
Alzheimer’s Disease, Inflammation, and the Role of Antioxidants
Abstract
The World Health Organization refers to Alzheimer’s disease (AD) as a global health priority. As the average age of the world’s population is increasing, so too is the rate of AD. There are an estimated 47 million people globally who have been diagnosed with AD dementia, and researchers have yet to figure out the root cause. All misfolded aggregate proteins that are involved in neurodegenerative disorders (amyloid-β, Huntington’s tau, α-synuclein) induce oxidative stress. It is that oxidative stress that leads to inflammation and, in conjunction with amyloid protein and tau hyperphosphorylation, progresses to and exacerbates AD. The consumption of antioxidants and nutrients, specifically vitamin E, caffeine, and turmeric, may slow the progression of AD and can be found in a wide variety of dietary foods. This review explores the role of inflammation on AD, the antioxidants that can potentially combat it, and future directions of how the treatment of the disease can be better understood.
EPIDEMIOLOGY OF ALZHEIMER’S DISEASE
Alzheimer’s disease (AD) is growing into one of the world’s largest cognitive diseases. According to the Alzheimer’s Association, someone develops AD every 65 seconds in the United States [1]. In fact, there are currently 5.8 million people in the United States living with AD, and that number is projected to increase to 14 million in 2050 [1]. Globally, it is estimated that about 47 million people have AD dementia with the highest rates in Finland, United States, Canada, Sweden, and Norway [2]. It is currently the sixth leading cause of death in the United States [3]. Therefore, it is imperative for physicians to be armed with the knowledge to help an ever-aging patient population to reduce the likelihood of developing AD or limiting cognitive decline in those already diagnosed.
PATHOGENESIS OF ALZHEIMER’S DISEASE
Although the development of AD is a multifactorial process, it has two known pathologies. The first being the hyperphosphorylation of tau proteins, and the second mechanism is an increased aggregation of amyloid plaques. Tau is a microtubule that works as a scaffold protein in axons. When these proteins become hyperphosphorylated and impaired in AD patients, it leads to an aberrant skeletal framework, a disruption cell operation, neuron dystrophy, and cause an injury to axonal transport [4, 5]. AD is also associated with amyloid-β (Aβ) consistently building up with a high resistance to protease degradation. These beta-pleated sheets aggregate and form amyloid plaques in the brain, causing neurodegeneration and injury [4]. In vitro studies from AD brains applied to neurons showed decreased long-term potentiation, synaptic dysfunction, and damage dendritic spines, which leads to neuronal death [5]. The misfolded proteins that aggregate all induce oxidative stress [6]. While it is evident that these two mechanisms contribute to the development of AD, there is another pathology, such as inflammation, to consider.
METHODOLOGY
This review highlights basic explanatory research from secondary data. A search of PubMed with the following MeSH terms was done: Alzheimer’s Disease, Antioxidants, Inflammation, Vitamin E, Tumeric, Estrogen, Caffeine. The search was limited to publications within the last 25 years, as the topic has not been extremely well researched, particularly the effect of antioxidants on AD.
INFLAMMATION AND ALZHEIMER’S DISEASE
Growing evidence has shown that inflammation could be a hallmark contributor to AD development and exacerbation. Pro-inflammatory cytokines like TNF-α, IL-1β, and IL-6 are upregulated in brains of individuals with AD, which leads to an accumulation of Aβ plaque aggregates and tau hyperphosphorylation resulting in neuronal loss [7–9]. Inflammation in the brain is mediated by astrocytes and microglia. Microglia have multiple functions, but one important role established are being the scavenger cells of the brain responding to disuse damage and apoptosis. Neuronal injury from amyloid plaques are increased by the production of neurotoxic Aβ in response to cytokines and inflammatory stimuli [9]. Deposition of Aβ in the brain results in neuroinflammation by triggering microglia, which is stated to be a major source of pro-inflammatory cytokines in AD [10]. After microglia phagocytize the amyloid, they decrease the clearance process of amyloid over time, since amyloid is difficult to proteolyze. Microglia then become compromised and less efficient in breaking down amyloid plaques. They release more pro-inflammatory cytokines, which may further exacerbate AD progression.
Mechanisms proposed for microglia degeneration include TREM2, CX3CR1, GABA, and other inflammatory cytokine mediators. TREM2 is an innate immune receptor expressed on the cell surface of microglia that causes macrophages to activate phagocytosis causing the release of reactive oxygen species [11]. A study done on mice has shown that TREM2 alters phagocytosis and lipid metabolism exacerbating AD [9]. Numerous studies demonstrate that the CX3CL1/CX3CR1 receptor ligands, another transcription factor and chemokine, can dose-dependently suppress the death of neurons by activating microglia and decrease levels of nitric oxide, IL-6, and TNF-α after stimulation of lipopolysaccharide [9]. However, postmortem studies in AD patients showed decreased levels of CX3CL1 when compared with age-matched controls [9]. In addition, the aforementioned study done on mice stated that genetic deletion of CX3CR1 changes the inflammatory process, resulting in higher Aβ phagocytosis in microglia [9, 12]. CX3CL1/CX3CL1 affects not only microglia, but tau hyperphosphorylation as well. AD transgenic mice with a CX3CR1 deletion led to increased tau phosphorylation, accumulation, microglial activation, and an increased deficit in hippocampal learning, exacerbating AD [9, 13].
Microglia also release pro-inflammatory cytokines, such as IL-6, TNF-α, and IL-1β. These cytokines play a central role in the regulation and beginning phases of inflammation, allowing leukocytes and immune cells to migrate to sites by increasing vascular and endothelial adhesion molecules. IL-1β, synthesized by activated microglia and astrocytes, regulates the formation of amyloid-β protein precursor from glial cells, which is a precursor to amyloid plaques. One study showed that IL-1β is overexpressed by microglia and astrocytes at Aβ plaques, and they are present in AD brains and animal models of AD when compared to age controls [14, 15]. This study provides evidence that overexpression of IL-1β exacerbates tau phosphorylation and neurofibrillary tangles by activation of different kinases [16].
Astrocytes, also considered as a supportive cell, play an integral role in AD. Astrocytes are involved in cell signaling, synaptic remodeling, and management of oxidative stress. When they become reactive, astrocytes express inflammatory mediators. High levels of extracellular S100B, which is released from astrocytes via IL-1β due to pro-inflammatory mediators and other triggers, can act as a cytokine and have been increased in clinical conditions that include brain trauma, ischemia and neurodegenerative, and inflammatory and psychiatric diseases [17, 18]. Researchers at Northwestern University concluded the protein S100B, a protein that induces neurite proliferation, is overexpressed in AD brains [17, 19]. These effects can increase brain inflammation and escalate amyloid plaques from amyloid-β protein precursor. In addition, exposure of cultured astrocytes to Aβ significantly upregulate IL-1β, TNF-α, IL-6, and NO production, therefore intensifying inflammation and progression of AD [19].
Increasing research shows that oligodendrocyte, the myelin forming cells in the central nervous system, can contribute to oxidative stress. One proposed mechanism for oxidative stress previously shown in rat models, is that oligodendrocytes have a greater decreased glutathione content than any other cell in the brain and an increased iron content, which make them more vulnerable to oxidative stress and damage [19]. Oligodendrocytes can lead to increased compliment in AD brains, exacerbating inflammation. Aβ is also potentially toxic to oligodendrocytes. According to a study done in mice, oligodendrocytes exposed with Aβ oxidative stress can lead to cell death, resulting in demyelination. This demyelination decreases the neuron action potential time and increases cognitive impairment in AD [20]. This evidence suggests that neurons in the nervous system play a role in increasing inflammation and stress, thereby increasing the progression of AD.
It is vital to have a balance between oxidants and antioxidants. When unbalanced, the overproduction of reactive oxygen species (ROS) or lack of antioxidants leads to oxidative stress. Free radicals are generated from both endogenous and exogenous sources, such as, inflammation, ischemia, cancer, aging, environmental pollutants, cigarette smoke, alcohol, and heavy metals, such as mercury, and iron [21]. Oxidants are generated in our bodies from cellular by-products of metabolism or external factors such as ionizing radiation [22].
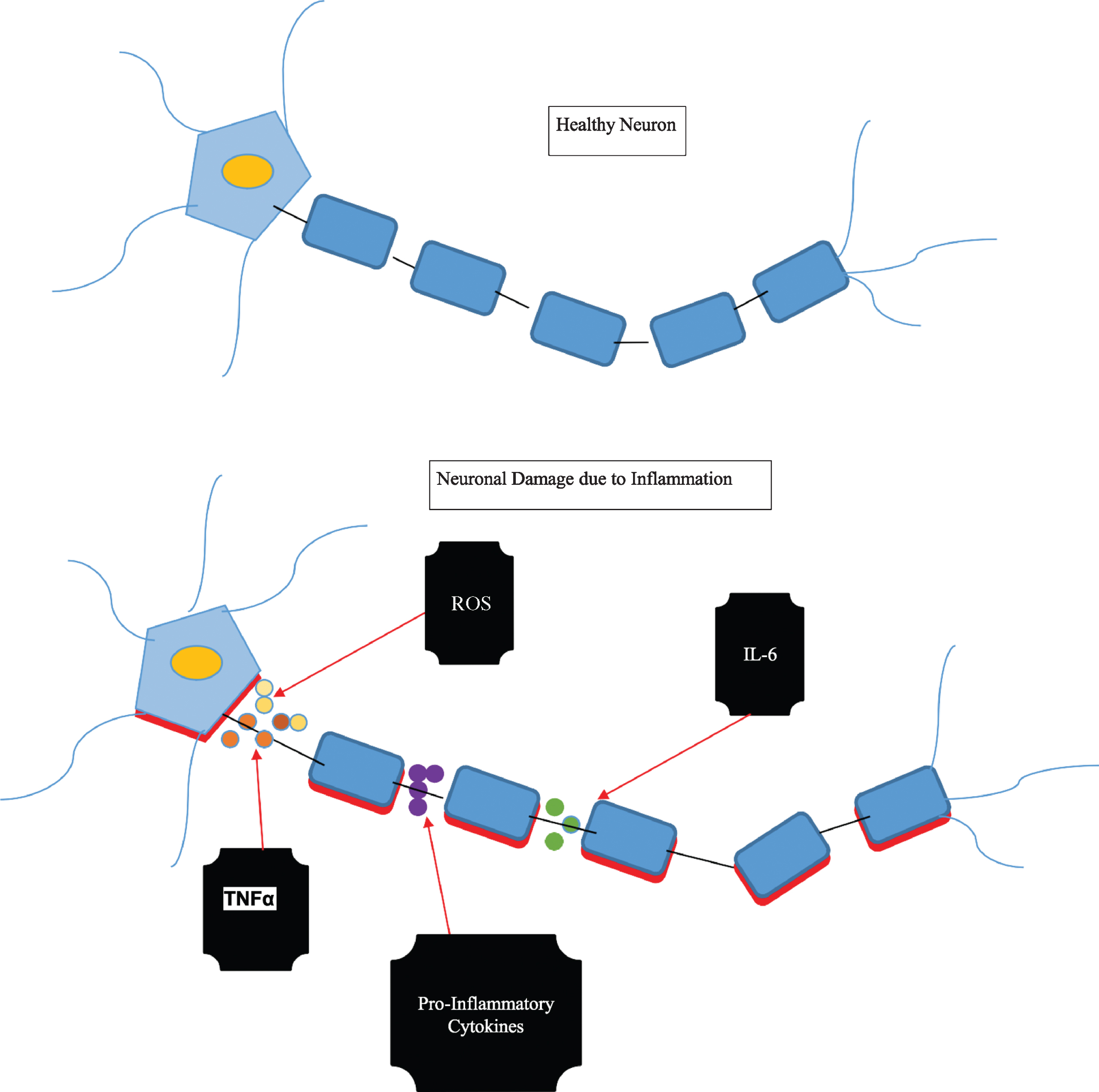
However, in AD, the activity of antioxidant enzymes is altered, which leads to the free buildup of oxidative damage [23]. These oxidants can be detrimental to our cellular structures that provide the scaffolding and integrity in AD brains through DNA oxidation products, hydroxyl radical pairing with DNA bases, and lipid peroxidation. Oxidant levels that are too high can result in cell damage (see Fig. 1). Oxidative species is a characteristic of AD brains, especially in the pathology of senile plaques [24]. Before the presence of Aβ pathology and clinical symptoms, there is evidence that the production of ROS increases due to mitochondrial damage [23]. Antioxidants decrease free-radical-mediated damage in neuronal cells through detoxification, and if the balance between antioxidants/oxidants are unbalanced unfavorably, there can be detrimental effects, especially in the AD brain.
Fig. 1
Pro-inflammatory mediators acting on a healthy neuron resulting in inflammation and damage.
THE ROLE OF ANTIOXIDANTS IN ALZHEIMER’S DISEASE TREATMENTS
Vitamin E
Antioxidants are both endogenous and exogenous compounds that prevent or slow the damage to cells caused by free radicals. The combative capacity of vitamin E for peroxyl radicals is the highest among antioxidants [25]. Vitamin E and vitamin C can possibly ensnare the dementia pathogenesis in AD. Vitamin C acts acting as a reducing agent, by reducing metals like iron and copper in the body, and can be oxidized by most reactive oxidative/nitrosative species. This results in neuroprotection and may improve cognition and prevent aggregation of Aβ protein [22]. Vitamin E acts on lipid fat soluble membrane lipoproteins, and low density lipoprotein [15]. It is responsible for the elimination of free radicals in the red blood cell membrane and stops the spread of lipoperoxidation, thereby acting to prevent hemolysis [26]. Vitamin E has the power to inhibit neuronal death caused by inflammatory processes.
Although there are many forms of vitamin E, α-tocopherol is the most abundant and bioavailable antioxidant form of vitamin E in human tissues [27, 28]. The recommended daily dose for men and women for vitamin E is 15 mg, or 22 IU of vitamin E according to the U.S. National Academy of Sciences Food and Nutrition Board [29]. Vitamin E is transported across the brain through a unique transporter called the alpha-tocopherol transfer protein (alpha-TTP), which is found in high concentration in the cerebellum and astrocytes [29]. It is understood that if an aberrant mutation or alteration to the gene that encodes for alpha-TTP can develop neurodegenerative diseases, loss of reflexes and proprioception, and very low vitamin E [29]. It has been shown that alpha-TTP expression is increased in brains of patients with neurodegenerative diseases, which illustrates that vitamin E is an essential antioxidant factor for neuroprotection [29]. After reviewing 80 micronutrients on the effect of AD, a 2014 meta-analysis portrayed that vitamin E was displayed in lower plasma levels in patients with AD [30]. Four years later, another meta-analysis was done in 2018 and concluded that AD is associated with a low concentration of serum vitamin E [31].
One of the longest AD treatment trials, designed by TEAM–AD, discussed the efficacy of α-tocopherol, memantine, or their combination in delaying clinical progression of AD in patients taking an acetylcholinesterase inhibitor. The authors found that 2000 IU/day of vitamin E significantly delayed the clinical progression of AD symptoms [30, 32]. A study done with transgenic mice models with the AD-phenotype reported vitamin E decreased the oxidation of lipids in the brain and reduced Aβ levels and plaque deposition when it was given in the early stages of AD [15]. One finding worth noting from this study is that when plaques were already deposited in the brain; vitamin E would have no effect reduction of brain stress caused by AD [15]. An additional study performed on AD patients examined the effects if vitamin E and Selegene and found that when given 1000 IU/twice per day for two years, there was a delay in deterioration of daily life and a decreased need for care [33]. Additional evidence suggests that giving vitamin E to Drosophila suppressed tau-induced neurotoxicity [15]. Consuming vitamin E rich foods decreases long-term risk of dementia and AD after adjusting for age, education, sex, race, APOE ɛ4 allele, and length of follow-up [15]. One important point to note is that these results were only significant in patients without the APOE ɛ4 allele [15, 34]. The Rotterdam study [34], a large-scale study in the Netherlands, found that high intake of vitamin C and vitamin E may be associated with a lower incidence of AD. It is important to note that there were positive findings in patients who had higher intake in vitamin E rich foods. In addition, the average vitamin E intake did not develop a lower risk of dementia [35]. From the research that has been done thus far, there is strong evidence that vitamin E can be beneficial for those in the early stages of AD by delaying its clinical progression, which could potentially result in a decreased need for care and better quality of life.
Estrogen
Estrogen has emerged to function as an antioxidant in some in vivo and in vitro studies, which is significant because 15% of women over the age of 65 suffer from AD [36]. AD neuropathology is known to have an oxidative stress component with the idea proposed that polyunsaturated fatty acids of the membranes of neurons increase the oxidative damage susceptibility of lipids. AD was found to be less frequent among postmenopausal women who used estrogen replacement therapy. A study carried out in Italian municipalities examined 2,816 women and found that estrogen replacement therapies are associated with a decreased prevalence in AD in postmenopausal women according to the NINCDS-ADRDA criteria [37]. Another study done in Rochester, Minnesota assessed 222 patients and found that estrogen replacement therapy is associated with a reduced risk of AD in postmenopausal women [38, 39]. A study done at the University of Milan showed that estrogen has various benefits related to combating neurodegeneration, including preventing neural apoptosis and inhibition of the production of Aβ [40]. One mechanism proposed for estrogen inhibiting the production of Aβ is estrogen downregulating the expression of MMP-9. When MMP-9 is expressed, it is associated with chronic inflammatory and degradative diseases, and it is involved in the destruction of proteases, such as Aβ [41]. The evidence suggests that giving estrogen to high-risk patients can potentially decrease neuronal inflammation and AD in older women. However, giving unopposed estrogen in postmenopausal women has a higher link to breast cancer, blood clots, and ovarian cancer [42].
OTHER VARIOUS SUPPLEMENTS AND ALZHEIMER’S DISEASE
Caffeine
Humans with the APOE ɛ4 allele have an increased chance of developing AD. APOE ɛ4 has been linked to increased cholesterol levels due to the aid of transport of cholesterol across the blood-brain barrier to neurons [43]. Researchers have proposed that increased cholesterol raises the risk of AD in people with the APOE ɛ4 allele, and increased cholesterol has been associated with more oxidative stress through ROS [43]. Caffeine has been shown to reduce brain Aβ levels for early-onset familial AD in mice [15]. One study concluded that caffeine treatment with APP mice reduced Aβ deposition in the hippocampus (associated with memory function) by 40% and part of the temporal lobe (primary auditory cortex) by 46% [43]. In addition, another study found that caffeine treatment at 292 mg/d suppressed Aβ and decreased plaque numbers [44]. The FDA has cited about 400 milligrams a day or equivalent to roughly 5 cups a day as an amount not generally associated with dangerous or negative effects [45]. Caffeine decreased amyloid accumulation and decreased cholesterol induced phosphorylation of tau proteins after feeding rabbits 2% more cholesterol in their diet, creating a greater production of amyloid [46]. In the CAIDE study, [47] drinking 3–5 cups of coffee per day was associated with a decreased risk of dementia and AD by about 65% later in life. However, too much caffeine consumption can cause adverse side effects such as tremors, palpitations and arrhythmias. In an experiment conducted with human neuroblastoma cells, caffeine lowered reactive oxygen species production cultured cells with Aβ [44]. In addition, the study also illustrated an increased superoxide dismutase levels, and lowered malondialdehyde, a marker for oxidative stress levels significantly [43].
Turmeric
Turmeric has been known to have anti-inflammatory and antioxidant effects in the body.
Turmeric is able to cross the blood-brain barrier and exhibit therapeutic potential via the inhibition of peroxidases, and decreases aggregation of Aβ and reduction of neuroinflammation [48]. A study conducted at UCLA demonstrated that a combination of macrophages and turmeric showed an improved uptake and ingestion of Aβ plaque in AD patients [49]. Turmeric has been found to reduce the release of ROS by stimulating neutrophils and inhibiting the activation of the pro-inflammatory cytokines TNF-α, IL-6, and IL-1β. This leads to a decrease in the main mechanisms for the inflammation of cytokines. A study done in-vitro showed increased production of the anti-inflammatory cytokine IL-4 in cultured microglia from mice that were treated with turmeric extract [50]. IL-4 is an anti-inflammatory cytokine that has been shown to reduce the production of inflammatory mediators in microglia such as TNF-α, and inhibit microglia activation [50]. Meta-analysis show that turmeric significantly reduces concentration of circulating IL-6, a pro inflammatory cytokine, with more apparent effects in patients with a higher degree of systemic inflammation [51]. A 40% reduction of Aβ was seen in AD mice that were given low doses of turmeric compared to those that were not treated with turmeric [15, 52]. Turmeric exposure also decreased pro-inflammatory cytokines IL-1, and TNF-α [53]. Turmeric has been shown to directly bind small Aβ species to block aggregation and fibril formation in vitro and in vivo [54]. Evidence shows that turmeric has anti-inflammatory and antioxidant potential to alleviate inflammation.
DISCUSSION
There is growing evidence indicating that there is an inflammatory component to AD [7, 9]. The release of inflammatory cytokines by activated microglia and astrocytes exacerbate inflammation in the AD brain [10]. In turn, this leads to not only increased Aβ aggregation, but also increased tau hyperphosphorylation [8, 9]. Astrocytes and oligodendrocytes have also been shown to intensify the role of inflammation [19, 20]. It has become clearer that inflammation is a pathogenesis for AD, and one method for decreasing inflammation is the role of antioxidants and nutrients. Vitamin E has been shown to decrease the clinical progression of early AD symptoms, [30, 32] and a high intake of vitamin C and vitamin E from food may be associated with a lower incidence of AD [34]. Estrogen may show protective effects on the brain by inhibition of MMP-9 and downregulation of apoptosis and Aβ production [31]. Caffeine has been shown to reduce Aβ plaques in mice models [15]. In addition, 3–5 cups of coffee per day is associated with a decreased risk of dementia and AD by about 65% later in life [47]. Turmeric has antioxidant properties, which have shown to ease the aid of macrophages digesting Aβ plaques [49]. Turmeric binds directly to Aβ aggregated proteins, and blocks fibril formation via an increase in TH2 cytokines, and anti-inflammatory mediators IL-4, and IL-2 leading to a decrease in inflammatory mediators [50, 54].
As Socrates once said, “Let food be thy medicine, and let medicine be thy food.” As such, physicians should encourage their patients to inquire about their diet and lifestyle and reinforce healthy habits. Foods such as berries (strawberries, raspberries, and blueberries), kale, dark chocolate, and pecans, which are rich in antioxidants, should be explored in moderation. Similarly, consumption of foods rich in vitamin E such as various nuts, seeds, vegetable oils, and green leafy vegetables could also be encouraged. Additionally, studies have shown that the MIND diet (Mediterranean- DASH diet) has neuroprotective benefits and has been found to reduce the risk of AD, with clinical trials being evaluated in the United States [55, 56]. This diet has also been recommended by the Alzheimer’s Association to help reduce the risk of heart disease and dementia [57]. Although the USPSTF currently has no guidelines on the quantity of antioxidants one should consume, physicians should counsel their patients to increase their consumption of antioxidant rich foods to enhance their diet.
CERTAIN LIMITATIONS
While in vitro animal and laboratory studies have shown that antioxidant rich nutrients can protect the brain from oxidative and inflammatory damage, there is limited data available from epidemiological studies and human clinical trials on the role of antioxidants on inflammation in AD. Additionally, while age is the most important risk factor for AD, there is limited information on the modifiable risk factors for AD such as lower educational status [58]. AD research should focus on lifestyle medicine in conjunction with cognitive status and genomic testing form a complete portrait of the AD brain, antioxidants, and inflammation.
FUTURE DIRECTIONS
Future research should explore more precise and definitive mechanisms that trigger inflammation and solidify its role on AD, so adequate treatment can be targeted. One mechanism that has been proposed is immunizations with antibodies that bind Aβ; however, these immunizations have failed in phase three of clinical trials due to multiple issues [59]. One possible reason for this could have been the treatment was initiated too late in the disease process [59]. Personalized gene therapy with peptide modification and sequencing can be beneficial to treating AD [60, 61]. In 2012, the NIH began sequencing the genomes of thousands of volunteers with AD [62]. The results of sequencing can hopefully pave the way for new and innovative treatments. Another therapy to further research is the role of other antioxidants and their effect of decreasing inflammation on the brain in AD patients. Inflammation and AD are inevitably linked, with more research needed to solidify the mechanisms associated with them. Future research can elucidate the role of the intake of antioxidants, which can provide more insight on how to live a healthier, and hopefully, neurodegenerative-free lifestyle.
CONFLICT OF INTEREST
None of the authors have any financial interests, relationships or affiliations relevant to the subject of this manuscript.
ACKNOWLEDGMENTS
HCA Healthcare supported this research (in whole or in part) and/or an HCA Healthcare affiliated entity. The views expressed in this publication represent those of the author(s) and do not necessarily represent the official views of HCA Healthcare or any of its affiliated entities.
REFERENCES
[1] | Alzheimer’s Association ((2019) ) Alzheimer’s disease facts and figures. Alzheimers Dement 15: , 321–387. |
[2] | Newcombe EA , Camats-Perna J , Silva ML , Valmas N , Huat TJ , Medeiros R ((2018) ) Inflammation: the link between comorbidities, genetics, and Alzheimer’s disease. J Neuroinflammation 15: , 276. |
[3] | National Institute on Aging (2019) Alzheimer’s Disease Fact Sheet. |
[4] | Sanabria-Castro A , Alvarado-Echeverría I , Monge-Bonilla C ((2017) ) Molecular pathogenesis of Alzheimer’s disease: an update. Ann Neurosci 24: , 46–54. |
[5] | Lane CA , Hardy J , Schott JM ((2017) ) Alzheimer’s disease. Eur J Neurol 25: , 59–70. |
[6] | Angelova PR , Abramov AY ((2018) ) Role of mitochondrial ROS in the brain: from physiology to neurodegeneration. Lett 592: , 692–702. |
[7] | Wang WY , Tan MS , Yu JT , Tan L ((2015) ) Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann Transl Med 3: , 136. |
[8] | Von Bernhardi R , Tichauer JE , Eugenin J ((2010) ) Aging-dependent changes of microglial cells and their relevance for neurodegenerative disorders. J Neurochem 112: , 1099–1114. |
[9] | Kinney JW , Bemiller SM , Murtishaw AS , Leisgang AM , Salazar AM , Lamb BT ((2018) ) Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement 4: , 575–590. |
[10] | Prinz M , Priller J , Sisodia SS , Ransohoff RM ((2011) ) Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat Neurosci 14: , 1227–1235. |
[11] | Boutajangout A , Wisniewski T ((2013) ) The innate immune system in Alzheimer’s disease. Biol 2013: , 1–7. |
[12] | Das Sarma J ((2013) ) Microglia-mediated neuroinflammation is an amplifier of virus-induced neuropathology. J Neurovirol 20: , 122–136. |
[13] | Bhaskar K , Konerth M , Kokiko-Cochran ON , Cardona A , Ransohoff RM , Lamb BT ((2010) ) Regulation of tau pathology by the microglial fractalkine receptor. Neuron 68: , 19–31. |
[14] | Forlenza OV , Diniz BS , Talib LL , Mendonca VA , Ojopi EB , Gattaz WF , Teixeira AL ((2009) ) Increased serum IL-1beta level in Alzheimer’s disease and mild cognitive impairment. Dement Geriatr Cogn Disord 28: , 507–512. |
[15] | Feng Y , Wang X ((2012) ) Antioxidant therapies for Alzheimer’s disease. Oxid Med Cell Longev 2012: , 1–17. |
[16] | Ghosh S , Wu MD , Shaftel SS , Kyrkanides S , LaFerla FM , Olschowka JA , O’Banion MK ((2013) ) Sustained interleukin-1β overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J Neurosci 33: , 5053–5064. |
[17] | Leclerc E , Sturchler E , Vetter SW ((2010) ) The S100B/RAGE axis in Alzheimer’s disease. Cardiovasc Psychiatry Neurol 2010: , 549581. |
[18] | de Souza DF , Leite MC , Quincozes-Santos A , Nardin P , Tortorelli LS , Rigo MM , Gottfried C , Leal RB , Goncalves CA ((2008) ) S100B secretion is stimulated by IL-1beta in glial cultures and hippocampal slices of rats: likely involvement of MAPK pathway. J Neuroimmunol 206: , 52–57. |
[19] | Wyss-CorayT, RogersJ ((2012) ) Inflammation in Alzheimer disease – A brief review of the basic science and clinical literature. Cold Spring Harb Perspect Med 2: , a006346. |
[20] | Nasrabady SE , Rizvi B , Goldman JE , Brickman AM ((2018) ) White matter changes in Alzheimer’s disease: a focus on myelin and oligodendrocytes. Acta Neuropathol Commun 6: , 22. |
[21] | Pizzino G , Irrera N , Cucinotta M , Pallio G , Mannino F , Arcoraci V , Squadrito F , Altavilla D , Bitto A ((2017) ) Oxidative stress: harms and benefits for human health. Oxid Med Cell Longev 2017: , 8416763. |
[22] | Kurutas EB ((2016) ) The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: current state. Nutr J 15: , 71. |
[23] | Tönnies E , Trushina E ((2017) ) Oxidative stress, synaptic dysfunction, and Alzheimer’s disease. J Alzheimers Dis 57: , 1105–1121. |
[24] | Moneim AE ((2015) ) Oxidant/antioxidant imbalance and the risk of Alzheimer’s disease. Curr Alzheimer Res 12: , 335–349. |
[25] | Lloret A , Esteve D , Monllor P , Cervera-Ferri A , Lloret A ((2019) ) The effectiveness of vitamin E treatment in Alzheimer’s disease. Int J Mol Sci 20: , E879. |
[26] | Teixeira JP , de Castro AA , Soares FV , da Cunha EFF , Ramalho TC ((2019) ) Future therapeutic perspectives into the Alzheimer’s disease targeting the oxidative stress hypothesis. Molecules 24: , E4410. |
[27] | Joshi YB , Pratico D ((2012) ) Vitamin E in aging, dementia, and Alzheimer’s disease. BioFactors 38: , 90–97. |
[28] | Rigotti A ((2007) ) Absorption, transport, and tissue delivery of vitamin E. Mol Aspects Med 28: , 423–436. |
[29] | Institute of Medicine (US) Panel on Dietary Antioxidants and Related Compounds (2000) Dietary Reference Intakes for Vitamin C, Vitamin E, Selenium, and Carotenoids. |
[30] | La Fata G , Weber P , Mohajeri MH ((2014) ) Effects of vitamin E on cognitive performance during ageing and in Alzheimer’s disease. Nutrients 6: , 5453–5472. |
[31] | Dong Y , Chen X , Liu Y , Shu Y , Chen T , Xu L , Li M , Guan X ((2018) ) Low-serum vitamin E levels increase the risk of Alzheimer disease in older people? Evidence from a meta-analysis of case-control studies. Int J Geriatr Psychiatry 33: , e257–e263. |
[32] | Dysken MW , Sano M , Asthana S , Vertrees JE , Pallaki M , Llorente M , Love S , Schellenberg GD , McCarten JR , Malphurs J , Prieto S , Chen P , Loreck DJ , Trapp G , Bakshi RS , Mintzer JE , Heidebrink JL , Vidal-Cardona A , Arroyo LM , Cruz AR , Zachariah S , Kowall NW , Chopra MP , Craft S , Thielke S , Turvey CL , Woodman C , Monnell KA , Gordon K , Tomaska J , Segal Y , Peduzzi PN , Guarino PD ((2014) ) Effect of vitamin E and memantine on functional decline in Alzheimer disease: The TEAM-AD VA Cooperative Randomized Trial. JAMA 311: , 33–44. |
[33] | Sano M , Ernesto C , Thomas RG , Klauber MR , Schafer K , Grundman M , Woodbury P , Growdon J , Cotman CW , Pfeiffer E , Schneider LS , Thal LJ ((1997) ) A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. The Alzheimer’s Disease Cooperative Study. N Engl J Med 336: , 1216–1222. |
[34] | Morris MC , Evans DA , Bienias JL , Tangney CC , Bennett DA , Aggarwal N , Wilson RS , Scherr PA ((2002) ) Dietary intake of antioxidant nutrients and the risk of incident Alzheimer disease in a biracial community study. JAMA 287: , 3230–3237. |
[35] | Devore EE , Grodstein F , van Rooij FJ , Hofman A , Stampfer MJ , Witteman JC , Breteler MM. ((2010) ) Dietary antioxidants and long-term risk of dementia. Arch Neurol 67: , 819–825. |
[36] | Kong D , Yan Y , He X , Yang H , Liang B , Wang J , He Y , Ding Y , Yu H ((2019) ) Effects of resveratrol on the mechanisms of antioxidants and estrogen in Alzheimer’s disease. Biomed Res Int 2019: , 1–8. |
[37] | Baldereschi M , Di Carlo A , Lepore V , Bracco L , Maggi S , Grigoletto F , Scarlato G , Amaducci L ((1998) ) Estrogen-replacement Therapy and Alzheimer’s Disease in the Italian Longitudinal Study on Aging. Neurology 50: , 996–1002. |
[38] | Waring SC , Rocca WA , Petersen RC , O’Brien PC , Tangalos EG , Kokmen E ((1999) ) Postmenopausal estrogen replacement therapy and risk of AD: a population-based study. Neurology 52: , 965–970. |
[39] | Baldereschi M , Di Carlo A , Lepore V , Bracco L , Maggi S , Grigoletto F , Scarlato G , Amaducci L ((1998) ) Estrogen-replacement therapy and Alzheimer’s disease in the Italian Longitudinal Study on Aging. Neurology 50: , 996–1002. |
[40] | Aliev G , Obrenovich ME , Reddy VP , Shenk JC , Moreira PI , Nunomura A , Zhu X , Smith MA , Perry G ((2008) ) Antioxidant therapy in Alzheimer’s disease: theory and practice. Mini Rev Med Chem 8: , 1395–1406. |
[41] | Vegeto E , Bonincontro C , Pollio G , Sala A , Viappiani S , Nardi F , Brusadelli A , Viviani B , Ciana P , Maggi A ((2001) ) Estrogen prevents the lipopolysaccharide-induced inflammatory response in microglia. J Neurosci 21: , 1809–1818. |
[42] | Beral V , Gaitskell K , Hermon C , Moser K , Reeves G , Peto R ((2015) ) Menopausal hormone use and ovarian cancer risk: individual participant meta-analysis of 52 epidemiological studies. Lancet 385: , 1835–1842. |
[43] | Liu CC , Liu CC , Kanekiyo T , Xu H , Bu G ((2013) ) Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol 9: , 106–118. |
[44] | Kolahdouzan M , Hamadeh MJ ((2017) ) The neuroprotective effects of caffeine in neurodegenerative diseases. CNS Neurosci Ther 23: , 272–290. |
[45] | US Food and Drug Administration (2019) Spilling the Beans: How Much Caffeine is Too Much? |
[46] | Prasanthi JR , Dasari B , Marwarha G , Larson T , Chen X , Geiger JD , Ghribi O ((2010) ) Caffeine protects against oxidative stress and Alzheimer’s disease-like pathology in rabbit hippocampus induced by cholesterol-enriched diet. Free Radic Biol Med 49: , 1212–1220. |
[47] | Eskelinen MH , Kivipelto M ((2010) ) Caffeine as a protective factor in dementia and Alzheimer’s disease. J Alzheimers Dis 20: , S167–174. |
[48] | Reddy PH , Manczak M , Yin X , Grady MC , Mitchell A , Tonk S , Kuruva CS , Bhatti JS , Kandimalla R , Vijayan M , Kumar S , Wang R , Pradeepkiran JA , Ogunmokun G , Thamarai K , Quesada K , Boles A , Reddy AP ((2018) ) Protective effects of Indian spice curcumin against amyloid-β in Alzheimer’s disease. J Alzheimers Dis 61: , 843–866. |
[49] | Mishra S , Palanivelu K ((2008) ) The effect of curcumin (turmeric) on Alzheimer’s disease: an overview. Ann Indian Acad Neurol 11: , 13–19. |
[50] | Lyons A , Griffin RJ , Costelloe CE , Clarke RM , Lynch MA ((2007) ) IL-4 attenuates the neuroinflammation induced by amyloid-beta in vivo and in vitro. J Neurochem 101: , 771–81. |
[51] | Wang J , Song Y , Chen Z , Leng SX ((2018) ) Connection between systemic inflammation and neuroinflammation underlies neuroprotective mechanism of several phytochemicals in neurodegenerative diseases. Oxid Med Cell Longev 2018: , 1972714. |
[52] | Shytle RD , Tan J , Bickford PC , Rezai-Zadeh K , Hou L , Zeng J , Sanberg P , Alberte RS , Fink RC , Roschek B Jr ((2012) ) Optimized turmeric extract reduces β-amyloid and phosphorylated tau protein burden in Alzheimer’s transgenic mice. Curr Alzheimer Res 9: , 500–506. |
[53] | Rahardjo B , Widjajanto E , Sujuti H , Keman K ((2014) ) Curcumin decreased level of proinflammatory cytokines in monocyte cultures exposed to preeclamptic plasma by affecting the transcription factors NF-κB and PPAR-γ. Biomark Genom Med 6: , 105–115. |
[54] | Yang F , Lim GP , Begum AN , Ubeda OJ , Simmons MR , Ambegoakar SS , Chen PP , Kayed R , Glabe CG , Frautschy SA , Cole GM ((2004) ) Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem 280: , 5892–5901. |
[55] | Morris MC , Tangney CC , Wang Y , Sacks FM , Barnes LL , Bennett DA , Aggarwal NT ((2015) ) MIND diet slows cognitive decline with aging. Alzheimers Dement 11: , 1015–1022. |
[56] | Morris MC , Tangney CC , Wang Y , Sacks FM , Bennett DA , Aggarwal NT ((2014) ) MIND diet associated with reduced incidence of Alzheimer’s disease. Alzheimers Dement 11: , 1007–1014. |
[57] | Alzheimer’s Association (2019) Adopt a Healthy Diet. https://www.alz.org/help-support/brain_health/adopt_a_he-althy_diet |
[58] | Luchsinger JA , Reitz C , Honig LS , Tang MX , Shea S , Mayeux R ((2005) ) Aggregation of vascular risk factors and risk of incident Alzheimer disease. Neurology 65: , 545–551. |
[59] | Musiek ES , Schindler SE ((2013) ) Alzheimer disease: current concepts & future directions. Mo Med 110: , 395–400. |
[60] | Baig MH , Ahmad K , Rabbani G , Choi I ((2018) ) Use of peptides for the management of Alzheimer’s disease: diagnosis and inhibition. Front Aging Neurosci 10: , 21. |
[61] | Nilsson P , Iwata N , Muramatsu S , Tjernberg LO , Winblad B , Saido TC ((2010) ) Gene therapy in Alzheimer’s disease - potential for disease modification. J Cell Mol Med 14: , 741–757. |
[62] | Vauhn P , Cire B , Benowitz S ((2014) ) NIH funds next step of cutting-edge research into Alzheimer’s disease genome. https://www.nih.gov/news-events/news-releases/nih-funds-next-step-cutting-edge-research-into-alzheimers-disease-genome |


