
Porphyromonas gingivalis LPS and Actinomyces naeslundii Conditioned Medium Enhance the Release of a Low Molecular Weight, Transcriptionally Active, Fragment of Glycogen Synthase-3 Kinase in IMR-32 Cell Line
Abstract
Background:
Glycogen synthase-3 kinase (GSK3) is one of the major contributors of tau hyperphosphorylation linked to neurofibrillary tangles in Alzheimer’s disease (AD).
Objective:
To determine a mechanism of GSK-3β activation by two periodontal bacteria consistently confirmed in AD autopsied brains.
Methods:
Porphyromonas gingivalis FDC381 and Actinomyces naeslundii ATCC10301 conditioned media were collected. IMR-32 cells were challenged for 48 h with the conditioned media alongside P. gingivalis (ATCC33277) ultrapurified lipopolysaccharide (LPS) designated Pg.LPS under established cell culture conditions either alone or combined. Gene expression and protein analyses for GSK-3β were carried out.
Results:
qPCR demonstrated that GSK-3β gene was overexpressed in IMR-32 cells treated with Pg.LPS with a 2.09-fold change (p = 0.0005), while A. naeslundii treated cells demonstrated 1.41-fold change (p = 0.004). Western blotting of the cells challenged with Pg.LPS (p = 0.01) and A. naeslundii conditioned medium (p = 0.001) demonstrated the 37 kDa band for each treatment with variable intensity across the medium control. Immunohistochemistry with the GSK-3β of the IMR-32 cells challenged with Pg.LPS and A. naeslundii alone demonstrated cytoplasmic and nuclear localization.
Conclusions:
Exposure to various bacterial factors upregulated the gene expression of GSK-3β. Western blotting for GSK-3β confirmed the presence of the cleaved fragment by Pg.LPS (37 kDa band p = 0.01) and A. naeslundii conditioned medium (37 kDa band p = 0.001). Immunostaining demonstrated both cytoplasmic and nuclear localization of GSK-3β. Therefore, Pg.LPS and an unknown factor from the A. naeslundii conditioned medium mediated GSK-3β activation via its transcriptionally active, cleaved, fragment. These virulence factors in the body appear to be detrimental to brain health.
INTRODUCTION
Porphyromonas gingivalis and Actinomyces naeslundii are bacteria associated with periodontal disease which have been shown to spread to the brain tissue of patients with Alzheimer’s disease (AD). Glycogen synthase-3 kinase (GSK-3) is a metabolic enzyme that regulates and controls multiple physiological processes in the human body including the inflammatory response triggered by bacteria.1,2 It has two isoforms, GSK-3α and GSK-3β. The GSK-3β form is abundant in the brain where it is found mainly in the neurons. Over-activity of GSK-3β in AD is associated with the death of neurons. GSK-3β belongs to a class of kinase enzymes that catalyze several substrates, which usually need to be pre-phosphorylated by other kinases.1 GSK-3β kinase is one of the major contributors of tau hyperphosphorylation linked to neurofibrillary tangle (NFT) formation in AD.3 AD is a neurodegenerative disorder, and the most prevalent example of dementia. AD can manifest in two forms either as familial (less common) or sporadic (most common). Individuals with AD clinically display behavioral and memory associated symptoms which are correlated with hallmark proteins in postmortem brain tissue sections.4,5 These hallmark lesions are amyloid-β (Aβ) plaques and abnormally phosphorylated tau protein binding to NFTs.6,7 The cause of this neurodegenerative disease remains unknown but a multi-domain etiology is implicated.8,9 Drug-based therapy to slow down deteriorating memory in the early stages of AD is emerging; however, researchers have yet to find a more adequate treatment for controlling the various aspects for this debilitating disease. Further investigations into the many risk factors involved with the etiology of AD are still necessary.10
A number of published articles have reported the detection of several oral bacteria related to periodontal disease including spirochetes, Porphyromonas gingivalis, and Actinomyces naeslundii in AD autopsy brain tissue11–14 with/without next generation sequencing methodologies; and to a lesser extent in age related control brains.15 P. gingivalis and lipopolysaccharide (LPS), located in its outer membrane, have also been detected in AD brains.16 The rationale for exploring the role of bacterial virulence factors (P. gingivalis, LPS, and A. naeslundii) cleaving GSK-3β comes from our ongoing laboratory investigations with specific interest in periodontal disease pathogens that have been documented in literature for their definitive detection in autopsied AD brains.11–15 Recently, there has been more acceptance amongst AD researchers for having an inflammatory cause8 with a possible infectious origin in the context of host’s dysbiotic microbiomes.17,18 An infectious component further correlates with peripheral inflammation (cytokines in blood) from pathogens like P. gingivalis and its LPS which negatively impact brain health during life.19,20 This increase in pro-inflammatory cytokines associates with blood-brain barrier damage during aging, which eventually contributes to overall cognitive decline.21 A. naeslundii is a Gram-positive, bacillus found typically in the oral biofilms of healthy individuals.22 This bacterium is largely seen as an avirulent saprophyte, gaining its nutrients from decaying organic material.23 This explains its prevalence in patients with poor oral health. A. naeslundii is also one of the early colonizers of the oral biofilm and is one of the few Gram-positive bacteria that have two different types of fimbriae. Type one fimbriae allow A. naeslundii to adhere to tooth surfaces. Type two fimbriae allow A. naeslundii to adhere to β linked galactose and galactosamine containing glycoproteins, which are typically found on bacterial and epithelial cell surfaces.24 A. naeslundii can also change the pH of its environment to hinder the growth of competing bacteria by releasing ammonia to control the acidic pH.25 Our in-house studies confirm that P. gingivalis benefits from the control in acidic pH as it prefers a neutral to slightly alkaline pH range. Another feature of A. naeslundii is that like P. gingivalis, it is able to become pathogenic (dysbiosed) and cause actinomycosis separately, and periodontitis under the influence of P. gingivalis. Noble et al.26 associated the serum IgG titers of A. naeslundii to be higher in AD patients’ blood serum. P. gingivalis on the other hand is a Gram-negative bacterium and is considered as the keystone pathogen of periodontitis27 and Actinomyces species are residents of this subgingival dysbiosed biofilm.28,29 This has helped to formulate the hypothesis that both P. gingivalis and A. naeslundii have the ability to co-aggregate in highly inflammophilic environments, which may be an explanation for both of these microbes to co-exist in the periodontal pockets and brains of AD patients. The present study aimed to widen the concept that virulence factors of oral bacteria may be detrimental to brain health; and that P. gingivalis infection alone may be insufficient to cause AD and that multispecies of oral microbes and/or their virulence factors alone may contribute to this complex degenerative disease. Thus, in the present study we introduce the dual role of P. gingivalis and A. naeslundii virulence in order to investigate the mechanism of GSK-3β activation by periodontal bacterial factors in vitro as a step towards a multispecies pathogenic bacterial co-operation under inflammophilic conditions contributing to direct and downstream chronic neuroinflammation in AD.
MATERIALS AND METHODS
P. gingivalis conditioned medium as a source of crude virulence factors
P. gingivalis (FDC381) was cultured in-house under anaerobic conditions to density of 5×109 per mL in Tryptone Soya Broth (TSB) supplemented with hemin (5 μg/mL final concentration, Sigma-Aldrich UK), and menadione (1 μg/mL final concentration). The liquid broth in 15 mL culture tubes (loosened lids) were degassed by placing them into anaerobic jars (Thermo Scientific™ Oxoid™ [PDF] AnaeroJar™ 2.5 L) with an anaerobic sachet (Thermo Scientific™ Oxoid™ AnaeroGen™ 2.5 L Sachet). The anaerobic jars were placed in an incubator set at 37°C for 24 h prior to need. The next day, the degassed TSB was directly inoculated with a single colony of P. gingivalis from a culture previously grown on a blood agar plate into separate degassed 10 mL aliquots. The broth cultures, with loosened lids, were placed into an anaerobic jar containing an anaerobic sachet (details above). The lids (on anaerobic jars) were speedily secured and placed in an incubator set at 37°C for 48 h. Following growth, the liquid culture was centrifuged at 20,238 g at 4°C for 30 min to pellet the bacterial cells. The supernatant containing the virulence factors was collected and aliquoted for storage at –80°C until needed for exposure to cells (IMR-32 cells) under cell culture conditions.
A. naeslundii conditioned medium as a source of crude virulence factors
A. naeslundii (ATCC10301) culture on Vegitone agar plates was obtained as a gift from the University of Central Lancashire, Preston, UK, microbiological culture collection. A subculture was prepared by taking a loop-full of the inoculum (from an A. naeslundii colony) onto TSB-blood agar plates as for P. gingivalis preferred growth conditions and incubated at 37°C into anaerobic jars with an anaerobic sachet as for P. gingivalis liquid culture above for 48 h. Following the growth of A. naeslundii colonies on solid medium, a loopful of a single colony was taken and inoculated into sterile, degassed P. gingivalis preferred TSB liquid medium and incubated at 37°C under anaerobic (anaerobic sachet) conditions for 48 h. Following growth, the conditioned medium containing crude virulence factors was separated from the bacterial cells by centrifugation as mentioned above for P. gingivalis. The supernatant was then collected. Aliquots were prepared in pre-labelled sterile tubes and stored at –80°C until needed for their exposure to IMR-32 cells.
Cell culture: Exposure of cells to various treatments
Source of IMR-32 cell line
The IMR-32 cell line CCL-127™ was obtained from American Type Culture Collection (ATCC) (https://www.atcc.org/products/ccl-127). According to (https://www.atcc.org/products/ccl-127), the established IMR-32 cell line is from a confirmed neuropathology diagnosis of neuroblastoma occurring in a 13-month-old Caucasian male.
IMR-32 cells were cultured either in 6 well plates (for total RNA isolation) or flasks (T25) for cell lysates (Nunc, ThermoFisher) and on glass coverslips for immunostaining in six well plates. IMR-32 cells were cultured in the presence of Dulbecco’s Modified Eagle’s minimal essential medium (DMEM), supplemented with 10% fetal calf serum, 4 mM glutamine, 2 mM sodium pyruvate, and with and without 0.1 mM penicillin/streptomycin (Invitrogen). All flasks and 6 well plates were incubated at 37°C in a humidified atmosphere of 5% CO2, 95% air. Following an initial overnight growth in DMEM, the IMR-32 cells were cultured either under standard cell culture conditions or exposed to control medium (1 in 5 dilution of sterile TBS in DMEM), and P. gingivalis and A. naeslundii virulence factors alone (diluted 1 in 5 in penicillin/streptomycin antibiotic free DMEM). P. gingivalis 33277 ultrapure LPS was purchased from InVivogen Europe (https://www.invivogen.com/lps-pg). A stock solution of the ultrapure LPS was prepared in sterile water at 1 mg/mL. IMR-32 cells were exposed to the ultrapure LPS at 1 μg/mL final concentration diluted in penicillin/streptomycin antibiotic free DMEM. P. gingivalis LPS either alone or combined with P. gingivalis FDC381 conditioned medium and separately A. naeslundii conditioned medium combined with P. gingivalis LPS were also tested on IMR-32 cells for 48 h. Following treatments, the cells cultured in flasks were detached using cell scrapers and the liquid was transferred into a 15 mL centrifuge tube and centrifuged at 376 g for 5 min. The supernatant was discarded, and the pellets were retained to prepare total RNA extraction or cell lysates. Following treatments, the cells on glass coverslips were washed free of any cell culture related protein with sterile PBS (1×3 washes) and fixed in 10% formaldehyde at 4°C overnight and washed in fresh PBS over the weekend prior to immunostaining.
Gene expression of control and treated IMR-32 cells
Total RNA extraction
Total RNA from the control and treated IMR-32 cell line was extracted using the TRIzol reagent (Invitrogen) and purified by the RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. The on-column DNase digestion was performed with the DNase-free DNase set (Qiagen) to eliminate genomic DNA contamination. Total RNA was suspended in 30 μl RNase-free water and the quantity and quality of RNA was evaluated by a Nanodrop One spectrophotometer (Thermo Scientific) and an Agilent Bioanalyzer.
Primer sets used for qPCR analysis
The primers used for the qPCR analysis were previously published30–32 and are listed in Table 1.
Table 1
Primer sets used for qPCR analysis for the genes of interest were previously published and are cited accordingly
| Gene and the references used for the primers for qPCR analysis | Forward: 5′-3′ | Reverse: 5′-3′ |
| ACTB (beta-actin) Cavalcanti et al.29 | 5′-TTCCTTCCTGGGCATGGAGT-′3 | 5′-TACAGGTCTTTGCGGATGTC-′3 |
| B2M (b2microglobulin) Shrout et al.30 | 5′-GCCGTGTGAACCATGTGACTTT-3′ | 5′-CCAAATGCGGCATCTTCAAA-3′ |
| GSK-3β Chen et al.31 | 5′-ATTTTCCAGGGGATAGTG GTGT-3′ | 5′-GGTCGGAAGACCTTAGTCCAAG-3′ |
Quantitative or real time polymerase chain reaction (qPCR)
One microgram of total RNA was used to generate complementary DNA (cDNA) in a 20 μl reaction by the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). The cDNA was diluted 1 : 5 and 2 μl used in the qPCR reaction with 600 nM of each specific primer and 5 μl of PowerUp SYBR Green Master Mix (Applied Biosystems) in a 10 μl volume reaction per each gene separately. The qPCR was performed using a ViiA7 Real-time PCR System (Applied Biosystems) and data were analyzed by QuantStudio Real-Time PCR software The expression levels determined by qPCR are based on relative quantification; of two reference genes, beta-actin (ACTB) and beta2-microglobulin (B2M).
The specificity of each primer set was evaluated by a melt curve that revealed a single peak which confirmed a single product was amplified. Each sample was analyzed in triplicates and the expression of GSK-3β was determined using the 2-ΔΔCt method.33 Using this method, the fold-changes were obtained in gene expression, which were normalized to an internal control gene (B2M), relative to a control IMR-32 sample designated medium control.
Cell lysate preparation
The medium of treated cells in flasks (T25) was withdrawn and the cells adhered to the flasks were washed twice with 5 mL phosphate buffered saline (PBS pH 7.5), detached using cell scrapers, suspended in PBS and then transferred into 15 mL sterile, Falcon tubes and centrifuged at 376 g for 10 min. After draining off excess PBS, the cell pellet was lysed in 250 μL volume of lysis buffer RIPA buffer, pH 8.0: containing 50 mM Tris, 150 mM NaCl, 5 mM EDTA, 0.5% Sodium deoxycholate, 0.5% (v/v) NP-40 and 1% sodium dodecyl sulphate. 1/100 final of phenylmethanesulphonyl or PMSF and 5 mM dithiothreitol, 5% protease inhibitor cocktails 2 and 3 from Sigma-Aldrich, UK. The cells were vortex mixed and incubated on ice. The lysed cell mixture was transferred into pre-labelled sterile Eppendorf tubes and centrifuged at 20,238 g for 20 min (Sigma 1–14 microfuge). The liquid phase was withdrawn, transferred into new pre-labelled 1.5 mL Eppendorf centrifuge tubes, and used to determine total protein following the Bradford protein assay.34 All cell lysates were stored at –80°C until needed for electrophoresis and western blotting.
Protein assay
The total protein concentration was determined using the Coomassie Blue protein Assay (Sigma-Aldrich, UK).34 Briefly, protein concentration was obtained from a standard curve prepared using 100–400 μg/mL BSA diluted in lysis buffer. Following the addition of the Coomassie Blue reagent to all standards and test samples, absorbance was measured at 595 nm wavelength using a Jenway 7315 spectrophotometer. The unknown concentration of the samples was calculated by comparing the absorbance values with the standard curve. The lysates were stored at –80°C until needed for western blotting.
Western blot analysis of cell lysates
All lysates from the IMR-32 cell line (under standard cell culture conditions and exposure to control medium and other treatments (1 in 5 dilution of sterile TBS, and P. gingivalis, A. naeslundii virulence factors, purified P. gingivalis LPS (1 μg/mL), and combined P. gingivalis LPS with P. gingivalis FDC381 conditioned medium and separately with A. naeslundii conditioned medium) were separated by SDS-PAGE on precast 12% mini-protean TGX stain-free linear gels (BioRad Laboratories, USA). Protein ladder, (PageRuler Plus, 26619, from Thermo Scientific) was loaded in the first well of each gel. All samples (10 μg) of total protein in Laemmli reducing sample buffer containing 0.3% mercaptoethanol (Alfa Aesar) was electroblotted onto polyvinylidine difluoride (PVDF) membranes, as previously described by Poole et al.16 and Kanasingam et al.35 The membranes were incubated overnight in rabbit anti-GSK-3β Abcam UK[Y174] (ab32391) antibody diluted 1/5000 in PBS/5% milk overnight on a rotary device at 4°C. The next day, membranes were washed (3×15 min) in PBS containing 0.25% tween 20 and then were incubated in the secondary detection antibody goat anti-rabbit Abcam UK (ab205718) conjugated to horseradish peroxidase diluted 1/10 000 in 5% w/v skimmed milk/PBS for 2 h at room temperature. After the incubation, the membranes were washed again (3 × 15 min) in PBS containing 0.25% tween 20 followed by the detection of any positive bands using the enhanced super signal west Pico Plus® chemiluminescent substrate reagent (Thermo Scientific), as per supplier’s instructions. The specific signal from the protein on the membranes was visualized using a ChemicDoc® (Bio-Rad, UK) and images were captured with Image Lab® Software Version 6.0.1. A final step included staining the membrane with India ink (Windsor & Newton) to determine the amount of protein transferred onto the membrane(s) as a loading control as previously reported.16,35 Densitometry was carried out on the bands using the Image J software, and the resulting data was normalized to the loading control.
IMR-32 cells: GSK-3β immunohistochemistry
Following formalin stabilization of endogenous proteins (10% formaldehyde at 4°C overnight, washes in PBS), peroxidase activity was quenched using 0.3% H2O2 in 0.1 M PBS, pH 7.2, for 30 min. No antigen retrieval was carried out. Following thorough washings of cells in PBS, non-specific antibody binding was controlled by 30 min incubation in blocking buffer solution containing 0.1% normal goat serum (Vectastain kit, PK 4002) in 1× PBS containing 0.02% tween 20.
Negative controls
The negative control included omission the primary antibody by substituting it with the block buffer solution.
Test coverslips
The primary antibody rabbit anti-GSK-3β purchased from Abcam UK [Y174] (ab32391) was diluted 1/5000 in block solution and applied to the cells (all treatments and controls). All coverslips were incubated overnight at 4°C in a humidity chamber. The next day, the coverslips were washed (1 × 3) for 5 min each in PBS before re-incubating the cells in the secondary detection antibody from Vectastain kit for rabbit peroxidase IgG kit (PK 4002) (Vector Laboratories, UK), according to the suppliers’ instructions. The detection was completed using the DAB Peroxidase Kit (SK 4100). Except for the control cells, which were lightly counterstained in hematoxylin to make cells visible for imaging reasons, the treated cells were not counterstained. This was to be able to establish any nuclear staining of GSK-3β within the IMR-32 cells. The coverslips with cells were mounted onto prelabelled microscope glass slides. Examination of the cells and image capture were carried out using the Nikon Eclipse E200 Microscope and DS-L2 v.441 Software (Nikon, UK).
Statistical analysis
Fold change in the expression of GSK-3β by qPCR analysis of IMR-32 cells across controls IMR-32 std cell culture conditions (std TC), medium control, and test conditions P. gingivalis FDC381 conditioned medium alone (Pg.381), A. naeslundii conditioned medium alone, P. gingivalis 33277 ultrapure LPS alone (Pg.LPS), Pg.381+A. naeslundii combined (Pg.381+An) and P. gingivalis 33277 LPS+A. naeslundii combined (Pg.LPS+An). A two variant T-test (T-Test) was carried out using the Excel program. A p value, less than or equal to≤0.05 was considered significant.
For the densitometry of western blotting bands (47 kDa and 37 kDa), the data was evaluated using the Statistical Package for the Social Sciences (SPSS version 29.0.1.0). The ANOVA test was conducted based on the null hypothesis (where a p value, less than or equal to≤0.05 was considered significant) that there was no difference between the band densities.
RESULTS
Molecular biology: GSK-3β gene expression
The expression level of B2M gene was the most stable when compared to ACTB gene within all samples and therefore B2M was selected as a reference gene for the qPCR analysis of GSK-3β in IMR-32 cells across the various treatments (Fig. 1).
Fig. 1
GSK-3β q-PCR analysis. q-PCR analysis: There is a statistically significant difference in the IMR32 mRNA fold change (number within each black bar represents fold change) in the expression of GSK-3β by q-PCR analysis (N = 3) across the medium control and IMR-32 A. naeslundii (p = 0.004) and IMR-32 PgLPS (p = 0.0005) as analyzed by the two variant T-Test.
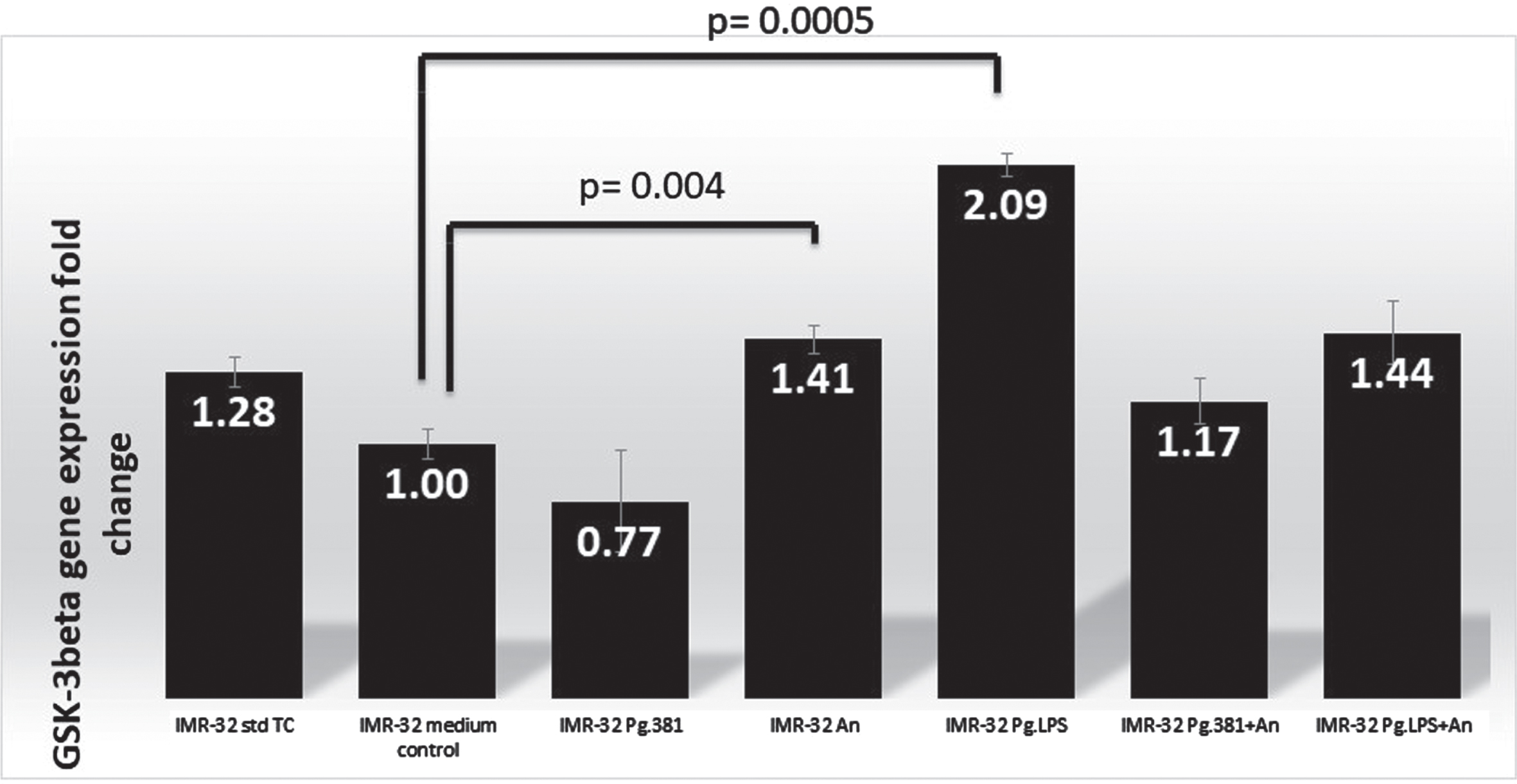
The IMR-32 cells treated with P. gingivalis conditioned medium (bar labeled as IMR32-Pg381) showed GSK-3β gene expression was downregulated by qPCR analysis as compared with the expression of the B2M gene. GSK-3β was marginally over-expressed in IMR-32 A. naeslundii conditioned medium treated cells (labelled as IMR-32 An, fold-change 1.41) over both control conditions (IMR-32 std TC), and in comparison, with the med control. IMR-32 Pg.LPS showed GSK-3β gene expression was upregulated by qPCR analysis with a 2.09-fold change (Fig. 1). The fold changes (up/downregulated) identified by qPCR as compared with the B2M gene were statistically significant as a result of the P. gingivalis conditioned medium alone treated cells (IMR-32 Pg381, p = 0.025) and A. naeslundii conditioned medium alone (IMR-32 An, p = 0.004); Pg.LPS treated cells p = 0.0005 and P. gingivalis conditioned medium combined with A. naeslundii conditioned medium (IMR-32 Pg381+An, p = 0.005) and Pg.LPS combined with A. naeslundii conditioned medium treated cells (IMR-32 Pg.LPS+An p = 0.02) over the med control treatment according to the two variant T-Test (Fig. 1).
Western blotting: Rabbit anti-GSK-3β antibody
Western blot analysis in samples from controls (lanes 1–2 labelled IMR-32 std TC and IMR-32 TSB) and treatment lane 3 labelled IMR-32 Pg381 (Fig. 2A), demonstrated an abundant and a very strong band around the 47 kDa molecular weight size corresponding to the native GSK-3β protein. This band was moderately strong in the lanes loaded with A. naeslundii conditioned medium (IMR-32 An), and in lanes labelled as IMR-32 Pg381+An and IMR-32 Pg.LPS+An. The band corresponding to 47 kDa size was very weak in the lane loaded with IMR-32 Pg.LPS.
Fig. 2A
IMR-32 cell lysate western blotting: GSK-3β antibody. Immunoblot of the IMR-32 cell lysate (protein at 10 μg per lane) with anti-GSK-3β antibody. Distinct bands around the 47 kDa molecular weight size corresponding to GSK-3β in lanes with the prefix IMR-32 std TC (std cont) and then IMR-32 TSB (med cont), IMR-32 Pg381, IMR-32 An, IMR-32 Pg.LPS, IMR-32 Pg381+An and IMR-32 Pg.LPS+An. A 37 kDa was observed more clearly in lanes with the prefix IMR-32 Pg381, IMR-32 An, IMR-32 Pg.LPS, IMR-32 Pg381+An and IMR-32 Pg.LPS+An.
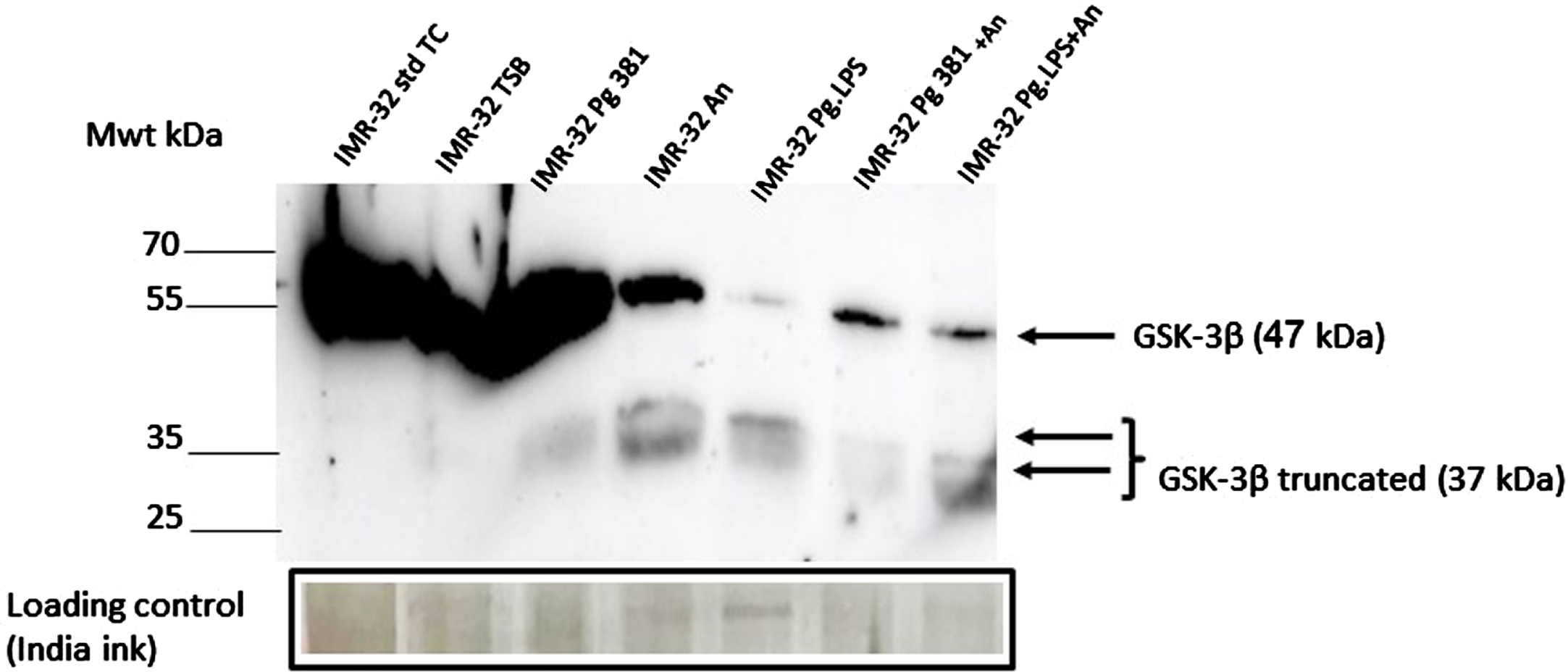
Distinct low molecular weight bands around the 37 kDa molecular weight size were observed albeit weak in lanes labelled P. gingivalis 381, but more strongly in lanes A. naeslundii, and P. gingivalis 33277 LPS, and P. gingivalis 33277 LPS (Fig. 2A).
GSK-3β densitometry
There was no statistical difference between the control cultures (standard cell culture designated IMR-32 std TC, and medium control designated IMR-32 TSB (p = 0.816).
47 kDa band
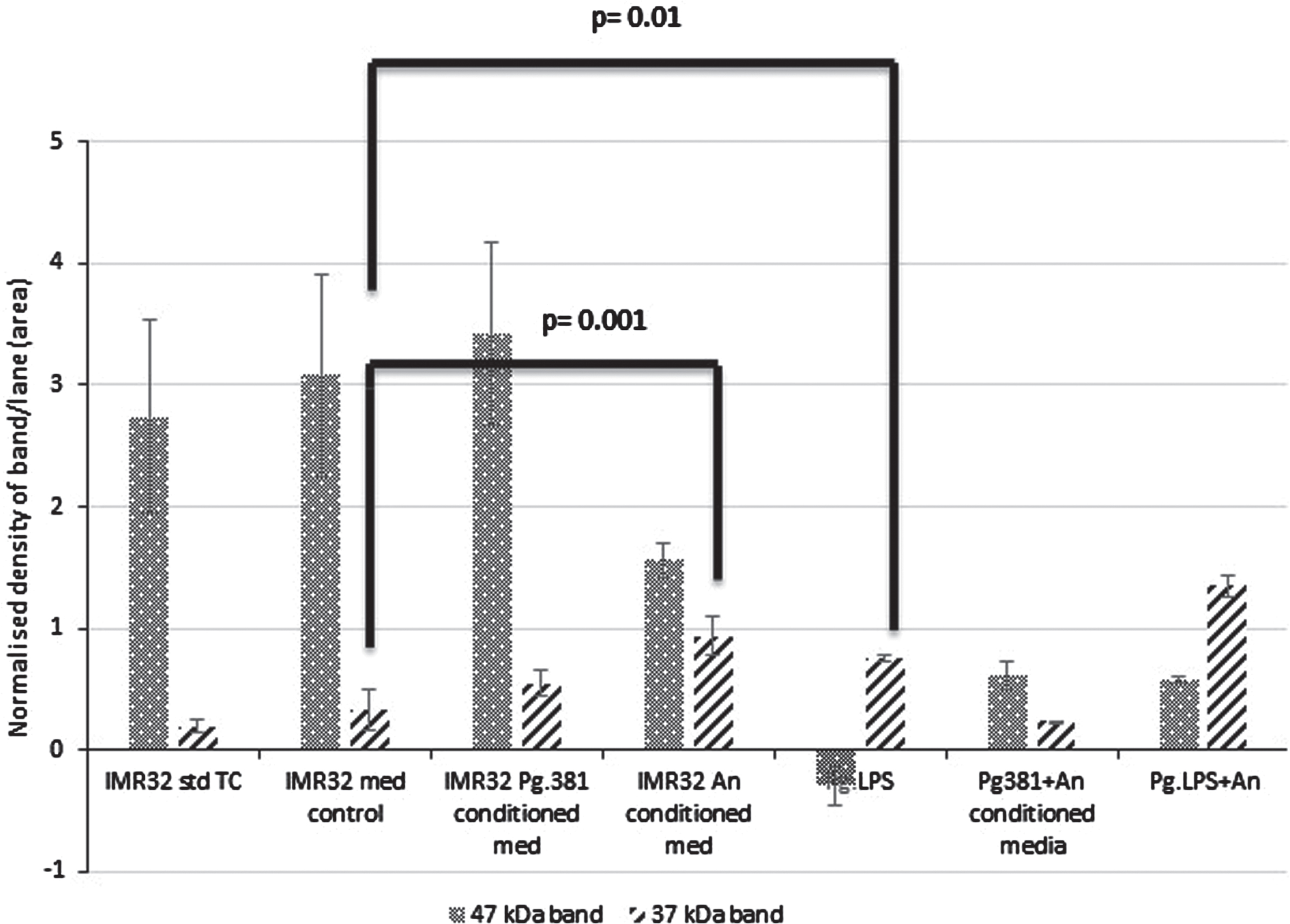
The 47 kDa band: across med control (IMR-32 TSB) versus IMR-32 Pg381 conditioned medium was not statistically significant (p = 0.692) (Fig. 2B). All other treatments including IMR-32 A. naeslundii conditioned medium (p = 0.001); IMR-32 Pg.LPS (p = 0.001); IMR-32 Pg381+An (p = 0.01); Pg.LPS+A. naeslundii (p = 0.01) compared to IMR-32 TSB were statistically significant at the≤0.05 level.
Fig. 2B
Densitometric analysis of the cell lysate immunoblotted with anti-GSK-3β antibody. All error bars represent standard error of mean. Statistical analysis of the results for the 47 and 37 kDa bands. There was no statistical difference between the control cultures prefixed with IMR-32 std cont (std TC) and IMR-32 med cont (TSB) (p = 0.816). The 47 kDa band: across med control versus all treatment conditions IMR-32 Pg381 conditioned med (p = 0.692); IMR-32 A. naeslundii conditioned medium (p = 0.001); IMR-32 Pg.LPS (p = 0.001); IMR-32 Pg381+An (p = 0.01); Pg.LPS+A. naeslundii (p = 0.01). The 37 kDa band: across med control versus all treatment conditions IMR-32 Pg381 conditioned med (p = 0.369); IMR-32 A. naeslundii conditioned medium (p = 0.001); IMR-32 Pg.LPS (p = 0.01); IMR-32 Pg381+An (p = 0.035); and, Pg.LPS+An (p = 0.001).
37 kDa band
Statistically the results for the GSK-3β 37 kDa band across medium control (IMR-32 TSB), versus al IMR-32 Pg381 conditioned medium (p = 0.369) was not significant. The IMR-32 A. naeslundii conditioned medium (p = 0.001), IMR-32 Pg.LPS (p = 0.01), IMR-32 Pg381+An combined (p = 0.035), and Pg.LPS+An combined (p = 0.001) were statistically significant (Fig. 2B).
Immunohistochemistry
The negative controls where the primary antibody was omitted remained negative in all variables (Fig. 3A).
Fig. 3
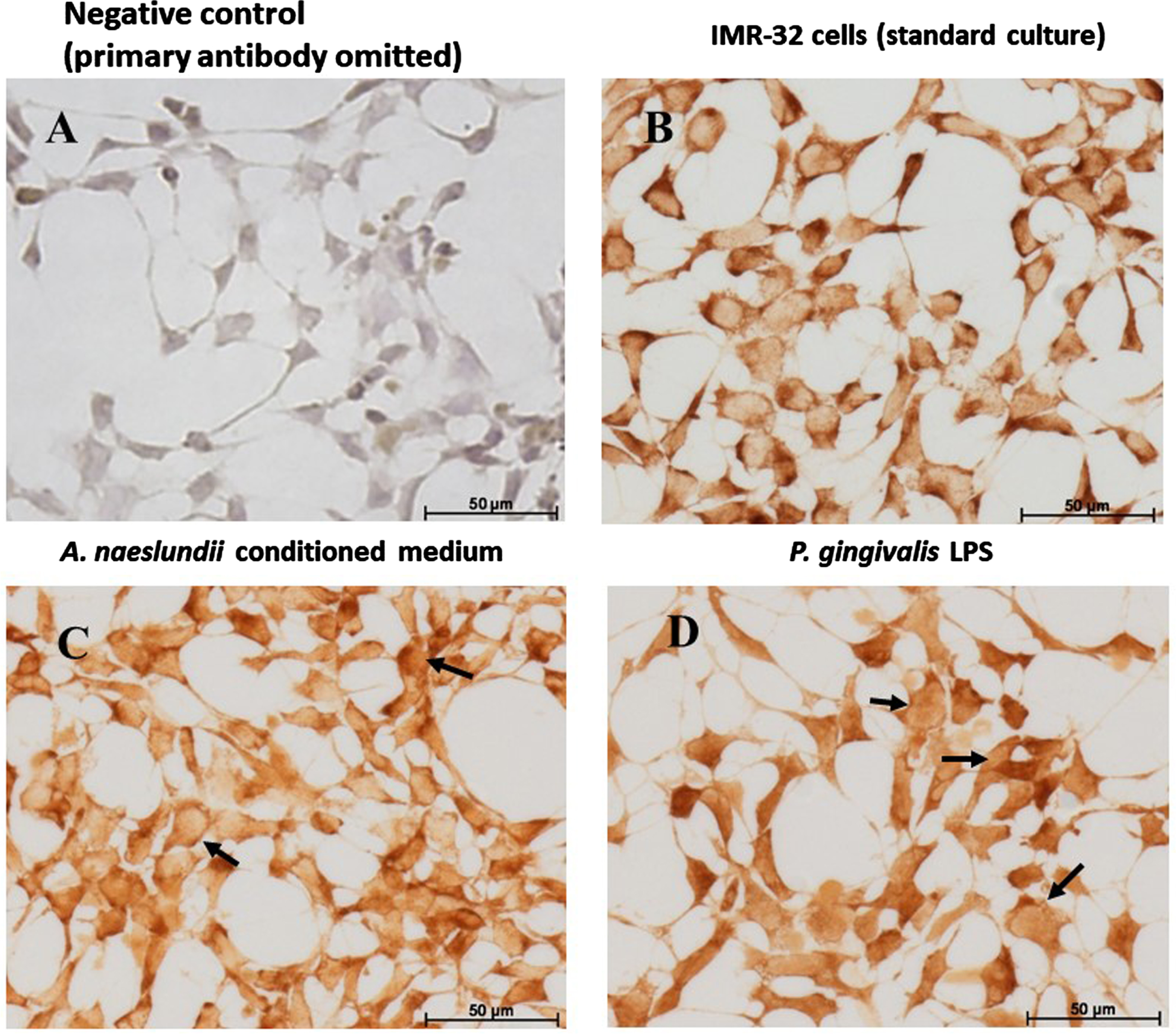
IMR 32 cells: GSK-3β Immunohistochemistry. GSK-3β immunostaining of IMR-32 cells: Comparing with the negative control (A), GSK-3β is expressed by IMR-32 cells under control and all treatment conditions. Under standard culture conditions (B), IMR-32 showed strong cytoplasmic localization of GSK-3β. A. naeslundii virulence factors demonstrated strong GSK-3β immunostaining in the cytoplasm (C) with a hint of nuclear staining in some cells (C arrows). Cells treated with Pg.LPS (D) predominantly demonstrated nuclear staining (D, arrows) and weaker cytoplasmic staining.
Test coverslips
IMR-32 cells GSK-3β immunostaining
Comparing with the negative control (Fig. 3A) with GSK-3β immunostaining, the protein was expressed by IMR-32 cells under all treatment conditions. Under standard culture conditions, IMR-32 cells demonstrated strong intracellular cytoplasmic localization of GSK-3β (Fig. 3B). Cells exposed to A. naeslundii conditioned medium alone demonstrated both cytoplasmic and weaker nuclear immunolocalization (Fig. 3C arrows). Cells treated with Pg.LPS (Fig. 3D) demonstrated weaker cytoplasmic staining compared to standard cell culture conditions, but the GSK-3β staining was also observed within the nucleus (Fig. 3D arrows).
DISCUSSION
The present study examined the gene expression of GSK-3β at transcription level by qPCR. This was followed by examining the translation of GSK-3β at the protein level by western blotting and its cellular localization by immunohistochemistry in IMR 32 cells. The qPCR results indicated that the ultrapure P. gingivalis LPS and an unknown factor from A. naeslundii conditioned medium upregulated the GSK-3β gene expression. Western blotting indicated that the native 47 kDa band size GSK-3β was being transcribed. However, the effect of the P. gingivalis LPS for example, as observed on the native GSK-3β was a cleavage product predominantly around 37 kDa band size. Immunohistochemistry of the GSK-3β failed to demonstrate any nuclear staining in the cells cultured under standard cell culture or the medium control conditions. However, P. gingivalis LPS treated IMR-32 cells demonstrated both intracellular and nuclear localization of the protein suggesting that the cleaved fragment of GSK-3β was responsible for its nuclear localization. A previous report of qPCR data from the same P. gingivalis LPS treatment of IMR-32 cells19 had indicated that the metabolic enzyme GSK-3β was not only upregulated, but was also activated, which in turn, had initiated the transcription of a number of downstream transcription factors (FOXO1, STAT1, STAT3, CREB1, EGR2, IRF1, FOS, RELA, NFKB1), kinases (AKT1, PIK3R1, GSK3B, PCK1, CSF1 R, IRAK1, JAK2, MAPK3K1, IKBKB, INSR), and other receptors and associated proteins (VCAM1, MYD88, CD40, TNFRSF1A, IGF1 R, PTGER1, HRH3), and cytokines (TNFA, IL1B, CSF1, CSF2, IL6, IL8, and IL17A). The present study agreed with the increased expression of the GSK-3β, but the main differences were the appearance of a native and a cleaved protein of the GSK-3β by western blotting. Furthermore, immunohistochemical analysis demonstrated the truncated protein entered the nucleus leading to the suggestion that it was transcriptionally active for switching on downstream transcription factors, kinases and various proteins described elsewhere.19
This implies that the GSK-3β enzyme is adversely and directly, effected by the P. gingivalis LPS virulence factor and possibly by an unknown factor of A. naeslundii. Bacterial factors playing a role in altering the pathophysiology of brain cells in additional ways to just being potent immune modulators of inflammatory cells36 in the body is an important concept. This widens the concept that P. gingivalis infection alone is not sufficient to cause AD and that multispecies of microbes and their virulence factors contribute to this complex neurodegenerative disease process. This information is another step towards establishing a multispecies pathogenic bacterial co-operation under inflammophilic conditions that is contributing to direct and downstream chronic neuroinflammation in AD via GSK-3β gene activation.
P. gingivalis is the most widely studied periodontal disease bacterium in the laboratory. To this end P. gingivalis LPS exists in at least two known forms, O-LPS and A-LPS. The A-LPS shows heterogeneity in which two isoforms of LPS differentiated by LPS (1435/1449) and LPS (1690) , which appear responsible for tissue specific immune signaling pathways activation and increased virulence.37–39 P. gingivalis can subvert hosts’ innate immune system via its ability to change its LPS1435/1449 and/or LPS1690 composition. This may enable P. gingivalis to overcome the local pro-inflammatory environment of the AD brain. P. gingivalis virulence factors play a role in AD pathophysiology of hallmark protein deposition (Aβ plaques and NFTs) as previously reported by Dominiy et al.14 and Illievski et al.40 and in innate immune activation as reported by Poole et al.41
P. gingivalis has a plethora of virulence factors42 of which LPS, gingipains, fimbriae, hemagglutinins, and outer membrane vesicles are of major importance. LPS is located in the outer membrane of Gram-negative bacteria and is a potent stimulator of host’s innate immune signal transduction pathways in a tissue/cell specific manner.43 Gingipains overall constitutes a much higher proportion of the virulence factors in conditioned medium followed by LPS.16 Since gingipains is a bacterial enzyme known to cleave proteins like tau,14 which in turn, is highly associated with the formation of NFTs ex vivo as demonstrated by Kanasingham et al.44 The present study eliminates a role for gingipains activity in cleaving GSK-3β for two reasons. Firstly, if this was the case, a more definitive 37 kDa band of GSK-3β would have been expected in the western blot for IMR-32 cells challenged with Pg381 conditioned medium alone. Secondly, despite the gingipains and LPS antibody epitopes being close together on the outer membrane of the bacterium, both ultrapure and regular P. gingivalis LPS that are available commercially are free of gingipains catalytic activity.
The present study demonstrated that the dual bacterial virulence factors were responsible for cleaving the GSK-3β protein and this was statistically significant for A. naeslundii conditioned medium and P. gingivalis LPS. GSK-3β is a metabolic enzyme that regulates and controls multiple physiological processes in the human body and one of these could be to protect hosts’ immune responses from being subverted by pathogenic bacteria2. For the bacteria, this could be a strategy to dampen the host’s innate immune response for survival of the infecting microbe.
The present study suggests that the cleavage process is a mechanism for GSK-3β to enter the nucleus and thereby mediate transcription of the GSK-3β gene. GSK-3β gene activation was documented by Bahar et al.20 in their P. gingivalis orally infected obese and diabetic mouse model, but at the time, its mechanism was not investigated. The present study has helped to understand the mechanism of GSK-3β gene activation and was confirmed by the statistically significant, 2-fold change in its increased expression as identified by qPCR for P. gingivalis LPS as compared with the B2M gene. This upregulation appears to show two bands one at the expected 47 kDa band size and a truncated 37 kDa. This is illustrated by Fig. 2, whereby the Pg.LPS bar, shows a weak 47 kDa band. With regard to the qPCR data where a 2-fold upregulation of the gene is recorded, it is plausible to suggest that the native 47 kDa band protein is being cleaved as fast as it is being manufactured by the host’s cell. However, in the Pg.LPS and A. naeslundii combined treated cells, GSK-3β 47 kDa band is very clearly present. This suggests there is a compensatory effect in the rate at which the GSK-3β is being cleaved when the virulence factors of these two microbes are combined. This may be a step towards a multispecies pathogenic bacterial co-operation under inflammophilic conditions contributing to direct and downstream chronic neuroinflammation in AD.
Literature suggests that A. naeslundii can play a coordinator role, for late and early dental biofilm colonizers due to them possessing receptors that allow for their adherence to other biofilm bacteria.22 Biofilms are defined as aggregations of microorganisms attached to each other and to surfaces enclosed in self-secreted extracellular polymeric substance (EPS).45–47 A. naeslundii is a strong candidate for co-aggregating48 and directly assisting in high mutualistic interactions with P. gingivalis. Under our laboratory conditions, A. naeslundii grew in the P. gingivalis preferred growth medium at anaerobic conditions and incubation temperature (37°C); and secreted extra EPS confirming their harmonious existence as biofilm bacteria. These results agree with Yailing et al.,49 who also found that 37°C and anaerobic conditions were standard when working with the vast majority of Actinomyces, including A. naeslundii. The ability to adapt to changing environments makes A. naeslundii a facultatively anaerobic bacterium.
P. gingivalis with A. naeslundii have been detected in AD brains,12,13 by next generation sequencing and by immunohistochemistry15 methodologies. Further investigations to observe their ability to live together within a natural biofilm are lacking. The present study confirms that P. gingivalis and A. naeslundii can grow together both in vitro and in clinical biofilms in the form of plaque and calculus, thereby causing periodontal disease.28,29 It is therefore, plausible to suggest that when oral health becomes inadequately managed, these two bacteria facilitate each other’s entry into the AD patients brain.13 Moreover, P. gingivalis in its keystone pathogen role, creates an inflammophilic environment for selection of its co-species and by doing so, exerts control over any competing microbial species.39
The rationale for choosing the IMR-32 cells was due to their innate immune responses being very close to those reported in the human brain.50 Nevertheless, they are an undifferentiated neuroblastoma cell line with two different phenotypes.51 Despite their undifferentiated status, in this study they have provided a simpler model over a complex in vivo animal model where much of the related work has been performed by us and others confirming that GSK-3β activation has been implicated in the NFT formation in mice.19,40 However, such in vivo studies have not clarified a specific bacterial product or the mechanism by which the GSK-3β becomes activated. In comparison this cell culture model has provided a clear explanation for a pathway to GSK-3β activation by bacterial virulence factors from A. Naeslundii and in particular P. gingivalis LPS.
Conclusions
The observation that A. naeslundii and P. gingivalis grew unaffected and confluently in P. gingivalis preferred growth medium at anaerobic conditions in the laboratory was taken in support of a temporal and spatial dwelling of these microbes within the sub-gingival biofilm. During dysbiosis, these two bacteria can act as putative pathogens and are known to be the etiological agents of periodontal disease.
P. gingivalis LPS and an unknown factor in A. naeslundii conditioned medium mediated GSK-3β activation via its transcriptionally active, cleaved, fragment. This highlights an important concept that the virulence factors of oral bacterial factors in the body may be detrimental to brain health. Better understandings of GSK-3β mediated inflammatory signaling and development of inhibitors for combating infection, could provide a new dawn of treatment for bacterial involvement in neurodegenerative diseases. In the meantime, maintaining adequate oral health is of vital importance throughout life and especially in old age.
AUTHOR CONTRIBUTIONS
Sim Singhrao (Conceptualization; Data curation; Funding acquisition; Investigation; Methodology; Project administration; Supervision; Validation; Writing – original draft; Writing – review & editing); Claudia Consoli (Formal analysis; Investigation); Sarah R. Dennison (Data curation); Shalini Kanagasingam (Funding acquisition; Supervision); Richard Welbury (Funding acquisition; Supervision).
ACKNOWLEDGMENTS
The work presented in this manuscript was partially authored by Jessica Inacio, as part of her MSc thesis in 2022. The MSc study was supervised by Dr. S. K. Singhrao, Dr. S. Kanagasingam, and Prof. R. Welbury. We thank Jessica for the western blot and the immunohistochemistry images. Subsequently, the molecular biology (Fig. 1) was from experiments conducted and statistically analyzed by Dr. C. Consoli, the densitometry on the western blot (Fig. 2A) was performed by Dr. S. K. Singhrao and the statistical analysis (Fig. 2B) was performed by Dr. S. R. Dennison.
FUNDING
This work was funded in part by two PreViser awards received from the Oral and Dental Research Trust (UK) awarded to SK and SKS in 2017 and again with RW and SKS in 2018.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY
The data supporting the findings of this study are available within the article and/or its supplementary material.
REFERENCES
1. | Beurel E , Grieco SF and Jope RS . Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol Ther (2015) ; 148: : 114–131. |
2. | Wang HH , Lamont RJ , Kumar A , et al. GSK3beta and the control of infectious bacterial diseases. Trends Microbiol (2014) ; 22: : 208–217. |
3. | Hanger D , Hughes K , Woodgett J , et al. Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau: Generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci Lett (1992) ; 147: : 58–62. |
4. | Braak H , Alafuzoff I , Arzberger T , et al. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol (2006) ; 112: : 389–404. |
5. | Hyman BT , Phelps CH , Beach TG , et al. National Institute on Aging– Alzheimer’s Association Guidelines for the Neuropathologic Assessment of Alzheimer’s Disease. Alzheimers Dement (2012) ; 8: : 1–13. |
6. | Dugger BN and Dickson DW . Pathology of neurodegenerative diseases. Cold Spring Harb Perspect Biol (2017) ; 9: : a028035. |
7. | Scheltens P , Blennow K , Breteler MM , et al. Alzheimer’s disease. Lancet (2016) ; 388: : 505–517. |
8. | Guerrero A , De Strooper B and Arancibia-Carcamo IL . Cellular senescence at the crossroads of inflammation and Alzheimer’s disease. Trends Neurosci (2021) ; 4: : 14–27. |
9. | Akushevicha I , Yashkina A , Ukraintsevaa S , et al. The construction of a multidomain risk model of Alzheimer’s disease and related dementias a Biodemography of Aging Research Unit. J Alzheimers Dis (2023) ; 96: : 535–550. |
10. | Baumgart M , Snyder HM , Carrillo MC , et al. Summary of the evidence on modifiable risk factors for cognitive decline and dementia: A population-based perspective. Alzheimers Dement (2015) ; 11: : 718–726. |
11. | Riviere GR , Riviere KH , Smith KS . Molecular and immunological evidence of oral Treponema in the human brain and their association with Alzheimer’s disease. Oral Microbiol Immunol (2002) ; 17: : 113–118. |
12. | Emery DC , Shoemark DK , Batstone TE , et al. 16S rRNA next generation sequencing analysis shows bacteria in Alzheimer’s post-mortem brain. Front Aging Neurosci (2017) ; 9: : 195. |
13. | Siddiqui H , Eribe E , Singhrao S , et al. High throughput sequencing detect gingivitis and periodontal oral bacteria in Alzheimer’s disease autopsy brains. Neurosci Res (2019) ; 1: : 3. |
14. | Dominy SS , Lynch C , Ermini F , et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv (2019) ; 5: : eaau3333. |
15. | Howard J and Pilkington GJ . Fibronectin staining detects micro-organisms in aged and Alzheimer’s disease brain. Neuroreport (1992) ; 3: : 615–618. |
16. | Poole S , Singhrao SK , Kesavalu L , et al. Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer’s disease brain tissue. J Alzheimers Dis (2013) ; 36: : 665–677. |
17. | Singhrao SK and Harding A . Is Alzheimer’s disease a polymicrobial host microbiome dysbiosis? Expert Rev Anti Infect Ther (2020) ; 18: : 275–277. |
18. | Bulgart HR , Neczypor EW , Wold LE , et al. Microbial involvement in Alzheimer disease development and progression. Mol Neurodegener (2020) ; 15: : 42. |
19. | Bahar B and Singhrao SK . An evaluation of the molecular mode of action of trans-resveratrol in the Porphyromonas gingivalis lipopolysaccharide challenged neuronal cell model. Mol Biol Rep (2021) ; 48: : 147–156. |
20. | Bahar B , Kanagasingam S , Tambuwala MM , et al. Porphyromonas gingivalis (W83) infection induces Alzheimer’s disease like pathophysiology in obese and diabetic mice. J Alzheimers Dis (2021) ; 82: : 1259–1275. |
21. | Ide M , Harris M , Stevens A , et al. Periodontitis and cognitive decline in Alzheimer’s disease. PLoS One (2016) ; 11: : e0151081. |
22. | Lamont RJ and Jenkinson HF . Life below the gum line: Pathogenic mechanisms of Porphyromonas gingivalis. Microbiol Mol Biol Rev (1998) ; 62: : 12441263. |
23. | Supriya BG , Harisree S , Savio J , et al. Actinomyces naeslundii causing pulmonary endobronchial Actinomycosis – A case report. Indian J Pathol Microbiol (2019) ; 62: : 326–328. |
24. | Tang G , Yip HK , Samaranayake LP , et al. Direct detection of cell surface interactive forces of sessile, fimbriated and non-fimbriated Actinomyces susing atomic force microscopy. Arch Oral Biol (2004) ; 49: : 727–738. |
25. | Takahashi N and Yamada T . Glucose and lactate metabolism by Actinomyces Naeslundii. Crit Rev Oral Biol Med (1999) ; 10: : 487–503. |
26. | Noble JM , Scarmeas N , Celenti RS , et al. Serum IgG antibody levels to periodontal microbiota are associated with incident Alzheimer disease. PloS One (2014) ; 9: : e114959. |
27. | Hajishengallis G , Darveau RP and Curtis MA . The keystone-pathogen hypothesis. Nat Rev Microbiol (2012) ; 10: : 717–725. |
28. | Joshi V , Matthews C , Aspiras M , et al. Smoking decreases structural and functional resilience in the subgingival ecosystem. J Clin Periodontol (2014) ; 41: : 1037–1047. |
29. | Socransky SS , Haffajee AD , Cugini MA , et al. Microbial complexes in subgingival plaque. J Clin Periodontol (1998) ; 25: : 134–144. |
30. | Cavalcanti MCO , Failling K , Schuppe HC , et al. Validation of reference genes in human testis and ejaculate. Andrologia (2011) ; 43: : 361–367. |
31. | Shrout J , Yousefzadeh M , Dodd A , et al. B2microglobulin mRNA expression levels are prognostic for lymph node metastasis in colorectal cancer patients. Br J Cancer (2008) ; 98: : 1999–2005. |
32. | Chen L , Zuo Y , Pan R , et al. GSK-3β regulates the expression of P21 to promote the progression of chordoma. Cancer Manag Res (2021) ; 13: : 201–214. |
33. | Livak KJ , Schmittgen TD . Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta-deltaC(T) method. Methods (2001) ; 25: : 402–408. |
34. | Bradford MM . A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem (1976) ; 72: : 248–254. |
35. | Kanagasingam S , von Ruhland C , Welbury R , et al. Porphyromonas gingivalis conditioned medium induces amyloidogenic processing of the amyloid precursor protein upon in vitro infection of SH-SY5Y cells. J Alzheimers Dis Rep (2022) ; 6: : 577–587. |
36. | Beutler B and Rietschel ET . Innate immune sensing and its roots: The story of endotoxin. Nat Rev Immunol (2003) ; 3: : 169–176. |
37. | Dixon DR and Darveau RP . Lipopolysaccharide heterogeneity: Innate host responses to bacterial modification of lipid a structure. J Dent Res (2005) ; 84: : 584–595. |
38. | Herath TD , Darveau RP , Seneviratne CJ , et al. Tetra- and penta-acylated lipid A structures of Porphyromonas gingivalis LPS differentially activate TLR4-mediated NF-κB signal transduction cascade and immuno-inflammatory response in human gingival fibroblasts. PLoS One (2013) ; 8: : e58496. |
39. | Olsen I and Singhrao SK . Importance of heterogeneity in Porphyromonas gingivalis lipopolysaccharide lipid A in tissue specific inflammatory signaling. J Oral Microbiol (2018) ; 10: : 1440128. |
40. | Ilievski V , Zuchowska PK , Green SJ , et al. Chronic oral application of a periodontal pathogen results in brain inflammation, neurodegeneration and amyloid beta production in wild type mice. PLoS One (2018) ; 13: : e0204941. |
41. | Poole S , Singhrao SK , Chukkapalli S , et al. Active invasion of an oral bacterium and infection-induced complement activation in ApoEnull mice brains. J Alzheimers Dis (2015) ; 43: : 67–80. |
42. | How KY , Song KP , Chan KG . Porphyromonas gingivalis: An overview of periodontopathic pathogen below the gum line. Front Microbiol (2016) ; 7: : 53. |
43. | Beutler B . Endotoxin, toll-like receptor 4, and the afferent limb of innate immunity. Curr Opin Microbiol (2000) ; 3: : 23–28. |
44. | Kanagasingam S , von Ruhland C , Welbury R , et al. Antimicrobial, polarising light and paired helical filament properties of fragmented tau peptides of selected putative gingipains. J Alzheimers Dis (2022) ; 89: : 1279–1291. |
45. | Marsh PD . Dental plaque: Biological significance of a biofilm and community life-style. J Clin Periodontol (2005) ; 32: : 7–15. |
46. | Kolenbrander PE . Oral microbial communities: Biofilms, interactions, and genetic systems. Ann Rev Microbiol (2000) ; 54: : 413–437. |
47. | Huang CB , Alimova Y , Myers TM , et al. Short- and medium-chain fatty acids exhibit antimicrobial activity for oral microorganisms. Arch Oral Biol (2011) ; 56: : 650–654. |
48. | Rosenberg M , Buivids IA and Ellen RP . Adhesion of Actinomyces viscosus to Porphyromonas (Bacteroides) gingivalis-coated hexadecane droplets. J Bacteriol (1991) ; 173: : 2581–2589. |
49. | Yaling L , Tao H , Jingyi Z , et al. Characterization of the Actinomyces naeslundii ureolysis and its role in bacterial acid uricity and capacity to modulate pH homeostasis. Microbiol Res (2006) ; 161: : 304–310. |
50. | Singhrao SK , Neal JW , Rushmere NK , et al. Human neurons are lysed by complement because they spontaneously activate the classical pathway and lack membrane bound complement regulators. Am J Pathol (2000) ; 157: : 905–918. |
51. | Begaud-Grimaud G , Battu S , Lazcoz P , et al. Study of the phenotypic relationship in the IMR-32 human neuroblastoma cell line by sedimentation field flow fractionation. Int J Oncol (2007) ; 31: : 883–892. |


