
The Bri2 and Bri3 BRICHOS Domains Interact Differently with Aβ42 and Alzheimer Amyloid Plaques
Abstract
Alzheimer’s disease (AD) is the most common form of dementia and there is no successful treatment available. Evidence suggests that fibril formation of the amyloid β-peptide (Aβ) is a major underlying cause of AD, and treatment strategies that reduce the toxic effects of Aβ amyloid are sought for. The BRICHOS domain is found in several proteins, including Bri2 (also called integral membrane protein 2B (ITM2B)), mutants of which are associated with amyloid and neurodegeneration, and Bri3 (ITM2C). We have used mouse hippocampal neurons and brain tissues from mice and humans and show Bri3 deposits dispersed on AD plaques. In contrast to what has been shown for Bri2, Bri3 immunoreactivity is decreased in AD brain homogenates compared to controls. Both Bri2 and Bri3 BRICHOS domains interact with Aβ40 and Aβ42 present in neurons and reduce Aβ42 amyloid fibril formation in vitro, but Bri3 BRICHOS is less efficient. These results indicate that Bri2 and Bri3 BRICHOS have different roles in relation to Aβ aggregation.
INTRODUCTION
Alzheimer’s disease (AD) is one out of about 40 known diseases in which specific proteins accumulate into insoluble amyloid deposits [1], and it is the most common form of dementia, affecting an increasing number of people worldwide [2]. AD is characterized pathologically by the accumulation of misfolded amyloid β-peptide (Aβ) in plaques, and of hyperphosphorylated tau in neurofibrillary tangles (NFTs), but also neuronal loss, neuroinflammation and brain atrophy is observed [3, 4]. According to the amyloid cascade hypothesis, Aβ generation, oligomerization and fibrillation are the main causes of AD [3]. Thus, many treatment strategies aim to halt Aβ aggregation and in particular the generation of Aβ oligomers [5]. The amyloid-β protein precursor (AβPP) is a type I transmembrane (TM) protein that undergoes posttranslational modifications and proteolytic cleavages by α-, β-, and γ-secretases. Cleavage by β-secretase, or β-site AβPP-cleaving enzyme 1 (BACE1), and further processing by γ-secretase is referred to as the “amyloidogenic” pathway, which gives rise to Aβ peptides that can range from 38–43 amino acids in length. Mainly the highly aggregation prone Aβ42, but also Aβ40, are found in amyloid plaques in AD post mortem brain [4, 6, 7].
The “anti-amyloid” chaperone domain, BRICHOS, is found in several distantly related protein families, among them the dementia-associated Bri2 [8, 9]. The BRICHOS domain of Bri2 on its own inhibits Aβ40 and Aβ42 fibril formation in vitro [9, 10] by affecting both the secondary nucleation and elongation steps of Aβ aggregation, thereby supposedly decreasing the amount of toxic oligomers formed [11]. Bri2 BRICHOS furthermore reduces Aβ42 toxicity in vivo in a Drosophila model of AD and ex vivo in mouse hippocampal slices [12].
Human Bri2 is ubiquitously expressed, and in the brain, it is particularly abundant in the CA1 pyramidal neurons [13, 14]. Figure 1 shows a schematic representation of the different parts of Bri2 and Bri3. Bri2 is processed in several steps; furin or furin-like proteases release a 23 amino acid residue C-terminal peptide (Bri23) generating the membrane bound mature Bri2 (mBri2), which can be further processed by ADAM10, releasing the BRICHOS domain, and finally the signal peptide peptidase-like 2b (SPPL2b) cleaves mBri2 intramembranously [15, 16]. Mutations in Bri2 give rise to release of extended 34-residue C-terminal peptides, ABri or ADan, which are deposited in the CNS in familial British dementia (FBD) and familial Danish dementia (FDD) patients, respectively [17, 18]. Furthermore, Bri2 has been shown to interact with AβPP, thereby decreasing the secretion of AβPP processing products, including Aβ40 and Aβ42 in both cell and animal models. It has been suggested that Bri2-AβPP binding blocks the secretase cleavage sites and/or interferes with BACE1, thereby affecting the AβPP processing [19–22]. Increased levels of Bri2 and altered levels of its processing enzymes have been detected in postmortem hippocampus of AD cases, and Bri2 was found to be associated with amyloid plaques [23]. Recently, it was shown that Bri2 affects neurite outgrowth and that this effect is regulated by phosphorylation [24].
Fig.1
Bri2 and Bri3 architecture and antibody reactivity. A) SDS-PAGE and Coomassie staining of recombinant Bri2 BRICHOS (residues 113–231) (left) and Bri3 BRICHOS (residues 112–230) (right) domains. The lines to the left show migration of size markers with masses in kDa. B) Bri2 and Bri3 are characterized by a similar architecture: an intracellular domain (IC) (yellow), a transmembrane (TM) region (green), a linker region (light blue), the BRICHOS domain (red) and a C-terminal segment (Bri23 or CT, blue). The sequence positions of the different domains in Bri2 and Bri3, respectively, are indicated. Antibodies, used in this study, were raised against the indicated segments of Bri2 and Bri3. C) Western blots of recombinant Bri2 and Bri3 BRICHOS proteins (corresponding to residues 113–231 of Bri2, residues 90–236 of Bri2 and residues 112–230 of Bri3) using the anti-Bri2 linker; the anti-Bri2 BRICHOS; and the anti-Bri3 BRICHOS antibodies. The upper band for Bri2 BRICHOS (113-231) corresponds to a dimer. Size markers (kDa) are shown to the right of the blots.
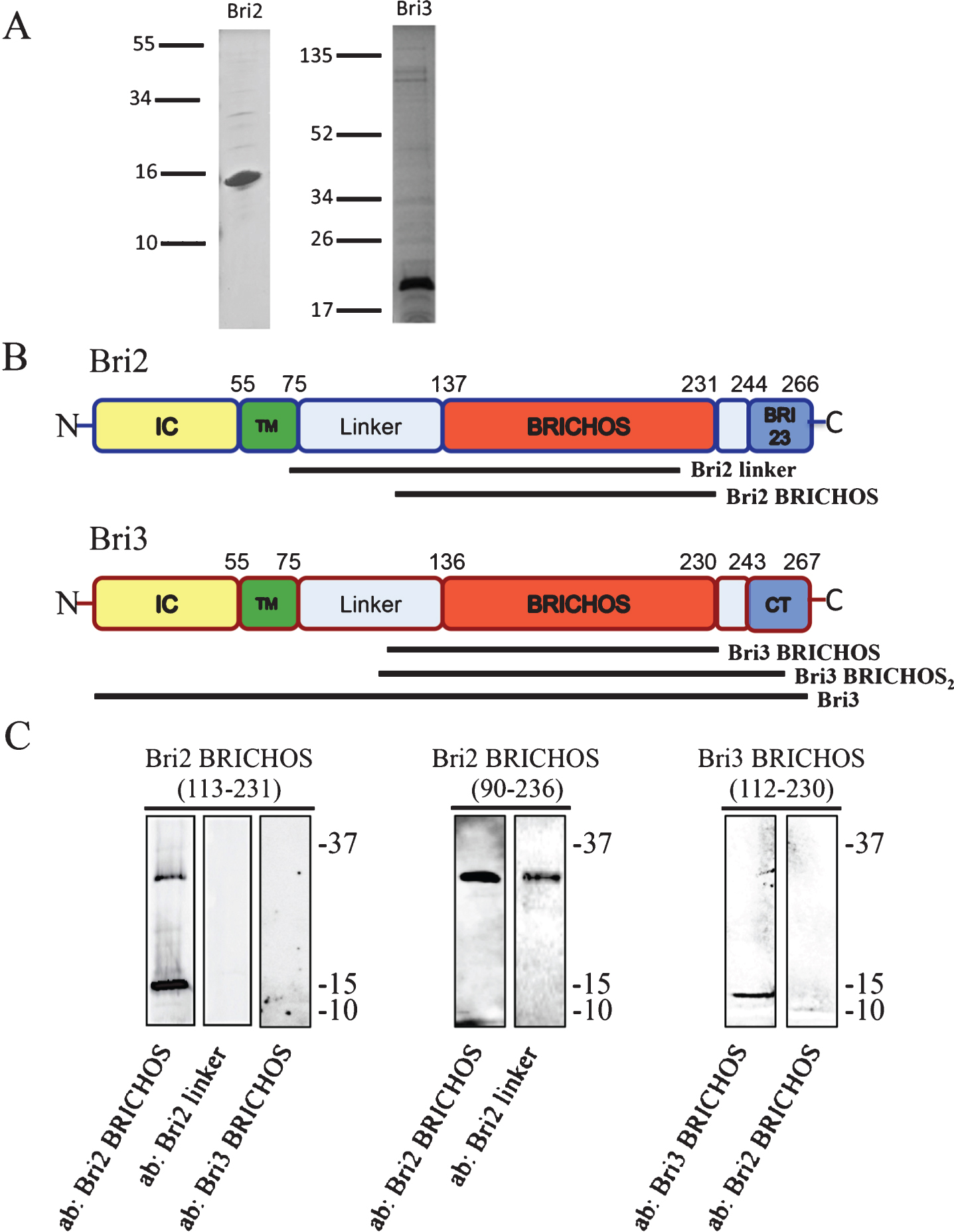
Bri3 contains a BRICHOS domain and is expressed exclusively in the CNS, with high levels in the hippocampus and cerebral cortex [25], but it has so far been less studied than Bri2. Using transfected cell lines, it has been shown that Bri3 is processed by furin, releasing a 25 amino acids long C-terminal peptide [26] and that Bri3, like Bri2, can be phosphorylated [24]. Bri3 has been concluded to bind AβPP, thereby downregulating Aβ40 and Aβ42 secretion by similar mechanisms as those proposed for Bri2 [25–27]. However, whether the BRICHOS domain of Bri3 affects Aβ42 aggregation and how it interacts with Aβ and AβPP remain unknown.
In this study, we compared the interactions of the BRICHOS domains of Bri2 and Bri3 with Aβ and AβPP in mouse primary neurons as well as in brain tissue from mice and from AD cases and controls. We observed that Bri3 levels are decreased in AD brain, and found that Bri2 and Bri3 BRICHOS domains interact with neuronal Aβ40 and Aβ42.
METHODS
Postmortem brain tissue
For western blot analysis, human postmortem hippocampal and temporal brain tissue from non-demented controls (hippocampus: n = 7; cortex: n = 8) and AD cases (hippocampus: n = 8; cortex: n = 5) was obtained from the Netherlands Brain Bank (Amsterdam, The Netherlands). Groups were matched for age and sex. Pathological diagnosis and neuropathological evaluation was performed on formalin-fixed, paraffin-embedded tissue from different brain areas as previously described [23]. Staging of AD pathology was evaluated according to modified assessment of Braak and Alafuzoff [28]. Clinical and pathological diagnosis, sex, age, postmortem delay, brain area, and the Braak scores for NFTs and amyloid load of all analyzed cases (n = 16) are listed in Supplementary Table 1. Of the total 16 cases, hippocampal and temporal cortical areas were available for 12 samples, only hippocampal areas for 3 samples and only temporal cortical areas for 1 sample. All donors or their next of kin provided written informed consent for brain autopsy and use of tissue and medical records for research purposes.
For immunohistochemical analysis human postmortem brain tissue samples from frontal cortex were obtained from the Brain Bank at Karolinska Institutet. Patients with sporadic AD, early onset familial AD caused by the Swedish AβPP double-mutation KM670/671NL (AβPPswe), patients with the Arctic AβPP mutation E693G (AβPParc) and brain tissue from control subjects without neurodegenerative disease were included (n = 2/group).
Recombinant proteins
Human Bri2 and Bri3 BRICHOS domains, corresponding to amino acid residues Bri2(90-236 or 113-231) (NP_068839.1) and Bri3(112-230) (NP_001274170.1) as fusion proteins with an N-terminal tag of Thioredoxin, 6 x Histidine, and S-tag, were expressed at 30°C in E. coli Shuffle T7 cells. Cells were grown in LB medium containing 100 μg/ml ampicillin. Expression was induced at an OD600 = 0.6–0.8 by adding 0.4 mM IPTG and the cells were then grown for another 4 h. Cells were harvested and suspended in 0.15 M NaCl, 20 mM sodium phosphate buffer (PBS), pH 7.4, frozen at –20°C, and lysed by sonication for 2 min and centrifuged at 6000xg for 20 min, 4°C. The pellet was suspended in 2 M urea in PBS pH 7.4, sonicated for 2 min, and centrifuged at 15000xg for 30 min, 4°C. The supernatant was filtered through a 5 μm filter (CA membrane, Sartorius stedim biotech) and kept at –20°C. After thawing, the sample was poured onto a 2.5 ml IMAC column (Ni Sepharose™ 6 Fast Flow, GE Healthcare) equilibrated with 2 M urea in PBS, pH 7.4. The column was washed with 2 M urea in PBS, followed by 1 M urea, and finally with 50 mM imidazole in PBS, pH 7.4. Target fusion protein was eluted with 300 mM imidazole in PBS, pH 7.4 and dialysed (regenerated cellulose RC, 6–8 kDa membrane; Spectrum Lab) against PBS, pH 7.4 containing thrombin (Merck) (enzyme/substrate weight ratio of 0.002) for 16 h at 4°C, re-applied onto the IMAC column, and BRICHOS domains were eluted with PBS, pH 7.4. See Fig. 1A for SDS-PAGE analyses of purified Bri2 and Bri3 BRICHOS; using 12–13.5% polyacrylamide gels, the samples were run under reducing conditions and proteins were visualized with Coomassie blue staining. Recombinant Bri2 BRICHOS(90–236) migrates on SDS-PAGE corresponding to a mass of about 30 kDa, while the mass determined by electrospsray ionization mass spectrometry is 20397±2 Da, in good agreement with the mass calculated from the amino acid sequence (20400 Da) [10]. This rules out the possibility that the aberrant migration on SDS-PAGE can be explained by mutations, N-terminal truncation or other inadvertent covalent modifications.
Recombinant human Aβ42 peptide with an N-terminal methionine was expressed and purified as described elsewhere [29]. Aβ42 peptide was expressed in E. coli BL21 and purified in batch format as described before [30], using ion exchange and a Superdex 75 10/300 GL size exclusion column (GE Healthcare Life Sciences) to ensure a monomeric state of the starting material. The extinction coefficient for Aβ42 at 280 nm is 1400 M–1cm–1.
Thioflavin T (ThT) aggregation assay
Fibril formation kinetics was studied by recording the ThT fluorescence over time in a plate reader (FLUOStar Galaxy from BMG Labtech, Offenberg, Germany). The fluorescence was measured using bottom optics in half-area 96-well polyethylene glycol-coated black polystyrene plates with clear bottom (Corning Glass, 3881) using a 440 nm excitation filter and a 490 nm emission filter. 3 μM of Aβ42 solution, with or without 0.6 or 1.5 μM of Bri2 or Bri3 BRICHOS protein, was supplemented with 10 μM ThT, 80 μl was added to each well, and the plate was sealed and immediately placed in the fluorescence reader at room temperature (RT), and incubated under quiescent conditions with readings made every 5 min.
Cell culture
Brains from embryonic day 16-17 C57BL6 mouse embryos were used to dissect out hippocampal and cortex tissue, to which neurobasal medium with 2% B27 and 1% L-glutamine was added and single cells were separated by pipetting. FACS analysis show that the cell population consists of about 82% neurons (TUJ-1 staining), 7% astrocytes (GFAP staining), and the remaining cells are microglia and oligodendrocytes [31]. Cells were seeded in chamber slides (NuncTM), cultured for 6-7 days and analyzed by proximity ligation assay (PLA) and confocal microscopy.
Antibodies
See Supplementary Table 2 for an overview of antibodies used in this study. For confocal microscopy of cells, dilutions, primary antibodies (and antigens) were (see Fig. 1B and C for Bri2 and Bri3 structures and antibody reactivities): 1:5000 rabbit anti-Bri3 BRICHOS (Bri3 residues 112–230, Capra Science); 1:200 of a second rabbit anti-Bri3 antibody, referred to as anti-Bri3 BRICHOS2 (Bri3 residue 110–258, Abcam Cat# ab101389, RRID:AB_10710084); 1:100 mouse anti-Bri3 (Bri3 residues 1–267, Sigma-Aldrich Cat# SAB1408047, RRID:AB_10745686); 1:40 rabbit anti-Bri2 linker (Bri2 residues 74–224, Sigma-Aldrich Cat# HPA029292, RRID:AB_10601917); 1:1000 goat anti-Bri2 BRICHOS (Bri2 residues 113–231, Capra Science); 1:1000 mouse anti-AβPP/Aβ, 4G8 (BioLegend Cat# 800701, RRID:AB_2564633); 1:200 mouse anti-Aβ42 (Millipore Cat# MABN12, RRID:AB_10562244); 1:200 of a second anti-Aβ42 antibody (rabbit), referred to as anti-Aβ42-2 (Thermo Fisher Scientific Cat# 44–344, RRID:AB_2313572); 1:300 mouse anti-Aβ40 (BioLegend Cat# 805401, RRID:AB_2564680); 1:400 mouse anti-N-terminal AβPP, 22C11 (Millipore Cat# MAB348-100UL, RRID:AB_2056583). Secondary antibodies for PLA were PLUS or MINUS oligonucleotide-conjugated (Duolink, Olink Bioscience, Sigma-Aldrich), chosen for appropriate species of origin of target antibodies, excitation and emission wavelengths (see below). For western blot, primary antibodies and dilutions used were: 1:2000 rabbit anti-Bri3 BRICHOS, 1:1000 rabbit anti-Bri3 BRICHOS2 ; 1:2000 goat anti-Bri2 BRICHOS, and 1:500 rabbit anti-Bri2 linker. Secondary antibodies conjugated with horseradish peroxidase (HRP), (anti-rabbit, GE Healthcare) (anti-goat, Life Technologies) were diluted 1:5000. In PLA experiments of transgenic mouse tissue: primary antibodies 1:1000 mouse anti-AβPP/Aβ, 6E10 (Covance Research Products Inc. Cat# SIG-39320-1000, RRID:AB_10175145); 1:1000 goat anti-Bri2 BRICHOS; 1:200 rabbit anti-Bri3 BRICHOS2. For immunohistochemistry of human brain tissue: rabbit anti-Bri3 BRICHOS and mouse anti-AβPP/Aβ (6E10) primary antibodies were diluted 1:500 and 1:1000, respectively.
SDS-PAGE and western blotting
Human postmortem tissue was homogenized using Tissue Protein Extraction Reagent (T-PER, 0.1 g/mL, Thermo Scientific, Waltham, USA) containing EDTA-free protease and phosphatase inhibitor cocktails (1:25 and 1:10 respectively, Roche, Basel, Germany). Samples were centrifuged at 10,000 g for 15 min at 4°C. Protein content in the supernatant was quantified using Bio-Rad protein assay (Bio-rad, Hercules, USA). Human postmortem homogenates (15 μg) were prepared in samples buffer (2% SDS, 0.03 M Tris, 5% 2-Mercaptoethanol, 10% glycerol, bromophenol blue) and heated 5 min at 95°C. Electrophoresis of postmortem samples was carried out using pre-cast NuPAGE Bis-Tris Mini Gels 4–12% (1.5 mm, 4–12%; Invitrogen, Carlsbad, USA). After blotting, the membranes were blocked in 5% milk/PBS for 1 h, followed by incubation with primary antibody in 5% milk, 0.1% Tween/PBS for 1 h at RT. The membrane was washed three times in 0.1% Tween/PBS and secondary antibodies in 5% milk, 0.1% Tween/PBS were added for 1 h at RT. After washing, enhanced chemoluminescence detection reagent (GE Healthcare) was added according to the manufacturer protocol and images were acquired using a CCD camera (LAS-3000).
Transgenic mice
Generation of transgenic mice expressing human AβPP with the Arctic mutation (E693G) has previously been described [32, 33]. Briefly, the TgAβPParc mice were generated on a mixed CBA/C57BL/6 background by microinjection of human AβPP (695 isoform) gene with the Arctic mutation under control of the Thy1.2 promoter. Mice were sacrificed by cervical dislocation and the brains were immediately removed and hemispheres were fresh-frozen on dry ice and stored at –80°C. Brain tissue from AβPP–/– mice was kindly provided by Dr Hui Zheng, Baylor College of Medicine [34].
PLA
PLA was performed according to Duolink II kit manufacturer’s instructions (OlinkBioscience). Briefly, cells were grown on chamber slides (NuncTM), fixed in 4% paraformaldehyde/PBS (Santa Cruz Biotech) solution for 15 min at RT, washed twice in PBS and permeabilized with 0.2% Triton X-100 in PBS for 15 min on ice, then blocked with Olink block for 30 min at 37°C. Primary antibodies were added and incubated over night at 4°C, and after washing secondary complementary PLUS and MINUS oligonucleotide-conjugated antibodies against one or two species, depending on the type of analysis, were added for 1 h at 37°C. Thereafter, ligation, amplification and washing steps were performed according to the manufacturer’s protocol. For detecting protein-protein interactions, two primary antibodies from different species (rabbit, goat or mouse) were used, and the secondary antibodies against those two species were added for detection. For detection of single proteins, one primary antibody and the respective PLUS and MINUS complementary secondary antibodies were used. Alexa Flour® Phallodin-488 nm were used for staining of actin.
Brain tissue from 9 months old homozygous transgenic mice with AβPP Arctic mutation (TgAβPParc), and from AβPP knock-out mice (AβPP–/–), was washed in PBS, fixed in 4% paraformaldehyde/PBS for 5 min and permeabilized with 0.2% Triton X-100/PBS for 15 min on ice, and blocked with Rodent Block M (biocare medical) for 30 min at RT. Primary antibodies were added and incubated at 37° C for 1 h. Cell nuclei were stained with DAPI (OlinkBioscience), mounted and analyzed by a Zeiss confocal microscope (Axiovert 200M). The far-red fluorophore for the PLA signals was excited at 633 nm and emission was detected above 650 nm. The DAPI was excited at 405 nm and emission was read at 420–480 nm. The 488 nm flourophore was excited at 488 nm and emission was read at 505–550 nm. Images were acquired at one confocal plane at 40X magnification.
Quantification of fluorescence intensity
Fluorescence quantification was done by first splitting the fluorescence signals from PLA, DAPI, and phalloidin. Measurements were then made for the PLA signal by integrating the fluorescence densities of manually encircled cell areas using Image J. Then the corrected total cell fluorescence was determined by subtracting the area-corrected mean background fluorescence (measured from the PLA signals from an area outside the cells) from the integrated cellular PLA fluorescence densities. The same procedure was repeated for the control cells, which were treated by adding only one of the primary antibodies plus both the PLUS and MINUS secondary antibodies. Statistical analysis was based on comparisons with student’s t-test between at least three positive cells and controls. Variability was reported as standard deviation (S.D).
Immunohistochemistry on human postmortem brain tissue
Immunohistochemical staining for detection of Bri3 and Aβ were performed on 5 μm thin sections from formalin fixed paraffin embedded tissue. Brain sections were de-paraffinized in xylene and rehydrated through graded alcohol series from 99% to 70%. Sections were autoclaved in a Decloaking Chamber (Biocare Medical) in DIVA decloaker solution (Biocare Medical, Concord, USA) for 30 min. Sections were washed with Tris-buffered saline containing 0.05% tween 20 (TBST), and incubated first with peroxidase blocking solution (Dako) for 5 min, washed in TBST and then incubated for 10 min with Background punisher (Dako). Double stained slides were incubated for 45 min at RT with an antibody cocktail containing: mouse anti-AβPP/Aβ (6E10, 1:1000) and rabbit anti-Bri3 BRICHOS (1:500) diluted in Dako antibody diluent (Dako). Negative control slides were incubated with the Dako antibody diluent only. Slides were washed in TBST and incubated for 30 min at RT with Mach 2 Double Stain1 containing conjugated secondary anti-mouse HRP and anti-rabbit-AP antibody (Biocare Medical). Horseradish peroxidase staining was visualized with the permanent green (Biosite) and AP staining was visualized with permanent red (Biosite) solutions. Slides were counterstained in hematoxylin, dehydrated through graded alcohols, cleared in xylene and then mounted with Permount. Images were acquired at 40X magnification.
Ethical approvals
All procedures performed in studies involving human or animal tissue material were in accordance with the ethical standards of the institutional and/or national research committees. Swedish ethical research committees diary numbers: 2011/962-31/1; 2013/118-31/2; S151-10; S75-13; S53-14. VU Medical centrum, The Netherlands: 2009/148.
RESULTS
Bri3 levels and amyloid plaque interactions in AD patients and healthy controls
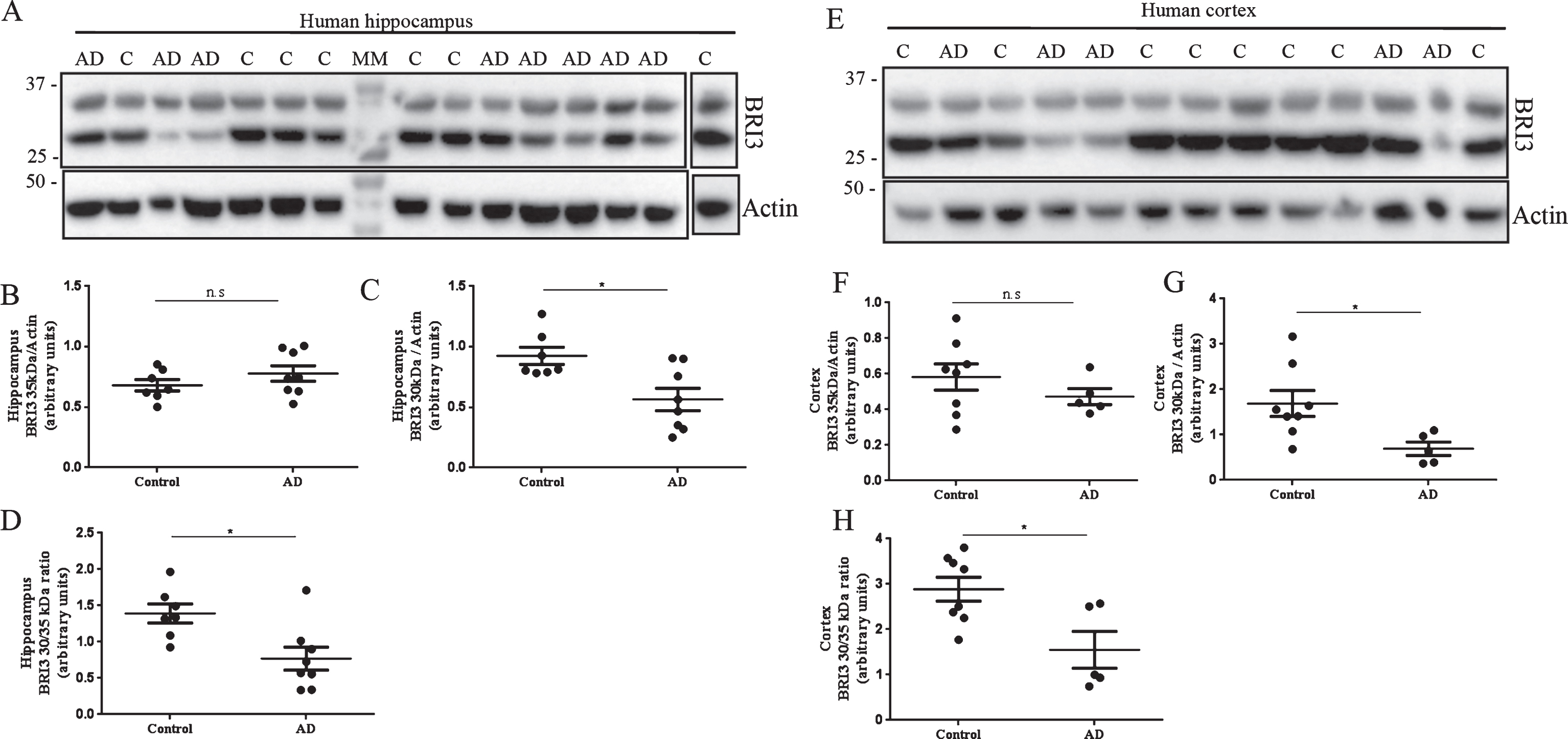
Western blot analysis of postmortem hippocampal and temporal cortex homogenates from AD cases and non-demented controls (Fig. 2) revealed two different bands at 35 and 30 kDa. It has been shown that full-length and processed forms of Bri2 and Bri3 migrate slower on SDS-PAGE than expected from their masses [15, 23, 26]. The identities of the 35 and 30 kDa bands in Fig. 2 are thus difficult to assign with certainty, but they likely represent full-length Bri3 and the furin-processed Bri3 lacking the 4 kDa C-terminal peptide, respectively [26]. Data analysis revealed no changes in the levels of 35 kDa Bri3 between controls and AD cases (Fig. 2A, B, E, F). However 30 kDa Bri3 immunoreactivity was specifically decreased in both hippocampus and temporal cortex in AD cases compared to controls (Fig. 2A, C, E, G). The 30 to 35 kDa band intensity ratio was also decreased in AD compared to controls in both brain areas (Fig. 2 D, H).
Fig.2
Bri3 is decreased in postmortem hippocampus and temporal cortex of AD cases. Western blots analysis using the Bri3 BRICHOS2 antibody against postmortem human hippocampus (A-D) and temporal cortex (E-H) from non-demented controls (hippocampus: n = 7; cortex: n = 8) and AD cases (hippocampus: n = 8; cortex: n = 5). Bri3 BRICHOS2 reactivity was quantified for the 35 kDa (B,F) and 30 kDa (C,G) bands and normalized to actin levels. The 30 to 35 kDa ratio was calculated for each sample and compared between groups (D,H). ns, not significant; *p < 0.05.
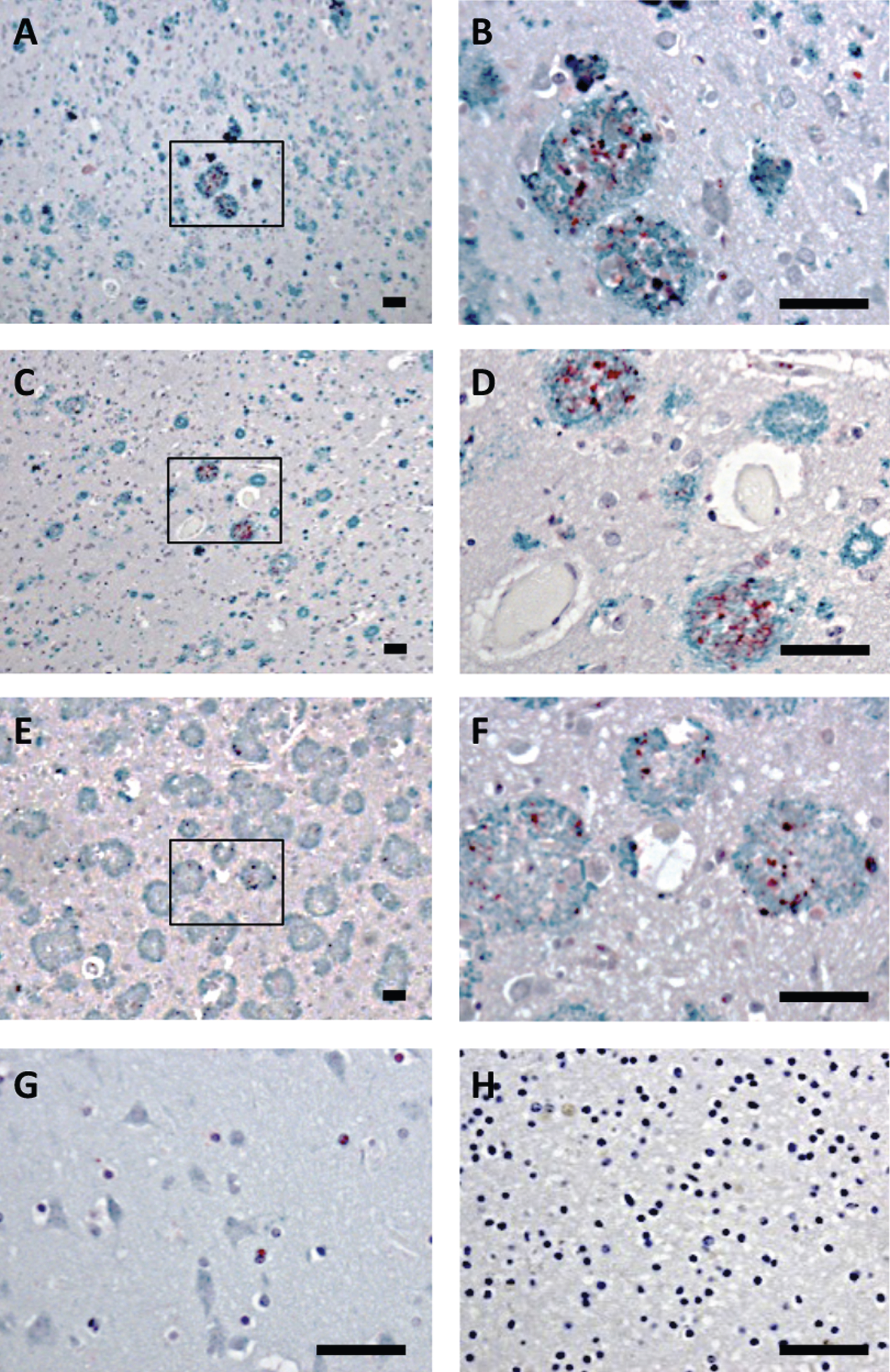
Brain tissue from the frontal cortex of patients with sporadic AD or AD due to AβPPswe or AβPParc mutations, as well as non-demented controls were analyzed by double-immunohistochemistry, using antibodies against Bri3 BRICHOS and Aβ. All individuals with AD showed plaques that were immunoreactive with the Aβ antibody, while in the control tissue no plaques were found (Fig. 3A-H and Supplementary Figure 1). In addition, the AD patients showed Bri3 immunoreactivity in a deposit-like pattern which co-localized with the Aβ plaques, but not every plaque was stained for Bri3 and only parts of the plaques showed Bri3 deposition. The reasons for the Bri3 staining pattern remain to be determined. No obvious differences could be detected in the co-localization pattern between the sporadic and familial cases of AD (Fig. 3A-F). Bri3 immunoreactivity in the control tissue showed diffuse staining in some cell somata (Fig. 3G). Control experiments in which only secondary antibodies were added to the human tissue sections showed no staining above background (Fig. 3H and Supplementary Figure 1G, H).
Fig.3
Bri3 co-localizes with AD amyloid plaques. Human brain sections stained with anti-Aβ/AβPP (6E10, green) and rabbit anti-Bri3 BRICHOS (red) antibodies from (A, B) sporadic AD case; (C, D) AD case with AβPPswe mutation; (E, F) AD case with AβPParc mutation; (G) healthy control; and (H) sporadic AD case stained using only the secondary antibody. The rectangles in A, C, and E indicate the areas shown in B, D, and F, respectively. All samples were counterstained with hematoxylin. Scale bars 50 μm. Images are representative of three independent experiments.
The finding that Bri3 co-localizes with AD plaques is analogous to what previously has been observed for Bri2 [23]. The Bri3 levels in AD, however, are changed in the opposite direction to what previously has been reported for Bri2 [23], suggesting significant differences in the ways the two BRICHOS proteins interact with Aβ in AD.
Bri3 BRICHOS inhibits Aβ42 fibril formation in vitro
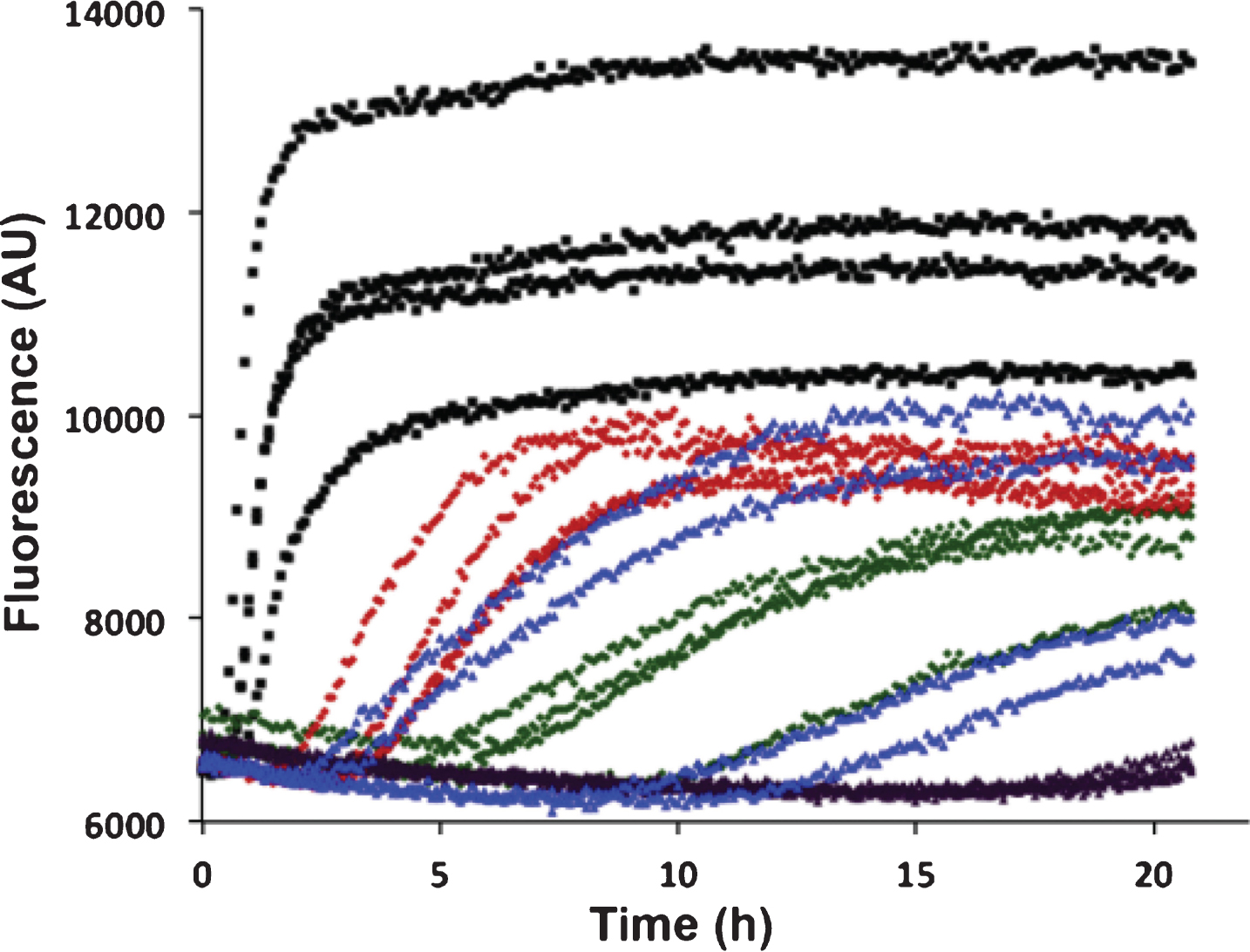
Aβ42 fibril formation alone or with recombinant BRICHOS domains of Bri2 and Bri3 was studied in vitro using ThT flurorescence as a reporter. Using 0.2 or 0.5 molar equivalents of isolated recombinant human Bri3 BRICHOS compared to Aβ42, a concentration dependent increase of Aβ42 aggregation half time (t1/2) was observed, as a result of prolonged lag phase and decreased fibrillation rate (Fig. 4). For 0.2 equivalents of Bri3 BRICHOS the median t1/2 was prolonged 3 times and for 0.5 equivalents it was prolonged about 6 times. The Aβ42 t1/2 in the presence of 0.2 equivalents of Bri2 BRICHOS varies, probably as a result of that small changes in BRICHOS concentrations give marked changes in Aβ kinetics around this ratio, but it is prolonged at least 5 times, and in the presence of 0.5 equivalents of Bri2 BRICHOS it is prolonged >10 times (Fig. 4). This shows that Bri3 BRICHOS interferes with Aβ42 fibril formation in vitro, but the Aβ42 aggregation half time is prolonged less in the presence of Bri3 BRICHOS compared to Bri2 BRICHOS.
Fig.4
In vitro effects of Bri2 and Bri3 BRICHOS on fibril formation of Aβ42. ThT fluorescence traces of 3 μM Aβ42 alone (black), and with addition of 0.2 molar equivalents of Bri3 BRICHOS (red), 0.5 molar equivalents of Bri3 BRICHOS (green), 0.2 molar equivalents of Bri2 BRICHOS (blue), or 0.5 molar equivalents of Bri2 BRICHOS (purple). The traces for each of four replicates for all samples are shown. The results are representative of at least five independent experiments.
Interactions between endogenous BRICHOS and Aβ in hippocampal neurons
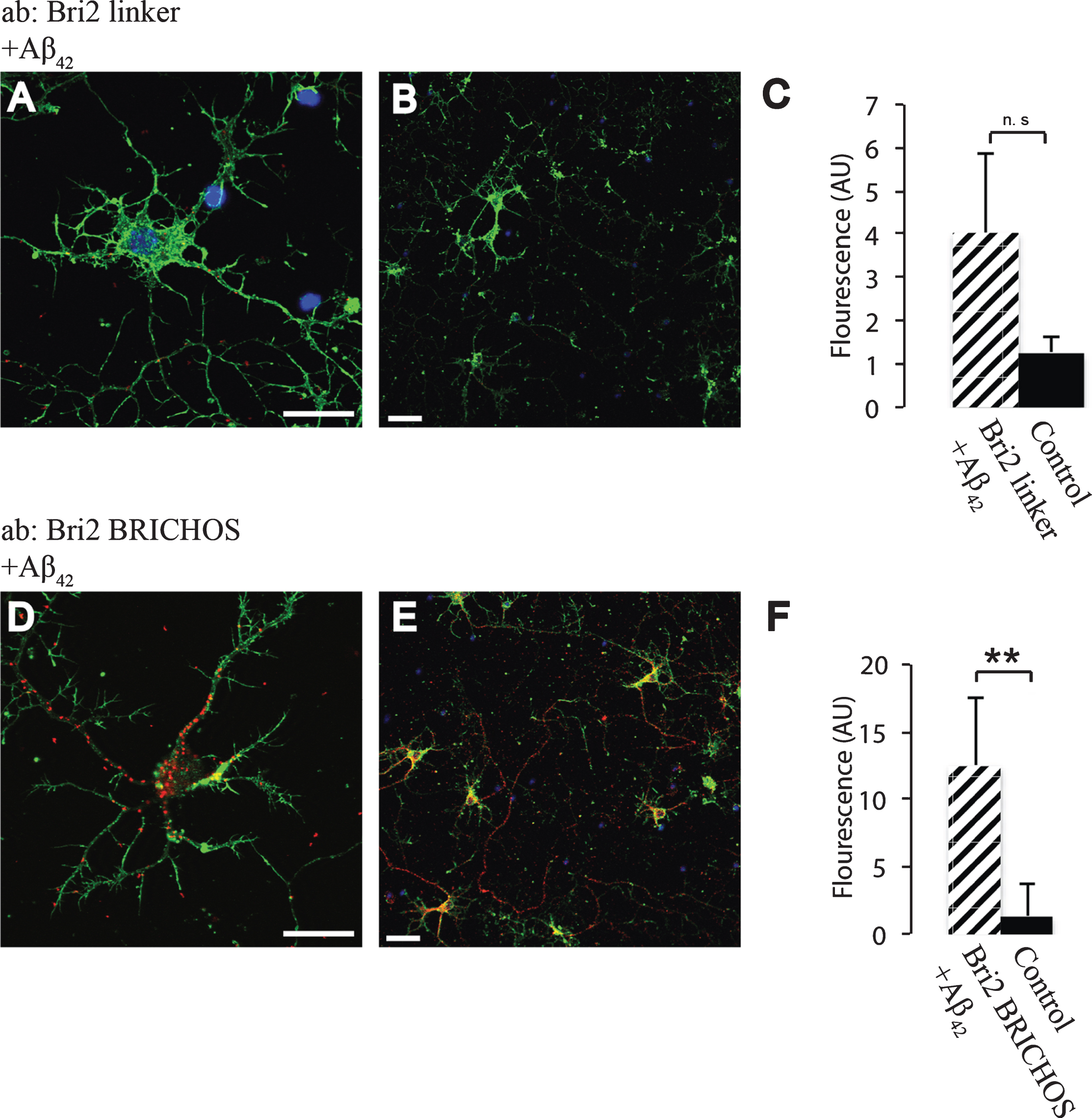
In order to study whether the BRICHOS domains of Bri2 and Bri3 interact with Aβ under physiological conditions, primary wt mouse hippocampal neurons were analyzed using PLA. To determine if the Bri2 BRICHOS domain as such interacts with Aβ42 we used two different Bri2 antibodies (Fig. 1B), one (the Bri2 linker antibody) that recognizes the linker region of Bri2, but not the BRICHOS domain, and another antibody (the Bri2 BRICHOS antibody) that recognizes the Bri2 BRICHOS domain (Fig. 1C). A significant number of signals indicative of protein-protein interactions were detected using the Aβ42 antibody (which specifically recognizes Aβ42) in combination with the Bri2 BRICHOS antibody, but when using the Bri2 linker antibody, no interactions with Aβ42 above control levels could be detected (Fig. 5). Abundant interactions with Bri2 BRICHOS were also seen using a different Aβ42 specific antibody (Aβ42-2) (Supplementary Figure 2). This indicates that interactions between Aβ42 and Bri2 BRICHOS require that the BRICHOS domain has been proteolytically separated from the linker region, and these interactions are most likely mediated by the BRICHOS domain as such.
Fig.5
Interactions between Aβ42 and Bri2 BRICHOS. PLA signals (red) for protein interactions with (A,B) anti-Bri2 linker and anti-Aβ42 antibodies; (D,E) anti-Bri2 BRICHOS and anti-Aβ42 antibodies. Staining of nuclei is performed with DAPI (blue) and filamentous actin is stained with phalloidin (green). Scale bar 20 μm. (C,F) Fluorescence intensity quantification for negative controls (black bars) and Bri2 linker plus Aβ42 (hatched bar), n = 4 (C), and negative controls and Bri2 BRICHOS plus Aβ42 (hatched bar), n = 3 (D). ns, p > 0.05 and.**p < 0.01. Average values and errors represent standard deviations. The results are representative of at least three independent experiments.
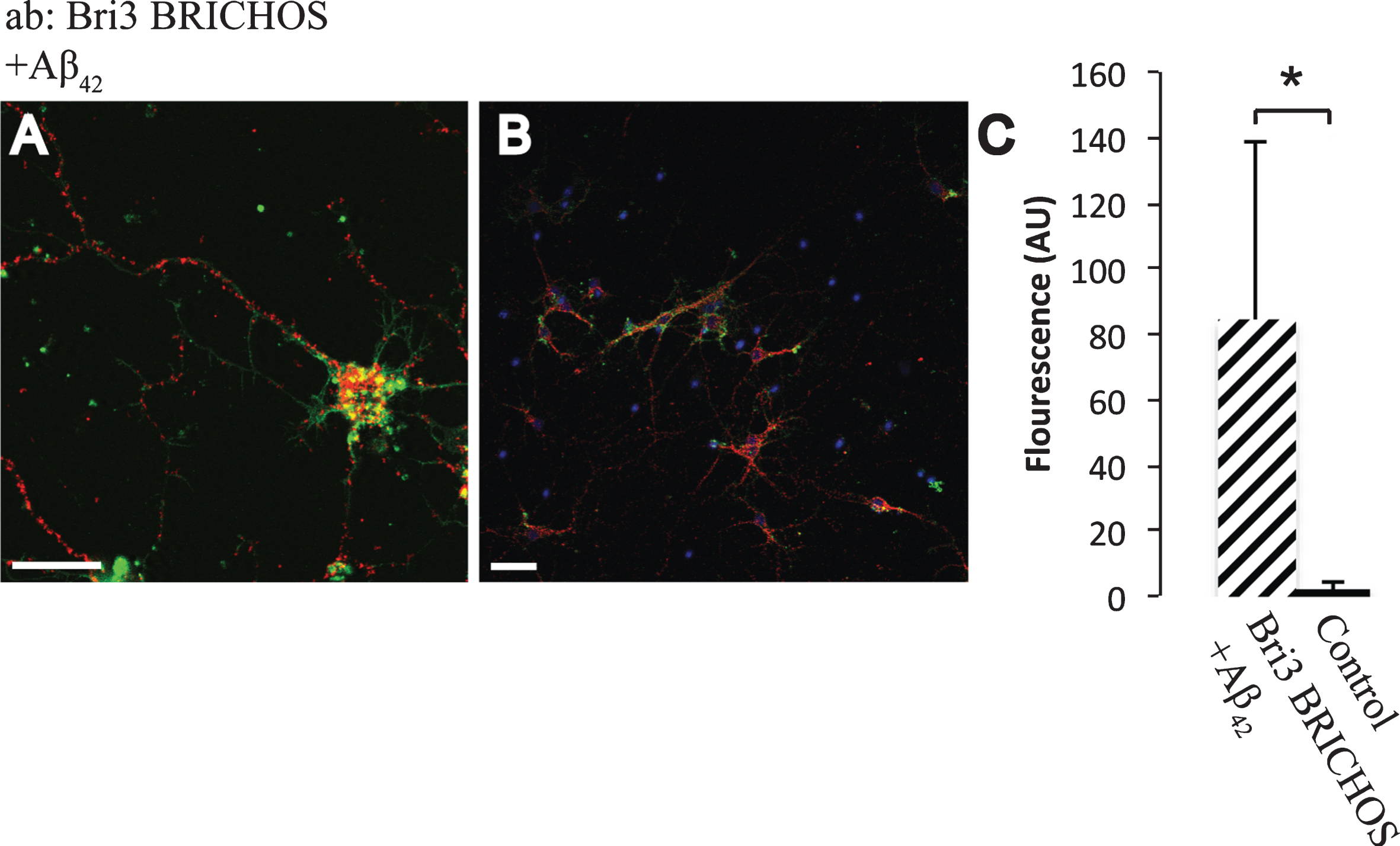
Using an antibody raised against recombinant Bri3 BRICHOS, abundant interactions with Aβ42 were detected by PLA and a significantly higher number of interaction signals were detected compared to in the negative control (Fig. 6). Similar results were obtained using a different Aβ42 specific antibody (Aβ42-2) and another Bri3 antibody (raised against Bri3 residues 1–267) (Supplementary Figure 2). Furthermore, significant interactions between both Bri2 and Bri3 BRICHOS and Aβ40 were detected using an Aβ40 specific antibody (Supplementary Figure 3). Due to lack of primary antibodies that are specific for the Bri3 linker region, it was not possible to investigate whether the linker region needs to be removed to allow Bri3 BRICHOS to interact with Aβ42.
Fig.6
Interactions between Aβ42 and Bri3. A,B) PLA signals (red) for protein interactions with anti-Bri3 BRICHOS and anti-Aβ42 antibodies. Staining of nuclei is performed with DAPI (blue) and filamentous actin is stained with phalloidin (green). Scale bar 20 μm. C) Fluorescence intensity quantification for negative controls (black bar) and Bri3 BRICHOS plus Aβ42 (hatched bar), n = 5; *p < 0.05. Average values and errors represent standard deviations. The results are representative of at least three independent experiments.
To verify that Bri2 interacts with AβPP at endogenous levels, we used the Bri2 linker antibody and an antibody recognizing AβPP and Aβ (4G8), and abundant signals were detected (Supplementary Figure 4). Since the combination of Aβ42 antibody and Bri2 linker antibody does not give PLA signals (Fig. 5A-C) the signals in Supplementary Figure 4 are concluded to arise from AβPP-Bri2 interactions. Similar as for Bri2, PLA signals were detected using antibodies against Bri3 BRICHOS (residue 110–258) in combination with an antibody recognizing the N-terminal part of AβPP (Supplementary Figure 4). Negative controls for all combinations of secondary antibodies used for each primary antibody confirmed that no unspecific signals were generated (Supplementary Figure 5).
AβPP/Aβ interactions with Bri2 and Bri3 in the hippocampal CA1 region in transgenic mice
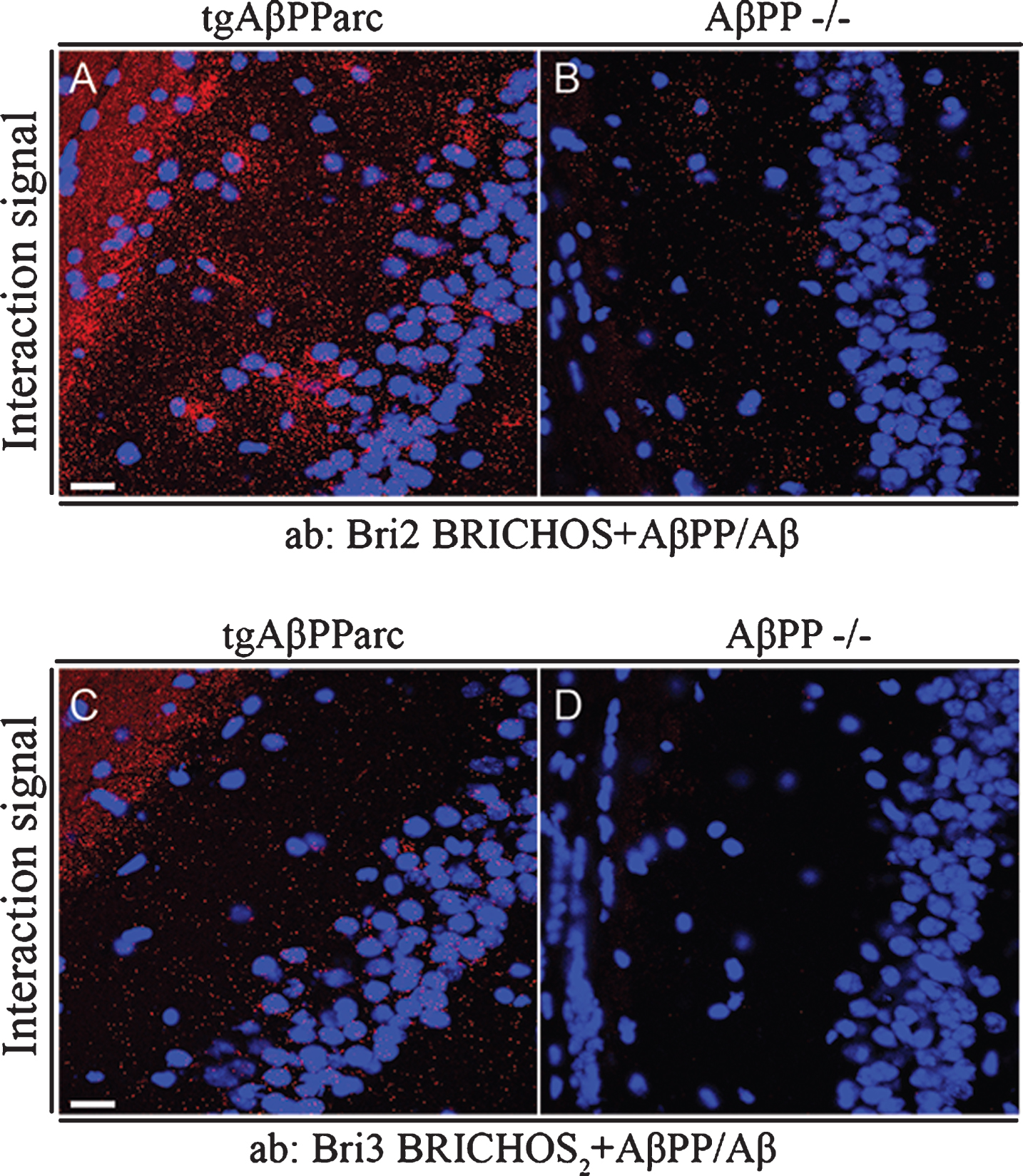
Interactions between Bri2 and Bri3 and AβPP/Aβ were furthermore analyzed in brain tissue from mice transgenic for the AβPP arctic mutation (E693G) [32] using PLA and confocal microscopy, and as a negative control, brain tissue from AβPP–/– was used [34]. Both endogenous Bri2 and Bri3 showed abundant interactions with transgenically overexpressed AβPP and/or Aβ in the CA1 region (Fig. 7A, C), but much fewer background PLA signals were detected in brain tissue sections from AβPP–/– mice (Fig. 7B, D). The interaction signals show no accumulation around plaques, which may suggest that the signals observed are dominated by interactions between Bri2/Bri3 and soluble Aβ/AβPP rather than by interactions with Aβ in amyloid deposits.
Fig.7
Bri2 and Bri3 interactions with Aβ/AβPP in CA1 region of hippocampus from tgAβPParc and AβPP –/– mice. PLA (red dots) for protein-protein interactions with (A-B) anti-Bri2 BRICHOS and anti-AβPP/Aβ antibodies (6E10); and (C-D) anti-Bri3 BRICHOS2 and anti-AβPP/Aβ antibodies (6E10). Images are representative of two to three independent experiments. Staining of nuclei is performed with DAPI (blue). Scale bars 20 μm.
DISCUSSION
The Bri2 BRICHOS domain is an efficient inhibitor of Aβ40 and Aβ42 aggregation in vitro and in in vivo models [11, 12], increased Bri2 levels are found in AD brain, and both Bri2 and Bri3 interfere with AβPP processing as shown in transgenic cell lines and mouse models [23, 25–27]. However, interactions between Bri2 or Bri3 BRICHOS and Aβ42 or Aβ40 in primary neurons have not been studied before, and Bri3 has not been analyzed in AD brain tissue previously.
In this study, we found that Bri3 BRICHOS is associated with Aβ plaques (Fig. 3). We have additionally observed that the soluble levels of mature Bri3 are decreased in AD cases compared to non-demented controls (Fig. 2). We did not observe any changes in the levels of immature Bri3, which may suggest that degradation events rather than processing from immature to mature Bri3 is perturbed in AD, but more studies are required before conclusions can be drawn. The now observed Bri3 decrease is in sharp contrast to the situation for Bri2, which showed increased levels in AD [23]. The isolated BRICHOS domain of Bri3 inhibits Aβ42 amyloid formation in vitro (Fig. 4), similar to Bri2 BRICHOS [11] but less efficiently, and Bri3 BRICHOS affects both the duration of the lag phase and the rate of fibril formation, indicating that it interferes with multiple steps in the Aβ42 fibril formation pathway. This experiment furthermore shows that BRICHOS domains of both Bri2 and Bri3 can interact directly with Aβ42, but the opposite changes in soluble Bri2 and Bri3 levels in AD suggest that they act in different ways in vivo.
We used PLA to study physical interactions between Bri2 or Bri3 BRICHOS and Aβ42 or AβPP, in primary hippocampal neurons. We show that both Bri2 and Bri3 interact with AβPP at endogenous levels (Supplementary Figure 4), which is supported by previous findings in transgenic cells and mice [19, 20, 22, 26, 27]. Bri2 BRICHOS interacts with both Aβ40 (Supplementary Figure 3A, C) and Aβ42 (Fig. 5D-F), however no interaction between the linker region of Bri2 and Aβ42 was found (Fig. 5A-C), strongly suggesting that binding to Aβ42 requires that the BRICHOS domain is shed from the Bri2 proprotein. The extracellular part of Bri2 is shed by ADAM10 [16], and it is therefore possible that Bri2 BRICHOS - Aβ42 interactions mainly occur in the extracellular space, after both proteins have been released from neurons. The PLA approach now used does not allow a distinction between intracellular interactions and interactions taking place at the plasma membrane. Whether the cellular interactions detected herein between Bri2 BRICHOS and Aβ involve Bri2 BRICHOS generated outside and/or inside cells need to be studied further. Bri2 BRICHOS is secreted from cells in culture but Bri3 BRICHOS could not be detected in the medium from transgenic cell lines [15, 16]. The divergence in proteolytic release of the BRICHOS domain in Bri2 and Bri3 suggest that Bri2 BRICHOS is an extracellular chaperone while Bri3 mainly performs its action intra- and/or juxta- (e.g., at the plasma membrane) cellularly. Thus, the deposition of Bri2 in and around Aβ plaques, may lead to reduced levels of extracellular Bri2 BRICHOS, thereby reducing the anti-amyloid activity and facilitating further fibrillation of Aβ, as previously suggested [23].
Bri3 including its BRICHOS domain interacts with both Aβ42 (Fig. 6) and Aβ40 (Supplementary Figure 3B, D) in primary neurons. The Bri3 BRICHOS domain in isolation reduces Aβ42 fibril formation, and PLA using an antibody directed against the BRICHOS domain shows Bri3 – Aβ interactions (Figs. 4 and 6). Therefore, it is plausible that the Bri3 BRICHOS domain is responsible for the interactions with Aβ, although interactions mediated by other parts of Bri3 cannot be excluded at the present stage. Neurons produce more intracellular Aβ42 than other cell types [35], and expression of Bri3 is limited to the CNS, especially CA1 hippocampal neurons [14, 25]. Aβ is produced in several subcellular compartments [35] and Aβ42 can be generated in the secretory pathway [36–38]. Bri3 and AβPP are transmembrane proteins biosynthesized through the secretory pathway, and the BRICHOS domain as well as released Aβ end up on the luminal side of the secretory pathway. Inhibition of proteasomal activity alters the levels of AβPP processing products [39–41] and we recently showed, in transfected cell lines, that the proSP-C BRICHOS domain reduces aggregation of amyloidogenic polypeptides in the ER, which otherwise leads to proteasomal inhibition [42]. It is possible that the Bri3 BRICHOS domain performs a similar function towards Aβ in neurons, which could be an important protective mechanism since intracellular Aβ accumulation affects synaptic plasticity, precedes the formation of NFTs and plaques, and since oligomerization of intracellular Aβ can take place [43–46].
Taken together, currently available results indicate that the BRICHOS domains of Bri2 and Bri3 are important for interactions primarily with Aβ. Our results further indicate that the processed Bri2 BRICHOS could act intra- (after internalization) as well as extracellularly, while full-length Bri3 and its BRICHOS domain bind Aβ intra- and/or juxtacellularly. For both Bri2 and Bri3, the aberrant levels and association with plaques in AD may lead to an abnormal function, reducing their abilities to chaperone Aβ peptides. More research is warranted about physiological mechanisms of Bri2 and Bri3 in relation to Aβ production and aggregation.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
We thank Dr. Hui Zheng, Baylor College of Medicine, Houston, USA for kindly providing us with brain tissue sections from AβPP –/– mice.
This project was supported by the Center for Innovative Medicine, the Swedish Research Council, the Swedish Alzheimer foundation and the Internationale Stichting Alzheimer Onderzoek (ISAO) foundation.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/ADR-170051.
REFERENCES
[1] | Sipe JD , Benson MD , Buxbaum JN , Ikeda SI , Merlini G , Saraiva MJ , Westermark P ((2016) ) Amyloid fibril proteins and amyloidosis: Chemical identification and clinical classification International Society of Amyloidosis 2016 Nomenclature Guidelines. Amyloid 23: , 209–213. |
[2] | Winblad B , Amouyel P , Andrieu S , Ballard C , Brayne C , Brodaty H , Cedazo-Minguez A , Dubois B , Edvardsson D , Feldman H , Fratiglioni L , Frisoni GB , Gauthier S , Georges J , Graff C , Iqbal K , Jessen F , Johansson G , Jonsson L , Kivipelto M , Knapp M , Mangialasche F , Melis R , Nordberg A , Rikkert MO , Qiu C , Sakmar TP , Scheltens P , Schneider LS , Sperling R , Tjernberg LO , Waldemar G , Wimo A , Zetterberg H ((2016) ) Defeating Alzheimer’s disease and other dementias: A priority for European science and society. Lancet Neurol 15: , 455–532. |
[3] | Hardy J , Selkoe DJ ((2002) ) The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297: , 353–356. |
[4] | Selkoe DJ ((2001) ) Alzheimer’s disease: Genes, proteins, and therapy. Physiol Rev 81: , 741–766. |
[5] | Ankarcrona M , Winblad B , Monteiro C , Fearns C , Powers ET , Johansson J , Westermark GT , Presto J , Ericzon BG , Kelly JW ((2016) ) Current and future treatment of amyloid diseases. J Intern Med 280: , 177–202. |
[6] | Coburger I , Hoefgen S , Than ME ((2014) ) The structural biology of the amyloid precursor protein APP - a complex puzzle reveals its multi-domain architecture. Biol Chem 395: , 485–498. |
[7] | Sisodia SS , St George-Hyslop PH ((2002) ) gamma-Secretase, Notch, Abeta and Alzheimer’s disease: Where do the presenilins fit in? Nat Rev Neurosci 3: , 281–290. |
[8] | Sanchez-Pulido L , Devos D , Valencia A ((2002) ) BRICHOS: A conserved domain in proteins associated with dementia, respiratory distress and cancer. Trends Biochem Sci 27: , 329–332. |
[9] | Willander H , Presto J , Askarieh G , Biverstål H , Frohm B , Knight SD , Johansson J , Linse S ((2012) ) BRICHOS domains efficiently delay fibrillation of amyloid β-peptide. J Biol Chem 287: , 31608–31617. |
[10] | Peng S , Fitzen M , Jörnvall H , Johansson J ((2010) ) The extracellular domain of Bri2 (ITM2B) binds the ABri peptide (1-23) and amyloid β-peptide (Aβ1-40). Implications for Bri2 effects on processing of amyloid precursor protein and Aβ aggregation. Biochem Biophys Res Commun 393: , 356–361. |
[11] | Arosio P , Michaels TC , Linse S , Mansson C , Emanuelsson C , Presto J , Johansson J , Vendruscolo M , Dobson CM , Knowles TP ((2016) ) Kinetic analysis reveals the diversity of microscopic mechanisms through which molecular chaperones suppress amyloid formation. Nat Commun 7: , 10948. |
[12] | Poska H , Haslbeck M , Kurudenkandy FR , Hermansson E , Chen G , Kostallas G , Abelein A , Biverstal H , Crux S , Fisahn A , Presto J , Johansson J ((2016) ) Dementia-related Bri2 BRICHOS is a versatile molecular chaperone that efficiently inhibits Abeta42 toxicity in Drosophila. Biochem J 473: , 3683–3704. |
[13] | Baron BW , Pytel P ((2017) ) Expression pattern of the BCL6 and ITM2B proteins in normal human brains and in Alzheimer disease. Appl Immunohistochem Mol Morphol 25: , 489–496. |
[14] | Zeisel A , Munoz-Manchado AB , Codeluppi S , Lonnerberg P , La Manno G , Jureus A , Marques S , Munguba H , He L , Betsholtz C , Rolny C , Castelo-Branco G , Hjerling-Leffler J , Linnarsson S ((2015) ) Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347: , 1138–1142. |
[15] | Martin L , Fluhrer R , Haass C ((2009) ) Substrate requirements for SPPL2b-dependent regulated intramembrane proteolysis. J Biol Chem 284: , 5662–5670. |
[16] | Martin L , Fluhrer R , Reiss K , Kremmer E , Saftig P , Haass C ((2008) ) Regulated intramembrane proteolysis of Bri2 (Itm2b) by ADAM10 and SPPL2a/SPPL2b. J Biol Chem 283: , 1644–1652. |
[17] | Vidal R , Frangione B , Rostagno A , Mead S , Revesz T , Plant G , Ghiso J ((1999) ) A stop-codon mutation in the BRI gene associated with familial British dementia. Nature 399: , 776–781. |
[18] | Vidal R , Revesz T , Rostagno A , Kim E , Holton JL , Bek T , Bojsen-Moller M , Braendgaard H , Plant G , Ghiso J , Frangione B ((2000) ) A decamer duplication in the 3’ region of the BRI gene originates an amyloid peptide that is associated with dementia in a Danish kindred. Proc Natl Acad Sci U S A 97: , 4920–4925. |
[19] | Fotinopoulou A , Tsachaki M , Vlavaki M , Poulopoulos A , Rostagno A , Frangione B , Ghiso J , Efthimiopoulos S ((2005) ) BRI2 interacts with amyloid precursor protein (APP) and regulates amyloid beta (Abeta) production. J Biol Chem 280: , 30768–30772. |
[20] | Matsuda S , Giliberto L , Matsuda Y , Davies P , McGowan E , Pickford F , Ghiso J , Frangione B , D’Adamio L ((2005) ) The familial dementia BRI2 gene binds the Alzheimer gene amyloid-beta precursor protein and inhibits amyloid-beta production. J Biol Chem 280: , 28912–28916. |
[21] | Matsuda S , Giliberto L , Matsuda Y , McGowan EM , D’Adamio L ((2008) ) BRI2 inhibits amyloid beta-peptide precursor protein processing by interfering with the docking of secretases to the substrate. J Neurosci 28: , 8668–8676. |
[22] | Tsachaki M , Fotinopoulou A , Slavi N , Zarkou V , Ghiso J , Efthimiopoulos S ((2013) ) BRI2 interacts with BACE1 and regulates its cellular levels by promoting its degradation and reducing its mRNA levels. Curr Alzheimer Res 10: , 532–541. |
[23] | Del Campo M , Hoozemans JJ , Dekkers LL , Rozemuller AJ , Korth C , Muller-Schiffmann A , Scheltens P , Blankenstein MA , Jimenez CR , Veerhuis R , Teunissen CE ((2014) ) BRI2-BRICHOS is increased in human amyloid plaques in early stages of Alzheimer’s disease. Neurobiol Aging 35: , 1596–1604. |
[24] | Martins F , Rebelo S , Santos M , Cotrim CZ , da Cruz e Silva EF , da Cruz e Silva OA ((2016) ) BRI2 and BRI3 are functionally distinct phosphoproteins. Cell Signal 28: , 130–144. |
[25] | Vidal R , Calero M , Revesz T , Plant G , Ghiso J , Frangione B ((2001) ) Sequence, genomic structure and tissue expression of Human BRI3, a member of the BRI gene family. Gene 266: , 95–102. |
[26] | Wickham L , Benjannet S , Marcinkiewicz E , Chretien M , Seidah NG ((2005) ) Beta-amyloid protein converting enzyme 1 and brain-specific type II membrane protein BRI3: Binding partners processed by furin. J Neurochem 92: , 93–102. |
[27] | Matsuda S , Matsuda Y , D’Adamio L ((2009) ) BRI3 inhibits amyloid precursor protein processing in a mechanistically distinct manner from its homologue dementia gene BRI2. J Biol Chem 284: , 15815–15825. |
[28] | Braak H , Alafuzoff I , Arzberger T , Kretzschmar H , Del Tredici K ((2006) ) Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112: , 389–404. |
[29] | Walsh DM , Thulin E , Minogue AM , Gustavsson N , Pang E , Teplow DB , Linse S ((2009) ) A facile method for expression and purification of the Alzheimer’s disease-associated amyloid beta-peptide. FEBS J 276: , 1266–1281. |
[30] | Cohen SI , Arosio P , Presto J , Kurudenkandy FR , Biverstal H , Dolfe L , Dunning C , Yang X , Frohm B , Vendruscolo M , Johansson J , Dobson CM , Fisahn A , Knowles TP , Linse S ((2015) ) A molecular chaperone breaks the catalytic cycle that generates toxic Abeta oligomers. Nat Struct Mol Biol 22: , 207–213. |
[31] | Behbahani H , Rickle A , Concha H , Ankarcrona M , Winblad B , Cowburn RF ((2005) ) Flow cytometry as a method for studying effects of stressors on primary rat neurons. J Neurosci Res 82: , 432–441. |
[32] | Ronnback A , Sagelius H , Bergstedt KD , Naslund J , Westermark GT , Winblad B , Graff C ((2012) ) Amyloid neuropathology in the single Arctic APP transgenic model affects interconnected brain regions. Neurobiol Aging 33: , 831 e811–839. |
[33] | Ronnback A , Zhu S , Dillner K , Aoki M , Lilius L , Naslund J , Winblad B , Graff C ((2011) ) Progressive neuropathology and cognitive decline in a single Arctic APP transgenic mouse model. Neurobiol Aging 32: , 280–292. |
[34] | Zheng H , Jiang M , Trumbauer ME , Sirinathsinghji DJ , Hopkins R , Smith DW , Heavens RP , Dawson GR , Boyce S , Conner MW , Stevens KA , Slunt HH , Sisoda SS , Chen HY , Van der Ploeg LH ((1995) ) beta-Amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell 81: , 525–531. |
[35] | LaFerla FM , Green KN , Oddo S ((2007) ) Intracellular amyloid-beta in Alzheimer’s disease. Nat Rev Neurosci 8: , 499–509. |
[36] | Busciglio J , Gabuzda DH , Matsudaira P , Yankner BA ((1993) ) Generation of beta-amyloid in the secretory pathway in neuronal and nonneuronal cells. Proc Natl Acad Sci U S A 90: , 2092–2096. |
[37] | Cook DG , Forman MS , Sung JC , Leight S , Kolson DL , Iwatsubo T , Lee VM , Doms RW ((1997) ) Alzheimer’s Abeta(1-42) is generated in the endoplasmic reticulum/intermediate compartment of NT2N cells. Nat Med 3: , 1021–1023. |
[38] | Wild-Bode C , Yamazaki T , Capell A , Leimer U , Steiner H , Ihara Y , Haass C ((1997) ) Intracellular generation and accumulation of amyloid beta-peptide terminating at amino acid 42. J Biol Chem 272: , 16085–16088. |
[39] | Agholme L , Hallbeck M , Benedikz E , Marcusson J , Kagedal K ((2012) ) Amyloid-beta secretion, generation, and lysosomal sequestration in response to proteasome inhibition: Involvement of autophagy. J Alzheimers Dis 31: , 343–358. |
[40] | Almeida CG , Takahashi RH , Gouras GK ((2006) ) Beta-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. J Neurosci 26: , 4277–4288. |
[41] | Flood F , Murphy S , Cowburn RF , Lannfelt L , Walker B , Johnston JA ((2005) ) Proteasome-mediated effects on amyloid precursor protein processing at the gamma-secretase site. Biochem J 385: , 545–550. |
[42] | Dolfe L , Winblad B , Johansson J , Presto J ((2016) ) BRICHOS binds to a designed amyloid-forming beta-protein and reduces proteasomal inhibition and aggresome formation. Biochem J 473: , 167–178. |
[43] | Gouras GK , Tsai J , Naslund J , Vincent B , Edgar M , Checler F , Greenfield JP , Haroutunian V , Buxbaum JD , Xu H , Greengard P , Relkin NR ((2000) ) Intraneuronal Abeta42 accumulation in human brain. Am J Pathol 156: , 15–20. |
[44] | Oddo S , Caccamo A , Shepherd JD , Murphy MP , Golde TE , Kayed R , Metherate R , Mattson MP , Akbari Y , LaFerla FM ((2003) ) Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 39: , 409–421. |
[45] | Takahashi RH , Almeida CG , Kearney PF , Yu F , Lin MT , Milner TA , Gouras GK ((2004) ) Oligomerization of Alzheimer’s beta-amyloid within processes and synapses of cultured neurons and brain. J Neurosci 24: , 3592–3599. |
[46] | Walsh DM , Tseng BP , Rydel RE , Podlisny MB , Selkoe DJ ((2000) ) The oligomerization of amyloid beta-protein begins intracellularly in cells derived from human brain. Biochemistry 39: , 10831–10839. |


