
An Atypical Case of Creutzfeldt-Jakob Syndrome Presenting with Cacosmia and Amyloid Positivity
Abstract
This report presents a challenging case of Creutzfeldt-Jakob Disease (CJD), a rare and rapidly progressing neurological disorder. The patient exhibited diverse and progressive neuro-psychiatric symptoms, including memory impairment, behavioral changes, and hallucinations associated with cacosmia. The diagnosis of CJD is complicated due to its variable clinical presentation, limited awareness, and the need for tissue pathology confirmation. Diagnostic tests, particularly brain magnetic resonance imaging (MRI) and cerebrospinal fluid (CSF) analysis, played crucial roles in the evaluation. The MRI revealed characteristic cortical ribboning patterns. CSF analysis initially suggested Alzheimer’s disease pathology continuum. Repeated Real-time-quaking-induced assay testing (RT-QuIC) confirmed the diagnosis despite an initial negative result. This case underscores the significance of contemplating CJD in individuals exhibiting rapidly progressive dementia, even in the presence of atypical clinical features. Furthermore, it emphasizes the importance of recognizing that an initial negative result from the RT-QuIC test should not preclude consideration of CJD, particularly when characteristic MRI findings are present.
INTRODUCTION
Creutzfeldt-Jakob Disease (CJD) is a rare, rapidly progressive and uniformly fatal neurological disease.1 It represents the most common form of human prion diseases,1 with an annual incidence of 2 in 1,000,000 individuals worldwide.1 Most sporadic CJD (sCJD) cases occur in persons between the age of 60 and 80 years with an average age at death of about 67 years, with a slight predominance in the Caucasian population.1,2 The underlying pathology is believed to stem from the gradual replacement of normal prion proteins with abnormal infectious ones, ultimately leading to neurotoxicity. Consequently, a definitive diagnosis relies on standard neuropathological techniques that confirm the presence of protease-resistant PrP and/or the existence of scrapie-associated fibrils. Diagnosing sCJD can be challenging due to its heterogeneous clinical presentations.3 Moreover, making a definite diagnosis according to diagnostic criteria requires neuropathological confirmation, but this is rarely done now due to increased specificity of biomarkers in magnetic resonance imaging (MRI)/cerebrospinal fluid (CSF) giving high certainty of diagnosis during life. The clinical picture, laboratory analysis (including CSF), electroencephalography (EEG), and MRI are all required for fulfilling the international diagnostic criteria. Definitive diagnosis of sCJD is made by histopathologic examination through post-mortem autopsy or ante-mortem biopsy.4 In this case report, we outline the diagnostic challenges encountered in a patient presenting with rapidly progressive dementia and the unusual symptom of cacosmia that required multiple CSF collections to achieve the diagnosis of probable CJD.
CASE PRESENTATION
We report the case of a 57-year-old right-handed man who presented to our hospital with an 8-month history of episodic memory impairment, apathy, mood decline, irritability and fluctuating cacosmia not related to otolaryngological causes. He was otherwise in good health, living independently with his wife, and had an unremarkable medical history, except for mild hypercholesterolemia. He had no past history of corneal grafting, dura mater transplantation, or pituitary growth hormone treatment, and there was no family history of dementia or rapid cognitive decline. The patient reported being a former smoker who had quit smoking a few years ago and consumed alcohol socially in moderation. He worked as a security guard. His family history was negative for neurological diseases, except for his maternal grandmother, who exhibited parkinsonian symptoms late in life. Upon admission, the neurological examination revealed disorientation to situation, space, and towards people, a severe deficit in episodic memory, significant impairment in selective attention, ideomotor apraxia, and dyscalculia. No signs of pyramidal, extrapyramidal, cerebellar disfunctions or myoclonus were observed. His Mini-Mental State Examination (MMSE) was 11/30 and Global Clinical Dementia Rating scale (CDR) 2 (sum of boxes 12). A neuropsychological evaluation revealed a cognitive profile characterized by mixed dysfunction (multiple domain amnesic) with severe impairment of memory (z-score: –3.87), visuoperceptual (z-score: –6.34), and executive functions (z-score: –3.34). During hospitalizations, routine blood tests, oncomarker screening, onconeural antibody tests, and infectious disease tests were conducted, all yielding negative results. Brain MRI diffusion-weighted imaging (DWI) and fluid-attenuated inversion recovery (FLAIR) sequences revealed high-intensity areas in the temporal lobe, frontal lobe, parietal and occipital lobe cortex, and caudate nucleus (Fig. 1), raising suspicion of prion pathology.5,6 The EEG was normal. A lumbar puncture was performed and showed RBCs: 1 el/ul, (reference range < 10), WBCs: 1 el/ul (reference range < 5), Albumin: 23.6 mg/dl (n.v 13.9–24.6) and CSF biomarker analysis was conducted using Chemiluminensces assay (Lumipulse). It revealed amyloid beta 42 : 471 pg/ml (reference range > 599), tau: 606 pg/ml (reference range < 342), and phospho-tau: 39.7 pg/ml (reference range < 56). Most importantly, CSF testing for suspected imaging-based CJD using RT-QuIC assay yielded a negative result, not demonstrating the presence of pathogenic prion protein in the CSF. Based on this data, particularly radiological findings, the patient did not fulfill the criteria for early onset major neurocognitive disorder (DSM V) due to Alzheimer’s and concomitant suspected non Alzheimer’s pathologic change with dementia (NYA-AA 2018)7 because of the persistent high suspicion of prion disease. Two months after the initial evaluation, the patient was readmitted to our hospital due to the onset of delusions and hallucinations. For example, he was seen talking while standing in front of the mirror, possibly experiencing visual hallucinations. Additionally, he reported visions of insects. Moreover, despite the evidence of worsening global cognitive status during the serial assessments conducted on the patient, no additional neurological signs and symptoms emerged that would meet the epidemiological criteria for probable sCJD. A second EEG displayed a pattern of periodic discharges with triphasic waves. Another diagnostic lumbar puncture was performed and showed RBCs: 1 el/ul, (reference range < 10), WBCs: 1 el/ul (reference range < 5), Albumin: 22.8 mg/dl (n.v 13.9–24.6). Biomarker analysis confirmed cerebral amyloidosis and detected an elevation in p-tau levels compared to the previous examination but still in normal range: amyloid beta 42 : 372 pg/ml (reference range > 599), tau: 681 pg/ml (reference range < 342), and phosphotau: 52.10 pg/ml (reference range < 56). Due to the persistent suspicion of prion disease, RT-QuIC testing was repeated on CSF and olfactory mucosa, and both tests yielded conclusive results for CJD. In addition, genotyping of the PRNP gene using Sanger sequencing did not reveal any pathogenic mutations. Therefore, probable CJD was diagnosed based on CDC criteria.4
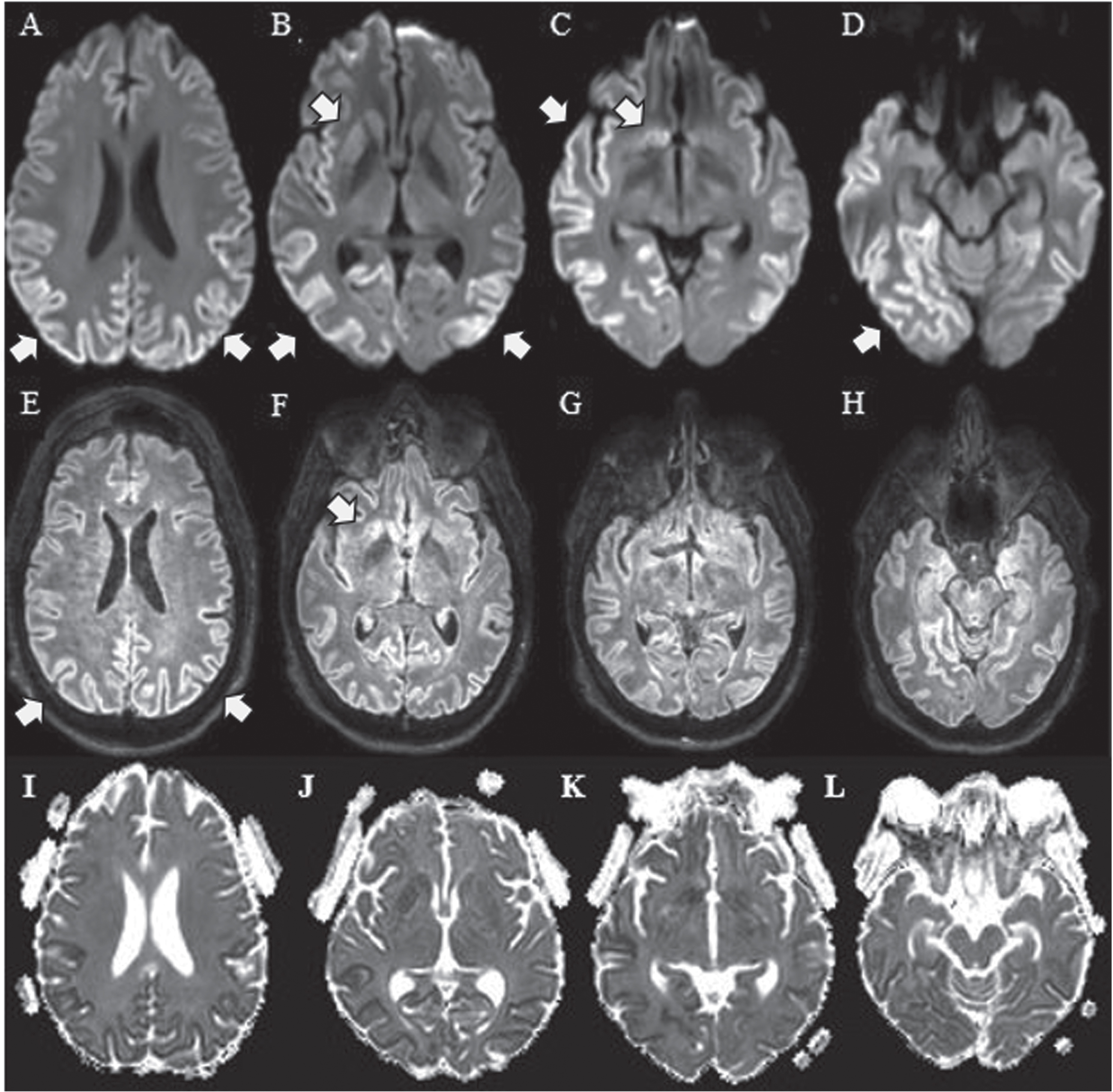
Fig.1
Multiple Axial DWI sequences from the patient’s brain MRI demonstrate abnormal signal hyperintensity (arrows) at several cortical locations (A, B, D) and in the caudate head on the diffusion-weighted sequence. (C) Less conspicuous signal hyperintensity is present at the corresponding locations on the fluid-attenuated inversion recovery (FLAIR) sequences (E, F, G, H). Apparent diffusion coefficient (ADC) corresponding sequences are shown in I, J, K and L.
DISCUSSION
The Center for Disease Control and Prevention (CDC) outlines two diagnostic pathways4 for probable sCJD (Table 1). The first pathway includes a neuropsychiatric disorder plus a positive RT-QuIC in CSF or other tissues. The second pathway requires three components: symptoms, supportive diagnostic studies, and exclusion of alternative diagnoses. The symptoms include rapidly progressive dementia and two findings of myoclonus, visual/cerebellar signs, pyramidal/extrapyramidal signs, and akinetic mutism. MRI, EEG, and CSF studies are common diagnostic modalities that help include and exclude a wide range of differential diagnoses of neurological diseases. In our case, the patient’s symptoms included rapidly progressive dementia associated with cacosmia. While most dysosmias reflect dynamic elements associated with degeneration of the olfactory epithelium, significant dysosmia has never been reported in CJD cases, despite being highly prevalent in multiple neurodegenerative disorders including Parkinson’s disease, Alzheimer’s disease (AD), Huntington’s disease,8,9 and amyotrophic lateral sclerosis.10 While a case of vCJD associated with anosmia has been described,11 there has never been a reported case of sCJD presenting with cacosmia. MRI is useful in the evaluation of patients with rapid progressive dementia, with 83% to 92% sensitivity and 87% to 95% specificity for the diagnosis of sCJD.5,6 It has characteristic findings of hyperintense signals on DWI and FLAIR sequences, and T2-weighted images, most commonly in the basal ganglia and thalamic area and a cortical ribboning pattern of the cerebral cortex. In our case, MRI exhibited characteristic findings of sCJD: cortical ribbon pattern involving the temporal lobe, frontal lobe, parietal lobe cortex, and an intense signal of caudate nucleus on DWI and FLAIR images decisively contributed to guiding our diagnostic suspicion.5,6 Our patient’s performed EEG at the first evaluation and two months later. Although historically the literature has defined abnormal EEG patterns as being detected in approximately 88% of CJD patients,12,13 current evidence indicates that abnormal EEG patterns, such as periodic sharp complexes attributed to CJD, offer less diagnostic value due to their high false negative potential. Recent studies have reported periodic complexes in only 11% of patients with probable sCJD and 22.5% of patients with definite CJD. Moreover, it is important to note that in the early stages of the disease, EEG findings often present as nonspecific, characterized by generalized or focal delta theta slowing and a decrease in reactivity. These early nonspecific changes highlight the importance of integrating EEG with other diagnostic modalities for a comprehensive assessment of sCJD.14 The third diagnostic examination was CSF assay. In the current case, despite elevated tau levels, they did not surpass the threshold of tau levels >1,150 pg/mL, which are associated with high accuracy and specificity in the diagnosis of CJD.15,16 However, we emphasize that cut-off values depend on the measurement technique, and it is unclear whether a total tau value of 606 pg/mL via Lumipulse would exceed 1,150 pg/mL on the NPDPSC assay. Moreover, the presence of CSF amyloid positivity in the absence of increased phospho-tau levels supported a classification of A + T–N+. This profile is consistent with AD pathology and concomitant suspected non-Alzheimer’s pathological changes within the context of the AD continuum.6 RT-QuIC in CSF is considered the most sensitive and specific diagnostic test for sporadic sCJD, with a test specificity of 100%.17 In terms of test sensitivity, the first-generation RT-QuIC shows figures ranging from 73% to 89%, while the second-generation RT-QuIC demonstrates a sensitivity ranging from 92% to 97%.17–20 Moreover, in one preliminary study that included 31 patients with CJD, RT-QuIC testing of olfactory epithelium obtained from nasal brushings was less sensitive (91% versus 97%) and similarly specific (100%) when compared with CSF testing.17 Diagnostic sensitivity is higher in common sCJD molecular subtypes (e.g., MM1 and MV1) and lower in less common subtypes (e.g., VV1, MM2-C, and sFI), short duration of disease or slow progressors.21,22 Nonetheless, typical MRI findings contributed to guiding our diagnostic suspicion after having excluded other potential causes that could result in cortical ribboning, such as autoimmune encephalitis, hypoxic/anoxic brain injury, osmotic demyelination, encephalitis, hepatic encephalopathy, and hypoglycemic encephalopathy.23,24 For this reason, a second lumbar puncture and a nasal brush with an RT-QuIC test were performed, and this time, they both confirmed the diagnostic suspicion of CJD. Historically, the reasons for the co-occurrence of cerebral amyloidosis and CJD have been debated in the literature.25,26 However, recent studies show no correlation between variables known to affect CJD pathology and those defining the pathological spectrum of cerebral amyloidosis/AD pathology.27,28 Therefore, an elevation in total tau and a reduced amyloid beta 42 ratio should not deter from a diagnosis of sCJD in the context of a positive MRI, particularly considering that amyloid co-pathology can be observed in sCJD and typically does not alter the disease course. In conclusion, our patient received a probable diagnosis of CJD using the former pathway, due to the lack of clinical supporting findings at the time of the initial evaluation. In particular, our patient exhibited key clinical features of rapidly progressive dementia and characteristic MRI findings and later characteristic EEG findings too. These features guided our diagnostic suspicion, despite atypical clinical findings, particularly cacosmia, an initial negative result on the CSF RT-QuIC test and the presence of AD co-pathology. Furthermore, despite the loss of the patient to follow-up after a few months, other colleagues who evaluated the patient subsequently reported further clinical deterioration with the onset of cerebellar problems and akinetic mutism. Even though there is no effective treatment for sCJD, obtaining an early diagnosis would provide clinical and emotional support to patients and their families.
Table 1
CDC Diagnostic Criteria for Creutzfeldt-Jacob Disease
| DEFINITE sCJD |
| Progressive neurological syndrome AND |
| Neuropathologically OR |
| Immunocytochemically OR biochemically confirmed |
| PROBABLE sCJD |
| I+2 of II and typical MRI brain scan |
| OR I + 2 of II and typical EEG |
| OR I + 2 of II and positive 14.3.3 |
| OR Progressive neurological syndrome and positive CSF RT-quIC in CSF |
| or other tissues |
| POSSIBLE sCJD |
| 1 + 2 of II and disease duration < 2 years |
| Exclusion of other cause in complete diagnostic work up |
| SYMPTOMS |
| I Rapidly progressive cognitive impairment |
| II Myoclonus |
| Visual or cerebellar problems |
| Pyramidal or extrapyramidal features |
| Akinetic mutism |
| SUPPORTIVE TESTS |
| MRI High signal in caudate/putamen on MRI brain scan OR≥2 |
| cortical regions on diffusion-weighted imaging or on FLAIR |
| EEG Periodic sharp waves complexes |
AUTHOR CONTRIBUTIONS
Alfredo Gabriele Nanni (Methodology; Project administration; Writing – original draft; Writing – review & editing); Daniele Urso (Conceptualization; Data curation; Methodology; Supervision; Writing – original draft; Writing – review & editing); Martina Caccamo (Data curation; Writing – review & editing); Valentina Gnoni (Writing – review & editing); Alessia Giugno (Writing – review & editing); Chiara Zecca (Data curation; Writing – review & editing); Maria Teresa Dell’Abate (Data curation; Writing – review & editing); Davide Vilella (Data curation; Writing – review & editing); Roberto De Blasi (Data curation; Writing – review & editing); Giancarlo Logroscino (Conceptualization; Data curation; Funding acquisition; Methodology; Supervision; Writing – review & editing).
ACKNOWLEDGMENTS
We would like to thank the patient and his family for allowing us to use their medical information for scientific publication.
FUNDING
This work has been supported with the funding of Regione Puglia and CNR for Tecnopolo per la Medicina di Precisione. D.G.R. n. 2117 of 21.11.2018 (CUPB84I18000540002) – C.I.R.E.M.I.C. (Research Center of Excellence for Neurodegenerative Diseases and Brain Aging) – University of Bari “Aldo Moro”.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
1. | Zerr I and Parchi P. Sporadic Creutzfeldt-Jakob disease, Handb Clin Neurol (2018) ; 153: : 155–174. |
2. | WHO Guidelines on Transmissible Spongiform Encephalopathies in relation to Biological and Pharmaceutical Products. Available from: https://www.who.int/publications/m/item/who-guidelines-on-transmissible-spongiform-encephalopathies (2024, accessed 26 April 2024). |
3. | Mead S and Rudge P. CJD mimics and chameleons, Pract Neurol (2017) ; 17: : 113–121. |
4. | Watson N , Hermann P , Ladogana A , et al. Validation of Revised International Creutzfeldt-Jakob Disease Surveillance Network Diagnostic Criteria for Sporadic Creutzfeldt-Jakob Disease, JAMA Netw Open (2022) ; 5: : e2146319. |
5. | Ukisu R , Kushihashi T , Kitanosono T , et al. Serial diffusion-weighted MRI of Creutzfeldt-Jakob disease, AJR Am J Roentgenol (2005) ; 184: : 560–566. |
6. | Murata T , Shiga Y , Higano S , et al. Conspicuity and evolution of lesions in Creutzfeldt-Jakob disease at diffusion-weighted imaging, AJNR Am J Neuroradiol (2002) ; 23: : 1164–1172. |
7. | Jack CR , Bennett DA , Blennow K , et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease, Alzheimers Dement (2018) ; 14: : 535–562. |
8. | Godoy MDCL , Voegels RL , Pinna F de R , et al. Olfaction in neurologic and neurodegenerative diseases: A literature review, Int Arch Otorhinolaryngol (2015) ; 19: : 176–179. |
9. | Dan X , Wechter N , Gray S , et al. Olfactory dysfunction in aging and neurodegenerative diseases, Ageing Res Rev (2021) ; 70: : 101416. |
10. | Viguera C , Wang J , Mosmiller E , et al. Olfactory dysfunction in amyotrophic lateral sclerosis, Ann Clin Transl Neurol (2018) ; 5: : 976–981. |
11. | Reuber M , Al-Din A , Baborie A , et al. New variant Creutzfeldt-Jakob disease presenting with loss of taste and smell, J Neurol Neurosurg Psychiatry (2001) ; 71: : 412–413. |
12. | Bortone E , Bettoni L , Giorgi C , et al. Reliability of EEG in the diagnosis of Creutzfeldt-Jakob disease, Electroencephalogr Clin Neurophysiol (1994) ; 90: : 323–330. |
13. | Steinhoff BJ , Räcker S , Herrendorf G , et al. Accuracy and reliability of periodic sharp wave complexes in Creutzfeldt-Jakob disease, Arch Neurol (1996) ; 53: : 162–166. |
14. | Shir D , Lazar EB , Graff-Radford J , et al. Analysis of clinical features, diagnostic tests, and biomarkers in patients with suspected Creutzfeldt-Jakob Disease, 2014–2021. JAMA Netw Open (2022) ; 5: : e2225098. |
15. | Hamlin C , Puoti G , Berri S , et al. A comparison of tau and 14-3-3 protein in the diagnosis of Creutzfeldt-Jakob disease, Neurology (2012) ; 79: : 547–552. |
16. | Forner SA , Takada LT , Bettcher BM , et al. Comparing CSF biomarkers and brain MRI in the diagnosis of sporadic Creutzfeldt-Jakob disease, Neurol Clin Pract (2015) ; 5: : 116–125. |
17. | Bongianni M , Orrù C , Groveman BR , et al. Diagnosis of human prion disease using real-time quaking-induced conversion testing of olfactory mucosa and cerebrospinal fluid samples, JAMA Neurol (2017) ; 74: : 155–162. |
18. | McGuire LI , Peden AH , Orrú CD , et al. Real time quaking-induced conversion analysis of cerebrospinal fluid in sporadic Creutzfeldt-Jakob disease, Ann Neurol (2012) ; 72: : 278–285. |
19. | Foutz A , Appleby BS , Hamlin C , et al. Diagnostic and prognostic value of human prion detection in cerebrospinal fluid, Ann Neurol (2017) ; 81: : 79–92. |
20. | Hermann P , Appleby B , Brandel JP , et al. Biomarkers and diagnostic guidelines for sporadic Creutzfeldt-Jakob disease, Lancet Neurol (2021) ; 20: : 235–246. |
21. | Sokhi D , Yakub F , Sharma K , et al. Heidenhain variant of sporadic Creutzfeldt-Jakob disease: First reported case from East Africa, Int Med Case Rep J (2021) ; 14: : 39–44. |
22. | Fiorini M , Iselle G , Perra D , et al. High diagnostic accuracy of RT-QuIC assay in a prospective study of patients with suspected sCJD, Int J Mol Sci (2020) ; 21: : 880. |
23. | Vitali P , Maccagnano E , Caverzasi E , et al. Diffusion-weighted MRI hyperintensity patterns differentiate CJD from other rapid dementias, Neurology (2011) ; 76: : 1711–1719. |
24. | Tschampa HJ , Kallenberg K , Urbach H , et al. MRI in the diagnosis of sporadic Creutzfeldt-Jakob disease: A study on inter-observer agreement, Brain (2005) ; 128: (Pt 9), 2026–2033. |
25. | Morales R , Moreno-Gonzalez I and Soto C. Cross-seeding of misfolded proteins: Implications for etiology and pathogenesis of protein misfolding diseases, PLoS Pathogens (2013) ; 9: : e1003537. |
26. | Falker C , Hartmann A , Guett I , et al. Exosomal cellular prion protein drives fibrillization of amyloid beta and counteracts amyloid beta-mediated neurotoxicity, J Neurochem (2016) ; 137: : 88–100. |
27. | Rossi M , Kai H , Baiardi S , et al. The characterization of AD/PART co-pathology in CJD suggests independent pathogenic mechanisms and no cross-seeding between misfolded Aβ and prion proteins, Acta Neuropathol Commun (2019) ; 7: : 53. |
28. | Hermann P , Haller P , Goebel S , et al. Total and phosphorylated cerebrospinal fluid tau in the differential diagnosis of sporadic Creutzfeldt-Jakob disease and rapidly progressive Alzheimer’s disease, Viruses (2022) ; 14: : 276. |


