
Revisiting the Heidenhain Variant of Creutzfeldt-Jakob Disease: Evidence for Prion Type Variability Influencing Clinical Course and Laboratory Findings
Abstract
The Heidenhain variant defines a peculiar clinical presentation of sporadic Creutzfeldt-Jakob disease (sCJD) characterized by isolated visual disturbances at disease onset and reflecting the early targeting of prions to the occipital cortex. Molecular and histopathological typing, thus far performed in 23 cases, has linked the Heidenhain variant to the MM1 sCJD type. To contribute a comprehensive characterization of cases with the Heidenhain variant, we reviewed a series of 370 definite sCJD cases. Eighteen patients (4.9%) fulfilled the selection criteria. Fourteen of them belonging to sCJD types MM1 or MM1+2C had a short duration of isolated visual symptoms and overall clinical disease, a high prevalence of periodic sharp-wave complexes in EEG, and a marked increase of cerebrospinal fluid proteins t-tau and 14-3-3 levels. In contrast, three cases of the MM 2C or MM 2+1C types showed a longer duration of isolated visual symptoms and overall clinical disease, non-specific EEG findings, and cerebrospinal fluid concentration below threshold for the diagnosis of “probable” CJD of both 14-3-3 and t-tau. However, a brain DWI-MRI disclosed an occipital cortical hyperintensity in the majority of examined cases of both groups. While confirming the strong linkage with the methionine genotype at the polymorphic codon 129 of the prion protein gene, our results definitely establish that the Heidenhain variant can also be associated with the MM 2C sCJD type in addition to the more common MM1 type. Likewise, our results highlight the significant differences in clinical evolution and laboratory findings between cases according to the dominant PrPSc type (type 1 versus type 2).
INTRODUCTION
Creutzfeldt-Jakob disease (CJD) belongs to the human transmissible spongiform encephalopathies or prion diseases, a group of neurodegenerative disorders characterized by tissue deposition of a misfolded form of the cellular prion protein (PrPSc). Despite their relative rarity, prion diseases show a wide spectrum of clinical and pathological phenotypes. Accordingly, the clinical presentation of sporadic CJD (sCJD) includes a wide range of neurological signs of cortical, sub-cortical, or cerebellar origin, either isolated or in various combinations [1–4]. Different strains of prions, likely enciphered by alternative conformations of PrPSc, are considered the main cause of this phenotypic diversity [5]. In addition, the host variability in the gene encoding PrP (PRNP), as determined in humans by polymorphisms or mutations, also modulates the disease phenotype [6, 7]. Current sCJD classification [3, 8] recognizes six major variants with distinctive clinic-pathological features largely correlating at the molecular level with the genotype at the polymorphic codon 129 (methionine, M or valine, V) in the prion protein gene and two PrPSc profiles (type 1 and type 2) distinguishable by the size of the proteinase K-resistant core of the protein (21 and 19 kDa, respectively). More precisely, with only minor exceptions, each phenotypic variant or “type” of sCJD results from a specific codon 129 genotype/PrPSc type combination (e.g., MM1, VV1, MM2, VV2, etc.). Most significantly, two types with distinctive histopathological features in the cerebral cortex and in the thalamus have been linked to the rare MM2 molecular combination and designated accordingly (MM2-cortical or MM 2C and MM2-thalamic or MM 2T). Finally, mixed types, comprising clinical-pathological features of two pure types, especially involving the co-existence of MM1 and MM 2C, have also been recognized [9, 10]. One of the most peculiar clinical presentations of CJD occurs in the so-called Heidenhain variant, which is characterized by isolated visual symptoms including poor vision, disturbed perception of colors or structures, and optical distortions as well as hallucinations without any ocular disease. The fact that visual symptoms may persist in isolation for weeks without cognitive decline or motor signs and sometimes causing diagnostic difficulty makes the Heidenhain variant of particular clinical interest. Indeed, affected patients sometimes present to ophthalmologists and are subjected to needless ocular interventions with risks of onward transmission [11]. Originally, Heidenhain ascribed to CJD the severe histopathological changes to include severe neuronal loss with glial reaction or status spongiosus found in the occipital cortex of two patients with a pre-senile form of rapidly progressive dementia presenting with prominent visual disturbances despite normal disc and fundi examination [12]. Since 1954, similar cases have been described, most often as small case series [11, 13–18] or individual case reports [19–37] and referred to as the Heidenhain variant of CJD [38]. To date, molecular and histopathological analyses have been performed in 23 cases with this clinical presentation, and the large majority (22 out of 23) belonged to the MM-MV1 sCJD type [11, 13, 16, 34, 35]. In a single case, however, the Heidenhain variant has also been linked to the MM 2C type, which has widened the molecular basis and histotype classification of this peculiar clinical phenotype [29]. To contribute a comprehensive clinical, histopathological, and molecular characterization of cases presenting with the Heidenhain variant, we here provide the results of a review of a series of 370 definite sCJD cases, including the detailed description of three peculiar cases of the Heidenhain variant belonging to the rare MM 2C and MM 2C+1 sCJD types.
MATERIAL AND METHODS
This study has been conducted with the highest respect for each participant according to the Declaration of Helsinki [39].
Patients
Three hundred and seventy cases with a definite diagnosis of sCJD were selected based on available medical records including a detailed description of the clinical evolution of symptoms and signs from onset. The case series included all sCJD phenotypic variants described to date in a relative proportion fairly representing their current reported incidence in the Caucasian population [3, 9, 40]. Three hundred and fifteen subjects were Italian patients referred to our laboratory for diagnosis from 2003 to 2013, while the remaining 55 were part of a previously published case series [9]. All the Italian cases were also part of the National CJD Registry in Rome, which collects data of all suspected CJD patients aiming to ascertain all cases with definite or probable disease in Italy. The presence of clinically suspected patients is notified to the reference center by their treating physicians, mainly neurologists. Most suspected patients are also examined by a neurologist of the surveillance unit at the reporting hospital, and copies of patients’ medical records are collected. For case selection, we combined the information recorded in the two available databases (Bologna and Rome) to search for cases presenting visual symptoms at onset. Subsequently, only patients suffering from isolated, non-transient, visual symptoms of cortical/subcortical origin at disease onset were diagnosed with the Heidenhain variant. Visual symptoms included at least one of the following: blurred vision, visual field restriction, vision loss up to cortical blindness, disturbed perception of colors or structures, optical hallucinations, and visual agnosia. Duration of visual symptoms was calculated based on the timing of appearance of associated neurological symptoms, as reported in the available medical records. Results of electroencephalographic (EEG) recordings were classified into three categories: periodic sharp-wave complexes (PSWCs), paroxysmal discharges (PDs), and diffuse non-specific slowing. When available, cerebrospinal fluid (CSF) biomarker assays included the measurement of total tau (t-tau) protein concentration by quantitative Elisa (INNOTEST ® hTAU Ag, Innogenetics) and the semiquantitative detection of protein 14-3-3 by western blotting, performed as previously described [41]. In particular, two CSF controls (with a weak or a strong 14-3-3 signal, respectively) were loaded in duplicate in every gel together with the CSF samples to be analyzed. The immunoreactivity signals were rated as negative, ambiguous, or positive on the basis of optical densitometric (OD) comparison with the weakly positive control as follows: the 14-3-3 signal was classified as negative when the 14-3-3 band OD was lower than the control; ambiguous (i.e., weakly positive) when the 14-3-3 OD was up to two times higher than the control, and positive when it was at least two times higher than the control. The latter decision point was chosen as optimal after an analysis of the test predictive value at different densitometry value ranges in a large series (>1000) of CSF samples from subjects with suspected CJD (unpublished data). Finally, cerebral magnetic resonance imaging (MRI) studies were reviewed when available. According to currently accepted clinical diagnostic criteria for sCJD [42], high signal abnormalities in caudate nucleus and putamen or at least two cortical regions (temporal-parietal-occipital) either in diffusion-weighted imaging (DWI) or fluid attenuated inversion recovery (FLAIR) sequences was considered “typical,” while non-specific findings included signal abnormalities suggesting gliosis, microvascular changes, or atrophy.
Molecular studies and CJD histotype classification
To exclude cases carrying mutations and to determine the genotype of PRNP polymorphic codon 129, we conducted a molecular analysis in all cases as previously described [40]. We performed PrPSc typing and sCJD histotype classification according to established methodology and consensus criteria [8, 43]. Specifically, we analyzed at least four brain regions (middle temporal gyrus, parietal cortex, occipital cortex, and thalamus) for PrPSc typing and at least eight (middle frontal gyrus, middle temporal gyrus, parietal cortex, occipital cortex, basal ganglia, hippocampus, thalamus, and cerebellum) for histotyping.
RESULTS
Among the 370 cases examined, 70 (18.9%) had visual symptoms at onset, but in many case, they were not isolated. Therefore, only 18 (4.9%) cases fulfilled the criteria for the Heidenhain variant (Tables 1 and 2). Molecular and histopathological analyses demonstrated that the cases belonged to four sCJD types: MM1 (10 cases), MM 1+2C (5 cases), MM 2+1C (2 cases), and MM 2C (1 case).
MM1
Mean age at the onset was 70.7 ± 7.8 years (range 57–80), mean duration of isolated visual symptoms was 1.6 ± 0.5 months (range 1-2), and mean total disease duration was 4.2 ± 2.1 months (range 2.5–10). According to frequency, visual symptoms included the following: disturbed perception of structures and/or colors (i.e., metamorphopsia, micropsia, dyschromatopsia) (80%), visual field restriction (60%), loss of vision (40%), blurred vision (30%), optical hallucinations, and optical anosognosia (20%). EEG examination revealed PSWCs in eight patients (80%), whereas PDs or a diffuse non-specific slowing were observed in one case each (10%). Fluid attenuated inversion recovery (FLAIR) and DWI brain MRI sequences were performed in four patients. They showed a typical (parietal and occipital, and/or basal ganglia) hyperintensity on FLAIR and/or DWI sequences in three (50%), a hyperintensity confined to the left parietal lobe in one, and were unremarkable in the other one. CSF 14-3-3 protein was positive, and t-tau markedly elevated (range 2335–2522 pg/ml) in all patients analyzed (five and two, respectively). Histopathological examination and PrPSc typing revealed the typical features of sCJD MM1 in all cases [44].
MM1+2C
Mean age at the onset of symptoms was 66.6 ± 9.4 years (range 54–80), mean duration of isolated visual symptoms was 1.5 ± 0.5 months (range 1–2.5), and mean disease duration was 4.1 ± 1 months (range 2.5–5). Cortical visual symptoms included blurred vision (80%), visual field restriction (60%), vision loss (60%), image distortion (40%), and optical hallucinations (40%). EEG recording revealed PSWCs in all patients (100%). Brain MRI with FLAIR and DWI sequences was positive in two subjects (66%) and non-specific in one (33%). CSF analysis of protein 14-3-3 was positive in four patients (80%) and ambiguous in one (20%), while t-tau was markedly elevated in all four patients (range 3990–7579 pg/ml) that were tested. Histopathological examination and PrPSc typing revealed the typical features of sCJD MM1 except for the presence of clusters of large vacuoles associated with perivacuolar and coarse PrP deposits mainly in the cerebral cortex or thalamus. Consistently, PrPSc type 1 was detected by western blotting in all areas analyzed, whereas PrPSc type 2 was only seen focally in the cerebral cortex and/or in the thalamus as a relatively minor co-occurring type.
MM2C+1
Patient #16, a 70-year-old female, presented with a subacute loss of visual acuity associated with visuo-spatial distortion and a mild campimetric deficit. After three months of isolated visual disturbances, she developed gait difficulties and a rapidly progressive cognitive impairment. Six months later, her difficulties were followed by cerebellar signs (ataxia, dysmetria), myoclonus, dysphagia, and dysphonia. EEG recording showed a non-specific generalized slowing, routine brain MRI was normal, while the 14-3-3 protein test was ambiguous. The patient progressed to akinetic mutism after 21 months of clinical course and died five months later. Histopathological examination and PrPSc typing revealed all the distinctive features of sCJD MM 2C+1. In particular, spongiform change comprised either large, confluent vacuoles correlating with perivacuolar and coarse PrP deposits or relatively small interspersed vacuoles associated with a synaptic pattern of PrP staining (Fig. 1A–E). As typically seen in sCJD MM 2C+1, the cerebral cortex was predominantly affected by the former type of spongiosis, whereas the cerebellum only showed the latter type (Fig. 1A,D). PrPSc typing revealed the co-occurrence of types 1 and 2 in all regions analyzed but the cerebellum (Fig. 1F). Consistent with histopathological findings, the amount of PrPSc type 2 was higher than that of type 1 in most samples from the cerebral cortex, whereas only type 1 was detected in the cerebellum (Fig. 1F).
Patient #17, a 54 year-old male, presented with six month history of visual hallucinations (he saw ladders, houses, or bikes while driving a digger), environmental agnosia (he failed to recognize the gate entrance of his house while driving), and dyscalculia. At this time, campimetry disclosed left homonymous hemianopsia. A brain MRI showed a diffused and bilateral hyperintensity in the occipital cortex with partial involvement of both parietal lobes and left temporal lobe in DWI (Fig. 2A–C) and FLAIR sequences. EEG was desynchronized and slow. Neuropsychological evaluation revealed a mild encoding memory deficit, while frontal lobe functions were normal. CSF examination disclosed 1070 pg/ml of t-tau, while the 14-3-3 protein test was ambiguous. The clinical course was slowly progressive; after two years, the patient became disoriented and developed a psychosis with paranoid delusions as well as pyramidal signs. He died after a prolonged lethargic state lasting approximately 15 months. At the neuropathologic examination, status spongiosus associated with severe gliosis and neuronal loss was the predominant finding in the cerebral cortex (Fig. 2D) and striatum. Typical spongiform change with vacuoles of intermediate or large size was only seen focally in relatively preserved areas of the cerebral cortex (Fig. 2F) and striatum as well as in the molecular layer of cerebellum (Fig. 2H). In the latter, however, PAS positive, unicentric, amyloid plaques were also noticed (Fig. 2I). PrP immunohistochemistry predominantly revealed a coarse granular pattern of staining in the cerebral cortex, striatum, thalamus, and cerebellum (Fig. 2E,G,L). In the latter, however, the coarse pattern was associated with a plaque-like type of PrP deposition predominantly involving the molecular layer and correlating with the anatomic distribution of amyloid plaques (Fig. 2L). In addition, foci of synaptic PrP deposits were visible (Fig. 2L). PrPSc typing revealed the presence of PrPSc type 2 in all areas analyzed. The relative amount of the protein was highest in the cerebral cortex of all lobes; lowest in the hippocampus, hypothalamus, and brainstem; and intermediate in the striatum, thalamus, and cerebellum (Fig. 2M). In addition, traces or a relatively low amount of PrPSc type 1 were detected in some samples from the cerebral cortex, brainstem, and cerebellum.
MM 2C
Patient #18, a 48-year-old obese and heavy smoker, presented with subacute visual complaints characterized by dazzling from lampposts at night. An ophthalmologic examination revealed an irregular campimetric deficit in the right eye, while visual acuity was normal. After two and a half months, he became disinhibited, sometimes unsettled, and the visual campimetric disturbance progressed to a right homonymous hemianopsia. Neurological examination disclosed optic apraxia and simultanagnosia, psychomotor slowing, and executive function impairment. A brain MRI revealed a hyperintense signal involving the occipital and parietal cortices in both DWI (Fig. 3A,B) and FLAIR (Fig. 3C) sequences. EEG showed a diffuse slowing of background activity. CSF assays revealed 1,140 pg/ml of t-tau, while protein 14-3-3 analyses yielded an ambiguous result. During the following two months, the patient developed cortical blindness, complete spatial and temporal disorientation, amnesia, severe dysarthria and dysphagia, paratonic rigidity, and recurring myoclonic jerks. The patient died of septic shock six months after the onset of visual symptoms. Histopathological examination and PrPSc typing were consistent with sCJD type MM 2C. Specifically, histopathologic examination revealed a striking difference in the degree of spongiform and neurodegenerative changes between the posterior and anterior cortices. While the occipital gray matter was entirely occupied by large confluent vacuoles, the fronto-temporal cortices only showed sparse foci of spongiform change (Fig. 3D,E), especially involving the middle layers. Outside the cerebral cortex, spongiform changes were mild and limited to small foci in the thalamus and molecular layer of cerebellum (Fig. 3H), while the striatum was spared (Fig. 3I). PrP immunohistochemistry revealed a perivacuolar pattern of protein deposition in all cortical areas (Fig. 3F,G). In addition, sparse foci of coarse PrP deposits were present in the thalamus, whereas no immunodeposition was seen in the cerebellum (data not shown). PrPSc typing revealed a type 2 pattern (Fig. 3L). PrPSc was more abundant in the occipital cortex than in the other cortical or subcortical areas where only traces or relatively low amounts of the protein were detected(Fig. 3L).
DISCUSSION
Heidenhain variant defines a peculiar clinical presentation of CJD characterized by isolated visual disturbances, reflecting the early and prevalent involvement of the occipital cortex by the degenerative changes.
In our large series of sCJD, the prevalence of Heidenhain variant was 4.9% . This number is quite consistent with that (3.7%) reported by Cooper et al. [11]. However, it is significantly lower than that reported by Kropp et al. (20%) [13] who applied less stringent clinical criteria for the definition of Heidenhain variant (e.g., visual symptoms at onset notwithstanding their being isolated for some time). sCJD is a highly heterogeneous disease caused by prion strains showing distinctive neuronal targeting [45]. Detailed studies of the regional distribution of lesions (the so-called lesion profile) across the spectrum of CJD types have shown that sCJD MM-MV1 (e.g., M1 strain) is associated with significant cortical pathology, and that this is often predominant in the occipital lobe [3, 46]. Accordingly, early molecular and histotype analyses performed in cases with the Heidenhain clinical presentation indicated that all these cases belonged to the MM-MV1 type [3, 11, 13]. More recently, however, a clinical onset with isolated visual symptoms has also been described in a case affected by MM 2C [29], a rarer sCJD type also characterized by prominent cortical targeting [3, 47]. In the present study, we definitely establish that the Heidenhain variant can be also associated with the MM 2C sCJD type, either isolated or co-occurring with the MM1 type, and highlight the significant differences in clinical evolution as well as laboratory findings between the two sCJD types. As expected, the majority of patients in our series (83%) were affected either by the MM1 (55.5%) or MM1+2C sCJD type (27.5%). In both instances, patients were clinically characterized by a short duration of isolated visual symptoms and overall disease course. EEG disclosed PSWCs in the majority of them, while 80% had a typical occipito-parietal hyperintensity on DWI or FLAIR MRI sequences. Finally, CSF assays, when performed, showed markedly increased t-tau levels (well above the 1200 pg/ml threshold) and above threshold levels of protein 14-3-3 (i.e., positive test) in virtually all cases. Interestingly, analyses of the timing of appearance of PSWCs in the EEG and of a “positive” CSF assay suggest that the latter might be more sensitive than the EEG early in the clinical course (i.e., when patients suffer from isolated visual symptoms). Indeed, while the time of appearance of PSWCs in our series ranged from one and a half to three months after onset, in two cases 14-3-3 and t-tau assays were already positive two weeks after onset (Table 2).
At variance with previous studies, three cases (17%) in our series demonstrated distinctive clinical features and an exclusive or predominant deposition of PrPsc type 2. Indeed, in these patients, the duration of isolated visual symptoms was significantly longer, and the clinical course was more slowly progressive than in MM1 or MM1+2C sCJD types. Two of them had a disease duration longer than two years, while in the third, the relatively mild histopathological changes were observed in all areas, but the occipital cortex also indicated a relatively slow progression. EEG findings, showing a diffuse slowing of background activity without PSWCs, were not distinctive in these patients. Similarly, both 14-3-3 and t-tau analyses revealed pathological values that were below the required threshold for the diagnosis of “probable” CJD [42]. In contrast, typical occipito-parietal hyperintensity on FLAIR and DWI sequences were clearly seen on brain MRI in both patients who underwent these analyses. Thus, the results of laboratory investigations significantly differ between the two sCJD types, which is in agreement with the known different pathophysiology of the disease process (e.g., earlier and more rapidly evolving neuronal dysfunction in MM1) and the significantly different transmission properties (e.g., significantly shorter incubation time and higher transmission rate of MM1 prions) [47–49] between the two sCJD types. Accordingly, CSF biomarkers (i.e., t-tau >1200 pg/ml and 14-3-3) and EEG abnormalities (i.e., PSWCs), reflecting either a subacute neurodegeneration or a rapidly evolving cortical and subcortical dysfunction, show a higher sensitivity in sCJD MM1. In contrast, the brain DWI-MRI increased signal, which instead correlates with the degree of spongiform change in the cerebral gray matter [50], has also a good sensitivity (or even higher according to our results) in MM2C, which shows a very distinctive type of spongiform change characterized by numerous large and confluent vacuoles. Although the present study focused on sCJD, it is worth mentioning that a CJD type with identical or very similar clinical-pathological features and transmission properties of sCJDMM1 has also been described in genetic CJD cases carrying the V210I-129M or the E200K-129 PRNP haplotype [7, 48, 51]. Thus, it is expected that cases with the Heidenhain variant may also occur in genetic CJD. Consistently, isolated visual symptoms at onset have been recently reported in two subjects carrying the V210I-129M PRNP haplotype, which further underlines the importance of genetic testing in all cases of possible and probable CJD, including those presenting with visual signs [54]. Despite its rarity, the Heidenhain variant of CJD remains of clinical relevance. A significant number of patients are initially referred to the ophthalmologist, and misdiagnoses are not uncommon, especially in the early clinical stages as well as in the clinical neurological setting. In addition to ocular diseases, differential diagnosis may include occipital stroke and other neurodegenerative conditions causing a posterior cortical atrophy (PCA) syndrome, including Alzheimer’s disease, dementia with Lewy bodies, and corticobasal degeneration [53, 54]. In agreement with the recent literature, which suggests that phenotypic differences (i.e., different visual symptoms) within PCA cases might most appropriately be judged to represent points on a continuum of variation, we failed to recognize a unitary clinical posterior cortical syndrome among sCJD cases that would justify the proposal of a distinct syndromic subtype of PCA. Since physical examination is unremarkable in most PCA cases, it is also unhelpful for the differential diagnosis. Nonetheless, the presence of symmetrical parkinsonism or rapid eye movement sleep behavior disorder (suggestive of Lewy body pathology) as well as asymmetric myoclonus/dystonia (suggestive of corticobasal degeneration) can give clues to the underlying cause of PCA [52]. When PCA is suspected, the clinical course, CSF biomarker analysis, brain DWI- or FLAIR-MRI, and, to a lesser extent, EEG recordings, remain the most important tools in the differential diagnosis. In this respect, the findings of the present study highlight the fact that, despite their relative rarity, CJD cases presenting with isolated visual symptoms may belong to different disease variants that are characterized by a distinct profile of laboratory findings. In particular, clinicians should be aware that the Heideinhain variant may also occur in atypical cases showing a relatively slow progression, negative or ambiguous CSF biomarkers results, and lack of specific EEG abnormalities.
ACKNOWLEDGMENTS
This work was financially supported by the Italian Ministry of Health, grant RF2011-02351092, and by the Gino Galletti Foundation. We wish to thank Barbara Polischi and Sabrina Boninsegna for their valuable technical assistance.
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/15-0668r2).
REFERENCES
[1] | Will RG , Matthews WB ((1984) ) A retrospective study of Creutzfeldt-Jakob disease in England and Wales 1970-79. I: Clinical features. J Neurol Neurosurg Psychiatry 47: , 134–140. |
[2] | Brown P , Cathala F , Castaigne P , Gajdusek DC ((1986) ) Creutzfeldt-Jakob disease: Clinical analysis of a consecutive series of 230 neuropathologically verified cases. Ann Neurol 20: , 597–602. |
[3] | Parchi P , Giese A , Capellari S , Brown P , Schulz-Schaeffer W , Windl O , Zerr I , Budka H , Kopp N , Piccardo P , Poser S , Rojiani A , Streichemberger N , Julien J , Vital C , Ghetti B , Gambetti P , Kretzschmar H ((1999) ) Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 46: , 224–233. |
[4] | Krasnianski A , Kaune J , Jung K , Kretzschmar HA , Zerr I ((2014) ) First symptom and initial diagnosis in sporadic CJD patients in Germany. J Neurol 261: , 1811–1817. |
[5] | Aguzzi A , Heikenwalder M , Polymenidou M ((2007) ) Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol 8: , 552–561. |
[6] | Gambetti P , Kong Q , Zou W , Parchi P , Chen SG ((2003) ) Sporadic and familial CJD: Classification and characterisation. Br Med Bull 66: , 213–239. |
[7] | Capellari S , Strammiello R , Saverioni D , Kretzschmar H , Parchi P ((2011) ) Genetic Creutzfeldt-Jakob disease and fatal familial insomnia: Insights into phenotypic variability and disease pathogenesis. Acta Neuropathol 121: , 21–37. |
[8] | Parchi P , de Boni L , Saverioni D , Cohen ML , Ferrer I , Gambetti P , Gelpi E , Giaccone G , Hauw JJ , Höftberger R , Ironside JW , Jansen C , Kovacs GG , Rozemuller A , Seilhean D , Tagliavini F , Giese A , Kretzschmar HA ((2012) ) Consensus classification of human prion disease histotypes allows reliable identification of molecular subtypes: An inter-rater study among surveillance centres in Europe and USA. Acta Neuropathol 124: , 517–529. |
[9] | Parchi P , Strammiello R , Notari S , Giese A , Langeveld JP , Ladogana A , Zerr I , Roncaroli F , Cras P , Ghetti B , Pocchiari M , Kretzschmar H , Capellari S ((2009) ) Incidence and spectrum of sporadic Creutzfeldt-Jakob disease variants with mixed phenotype and co-occurrence of PrPSc types: An updated classification. Acta Neuropathol 118: , 659–671. |
[10] | Cali I , Castellani R , Yuan J , Al-Shekhlee A , Cohen ML , Xiao X , Moleres FJ , Parchi P , Zou WQ , Gambetti P ((2006) ) Classification of sporadic Creutzfeldt-Jakob disease revisited. Brain 129: , 2266–2277. |
[11] | Cooper SA , Murray KL , Heath CA , Will RG , Knight RS ((2005) ) Isolated visual symptoms at onset in sporadic Creutzfeldt-Jakob disease: The clinical phenotype of the “Heidenhain variant”. Br J Ophthalmol 89: , 1341–1342. |
[12] | Heidenhain A ((1929) ) Klinische und anatomische Untersuchungen uber eine eigenartige organische Erkrankung des Zentralnervensystems im Praesenium. Zeitschrift für die gesamte Neurologie und Psychiatrie 118: , 49–114. |
[13] | Kropp S , Schulz-Schaeffer WJ , Finkenstaedt M , Riedemann C , Windl O , Steinhoff BJ , Zerr I , Kretzschmar HA , Poser S ((1999) ) The Heidenhain variant of Creutzfeldt-Jakob disease. Arch Neurol 56: , 55–61. |
[14] | Jacobs DA , Lesser RL , Mourelatos Z , Galetta SL , Balcer LJ ((2001) ) The Heidenhain variant of Creutzfeldt-Jakob disease: Clinical, pathologic, and neuroimaging findings. J Neuroophthalmol 21: , 99–102. |
[15] | Proulx AA , Strong MJ , Nicolle DA ((2008) ) Creutzfeldt-Jakob disease presenting with visual manifestations. Can J Ophthalmol 43: , 591–595. |
[16] | Appleby BS , Appleby KK , Crain BJ , Onyike CU , Wallin MT , Rabins PV ((2009) ) Characteristics of established and proposed sporadic Creutzfeldt-Jakob disease variants. Arch Neurol 66: , 208–215. |
[17] | Cartier LR , Quiroz G , Leiva MH , Vergara CR ((2012) ) Clinical and pathologic identification of different forms of Creutzfeldt Jakob disease in Chile. Rev Med Chil 140: , 161–168. |
[18] | Parker SE , Gujrati M , Pula JH , Zallek SN , Kattah JC ((2014) ) The Heidenhain variant of Creutzfeldt-Jakob disease–a case series. J Neuroophthalmol 34: , 4–9. |
[19] | Grünwald F , Pohl C , Bender H , Hartmann A , Menzel C , Ruhlmann J , Keller E , Biersack HJ ((1996) ) 18F-fluorodeoxyglucose-PET and 99mTc-bicisate-SPECT in Creutzfeldt-Jakob disease. Ann Nucl Med 10: , 131–134. |
[20] | Staffen W , Trinka E , Iglseder B , Pilz P , Homann N , Ladurner G ((1997) ) Clinical and diagnostic findings in a patient with Creutzfeldt-Jakob disease (type Heidenhain). J Neuroimaging 7: , 50–54. |
[21] | Finsterer J , Bancher C , Mamoli B ((1999) ) Giant visually-evoked potentials without myoclonus in the Heidenhain type of Creutzfeld-Jakob disease. J Neurol Sci 167: , 73–75. |
[22] | Deschauer M , Stephan M , Stuhlträger U , Holzhausen HJ , Duncker G , Zierz S ((2000) ) Bilateral inferior hemianopsia as an early symptom of Heidenhain type Creutzfeldt-Jakob disease. Klin Monbl Augenheilkd 216: , 227–231. |
[23] | Mathews D , Unwin DH ((2001) ) Quantitative cerebral blood flow imaging in a patient with the Heidenhain variant of Creutzfeldt-Jakob disease. Clin Nucl Med 26: , 770–773. |
[24] | Pachalska M , Kurzbauer H , MacQueen BD , Formińska-Kapuścik M , Herman-Sucharska I ((2001) ) Neuropsychological features of rapidly progressive dementia in a patient with an atypical presentation of Creutzfeldt-Jakob Disease. Med Sci Monit 7: , 1307–1315. |
[25] | Arruda WO , Bordignon KC , Milano JB , Ramina R ((2004) ) Creutzfeldt-Jakob disease, Heidenhain variant: Case report with MRI (DWI) findings. Arq Neuropsiquiatr 62: , 347–352. |
[26] | Tsuji Y , Kanamori H , Murakami G , Yokode M , Mezaki T , Doh-ura K , Taniguchi K , Matsubayashi K , Fukuyama H , Kita T , Tanaka M ((2004) ) Heidenhain variant of Creutzfeldt-Jakob disease: Diffusion-weighted MRI and PET characteristics. J Neuroimaging 14: , 63–66. |
[27] | Rizzo M , Bruni A , Barberio C , Magro G , Foncin JF ((2004) ) A Heidenhain variant of reutzfeldt-Jakob disease: Forensic implication. Forensic Sci Int 146: , 51–54. |
[28] | Fauquembergue M , Tilikete C , Perret-Liaudet A , Kopp N , Krolak-Salmon P , Vighetto A ((2005) ) Heidenhain’s variant of Creutzfeldt-Jakob’s disease. Rev Neurol (Paris) 161: , 578–581. |
[29] | Nozaki I , Hamaguchi T , Noguchi-Shinohara M , Ono K , Shirasaki H , Komai K , Kitamoto T , Yamada M ((2006) ) The MM2-cortical form of sporadic Creutzfeldt-Jakob disease presenting with visual disturbance. Neurology 67: , 531–533. |
[30] | Keyrouz SG , Labib BT , Sethi R ((2006) ) MRI and EEG findings in Heidenhain variant of Creutzfeldt-Jakob disease. Neurology 67: , 333. |
[31] | Foundas M , Donaldson MD , McAllister IL , Bridges LR ((2008) ) Vision loss due to coincident ocular and central causes in a patient with Heidenhain variant Creutzfeldt-Jakob disease. Age Ageing 37: , 231–232. |
[32] | Cornelius JR , Boes CJ , Ghearing G , Leavitt JA , Kumar N ((2009) ) Visual symptoms in the Heidenhain variant of Creutzfeldt-Jakob Disease. J Neuroimaging 19: , 283–287. |
[33] | Kher M , Rao MY , Acharya PT , Mahadevan A , Shankar SK ((2009) ) Heidenhain variant of Creutzfeldt-Jakob disease: An autopsy study from India. Ann Indian Acad Neurol 12: , 48–51. |
[34] | del Barrio-Manso I , Toribio-García A , Cordero-Coma M , Tuñón L , Baragaño E ((2010) ) Creutzfeldt - Jakob presenting with isolated visual symptoms: The Heidenhain variant. Arch Soc Esp Oftalmol 85: , 341–344. |
[35] | Prasad S , Lee EB , Woo JH , Alavi A , Galetta SL , Photo essay ((2010) ) MRI and positron emission tomography findings in Heidenhain variant Creutzfeldt-Jakob disease. J Neuroophthalmol 30: , 260–262. |
[36] | Verma R , Junewar V , Sahu R ((2013) ) Creutzfeldt-Jakob disease presenting with visual symptoms: A case of the ‘Heidenhain variant’. BMJ Case Rep 2013, pii: bcr2012008006. |
[37] | Vachalová I , Gindl V , Heckmann JG ((2014) ) Acute inferior homonymous quandrantanopia in a 71-year-old woman. J Clin Neurosci 21: , 683–685. |
[38] | Meyer A , Leigh D , Bagg CE ((1954) ) A rare presenile dementia associated with cortical blindness (Heidenhain’s syndrome). J Neurol Neurosurg Psychiatry 17: , 129–133. |
[39] | World Medical Association ((2013) ) World Medical Association Declaration of Helsinki: Ethical principles for medical research involving human subjects (2013). JAMA 310: , 2191–2194. |
[40] | Jansen C , Parchi P , Capellari S , Ibrahim-Verbaas CA , Schuur M , Strammiello R , Corrado P , Bishop MT , van Gool WA , Verbeek MM , Baas F , van Saane W , Spliet WG , Jansen GH , van Duijn CM , Rozemuller AJ ((2012) ) Human prion diseases in the Netherlands (1998-2009): Clinical, genetic and molecular aspects. PLoS One 7: , e36333. |
[41] | Castellani RJ , Colucci M , Xie Z , Zou W , Li C , Parchi P , Capellari S , Pastore M , Rahbar MH , Chen SG , Gambetti P ((2004) ) Sensitivity of 14-3-3 protein test varies in subtypes of sporadic Creutzfeldt-Jakob disease. Neurology 63: , 436–442. |
[42] | Zerr I , Kallenberg K , Summers DM , Romero C , Taratuto A , Heinemann U , Breithaupt M , Varges D , Meissner B , Ladogana A , Schuur M , Haik S , Collins SJ , Jansen GH , Stokin GB , Pimentel J , Hewer E , Collie D , Smith P , Roberts H , Brandel JP , van Duijn C , Pocchiari M , Begue C , Cras P , Will RG , Sanchez-Juan P ((2009) ) Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain 132: , 2659–2668. |
[43] | Parchi P , Notari S , Weber P , Schimmel H , Budka H , Ferrer I , Haik S , Hauw JJ , Head MW , Ironside JW , Limido L , Rodriguez A , Ströbel T , Tagliavini F , Kretzschmar HA ((2009) ) Inter-laboratory assessment of PrPSc typing in creutzfeldt-jakob disease: A Western blot study within the NeuroPrion Consortium. Brain Pathol 19: , 384–391. |
[44] | Parchi P , Strammiello R , Giese A , Kretzschmar H ((2011) ) Phenotypic variability of sporadic human prion disease and its molecular basis: Past, present, and future. Acta Neuropathol 121: , 91–112. |
[45] | Parchi P , Saverioni D ((2012) ) Molecular pathology, classification, and diagnosis of sporadic human prion disease variants. Folia Neuropathol 50: , 20–45. |
[46] | Parchi P , Capellari S , Gambetti P ((2000) ) Intracerebral distribution of the abnormal isoform of the prion protein in sporadic Creutzfeldt-Jakob disease and fatal insomnia. Microsc Res Tech 50: , 16–25. |
[47] | Bishop MT , Will RG , Manson JC ((2010) ) Defining sporadic Creutzfeldt-Jakob disease strains and their transmission properties. Proc Natl Acad Sci U S A 107: , 12005–12010. |
[48] | Parchi P , Cescatti M , Notari S , Schulz-Schaeffer WJ , Capellari S , Giese A , Zou WQ , Kretzschmar H , Ghetti B , Brown P ((2010) ) Agent strain variation in human prion disease: Insights from a molecular and pathological review of the National Institutes of Health series of experimentally transmitted disease. Brain 133: , 3030–3042. |
[49] | Kobayashi A , Teruya K , Matsuura Y , Shirai T , Nakamura Y , Yamada M , Mizusawa H , Mohri S , Kitamoto T ((2015) ) The influence of PRNP polymorphisms on human prion disease susceptibility: An update. Acta Neuropathol 130: , 159–170. |
[50] | Manners DN , Parchi P , Tonon C , Capellari S , Strammiello R , Testa C , Tani G , Malucelli E , Spagnolo C , Cortelli P , Montagna P , Lodi R , Barbiroli B ((2009) ) Pathologic correlates of diffusion MRI changes in Creutzfeldt-Jakob disease. Neurology 72: , 1425–1431. |
[51] | Nonno R , Bari MAD , Cardone F , Vaccari G , Fazzi P , Dell’Omo G , Cartoni C , Ingrosso L , Boyle A , Galeno R , Sbriccoli M , Lipp HP , Bruce M , Pocchiari M , Agrimi U ((2006) ) Efficient Transmission and Characterization of Creutzfeldt–Jakob Disease Strains in Bank Voles. PLoS Pathog 2: , e12. |
[52] | Imbriani P , Marfia GA , Marciani MG , Poleggi A , Pocchiari M , Puoti G , Caltagirone C , Pisani A ((2015) ) Heidenhain variant in two patients with inherited V210I Creutzfeldt-Jakob disease. Int J Neurosci, doi: 10.3109/00207454.2015.1047017 |
[53] | Victoroff J , Ross GW , Benson DF , Verity MA , Vinters HV ((1994) ) Posterior cortical atrophy. Neuropathologic correlations. Arch Neurol 51: , 269–274. |
[54] | Crutch SJ , Lehmann M , Schott JM , Rabinovici GD , Rossor MN , Fox NC ((2012) ) Posterior cortical atrophy. Lancet Neurol 11: , 170–178. |
Figures and Tables
Fig.1
Distinctive histopathology and regional distribution of PrPSc in patient #16 (MM 2C+1). (A) Spongiform change characterized by large, confluent vacuoles (H&E stain) and (B) perivacuolar and coarse PrP deposits (PrP immunohistochemistry) in the occipital cortex; (C) mixed type of spongiform change with large, confluent vacuoles intermixed with smaller vacuoles in the striatum (H&E stain); (D) spongiform change characterized by small, fine, microvacuoles (H&E stain) and (E) synaptic pattern of PrP deposition in the cerebellum (PrP immunohistochemistry). All pictures (A-E) have the same magnification (×100); F) PrPSc typing by western blot showing the co-occurrence of protein types 1 and 2 in all regions but the cerebellum, where only a relatively low amount of type 1 is seen. While the amount of PrPSc type 2 is higher than that of type 1 in most samples from the cerebral cortex, subcortical gray matter structures show either a similar amount of the two proteins or a predominant accumulation of type 1.
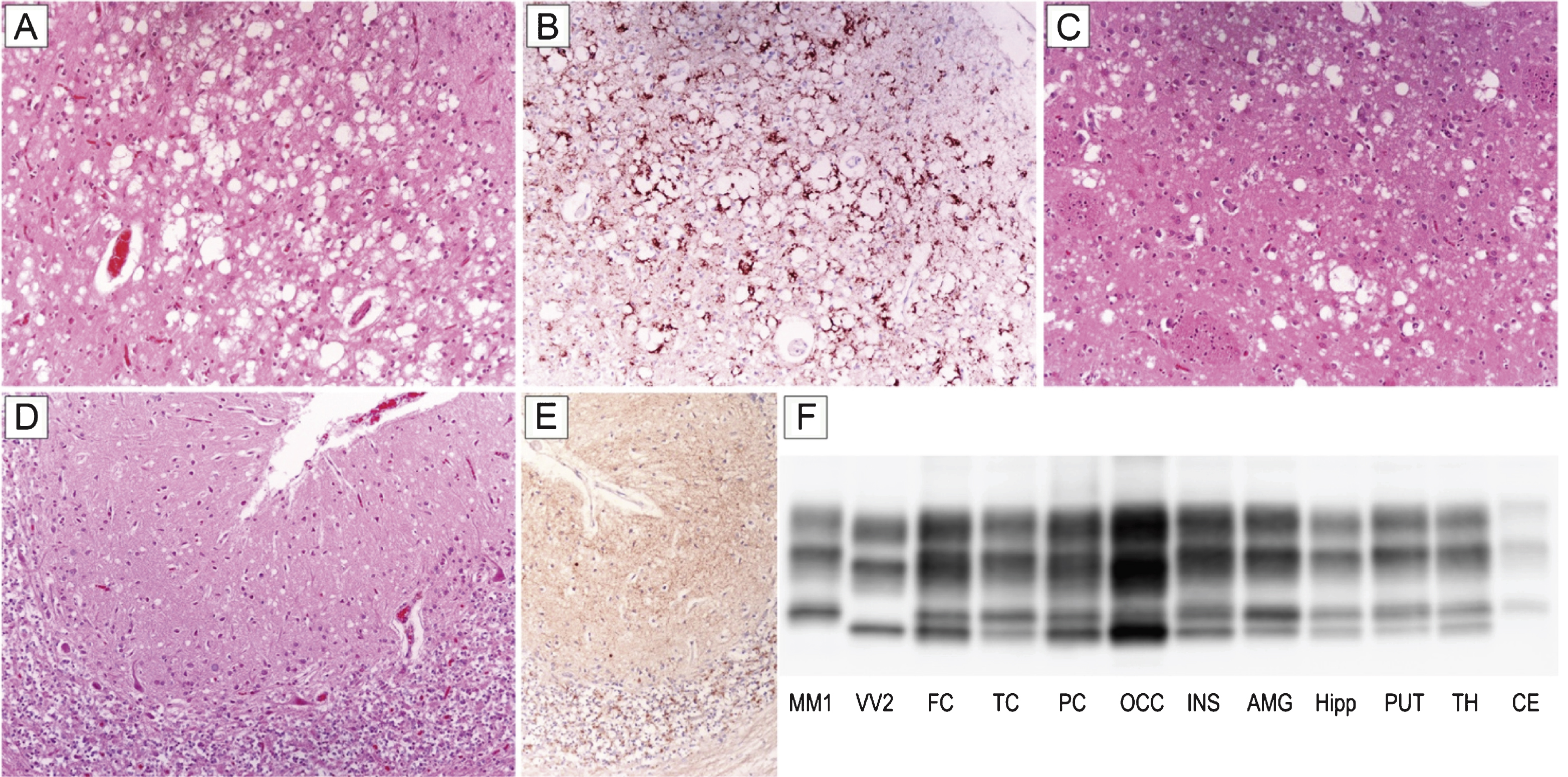
Fig.2
Results of neuroimaging, histopatological, and PrPSc studies in patient #17 (MM 2C+1). Brain DWI-MRIs at 12 (A), 16 (B), and 19 months (C) from clinical onset showing a prominent occipital hyperintensity extending to the parietal cortices during the clinical course. (D) Status spongiosus associated with severe gliosis and neuronal loss in the occipital cortex (H&E stain); (E) coarse granular pattern of PrP staining in the occipital cortex (PrP immunohistochemistry); (F) focal spongiform change with vacuoles of intermediate to large size in the temporal cortex (H&E stain); (G) coarse and perivacuolar PrP deposits in the frontal cortex (PrP immunohistochemistry); (H) focal mild spongiform change in the cerebellum (H&E stain); (I) PAS positive, unicentric, amyloid plaque in the molecular layer of cerebellum (PAS stain); (L) plaque-like type of PrP deposition predominantly involving the molecular layer in the cerebellum. All pictures (D-L) in the panel have the same magnification (×100) except for L (×40). (M) PrPSc typing by western blot showing PrPSc 2 in all areas analyzed co-occurring with relatively low amount of PrPSc type 1 in the cerebral cortex, brainstem, and cerebellum. Overall, the PrPSc amount is highest in the cerebral cortex; intermediate in the striatum, thalamus, and cerebellum; and lowest in the hippocampus, hypothalamus, and brainstem.
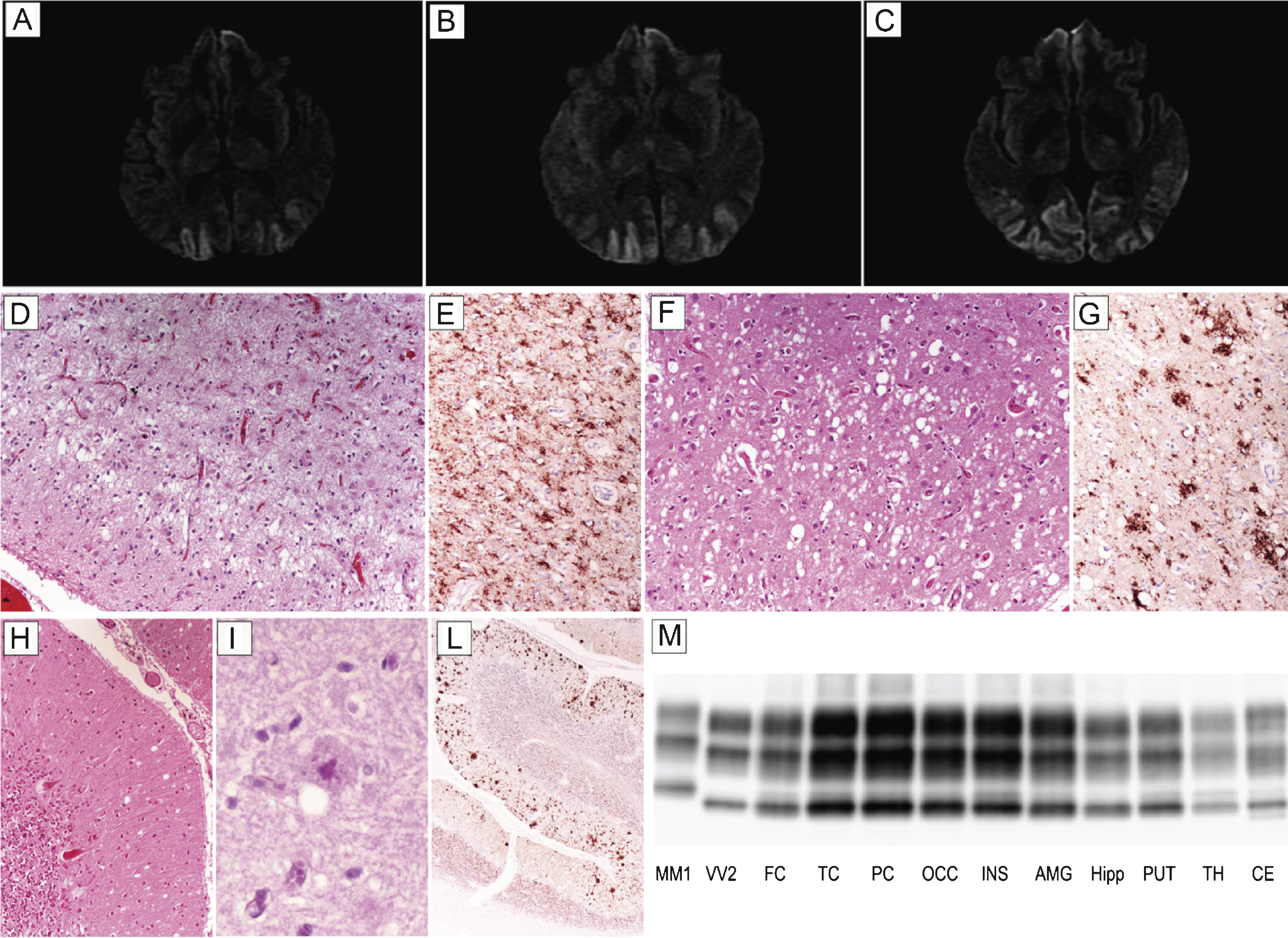
Fig.3
Results of neuroimaging, histopatological, and PrPSc studies in patient #18 (MM 2C). Hyperintensity of occipital cortex in DW- (A and B) and FLAIR- (C) sequences. (D) Spongiform change characterized by large, confluent vacuoles diffusely involving the gray matter (H&E stain, occipital cortex); (E) sparse foci of spongiform change especially involving the middle layers (H&E stain, frontal cortex); (F) perivacuolar pattern of PrP deposition in the occipital cortex; (G) sparse foci of coarse PrP deposits in the frontal cortex; (H) mild spongiform changes characterized by small vacuoles in the cerebellum (H&E stain); (I) lack of significant spongiform change in the putamen (H&E stain). Pictures (D-I) in the panel have the same magnification (×100). (L) PrPSc typing by western blotting revealing a type 2 pattern of electrophoretic mobility in all regions; PrPSc is overall more abundant in the cerebral cortex (especially in occipital cortex) than in subcortical areas where only traces or a relatively low amount of the abnormal protein are detected.
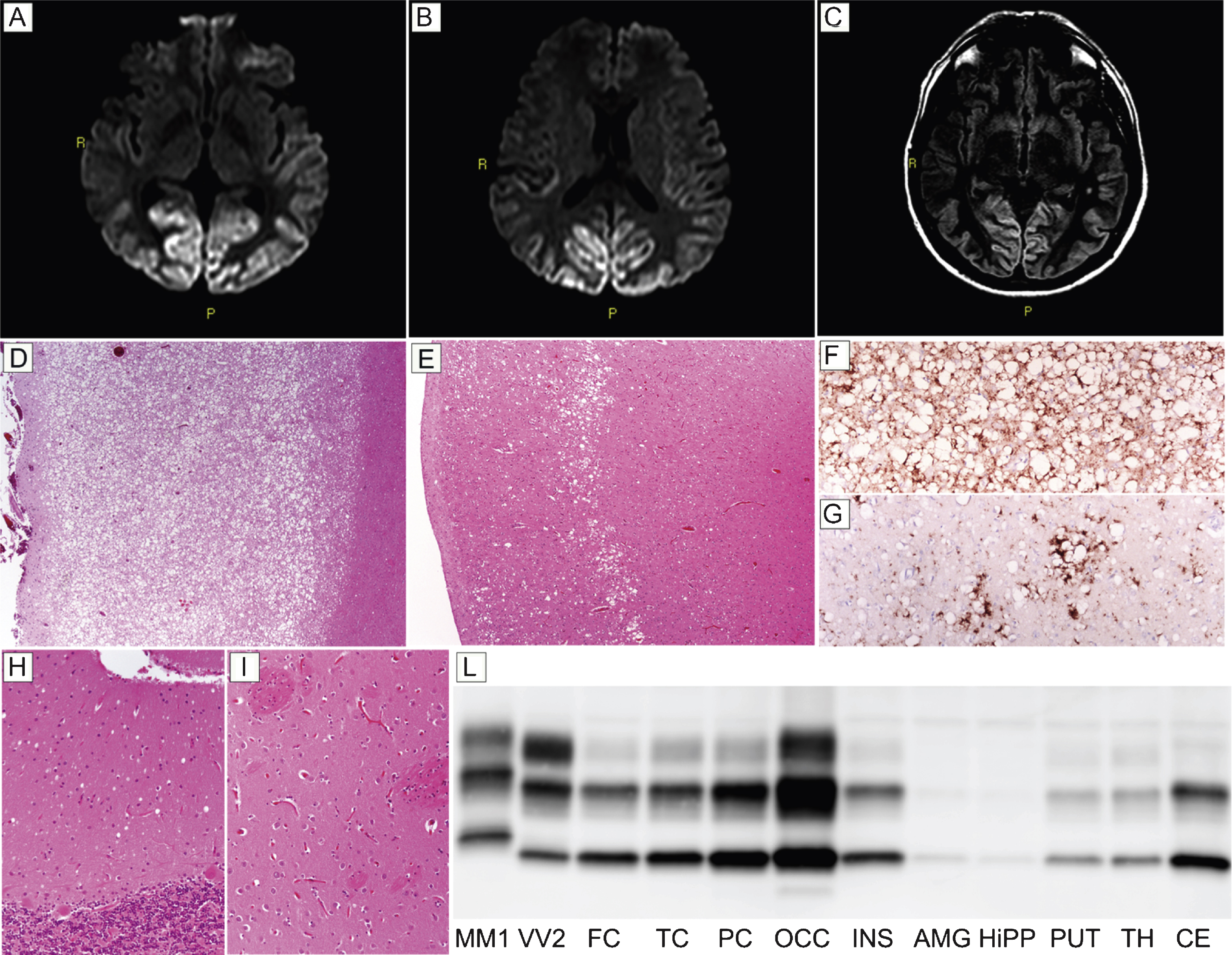
Table 1
Clinical features of patients with the Heidenhain variant of sCJD
| N° | Gender | Age | Visual symptoms (VS) | VS duration (mo) | Other symptoms | Disease duration (mo) |
| 1 | F | 54 | Visual field restriction, loss of vision, disturbed perception of colors or objects, hallucinations | 2.5 | Delirium, confusion, disorientation, dystonia, pyramidal signs, akinetic mutism | 5 |
| 2 | M | 63 | Blurred vision, visual field restriction, | 2 | Aphasia, attentive deficit, cerebellar signs, myoclonus | 4 |
| 3 | M | 67 | Blurred vision, visual field restriction, vision loss up to cortical blindness | 1.5 | Cognitive disturbances, abnormal behavior, myoclonus, tremor, pyramidal signs, akinetic mutism | 5 |
| 4 | M | 80 | Blurred vision, disturbed perception of colors or structures, hallucinations | 1 | Ataxia, disorientation | 4 |
| 5 | M | 69 | Blurred vision, vision loss | 1 | Dysarthria, myoclonus, cognitive and gait disturbances | 2.5 |
| 6 | F | 70 | Blurred vision, vision loss up to cortical blindness, disturbed perception of colors or structures | 2 | Ataxia, cognitive decline, myoclonus, akinetic mutism (several months with parenteral nutrition in vegetative state) | 10 |
| 7 | F | 80 | Disturbed perception of colors or objects, optical anosognosia | 1 | Cognitive decline, cerebellar and pyramidal signs, seizures, myoclonus, akinetic mutism | 5 |
| 8 | M | 78 | Disturbed perception of structures, optical hallucinations | 1.5 | Alien arm syndrome, apraxia, involuntary movements, ataxia | 3.5 |
| 9 | M | 69 | Visual field restriction, vision loss, optical hallucination (palinopsia) | 2 | Pyramidal signs, myoclonus, abrupt stupor, coma | 4 |
| 10 | F | 64 | Blurred vision, disturbed perception of objects (simultanagnosia) | 1.5 | Myoclonus, memory loss, disorientation, ataxic gait, akinetic mutism | 2.5 |
| 11 | F | 77 | Visual field restriction, vision loss up to cortical blindness, optical anosognosia | 1 | Myoclonus, ideomotor apraxia, disorientation | 3.5 |
| 12 | F | 63 | Visual field restriction, vision loss up to cortical blindness, disturbed perception of objects (distortion) | 2 | Memory loss, aphasia, weakness, myoclonus | 3.5 |
| 13 | M | 57 | Visual field restriction, blurred vision, disturbed perception of objects | 2 | Dementia, myoclonus, rigidity, seizures, coma | 3 |
| 14 | F | 79 | Disturbed perception of colors and objects, visual field restriction | 2 | Ataxia, myoclonus, dementia, aphasia, coma | 4 |
| 15 | F | 70 | Disturbed perception of colors or objects, visual field restriction | 1 | Ataxic gait, ideomotor apraxia, dystonia, memory loss, myoclonus, rigidity, apallic state | 3 |
| 16 | F | 70 | Vision loss, disturbed perception of colors or structures, optical anosognosia | 6 | Ataxia, dysphonia, dysphagia, cognitive disturbances, pyramidal signs | 26 |
| 17 | M | 54 | Optical hallucination, environmental agnosia | 12 | Discalculia, pyramidal signs, disorientation, psychosis | 44 |
| 18 | M | 48 | Blurred vision, visual field restriction, vision loss up to cortical blindness | 2.5 | Dysarthria, dysphagia, disorientation, myoclonus | 6 |
Table 2
Histotype classification and results of neurophysiologic, neuroimaging, and CSF studies of sCJD patients with the Heidenhain clinical variant
| N° | sCJD Type | Brain MRI | EEG (timing)b | CSF | ||
| 14-3-3 (test result) | t-tau (pg/ml) | Timingb | ||||
| 1 | MM 1+2C | Parietal and occipital hyperintensity on DWI | PSWCs (3)c | Ambiguous | 5600 | 2 |
| 2 | MM 1+2C | Normal (routine MRIa) | PSWCs (2.5)c | Positive | 6459 | 2.5 |
| 3 | MM 1+2C | Bilateral frontal atrophy (routine MRIa) | PSWCs (2) | Positive | NA | 2 |
| 4 | MM 1+2C | Parietal and temporal+BG hyperintensity on DWI | PSWCs (2) | Positive | 3990 | 2 |
| 5 | MM 1+2C | White matter signal alterations | PSWCs (1.5) | Positive | 7579 | 1.5 |
| 6 | MM1 | Parieto-occipital and basal ganglia hyperintensity on DWI | PSWCs (1.5)c | Positive | 2522 | 0.5 |
| 7 | MM1 | Non-specific (routine MRIa) | PSWCs (3)c | Positive | NA | 1.5 |
| 8 | MM1 | Non-specific | PDs (0.5) | Positive | 2335 | 0.5 |
| 9 | MM1 | Non-specific (routine MRIa) | PSWCs (1.5) | NA | NA | |
| 10 | MM1 | Parietal and occipital hyperintensity on FLAIR and DWI | PSWCs (1.5) | Positive | NA | 2 |
| 11 | MM1 | Normal (routine MRIa) | PSWCs (2) | Positive | NA | 2 |
| 12 | MM1 | NA | PSWCs (2) | NA | NA | |
| 13 | MM1 | Right parietal atrophy (routine MRIa) | PSWCs (2)c | NA | NA | |
| 14 | MM1 | Left parietal hyperintensity on FLAIR and DWI | Generalized slowing (3) | Positive | NA | 3 |
| 15 | MM1 | Non-specific vascular signs (routine MRIa) | PSWCs (2.5)c | NA | NA | |
| 16 | MM 2C+1 | Normal (routine MRIa) | Generalized slowing (9,13) | Ambiguous | NA | 9, 13 |
| 17 | MM 2C+1 | Parietal and occipital hyperintensity on FLAIR and DWI | Generalized slowing (6,8,11,13,20) | Ambiguous | 1070 | 6 |
| 18 | MM 2C | Parietal and occipital hyperintensity on FLAIR and DWI | Generalized slowing (3,5,6) | Ambiguous | 1140 | 3 |
aroutine MRI not including DWI and FLAIR sequences; bmonths after onset of symptoms; cEEG recording/s performed earlier in the clinical course did not show PSWCs; NA, not available; see text for other abbreviations.


