Effect of a Comprehensive Intervention on Plasma BDNF in Patients with Alzheimer’s Disease
Abstract
A comprehensive intervention (CI) on patients with Alzheimer’s disease was assessed by measuring plasmabrain-derived neurotrophic factor (pBDNF) and ADAS-Cog score (ADAS-Cogscore) before, immediately after (FU1), and 6 (FU2) and 24 months (FU3) after the CI. Baseline pBDNF was higher in patients with moderate AD (but not mild AD) than in healthy controls. At FU1, pBDNF and ADAS-Cogscore decreased significantly. At FU2 and FU3, patients’ cognitive status worsened and pBDNF further increased versus baseline, suggesting that CI interruption may be a stress event that prevents return to homeostasis. CI exerted positive short-term effects, but more information is needed on long-term consequences.
INTRODUCTION
Over 46 million individuals were estimated to suffer from dementia in 2015; their number is expected to soar to 74.7 million in 2030 and 131.5 million in 2050 [1]. Alzheimer’s disease (AD) accounts for 50–56% of all dementia cases [2]. Since no effective pharmacological treatments are available [3], non-pharmacological strategies are urgently needed.
Cognitive stimulation is a promising approach. Epidemiological studies have shown the beneficial effects of a cognitively active life, where stimulation is due to leisure activities [4–7] rather than to the complexity of occupational tasks [8–10]. Cognitive stimulation protocols have also been seen to improve patients’ mood and mental function [11–16]. However, the mechanisms underlying such benefits remain elusive.
Brain-derived neurotrophic factor (BDNF) may be involved in the effects of cognitive stimulation, since evidence from animal models of AD suggests that its modulation might contribute to the benefit achieved by environmental enrichment [17–20]. Testing these findings in humans requires a simple, non-invasive, and inexpensive biological matrix. Blood is the ideal candidate. Circulating BDNF comes from a number of peripheral sources [21–23], but may also derive from neurons and glial cells. Indeed, some experimental work suggests that the blood-brain barrier is permeable to the neurotrophin, although the agreement is not unanimous [24, 25]. A significant correlation has been documented in rats between BDNF concentrations in brain tissue and serum [26, 27]; more importantly, peripheral BDNF seems to reflect neural viability and integrity in healthy humans [28] as well as widespread brain amyloidosis [29], although Laske and colleagues [30] failed to find a correlation between BDNF concentration in serum and cerebrospinal fluid in AD patients.
The above findings prompted us to test the effects of a comprehensive intervention (CI) on plasma BDNF (pBDNF) in AD patients, to assess the potential involvement of the protein in the effects exerted by the CI and explore the possibility of its biomarker use. The CI aimed primarily at cognitive enhancement through the improvement of some cognitive functions and of patients’ motivation to remain active— for instance via adoption of mnemonic strategies to compensate for deficits and/or participation in leisure activities and socialization— and also provided advice on lifestyle and psychological support to patients and caregivers.
MATERIALS AND METHODS
Participants were 85 healthy subjects (HE) and 80 patients with early AD. The study protocol complied with the principles of the Declaration of Helsinki (code SC/12/301) and was approved by the local ethics committee (INRCA Bioethics Advisory Committee, Ancona, Italy). The main characteristics of the two populations are summarized in Supplementary Table 1. AD staging was based on the Clinical Dementia Rating Scale (CDR) [31]: only patients with mild or moderate dementia (CDR 1 and 2) were included. The criteria for AD diagnosis and patient inclusion/exclusion in the study are described in the Supplementary Material.
The CI consisted of 10 once weekly sessions of about 1 h. Its effect on pBDNF levels was tested immediately after the training sessions (FU1) and then at 6 (FU2) and 24 (FU3) months. Values were then compared to baseline.
The possible influence of acetylcholinesterase inhibitors, benzodiazepines, antidepressants, lipid-lowering medications, non-steroidal anti-inflamm-atory drugs, anticoagulants, antihypertensives, antidiabetics, and corticosteroids on pBDNF levels was taken into account. In particular, the use/non-use (dichotomous variable) of each drug class was established at baseline (HE group) and at baseline and FU1, FU2, and FU3 (AD patients). For each drug class, pBDNF levels were compared in subjects taking/not taking the medication. Smoking habits and alcohol consumption were assessed as described by Gagliardi et al. [32].
Patients were randomly assigned to the experimental group (EG, n = 38) or the control group (CG, n = 42). Any further bias was excluded by comparing EG and CG patients for age, gender, marital status, years of schooling, and their scores on the Mini-Mental State Examination (MMSE), Geriatric Depression Scale (GDS), Activities of Daily Living (ADL), Instrumental ADL (IADL), and CDR. Significant differences were found for age (EG versus CG, 78.67±0.92 versus 76.11±0.72 years, p = 0.034) and ADL (4.95±0.20 versus 5.53±0.11, p = 0.015), leading to data adjustment for the two variables in all analyses. The CI was applied as described previously [33]. Performance on the Alzheimer’s Disease Assessment Scale-Cog test corrected for schooling (ADAS-Cogscore) was the outcome measure.
Lithium heparin whole blood was collected at baseline and then at FU1, FU2, and FU3.
The pBDNF assay, CI, and statistical analyses are described in detail in the Supplementary Material.
RESULTS
Baseline pBDNF
Baseline pBDNF was significantly higher (p = 0.001) in AD than HE subjects (1.464 [1.25–1.72] versus 0.984 [0.83–1.16] [back-transformed mean and 95% confidence interval]), but the difference ceased to be significant after adjustment for age and schooling. When AD patients were divided by disease stage, pBDNF was significantly higher in CDR 2 (n = 38) than HE subjects, also after the adjustment for age and schooling (p < 0.001), whereas the difference between HE and CDR 1 (n = 42) subjects was not significant (Fig. 1A).
pBDNF exhibited a significant, positive correlation with the MMSE score (p = 0.002) in HE subjects (Fig. 1B) and with the ADAS-Cogscore (p < 0.001) in AD patients (Fig. 1C), whereas it was unrelated to age, schooling, GDS score, smoking habits, alcohol intake, and medications and, for AD patients, disease duration and age at disease onset.
Effect of the CI
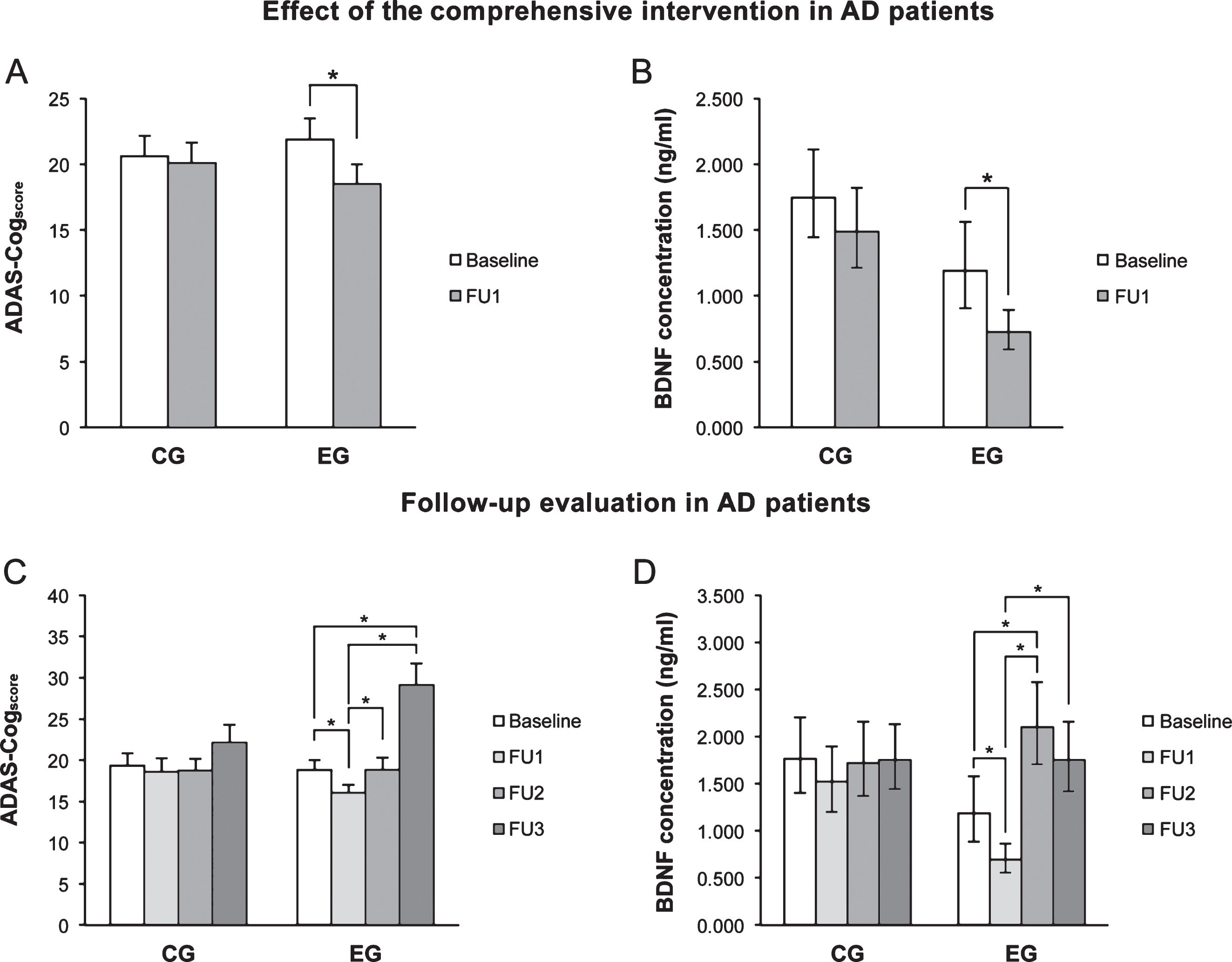
At FU1, the ADAS-Cogscore (Fig. 2A) and pBDNF (Fig. 2B) were significantly reduced in EG patients (both p < 0.001), whereas in controls the difference was not significant. A significant, negative correlation was found in EG patients between the baseline ADAS-Cogscore and pBDNF changes from baseline and FU1 (r = –0.460, p = 0.005). No significant correlation was found in controls, indicating that the phenomenon is training-induced.
Follow-up
In CG patients, the ADAS-Cogscore showed similar values from baseline through FU3, whereas in EG patients values fell significantly from baseline to FU1 (p = 0.010), they reverted to baseline at FU2 (p = 0.016 vs FU1), and then increased further at FU3 (p = 0.001 and p < 0.001 vs baseline and FU1, respectively) (Fig. 2C). In CG patients, pBDNF was unchanged from baseline to FU3, whereas in EG patients FU2 values were significantly higher compared to both baseline (p < 0.001) and FU1 (p < 0.001), while a slight reduction was seen at FU3 versus FU2 (p < 0.001 vs FU1) (Fig. 2D).
Medications did not influence pBDNF at any follow-up point.
DISCUSSION
The major findings of this study are i) that baseline pBDNF was significantly higher in patients with moderate AD than in healthy individuals; ii) that baseline pBDNF was not significantly different in patients with mild AD compared with healthy individuals; and iii) that the CI exerted a positive short-term and a potentially negative long-term effect irrespective of disease stage.
Reports of blood BDNF in AD are conflicting [34–45]. A possible explanation for the inconsistency is the lack of AD cohort overlap. The suggestion that changes in peripheral BDNF concentration may depend on AD stage [46] has not been widely explored; however, even the studies that have addressed the issue have applied non-uniform criteria to define disease severity (i.e., use of MMSE rather than CDR score), preventing comparison of their findings. The possible difference between plasma and serum BDNF may provide another source of bias [47]. Serum BDNF derives mainly from the thrombocyte storage pool, which is artificially released during experimental clotting [48, 49]. Platelets undergo several alterations during AD, including changes in membrane fluidity and cholesterol levels, abnormalities in cellular signal transduction, and atypical amyloid-β protein precursor metabolism [50]. By influencing thrombocyte functioning these phenomena may exert different, even opposite effects on serum and plasma BDNF, especially considering thrombocyte hyperactivation [51].
Although the inconsistent findings regarding circulating BDNF require further investigation, the present study provides information on the ability of pBDNF to be used to monitor AD progression, especially since it applied the CDR score for disease staging, excluded specific lifestyle influences, and investigated a broad medication panel including drugs that are commonly prescribed to the elderly but are not routinely evaluated, such as statins and antidiabetics [52, 53]. Although the mechanism underlying such stage dependence is unclear, it is conceivable that pBDNF increases as a consequence of advancing neuronal alteration. Support for the notion seems to come from the correlations, of different sign, detected between pBDNF level and cognitive status in our study participants, since higher pBDNF concentrations were associated to a better cognitive status in healthy controls and to a worse cognitive status in AD patients. The hypothesis has been advanced that increased circulating BDNF during physiological aging may be a neural reactive mechanism directed at preserving overall cognitive performance [54], whereas in AD it could reflect an attempt to counteract the deficits induced by amyloid-β accumulation [55, 56] and/or to provide a trophic stimulus to compensate for neuronal loss [57]. BDNF is known to play a key role in synaptic plasticity and neuronal survival [58] and to be involved in several functions that undergo age- and dementia-related impairment such as learning, memory, and stress adaptation [59–61]. In a recent study, gene delivery of BDNF in P301L transgenic mice has ameliorated several pathological features in this animal model of AD, suggesting a potential protective role for BDNF also in tauopathy [62]. It is thus reasonable to hypothesize that structural/functional impairment worsens as AD progresses, triggering a greater demand for BDNF, thus establishing a vicious circle.
As regards the CI, an improved cognitive status (a significantly decreased ADAS-Cogscore) and a significant pBDNF reduction were measured in AD patients immediately after the 10-week CI. Moreover, the significant negative correlation between baseline ADAS-Cogscore and pBDNF changes, detected in EG patients, suggests that the CI may exert a stronger effect on patients with more severe alterations, since higher baseline ADAS-Cogscore values were associated to a greater pBDNF reduction following the CI. Studies conducted in AD animal models show that environmental enrichment can reduce the amyloid-β burden, promote neurogenesis and cell survival, and improve synaptic transmission [63–66]. The CI tested in the present study may have induced similar effects in our patients. This would at least partly explain the pBDNF modulation exerted by the CI, since a positive effect on neural dysfunction may conceivably involve a reduced BNDF demand.
However, the positive effect of the CI was transient. The ADAS-Cogscore reverted to baseline at FU2 and exhibited a further increase at FU3, while pBDNF showed a rebound effect at FU2. These findings suggest that CI interruption may involve a risk. Indeed, whereas control patients showed constant ADAS-Cogscore and pBDNF values throughout the study, the experimental group suffered a deterioration of the cognitive status and a further increase in pBDNF concentration compared to baseline. The frailty of AD patients’ brain may at least partly explain these data, since a frail system exposed to a stressor usually fails to regain homeostasis after its end, and undergoes further deterioration [67]. CI interruption, and the default of the trigger that induced functional (ADAS-Cogscore) and molecular (pBDNF) changes at FU1 may have provided this type of stress event and have involved a worsening of the initial condition rather than a return to the previous equilibrium. This phenomenon, and the possible role of peripheral BDNF sources, deserve further investigation, since it cannot be excluded that the CI exerted secondary effects on circulating cells, which produce as well as store BDNF.
In conclusion, the present study showed that pBDNF, which is the “active” form of the molecule— i.e., the fraction that is available for crossing the blood-brain barrier through a saturable transport system [24, 68]— may serve as a peripheral biomarker to assess the effects of CIs in AD patients and indicated that more exhaustive information on the long-term consequences of CIs is needed before they can be introduced into clinical practice.
ACKNOWLEDGMENTS
The authors are grateful to the participants and their families and to Belinda Giorgetti and Moreno Solazzi for their technical assistance. The study was supported by “Ricerca Finalizzata” grant 154/GR-2009-1584108 (Italian Ministry of Health and Regione Marche).
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/16-1168r1).
Appendices
The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JAD-161168.
REFERENCES
[1] | Alzheimer’s Disease International. ((2015) ) World Alzheimer Report 2015: The Impact of Dementia, https://www.alz.co.uk/research/world-report-2015 |
[2] | Querfurth HW , LaFerla FM ((2010) ) Alzheimer’s disease. N Engl J Med 362: , 329–344. |
[3] | Schneider LS , Mangialasche F , Andreasen N , Feldman H , Giacobini E , Jones R , Mantua V , Mecocci P , Pani L , Winblad B , Kivipelto M ((2014) ) Clinical trials and late-stage drug development for Alzheimer’s disease: An appraisal from 1984 to 2014. J Intern Med 275: , 251–383. |
[4] | Wang HX , Karp A , Winblad B , Fratiglioni L ((2002) ) Late-life engagement in social and leisure activities is associated with decreased risk of dementia: A longitudinal study from Kungsholmen Project. Am J Epidemiol 155: , 1081–1087. |
[5] | Wilson RS , Bennett DA , Bienias JL , Aggarwal NT , Mendes De Leon CF , Morris MC , Schneider JA , Evans DA ((2002) ) Cognitive activity and incident AD in a population-based sample of older persons. Neurology 59: , 1910–1914. |
[6] | Wilson RS , Scherr PA , Schneider JA , Tang Y , Bennett DA ((2007) ) Relation of cognitive activity to risk of developing Alzheimer disease. Neurology 69: , 1911–1920. |
[7] | Sattler C , Toro P , Schonknecht P , Schroder J ((2012) ) Cognitive activity, education and socioeconomic status as preventive factors for mild cognitive impairment and Alzheimer’s disease. Psychiatry Res 196: , 90–95. |
[8] | Andel R , Crowe M , Pedersen NL , Mortimer J , Crimmins E , Johansson B , Gatz M ((2005) ) Complexity of work and risk of Alzheimer’s disease: A population-based study of Swedish twins. J Gerontol B Psychol Sci Soc Sci 60: , P251–P258. |
[9] | Karp A , Andel R , Parker MG , Wang HX , Winblad B , Fratiglioni L ((2009) ) Mentally stimulating activities at work during midlife and dementia risk after age 75: Follow-up study from the Kungsholmen. Project Am J Geriatr Psychiatry 17: , 227–236. |
[10] | Boots EA , Schultz SA , Almeida RP , Oh JM , Koscik RL , Dowling MN , Gallagher CL , Carlsson CM , Rowley HA , Bendlin BB , Asthana S , Sager MA , Hermann BP , Johnson SC , Okonkwo OC ((2015) ) Occupational complexity and cognitive reserve in a middle-aged cohort at risk for Alzheimer’s disease. Arch Clin Neuropsychol 30: , 634–642. |
[11] | Quayhagen MP , Quayhagen M , Corbeil RR , Hendrix RC , Jackson JE , Snyder L , Bower D ((2000) ) Coping with dementia: Evaluation of four nonpharmacologic interventions. Int Psychogeriatr 12: , 249–265. |
[12] | Spector A , Thorgrimsen L , Woods B , Royan L , Davies S , Butterworth M , Orrell M ((2003) ) Efficacy of an evidence-based cognitive stimulation therapy programme for people with dementia: Randomised controlled trial. Br J Psychiatry 183: , 248–254. |
[13] | Olazaran J , Muniz R , Reisberg B , Pena-Casanova J , del Ser T , Cruz-Jentoft AJ , Serrano P , Navarro E , Garcia de la Rocha ML , Frank A , Galiano M , Fernandez-Bullido Y , Serra JA , Gonzalez-Salvador MT , Sevilla C ((2004) ) Benefits of cognitive-motor intervention in MCI and mild to moderate Alzheimer disease. Neurology 63: , 2348–2353. |
[14] | Clare L , Linden DE , Woods RT , Whitaker R , Evans SJ , Parkinson CH , van Paasschen J , Nelis SM , Hoare Z , Yuen KS , Rugg MD ((2010) ) Goal-oriented cognitive rehabilitation for people with early-stage Alzheimer disease: A single-blind randomized controlled trial of clinical efficacy. Am J Geriatr Psychiatry 18: , 928–939. |
[15] | Maci T , Pira FL , Quattrocchi G , Nuovo SD , Perciavalle V , Zappia M ((2012) ) Physical and cognitive stimulation in Alzheimer disease. The GAIA Project: A pilot study. Am J Alzheimers Dis Other Demen 27: , 107–113. |
[16] | Ben-Sadoun G , Sacco G , Manera V , Bourgeois J , Konig A , Foulon P , Fosty B , Bremond F , d’Arripe-Lonqueville F , Robert P ((2016) ) Physical and cognitive stimulation using an exergame in subjects with normal aging, mild and moderate cognitive impairment. J Alzheimers Dis 53: , 1299–1314. |
[17] | Wolf SA , Kronenberg G , Lehmann K , Blankenship A , Overall R , Staufenbiel M , Kempermann G ((2006) ) Cognitive and physical activity differently modulate disease progression in the amyloid precursor protein (APP)-23 model of Alzheimer’s disease. Biol Psychiatry 60: , 1314–1323. |
[18] | Lahiani-Cohen I , Lourbopoulos A , Haber E , Rozenstein-Tsalkovich L , Abramsky O , Grigoriadis N , Rosenmann H ((2011) ) Moderate environmental enrichment mitigates taupathy in a neurofibrillary tangle mouse model. J Neuropathol Exp Neurol 70: , 610–621. |
[19] | Hu YS , Long N , Pigino G , Brady ST , Lazarov O ((2013) ) Molecular mechanisms of environmental enrichment: Impairments in Akt/GSK3β, neurotrophin-3 and CREB signaling. PLoS One 8: , e64460. |
[20] | Polito L , Chierchia A , Tunesi M , Bouybayoune I , Kehoe PG , Albani D , Forloni G ((2014) ) Environmental enrichment lessens cognitive decline in APP23 mice without affecting brain sirtuin expression. J Alzheimers Dis 42: , 851–864. |
[21] | Kerschensteiner M , Gallmeier E , Behrens L , Leal VV , Misgeld T , Klinkert WE , Kolbeck R , Hoppe E , Oropeza-Wekerle RL , Bartke C , Stadelmann C , Lassmann H , Wekerle H , Hohlfeld R ((1999) ) Activated human T cells, B cells, and monocytes produce brain-derived neurotrophic factor in vitro and in inflammatory brain lesions: A neuroprotective role of inflammation? J Exp Med 189: , 865–870. |
[22] | Nakahashi T , Fujimura H , Altar CA , Li J , Kambayashi J , Tandon NN , Sun B ((2000) ) Vascular endothelial cells synthesize and secrete brain-derived neurotrophic factor. FEBS Lett 470: , 113–117. |
[23] | Fujimura H , Altar CA , Chen R , Nakamura T , Nakahashi T , Kambayashi J , Sun B , Tandon NN ((2002) ) Brain-derived neurotrophic factor is stored in human platelets and released by agonist stimulation. Thromb Haemost 87: , 728–734. |
[24] | Pan W , Banks WA , Fasold MB , Bluth J , Kastin AJ ((1998) ) Transport of brain-derived neurotrophic factor across the blood-brain barrier. Neuropharmacology 37: , 1553–1561. |
[25] | Poduslo JF , Curran GL ((1996) ) Permeability at the blood-brain and blood-nerve barriers of the neurotrophic factors: NGF, CNTF, NT-3, BDNF. Brain Res Mol Brain Res 36: , 280–286. |
[26] | Karege F , Schwald M , Cisse M ((2002) ) Postnatal developmental profile of brain-derived neurotrophic factor in rat brain and platelets. Neurosci Lett 328: , 261–264. |
[27] | Sartorius A , Hellweg R , Litzke J , Vogt M , Dormann C , Vollmayr B , Danker-Hopfe H , Gass P ((2009) ) Correlations and discrepancies between serum and brain tissue levels of neurotrophins after electroconvulsive treatment in rats. Pharmacopsychiatry 42: , 270–276. |
[28] | Lang UE , Hellweg R , Seifert F , Schubert F , Gallinat J ((2007) ) Correlation between serum brain-derived neurotrophic factor level and an in vivo marker of cortical integrity. Biol Psychiatry 62: , 530–535. |
[29] | Hwang KS , Lazaris AS , Eastman JA , Teng E , Thompson PM , Gylys KH , Cole GM , Apostolova LG , Alzheimer’s Disease Neuroimaging, Initiative ((2015) ) Plasma BDNF levels associate with Pittsburgh compound B binding in the brain. Alzheimers Dement (Amst) 1: , 187–193. |
[30] | Laske C , Stransky E , Leyhe T , Eschweiler GW , Maetzler W , Wittorf A , Soekadar S , Richartz E , Koehler N , Bartels M , Buchkremer G , Schott K ((2007) ) BDNF serum and CSF concentrations in Alzheimer’s disease, normal pressure hydrocephalus and healthy controls. J Psychiatr Res 41: , 387–394. |
[31] | Hughes CP , Berg L , Danziger WL , Coben LA , Martin RL ((1982) ) A new clinical scale for the staging of dementia. Br J Psychiatry 140: , 566–572. |
[32] | Gagliardi C , Papa R , Postacchini D , Giuli C ((2016) ) Association between cognitive status and physical activity: Study profile on baseline survey of the My Mind Project. Int J Environ Res Public Health 13: , E585. |
[33] | Giuli C , Papa R , Lattanzio F , Postacchini D ((2016) ) The effects of cognitive training for elderly: Results from My Mind project. Rejuvenation Res 19: , 485–494. |
[34] | Angelucci F , Spalletta G , di Iulio F , Ciaramella A , Salani F , Colantoni L , Varsi AE , Gianni W , Sancesario G , Caltagirone C , Bassú P ((2010) ) Alzheimer’s disease (AD) and mild cognitive impairment (MCI) patients are characterized by increased BDNF serum level. Curr Alzheimer Res 7: , 15–20. |
[35] | Faria MC , Goncalves GS , Rocha NP , Moraes EN , Bicalho MA , Gualberto Cintra MT , Jardim de Paula , Jose’ Ravic de Miranda LF , Clayton de Souza Ferreira A , Teixeira AL , Gomes KB , Karvalho MD , Sousa LP ((2014) ) Increased plasma levels of BDNF and inflammatory markers in Alzheimer’s disease. J Psychiatr Res 53: , 166–172. |
[36] | Laske C , Stransky E , Leyhe T , Eschweiler GW , Maetzler W , Wittorf A , Soekadar S , Richartz E , Koehler N , Bartels M , Buchkremer G , Schott K ((2007) ) BDNF serum and CSF concentrations in Alzheimer’s disease, normal pressure hydrocephalus and healthy controls. J Psychiatr Res 41: , 387–394. |
[37] | Yasutake C , Kuroda K , Yanagawa T , Okamura T , Yoneda H ((2006) ) Serum BDNF, TNF-alpha and IL-1beta levels in dementia patients: Comparison between Alzheimer’s disease and vascular dementia. Eur Arch Psychiatry Clin Neurosci 256: , 402–406. |
[38] | Forlenza OV , Diniz BS , Teixeira AL , Ojopi EB , Talib LL , Mendonca A , Izzo G , Gattaz WF ((2010) ) Effect of brain-derived neurotrophic factor Val66Met polymorphism and serum levels on the progression of mild cognitive impairment. World J Biol Psychiatry 11: , 774–780. |
[39] | Ventriglia M , Zanardini R , Bonomini C , Zanetti O , Volpe D , Pasqualetti P , Gennarelli M , Bocchio-Chiavetto L ((2013) ) Serum brain-derived neurotrophic factor level in different neurological diseases. Biomed Res Int 2013: , 901082. |
[40] | Passaro A , Dalla Nora E , Morieri ML , Soavi C , Sanz JM , Zurlo A , Fellin R , Zuliani G ((2015) ) Brain-derived neurotrophic factor plasma levels: Relationship with dementia and diabetes in the elderly population. J Gerontol A Biol Sci Med Sci 70: , 294–302. |
[41] | O’Bryant SE , Hobson V , Hall JR , Waring SC , Chan W , Massman P , Lacritz L , Cullum CM , Diaz-Arrastia R , Texas Alzheimer’s Research, Consortium ((2009) ) Brain-derived neurotrophic factor levels in Alzheimer’s disease. J Alzheimers Dis 17: , 337–341. |
[42] | Ziegenhorn AA , Schulte-Herbruggen O , Danker-Hopfe H , Malbranc M , Hartung HD , Anders D , Lang UE , Steinhagen-Thiessen E , Schaub RT , Hellweg R ((2007) ) Serum neurotrophins–a study on the time course and influencing factors in a large old age sample. Neurobiol Aging 28: , 1436–1445. |
[43] | Woolley JD , Strobl EV , Shelly WB , Karydas AM , Robin Katelle RN , Wolkowitz OH , Miller BL , Rankin KP ((2012) ) BDNF serum concentrations show no relationship with diagnostic group or medication status in neurodegenerative disease. Curr Alzheimer Res 9: , 815–821. |
[44] | Sonali N , Tripathi M , Sagar R , Vivekanandhan S ((2013) ) Val66Met polymorphism and BDNF levels in Alzheimer’s disease patients in North Indian population. Int J Neurosci 123: , 409–416. |
[45] | Alvarez A , Aleixandre M , Linares C , Masliah E , Moessler H ((2014) ) Apathy and APOE4 are associated with reduced BDNF levels in Alzheimer’s disease. J Alzheimers Dis 42: , 1347–1355. |
[46] | Laske C , Strasnsky E , Leyhe T , Eschweiler GW , Wittorf A , Richartz E , Bartels M , Buchkremer G , Schott K ((2006) ) Stage-dependent BDNF serum concentrations in Alzheimer’s disease. J Neural Transm (Vienna) 113: , 1217–1224. |
[47] | Serra-Millas M ((2016) ) Are the changes in the peripheral brain-derived neurotrophic factor levels due to platelet activation? World J Psychiatry 6: , 84–101. |
[48] | Fujimura H , Altar CA , Chen R , Nakamura T , Nakahashi T , Kambayashi J , Sun B , Tandon NN ((2002) ) Brain-derived neurotrophic factor is stored in human platelets and released by agonist stimulation. Thromb Haemost 87: , 728–734. |
[49] | Lommatzsch M , Zingler D , Schuhbaeck K , Schloetcke K , Zingler C , Schuff-Werner P , Virchow JC ((2005) ) The impact of age, weight and gender on BDNF levels in human platelets and plasma. Neurobiol Aging 26: , 115–123. |
[50] | Catricala S , Torti M , Ricevuti G ((2012) ) Alzheimer disease and platelets: How’s that relevant. Immun Ageing 9: , 20. |
[51] | Canobbio I , Abubaker AA , Visconte C , Torti M , Pula G ((2015) ) Role of amyloid peptides in vascular dysfunction and platelet dysregulation in Alzheimer’s disease. Front Cell Neurosci 9: , 65. |
[52] | Zhang J , Mu X , Breker DA , Li Y , Gao Z , Huang Y ((2006) ) Atorvastatin treatment is associated with increased BDNF level and improved functional recovery after atherothrombotic stroke. Int J Neurosci 21: , 1–16. |
[53] | Allard JS , Perez EJ , Fukui K , Carpenter P , Ingram DK , de Cabo R ((2016) ) Prolonged metformin treatment leads to reduced transcription of Nrf2 and neurotrophic factors without cognitive impairment in older C57BL/6J mice. Behav Brain Res 301: , 1–9. |
[54] | Gunstad J , Benitez A , Smith J , Glickman E , Spitznagel MB , Alexander T , Juvancic-Heltzel J , Murray L ((2008) ) Serum brain-derived neurotrophic factor is associated with cognitive function in healthy older adults. J Geriatr Psychiatry Neurol 21: , 166–170. |
[55] | Kimura N , Takahashi M , Tashiro T , Terao K ((2006) ) Amyloid beta up-regulates brain-derived neurotrophic factor production from astrocytes: Rescue from amyloid beta-related neuritic degeneration. J Neurosci Res 84: , 782–789. |
[56] | Poon WW , Blurton-Jones M , Tu CH , Feinberg LM , Chabrier MA , Harris JW , Jeon NL , Cotman CW ((2011) ) β-Amyloid impairs axonal BDNF retrograde trafficking. Neurobiol Aging 32: , 821–833. |
[57] | Durany N , Michel T , Kurt J , Cruz-Sanchez FF , Cervas-Navarro J , Riederer P ((2000) ) Brain-derived neurotrophic factor and neurotrophin-3 levels in Alzheimer’s disease brains. Int J Dev Neurosci 18: , 807–813. |
[58] | Numakawa T , Suzuki S , Kumamaru E , Adachi N , Richards M , Kunugi H ((2010) ) BDNF function and intracellular signaling in neurons. Histol Histopathol 25: , 237–258. |
[59] | Rattiner LM , Davis M , Ressler KJ ((2005) ) Brain-derived neurotrophic factor in amygdala-dependent learning. Neuroscientist 11: , 323–333. |
[60] | Tyler WJ , Alonso M , Bramham CR , Pozzo-Miller LD ((2002) ) From acquisition to consolidation: On the role of brain-derived neurotrophic factor signaling in hippocampal-dependent learning. Learn Mem 9: , 224–237. |
[61] | Bennet MR , Lagopoulos J ((2014) ) Stress and trauma: BDNF control of dendritic-spine formation and regression. Prog Neurobiol 112: , 80–99. |
[62] | Jiao SS , Shen LL , Zhu C , Bu XL , Liu YH , Liu CH , Yao XQ , Zhang LL , Zhou HD , Walker DG , Tan J , Gotz J , Zhou XF , Wang YJ ((2016) ) Brain-derived neurotrophic factor protects against tau-related neurodegeneration of Alzheimer’s disease. Transl Psychiatry 6: , e907. |
[63] | Lazarov O , Robinson J , Tang YP , Hairston IS , Korade-Mirnics Z , Lee VM , Hersh LB , Sapolsky RM , Mirnics K , Sisodia SS ((2005) ) Environmental enrichment reduces Abeta levels and amyloid deposition in transgenic mice. Cell 120: , 701–713. |
[64] | Hu YS , Xu P , Pigino G , Brady ST , Larson J , Lazarov O ((2010) ) Complex environment experience rescues impaired neurogenesis, enhances synaptic plasticity, and attenuates neuropathology in familial Alzheimer’s disease-linked APPswe/PS1DeltaE9 mice. FESEB J 24: , 1667–1681. |
[65] | Herring A , Lewejohann L , Panzer AL , Donath A , Kroll O , Sachser N , Paulus W , Keyvani K ((2011) ) Preventive and therapeutic types of environmental enrichment counteract beta amyloid pathology by different molecular mechanisms. Neurobiol Dis 42: , 530–538. |
[66] | Li S , Jin M , Zhang D , Yang T , Koeglsperger T , Fu H , Selkoe DJ ((2013) ) Environmental novelty activates β2-adrenergic signaling to prevent the impairment of hippocampal LTP by Aβ oligomers. Neuron 77: , 929–941. |
[67] | Clegg A , Young J , Iliffe S , Rikkert MO , Rockwood K ((2013) ) Frailty in elderly people. Lancet 381: , 752–762. |
[68] | Komulainen P , Pedersen M , Hanninen T , Bruunsgaard H , Lakka TA , Kivipelto M , Hassinen M , Rauramaa TH , Pedersen BK , Rauramaa R ((2008) ) BDNF is a novel marker of cognitive function in ageing women: The DR’s EXTRA Study. Neurobiol Learn Mem 90: , 596–603. |
Figures and Tables
Fig.1
A) Baseline pBDNF was significantly higher in patients with moderate AD (CDR 2) than in healthy subjects (HE). B) In healthy subjects, a better cognitive status (higher MMSE score) was associated to a higher pBDNF. C) In AD patients, more impaired cognition (higher ADAS-Cogscore) was associated to higher pBDNF. In A, columns and error bars represent back-transformed means and 95% confidence interval. In B and C, a constant was added to the log-transformed data to avoid showing negative values.
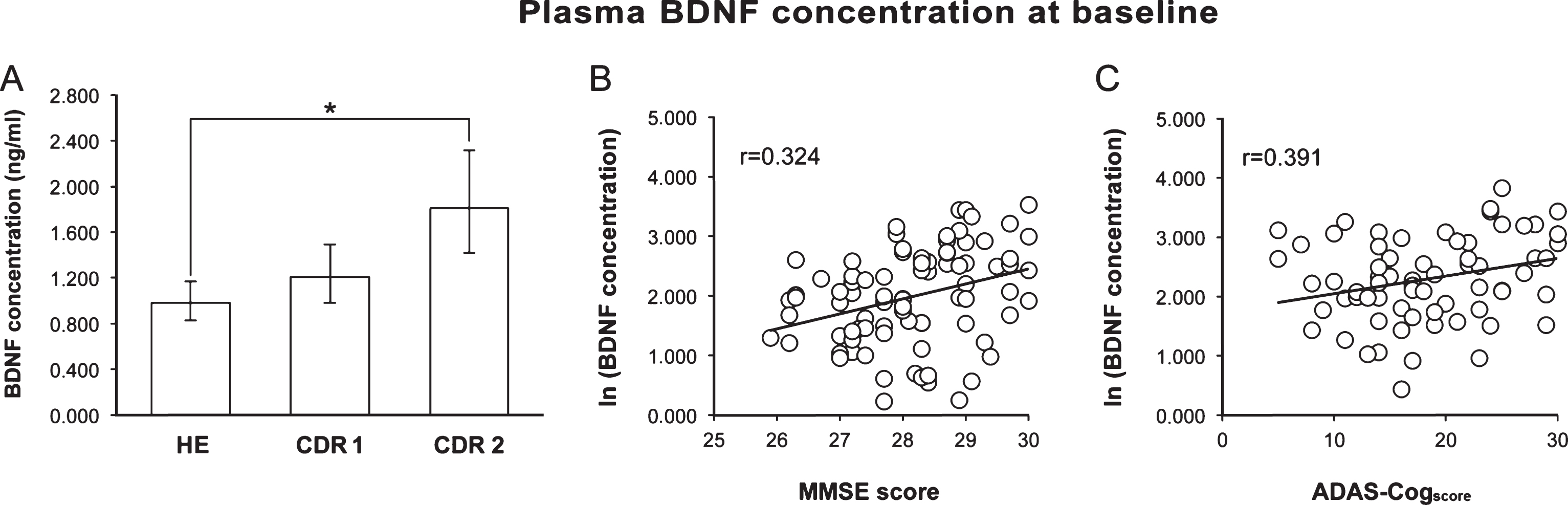
Fig.2
A, B) Immediately after the intervention (FU1), ADAS-Cogscore and pBDNF values fell significantly in the experimental group (EG) but were not significantly changed in control patients (CG). C, D) In EG patients, the ADAS-Cogscore reverted to baseline at 6 months (FU2) and showed a further significant increase at 24 months (FU3) compared to baseline, while pBDNF increased significantly at FU2 compared to FU1 as well as baseline and fell at FU3 versus FU2. In CG patients, ADAS-Cogscore and pBDNF values were virtually unchanged throughout follow-up. In A and C, columns and error bars represent means and standard error of the mean; in B and C, columns and error bars represent back-transformed means and 95% confidence interval. Data were adjusted for age and ADL.