D-x-G |
| 7: | 19.8639 | 114(114) | S-[LV]-R-H-D |
| 8: | 19.8593 | 106 (106) | R-Y-D-[AS]-G |
| 9: | 19.8593 | 114 (114) | L-R-H-D-[AS] |
| 10: | 19.8527 | 121 (121) | R-[HY]-D-H-G |
| 11: | 19.8527 | 102 (102) | R-[HY]-D-x-G-A |
| 12: | 19.8527 | 100 (100) | S-R-[HY]-D-x-G |
| 13: | 19.8527 | 103 (103) | R-[HY]-D-T-G |
| 14: | 19.8527 | 117 (117) | R-[HY]-D-x-G-F |
| 15: | 19.8503 | 135 (135) | S-x-R-H-D-[AL] |
| 16: | 19.8503 | 116 (116) | R-H-D-H-[AG] |
DISCUSSION
We have determined the specificity of 6E10 and 4G8 using an epitomic approach of immunoselection of random 12mer sequences from a random sequence library followed by deep sequencing to obtain thousands of sequences for each antibody. The top ranked patterns of amino acids that bound to the antibody were determined by analysis with the pattern recognition algorithm Pratt 2.1. We used the manufacturers recommended procedure of three successive pannings followed by phage amplification. We sequenced each of these three amplified panning rounds and also the unamplified phage eluted after the third panning. Perhaps surprisingly, the number of unique sequences does not change very much between the first and last panning although the total number of sequences observed increases markedly. This may be due to the fact that background sequences from the protein-A-coupled magnetic beads and non-specific IgG have been digitally subtracted from the total sequences. Phage replication also appears to be unnecessary for high throughput applications that do not require an exhaustive enumeration of all binding sequences as the number of unique sequences observed for 6E10 after amplification in the 3rd round (9,792) is not very different than for the unreplicated phage DNA (7,723). This would also eliminate potential artifacts caused by preferential replication of some phage sequences.
For both antibodies, the minimum sequence required for binding is shorter than previous studies have indicated [1]. For 4G8, the largest target sequence epitope we determined here is LVFFA (residues 17 and 21) compared to LVFFAED (residues 17–23) as determined by the overlapping spots technique [1]. Similarly, the largest target sequence epitope for 6E10 is RHD (residues 5–7) while the epitope determined by the overlapping spots method is FRHDSGY (residues 4–10) [1]. These results explain one of the known properties of 6E10 which is that it is specific for human Aβ, which has an R residue at position 5, which is a G in mouse Aβ. Since R5 is a critical position for 6E10 binding, the lack of this R residue explains why mouse Aβ does not bind.
The data also suggests an explanation why 4G8 binds aggregated forms of IAPP fibrils while 6E10 does not [10]. 4G8 does not bind monomeric IAPP, suggesting that its aggregation into β-sheet structure is necessary for antibody binding. One possibility is that since only two adjacent amino acids or alternating pairs of amino acids is the minimum required for binding is that it is likely that the same pairs may occur on the β-sheet amyloid structures of IAPP. In contrast, 6E10 requires three adjacent residues: RHD. For example, IAPP contains the sequence FGA that fits the major F-x-A binding pattern observed for 4G8. This corresponds to residues 23–25 of IAPP and the F and A residues would be adjacent and on the same side of the β sheet in both Aβ and IAPP fibrils. In contrast, the epitope for 6E10, RHD, does not occur in IAPP. Alpha synuclein does not contain any of the two amino acid patterns determined for 4G8, but 4G8 binds more weakly to alpha synuclein fibrils than either Aβ or IAPP fibrils so perhaps a different binding mode occurs with alpha synuclein fibrils [10].
We have also identified positions of the sequence where the identity of the amino acid either does not matter or only a restricted set of amino acids are allowed. This is useful information for understanding the specificity of the antibody and may have utility for understanding the binding mode of the peptide and antibody. In the case where the identity of an amino acid is not important, this suggests that this site does not make contact with the antibody antigen combining site. In the case where a restricted number of amino acids is allowed, this suggests that the antibody paratope is either large enough or flexible enough to accommodate the range of amino acid side chains upon binding. This information could be very useful for molecular modeling of the antibody-epitope complex in the case where the atomic structure of the antibody is known, but not of the complex.
Epitomic characterization of antibody binding using random sequence phage immunoselection followed by deep sequencing is a rapid, facile and inexpensive means of determining the antibody specificity. Besides the obvious utility in mapping the epitope recognized, it may also have considerable utility in understanding the mechanism of antigen-antibody interaction and determining the effect of in vitro manipulation of the binding site to improve specificity. Since a large number of sequences can be obtained, it may also be useful for characterizing the specificity of multiple antibodies in serum or plasma samples and for validating and authenticating the specificity of the immunoreactivity across multiple serum collections and animals, which is a particularly difficult problem in immunology.
ACKNOWLEDGMENTS
This project was funded by the Deanship of Scientific Research (DSR) at King Abdulaziz University, Jeddah, Saudi Arabia, under grant No. 3-141-36-HiCi. The authors, therefore, acknowledge with thanks DSR technical and financial support. Work at UC Irvine was supported by a grant from the Cure Alzheimer’s Fund. We thank Mr. Jeffrey Glabe for help with modifying and recompiling the Pratt program.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/18-0582r2).
REFERENCES
[1] | Hatami A , Albay R 3rd , Monjazeb S , Milton S , Glabe C ((2014) ) Monoclonal antibodies against Abeta42 fibrils distinguish multiple aggregation state polymorphisms in vitro and in Alzheimer disease brain. J Biol Chem 289: , 32131–32143. |
[2] | Rygiel K ((2016) ) Novel strategies for Alzheimer’s disease treatment: An overview of anti-amyloid beta monoclonal antibodies. Indian J Pharmacol 48: , 629–636. |
[3] | Nicoll JA , Wilkinson D , Holmes C , Steart P , Markham H , Weller RO ((2003) ) Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: A case report. Nat Med 9: , 448–452. |
[4] | Wisniewski T , Goni F ((2015) ) Immunotherapeutic approaches for Alzheimer’s disease. Neuron 85: , 1162–1176. |
[5] | Sevigny J , Chiao P , Bussiere T , Weinreb PH , Williams L , Maier M , Dunstan R , Salloway S , Chen T , Ling Y , O'Gorman J , Qian F , Arastu M , Li M , Chollate S , Brennan MS , Quintero-Monzon O , Scannevin RH , Arnold HM , Eng-ber T , Rhodes K , Ferrero J , Hang Y , Mikulskis A , Grimm J , Hock C , Nitsch RM , Sandrock A ((2016) ) The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 537: , 50–56. |
[6] | Kim KS , Miller DL , Sapienza VJ , Chen CMJ , Bai C , Grundke-Iqbal I , Currie CJ , Wisniewski HM ((1988) ) Production and characterization of monoclonal antibodies reactive to synthetic cerebrovascular amyloid peptide. Neurosci Res Commun 2: , 121–130. |
[7] | Kim KS , Wen GY , Bancher C , Chen CMJ , Sapienza VJ , Hong H , Wisniewski HM ((1990) ) Detection and quantita-tion of amyloid beta-peptide with 2 monoclonal antibodies. Neurosci Res Commun 7: , 113–122. |
[8] | Glabe CG ((2008) ) Structural classification of toxic amyloid oligomers. J Biol Chem 283: , 29639–29643. |
[9] | Kayed R , Canto I , Breydo L , Rasool S , Lukacsovich T , Wu J , Albay R , 3rd, Pensalfini A , Yeung S , Head E , Marsh JL , Glabe C ((2010) ) Conformation dependent monoclonal antibodies distinguish different replicating strains or con-formers of prefibrillar Abeta oligomers. Mol Neurodegener 5: , 57. |
[10] | Hatami A , Monjazeb S , Glabe C ((2016) ) The anti-amyloid-beta monoclonal antibody 4G8 recognizes a generic sequence-independent epitope associated with alpha-synuclein and islet amyloid polypeptide amyloid fibrils. J Alzheimers Dis 50: , 517–525. |
[11] | Sambrook J , Russell DW ((2006) ) Purification of nucleic acids by extraction with phenol: Chloroform. CSH Protoc (2006) , pdb.prot4455. |
[12] | Jonassen I , Collins JF , Higgins DG ((1995) ) Finding flexible patterns in unaligned protein sequences. Protein Sci 4: , 1587–1595. |
[13] | Devlin JJ , Panganiban LC , Devlin PE ((1990) ) Random peptide libraries: A source of specific protein binding molecules. Science 249: , 404–406. |
[14] | Felici F , Castagnoli L , Musacchio A , Jappelli R , Cesareni G ((1991) ) Selection of antibody ligands from a large library of oligopeptides expressed on a multivalent exposition vector. J Mol Biol 222: ,301–310. |
[15] | Jonassen I ((1997) ) Efficient discovery of conserved patterns using a pattern graph. Bioinformatics 13: , 509–522. |


