
The p110α/ΔNp63α complex mutations in triple-negative breast cancer: Potential targets for transcriptional-based therapies
Abstract
BACKGROUND:
Hotspot mutations occurring in the p110α domain of the PIK3CA gene, specifically p110αH1047R/L increase tumor metastasis and cell motility in triple-negative breast cancer (TNBC). These mutations also affect the transcriptional regulation of ΔNp63α, a significant isoform of the p53 protein involved in cancer progression. This study attempts to investigate the transcriptional impact of p110αH1047R/L mutations on the PIK3CA/ΔNp63α complex in TNBC carcinogenesis.
METHODS:
We performed site-directed mutagenesis to introduce p110αH1047R/L mutations and evaluated their oncogenic effects on the growth, invasion, migration, and apoptosis of three different TNBC cell lines in vitro. We investigated the impact of these mutations on the p110α/ΔNp63α complex and downstream transcriptional signaling pathways at the gene and protein levels. Additionally, we used bioinformatics techniques such as molecular dynamics simulations and protein-protein docking to gain insight into the stability and structural changes induced by the p110αH1047R/L mutations in the p110α/ΔNp63α complex and downstream signaling pathway.
RESULTS:
The presence of PIK3CA oncogenic hotspot mutations in the p110α/ΔNp63α complex led to increased scattering of TNBC cells during growth, migration, and invasion. Our in vitro mutagenesis assay showed that the p110αH1047R/L mutations activated the PI3K-Akt-mTOR and tyrosine kinase receptor pathways, resulting in increased cell proliferation, invasion, and apoptosis in TNBC cells. These mutations decreased the repressing effect of ΔNp63α on the p110α kinase domain, leading to the enhancement of downstream signaling pathways of PI3K and tyrosine kinase receptors and oncogenic transformation in TNBC. Additionally, our findings suggest that the physical interaction between the DNA binding domain of ΔNp63α and the kinase domain of p110α may be partially impaired, potentially leading to alterations in the conformation of the p110α/ΔNp63α complex.
CONCLUSION:
Our findings suggest that targeting the p110αH1047R/L mutations in TNBC could be a promising strategy for developing transcriptional-based therapies. Restoring the interaction between ΔNp63α and the p110α kinase domain, which is disrupted by these mutations, may provide a new approach to treating TNBC.
1Introduction
Metastatic breast cancer (MBC) is a life-threatening malignancy among women worldwide, resulting in over 41,000 deaths annually and a 5-year overall survival rate of 59% [1]. Among the subtypes of MBC, triple-negative breast cancer (TNBC) is a prevalent and highly recurrent subtype with a poor prognosis [1, 2]. The correlation between histopathological markers and mammographic and imaging characteristics can aid in the early detection and targeted treatment of TNBC patients [3].
The phosphatidylinositol 3-kinase (PI3K) family, a group of intracellular signal-transducing lipid kinases, plays a crucial role in TNBC metastasis through its involvement in cell differentiation, growth, proliferation, apoptosis, and survival [2]. Oncogenic point mutations in the PI3K gene, particularly in hotspot regions, have been identified as a significant contributor to the poor prognosis and resistance to standard therapies in patients with MBC [4]. These mutations, along with overexpression of human epidermal receptor 2 (HER2), are essential factors in the pathogenesis of the MBC [5]. Among the class, IA PI3K subunit p110α (PIK3CA), several oncogenic hotspot mutations have been reported in 33–40% of the MBC [5–7]. Most mutations in cancer affect the kinase domain of p110α, which is located in exon 20. Notably, two common mutations, p.H1047R, and p.H1047L have been identified in various types of cancers.
Tumor protein p63, a member of the p53 family, acts as a tumor suppressor protein and is involved in the pathogenesis of the MBC [8, 9]. The expression of TP63 results in two distinct isoforms, TAp63 and ΔNp63, that differ in their N-terminal transactivation domain which is present in the full-length form of the protein [9, 10]. ΔNp63α is the predominant isoform of tumor protein p63 and has been found to act as a crucial inhibitor of cancer metastasis. Its downregulation has been associated with increased metastasis, invasion, and migration in TNBC [10–13].
Gene expression analysis shows that the mutational inactivation of PI3Ks leads to decreased ΔNp63α expression and activity, potentially leading to genetic instability and cancer progression [11, 14–16]. A computational docking study shows that PIK3CA and ΔNp63α interact, activating PI3K kinase in TNBC. p110αH1047R/L in exon 20 inhibit ΔNp63α expression and are associated with a high mortality rate for patients with HER+ breast cancer [17, 18]. Despite extensive research on the functional and structural effects of the oncogenic hotspot mutation, the pathological significance of the oncogenic mutation of p110αH1047R/L at p110α/PIK3CA complex in the TNBC metastasis is still unknown [17, 18].
Emerging research has suggested the involvement of tyrosine kinase receptors in modulating the PI3K/ΔNp63α pathway in the TNBC [11, 19]. Upon activation, these receptors initiate the downstream PI3K signaling pathway, which subsequently activates Akt and mTOR. Additionally, these receptors have been observed to interact with the p110α/ΔNp63α complex, modulating its activity and promoting oncogenic transformation in TNBC [11, 20]. Considering the connection between tyrosine kinase receptors and the PI3K/ΔNp63α pathway in TNBC, targeting these receptors through therapeutic means could be a promising treatment approach for this particularly aggressive type of breast cancer [21].
This study aimed to investigate the impact of p110αH1047R/L mutations on the PIK3CA/ΔNp63α complex in the development of TNBC. By using in vitro models and evaluating the inhibitory function through homology modeling and protein-protein interaction, the study found that these mutations resulted in the destabilization of the interaction site between p110α and ΔNp63α in the PI3K/AKT-dependent pathway. The results suggest this destabilization of may play a role in TNBC development which may provide hints for developing new therapeutic strategies in TNBC.
2Materials and methods
2.1Ethical considerations
This study was approved by the Ethics Review Board of the Affiliated Hospital of Southwest Medical University (Approval No. KY2019041) following the Declaration of Helsinki (1983 Revision). Furthermore, all laboratory assessments were conducted according to the local guidelines of the Ethics Committee of the Oncology Department at the Affiliated Hospital of Southwest Medical University, Sichuan, China.
2.2Cell lines
The human breast ductal carcinoma cell line (BT-549), human breast adenocarcinoma cell line (MDA-MB-231), and non-metastatic human breast cancer cell line (MCF-7) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). The cells were grown in a combination of DMEM, Ham’s F-12 medium with reduced Ca2+ (0.04 mM; Invitrogen, Carlsbad, CA, USA), supplemented with 1% penicillin-G/streptomycin, epidermal growth factor (20 ng/mL), cholera toxin (100 ng/mL), insulin (10 μg/mL), hydrocortisone (500 ng/mL), and Chelex-treated horse serum (5%). The cells were cultured at 37°C with 5% CO2, and to assess their morphology the cells were plated at a low confluence, allowed to grow for 8–12 days, fixed with methanol, stained with 0.1% crystal violet in 70% ethanol, and observed under a light microscope.
2.3Plasmid construction and transfection
The human wild-type (WT) p110α and its mutant versions (p.H1047R and p.H1047L) were produced through PCR amplification using the templates pSG5-p110α-WT (V), pSG5-p110αH1047R, and pSG5-p110αH1047L (from Addgene, Watertown, MA, USA). The amplified products were then subcloned into a pLentim3-blasticidin vector [22]. A p110α-specific shRNA was created by inserting specific oligos into a pSG5-ampicillin vector obtained from Thermo Fisher Scientific (Table 1). The presence of the pSG5:p110α DNA construct was confirmed through restriction digestion and sequencing. To assess the transfection efficiency, BT-549 cells were transfected with pMD2.G, psPAX2 packaging plasmids, and the corresponding backbone plasmid using Lipofectamine 3000 as per the manufacturer’s instructions [23, 24]. At 48 hours post-transfection, the cells were harvested and analyzed through Western blot analysis after being incubated at 37°C.
Table 1
Primers and oligonucleotides used in this study
| Oligonucleotide | Sequence | Application |
| Human sh-p110α-1 | GAGTGGAGCCACTGAACTAT | shRNA |
| Human sh-p110α-2 | CTTCCATATCCCTTTGTTCTA | shRNA |
| Human shC | GAAGCAGCACGACTTCTTC | shRNA |
| ΔNp63-F | GATTCATATTGTAAGGGTCTCGG | qRT-PCR |
| ΔNp63-R | GGGCATTGTTTTCCAGGTA | qRT-PCR |
| ERK1/2-F | CCTGGAAGCCTCGAGAGATGTCTA | qRT-PCR |
| ERK1/2-R | TCCATTCAGCTCGAGTTCTGCACC | qRT-PCR |
| AKT-F | TCTATGGCGCTGAGATTGTG | qRT-PCR |
| AKT-R | CTTAATGTGCCCGTCCTTGT | qRT-PCR |
| p-ERK1/2-R | CTGGCAGGGTGAAGTTGG | qRT-PCR |
| GAPDH-F | GGCCTCCAAGGAGTAAGACC | qRT-PCR |
| GAPDH-R | AGGGGAGATTCAGTGTGGTG | qRT-PCR |
| Human sh-p110α-1 | GAGTGGAGCCACTGAACTAT | shRNA |
| Human sh-p110α-2 | CTTCCATATCCCTTTGTTCTA | shRNA |
| Human shC | GAAGCAGCACGACTTCTTC | shRNA |
| ΔNp63-F | GATTCATATTGTAAGGGTCTCGG | qRT-PCR |
| ΔNp63-R | GGGCATTGTTTTCCAGGTA | qRT-PCR |
| ERK1/2-F | CCTGGAAGCCTCGAGAGATGTCTA | qRT-PCR |
| ERK1/2-R | TCCATTCAGCTCGAGTTCTGCACC | qRT-PCR |
| AKT-F | TCTATGGCGCTGAGATTGTG | qRT-PCR |
| AKT-R | CTTAATGTGCCCGTCCTTGT | qRT-PCR |
2.4Generation of mutant p110α plasmids
The p110αH1047R/L mutants were created using the QuikChange® Site-Directed Mutagenesis Kit according to the manufacturer’s instructions [25]. The process involved PCR amplification to create the mutant versions of the p110 α gene. After PCR amplification, the product was treated with DpnI enzyme to eliminate the parental, non-mutated version of the p110α plasmid. Next, the mutated plasmids were transformed into XL1-Blue super-competent cells using a heat shock method on an LB agar plate. After overnight incubation at 37°C, the integrity of the newly generated plasmids was confirmed through sequencing using an upstream primer to ensure the successful incorporation of the desired mutations into the p110 α gene.
2.5Western blot analysis
The Western blotting procedure was carried out using conventional techniques following previously described protocols [26, 27]. Primary antibodies for p63, DPL, and ACTIN were procured from Santa Cruz Biotechnology, while Cell Signaling Technologies provided antibodies for Akt, p-Akt, Erk1/2, p-Erk1/2, and p110α.
2.6qRT-PCR
The RNA extraction procedure was conducted using the RNAeasy Plus Mini Kit (Qiagen), following the manufacturer’s guidelines. Reverse transcription of RNA into cDNA was carried out with the M-MLV First Strand Kit (Invitrogen). To determine ΔNp63 expression, qPCR was performed on a CFX96 real-time PCR system (Bio-Rad) using SoFast EvaGreen Supermix (Bio-Rad). The detailed qRT-PCR reaction plan was previously described [28–30].
2.7Transwell invasion assay
The Transwell invasion assay was conducted using 6.5 mm diameter polycarbonate filters (8μM pore size) covered with 35μL of high-concentration Corning Matrigel Matrix. Corning Costar 24-well cell culture inserts were used for the assay [31]. Initially, 5×105 cells were seeded on top of the chamber, containing 1% FBS, and allowed to invade for 24 hours at 37°C. Following the incubation period, the chambers were washed with PBS, and the cells were fixed with methanol for 20 minutes. Giemsa stain was used to stain the cells for 15 minutes. Five randomly selected fields were manually counted, and the assay was visualized through an inverted microscope. Representative samples of the assay were taken and images were captured.
2.8Tumor metastatic potential assay in vitro
The migration ability of BT-549 cells was evaluated using a wound-healing assay. BT-549 cells (5×105 cells/well) transfected with WT, V, or both p110αH1047R and p110αH1047L mutants were used for the assay as previously described [32]. Scratch wounds were created in the 90% confluent cell monolayer using a sterile 200-μL pipette tip. The cells were then rinsed with 3x PBS and incubated in a serum-free medium at 37°C for 24 hours. The migration of cells was observed at 0–24 hours after the scratch wound was created using a phase-contrast microscope (Olympus Cor., Tokyo, Japan), and the migratory differences were analyzed using ImageJ Wound Healing Tool plug-in software.
2.9Cell growth, migration, and apoptosis assays
Cell growth, migration, and apoptosis assays were performed to assess the effects of the p110αH1047R/L mutants on BT-549 cells. Real-time analysis of these cellular processes was conducted using a real-time cell analyzer (xCELLigence RTCA DP, Roche, Germany), which provided accurate and precise quantification of the parameters of interest [30, 33]. Standard flow cytometry with the FITC Annexin V Apoptosis Detection Kit (BD Biosciences, San Diego, CA, USA) was used to analyze the cell cycle and apoptosis according to the manufacturer’s instructions [26]. Flow cytometry data were acquired on a FACS Canto II flow cytometer and analyzed using FlowJo software (TOMY Digital Biology, Tokyo, Japan).
2.10Homology modeling, protein-protein interaction, and MD simulation
To generate the initial conformation, a crystal structure of the template protein (PDB entry: 2RD0) with 3.05 Å resolution was used. The PIK3CA sequence alignment was identified using the PDB’s BLASTp program. MODELLER 9.17 was used to construct 3D models of full-length PIK3CA, and hotspot mutations were generated in silico using Chimera software [34]. To verify the models’ quality, PROCHECK, ProSA-web, and QMEAN analysis were used. Clustal Omega was used to conduct multiple sequence alignments, and the phylogenetic tree analysis was performed to confirm the sequence similarity of mutations to p110α. Molecular docking with HADDOCK was used to evaluate the binding sites of the WT and mutant p110α models in complex with ΔNp63α [35]. The CPORT server was used to predict the active site for interaction between mutants and WT. Results were analyzed using LigPlot software and VMD [36]. Molecular dynamics (MD) simulations were performed to study the motion and fluctuations of three systems containing discrete particles under the influence of internal and external forces associated with hotspot mutant residues in human PIK3CA. The conformational diversity of p.H1047R/L mutations on the functional binding domain of p110α in complex with ΔNp63α (residues 114–359) was investigated through 50-ns MD simulations using GROMACS MD package (5.1.3) and GROMOS96 package (43a1) force field on a Centos Linux system. The simulations were designed to provide insight into the dynamics of the system and were performed using previously established methods described in [30, 37–39].
2.11Statistical analysis
The data were analyzed statistically using SPSS 21.0 software (Chicago, Illinois, USA), and graphs were created using GraphPad Prism 5.0 (GraphPad Software Inc., La Jolla, CA, USA). The biological assays were performed thrice, and the in vitro quantitative data were analyzed accordingly. Fisher’s exact test was used to determine the level of enrichment of mutations/aberrations affecting the pathway in the mutagenesis group in comparison to the WT or V control. A P-value of less than 0.05 was considered statistical significance threshold, and detailed information about the individual P-values and statistical analysis are provided in the figure captions.
3Results
3.1PIK3CA and ΔNp63α expression enhances scattered cell growth, migration, and invasion in TNBC cell lines
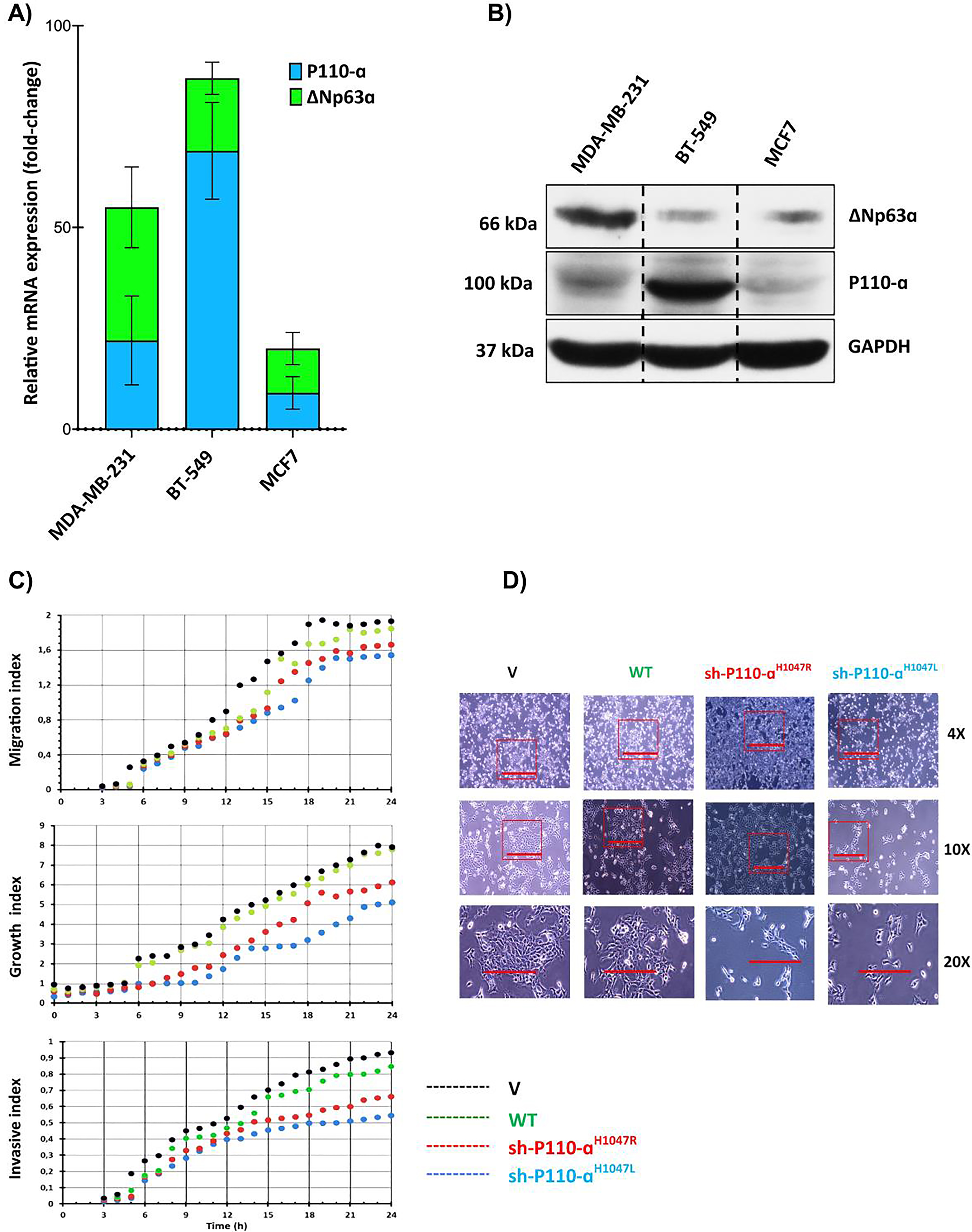
In previous studies, it was found that high expression of PIK3CA is inversely correlated with overall survival in the MBC [28]. To investigate the role of the PIK3CA/ΔNp63α complex and its association with malignancy in MBC, the expression levels of PIK3CA and ΔNp63α in both mRNA and protein levels were assessed in three MBC cell lines (BT-549 and MDA-MB-231) and one non-metastatic breast cancer cell line (MCF-7) (Fig. 1). The results indicate that the expression of PIK3CA was significantly higher in two advanced TNBC cell lines and one melanoma cell line when compared to MCF-7. However, there was no significant difference in PIK3CA expression observed in MDA-MB-231 cells. The advanced/metastatic TNBC cell line, BT-549, exhibited the highest expression levels of both PIK3CA and ΔNp63α. However, it is essential to exercise caution in interpreting these results, as the data from Fig. 1A and 1B do not conclusively demonstrate a direct association between elevated ΔNp63α levels and heightened disease severity or metastatic lesions in TNBC patients. The expression ratio of PIK3CA to ΔNp63α was highest in BT-549 and lowest in MDA-MB-231 cells. Previous research also found that PIK3CA hotspot mutations p. H1047R/L were significantly higher in the BT-549 TNBC cell line compared to other TNBC cell lines [40]. To evaluate the impact of p110αH1047R/L overexpression on MBC cell growth, migration, and invasion, pre-p110αH1047R/L were transiently transfected into BT-549 cells [41], respectively. Results showed that the expression of both p110αH1047L and p110αH1047R significantly inhibited cell proliferation in BT-549 cells (P < 0.05). The restoration of p110αH1047L led to reduced cell migration and invasion compared to p110αH1047R. In contrast, overexpression of p110αH1047R significantly enhanced cell growth, migration, and invasion (Fig. 1C). Meanwhile, after introducing WT or V p110α, there was little impact on cell migration and invasion. However, p110α removal in cells expressing p110αH1047L fully restored cell migration and invasion to levels seen in control cells. Furthermore, the findings revealed that BT-549 breast cancer cells with inhibited p110αH1047L demonstrated increased migration and invasion after 24 h of cell culture. The results in Fig. 1D show that control cells had normal morphology and formed compact colonies, while the expression of p110αH1047R/L led to scattered cell growth with spindle-like morphology. This suggests that p110αH1047R/L can cause changes in cellular behavior and may be involved in disease development. Further research is needed to understand the underlying mechanisms.
Fig. 1
Comparison and impact of p110α and ΔNp63α expression in breast cancer cells. (A) The relative mRNA expression levels of p110α and ΔNp63α in advanced breast cancer cell lines. (B) The protein levels of p110α and ΔNp63α in advanced breast cancer cell lines. (C) Cell migration, growth, and invasion were measured using a real-time cell analyzer. (D) Comparison of cell morphologies of BT-549 cells expressing p110αH1047R/L or vector control. Impact of p110αH1047R/L on Cell Migration and Invasion in BT-549 Cells. The BT-549 cells were transfected with shRNA-p110αH1047L and p110αH1047R plasmids for 24 hours.
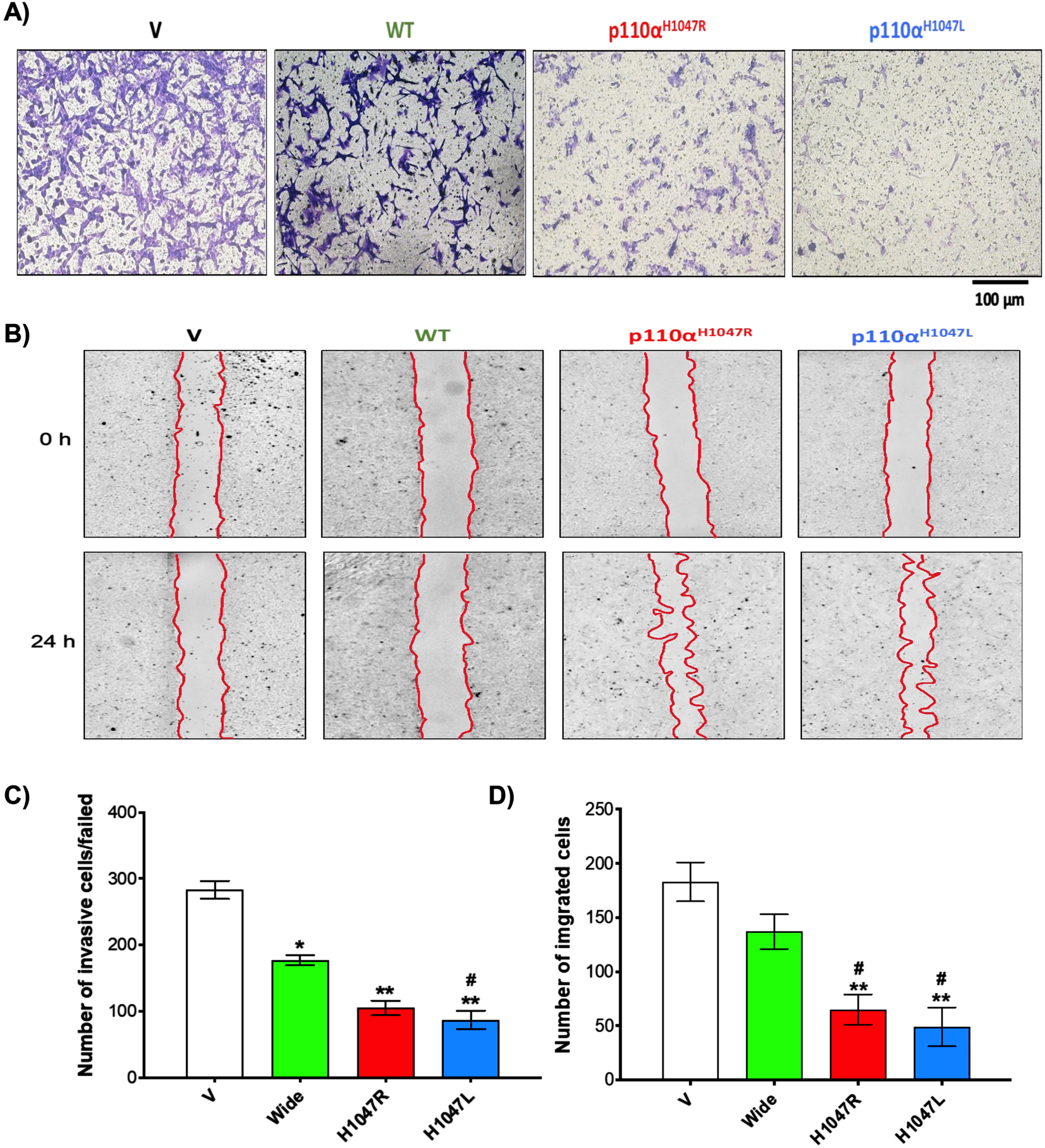
Our initial invasive screening results showed that p110αH1047R/L knockdown significantly reduced BT-549 cell proliferation/viability compared to WT (Fig. 2A). In addition, the number of invasive TNBC cells was significantly lower in the shp110αH1047R (150±11 cells/field) and shp110αH1047L groups (87±14 cells/field) compared to the Wide group (177±8 cells/field; p < 0.05) and V groups (283±14 cells/field; p < 0.001) (Fig. 2C). The results of the in vitro wound healing assay showed significant reductions in migration in both shp110αH1047R and shp110αH1047L groups (Fig. 2B). In addition, there was a notable decrease in TNBC cell migration in the p110αH1047L group, and p110αH1047R significantly inhibited cell proliferation in BT-549 cells. Remarkably, after 24 h of cell culture, p110αH1047L in BT-549 cells exhibited the greatest reduction in migration (Fig. 2D).
Fig. 2
Inhibitory effect of p110αH1047R/L on proliferation and invasion of TNBC cells. Analysis of cell invasion using a modified Boyden Chamber assay in BT-549 cell lines transfected with WT and p110αH1047L/R-shRNA. (A) Microscopic images of invaded cells, (B) bar graph representing several cells that invaded matrigel, (C) scratch wound-healing assay of BT-549 cells transfected with empty vector (V), shRNA (WT), or p110αH1047L/R-shRNA. The movement of cells into the wounds was observed at 0 and 24 hours after scratching (magnification: 10x). (D) Bar graph showing the number of cells that migrated into the scratched area. Values are presented as mean±SEM, n = 3, *P < 0.05, and **P < 0.01 for the V group; #P < 0.05, and ##P < 0.01 for the WT group.
3.2p110αH1047R/L inhibit ΔNp63α expression via Akt1-mediated inhibition and trigger the G2/M cell arrest
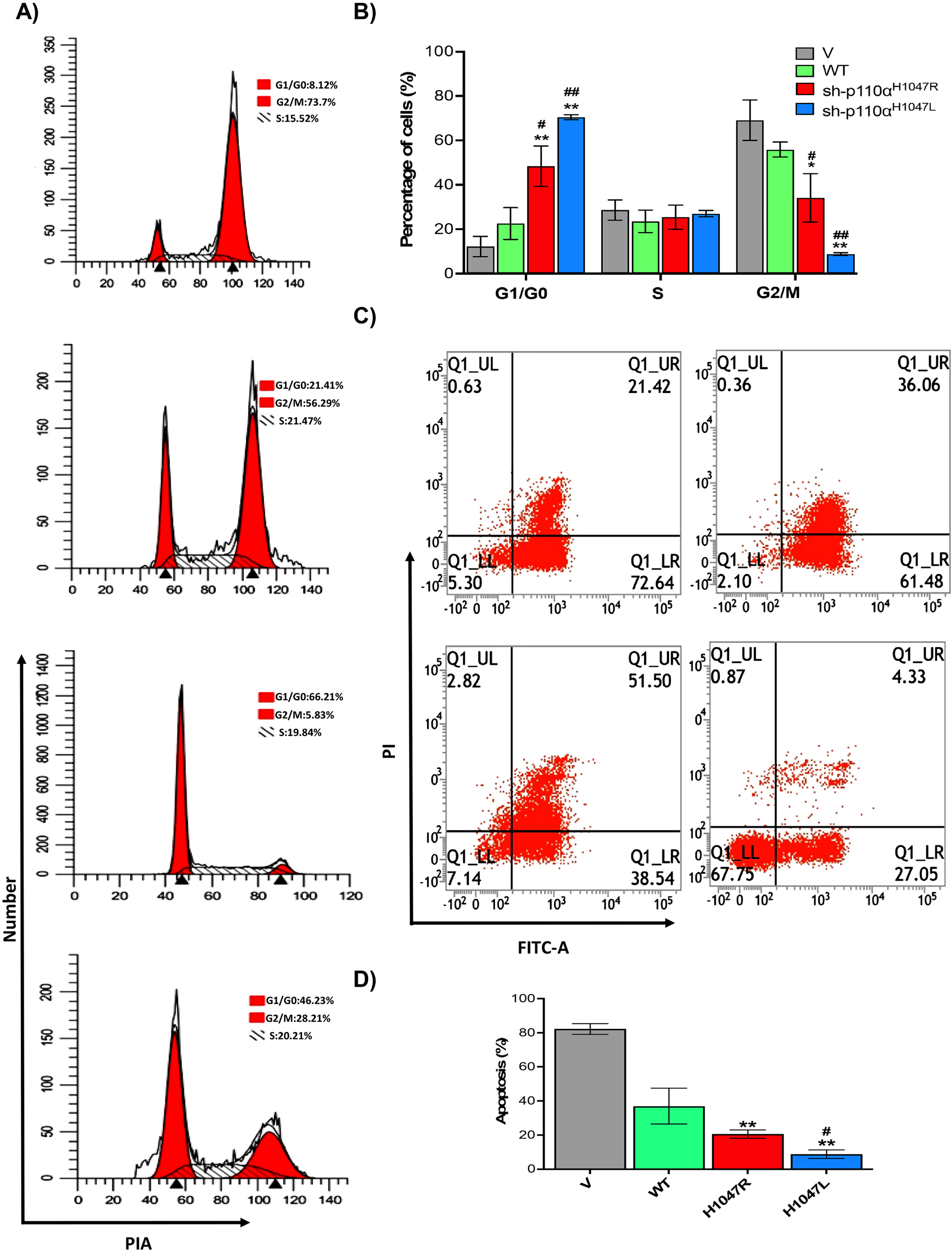
To assess the effect of p110αH1047R/L on BT-549 MBC cell apoptosis and cell cycle distribution, flow cytometry was used, as shown in Fig. 3A–D. The BT-549 cells expressing p110αH1047R/L showed a significant decrease in the proportion of cells in the G2/M phase and a significant increase in the proportion of cells in the G0/G1 phase, respectively. The p110αH1047L also caused progression from the G0/G1 to G2/M phase (Fig. 3A). The distribution of p110αH1047L -induced cells showed a lower proportion at the G2/M phase and a higher proportion at the G0/G1 phase (Fig. 3B). Additionally, the apoptosis rate decreased significantly in both p110αH1047R and p110αH1047L (Fig. 3C). The apoptosis rate was significantly lower in the shp110αH1047R (19.23.56±3.12%) and shp110αH1047L groups (15.01±3.09%) compared to the control group (79.21±5.66%; p < 0.001; Fig. 3D). The results suggest that p110αH1047R/L induces TNBC proliferation by suppressing mitotic arrest and apoptosis.
Fig. 3
Effect of p110αH1047R/L on cell cycle arrest and apoptosis of TNBC cells. (A) Flow cytometry assessed representative dot plots of cell cycle and (B) apoptotic cells after Annexin V-FITC/PI staining. (C) Percentage of cell cycle and (D) apoptotic cells with different concentrations of Anlotinib. Data are presented as mean±SD of three independent experiments. (*P < 0.05 and **P < 0.01 versus V group and #P < 0.05, and ##P < 0.01 versus WT group).
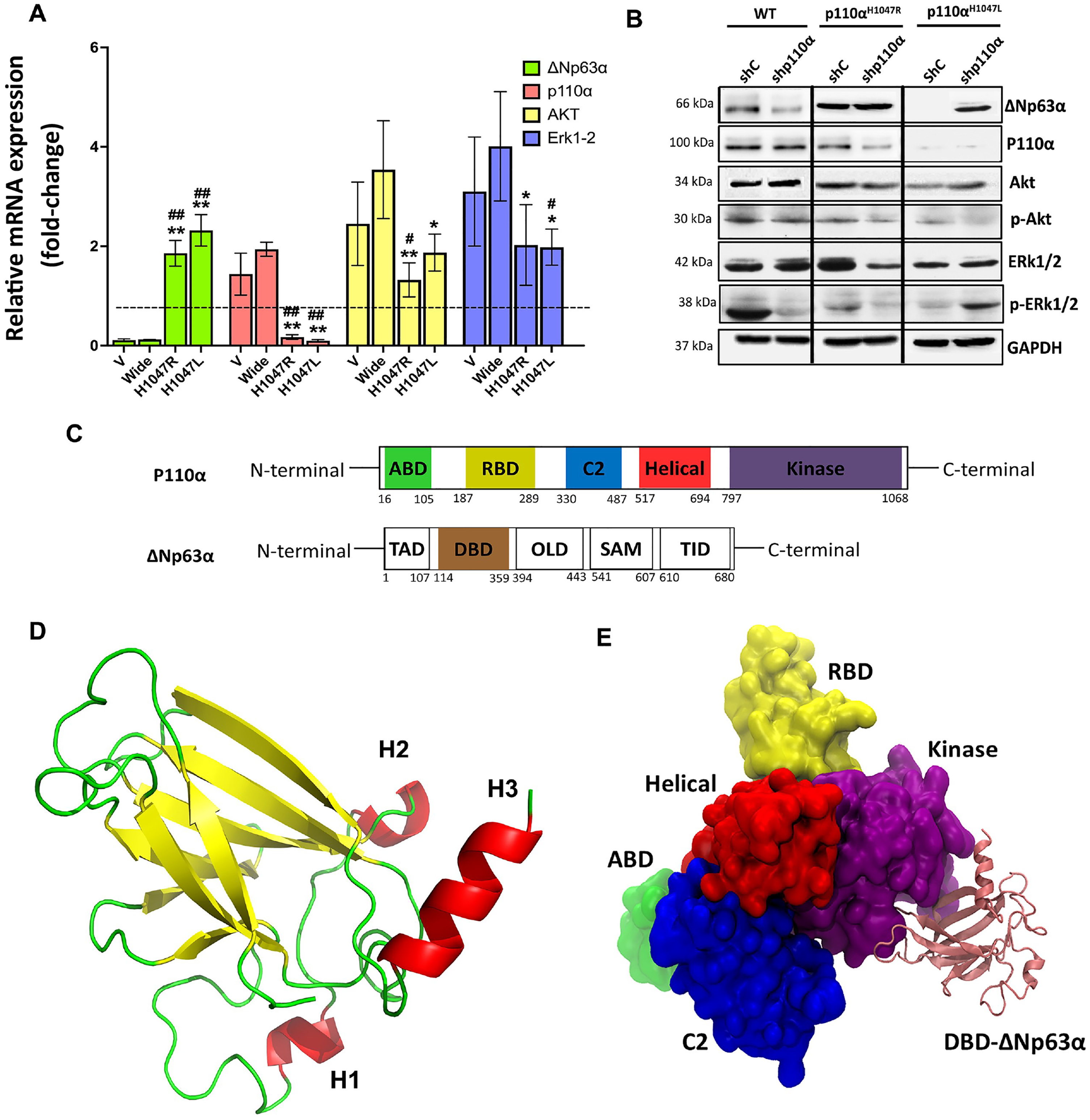
The impact of p110αH1047R/L on ΔNp63α expression and the Akt1-mediated pathway were assayed by the qRT-PCR (shown in Fig. 4A) and Western blot analysis (presented in Fig. 4B) utilized. The results showed that ΔNp63α expression was partially restored in p110αH1047L after knockdown, leading to decreased expression of AKT and p-AKT while ERK1/2 remained unchanged. ΔNp63α was found to be a common target in p110αH1047L-induced cell motility and tumor metastasis (Fig. 4A). Based on the study findings, it appears that the tyrosine kinase receptor Akt1 is instrumental in the inhibition of ΔNp63α expression, cell migration, and invasion mediated by p110αH1047R/L. Akt1 is known to be a downstream effector of the PI3K/Akt and tyrosine kinase receptors signaling pathway and is involved in various cellular processes such as cell survival, proliferation, and migration. The partial restoration of ΔNp63α expression after Akt1 knockdown in p110αH1047L cells indicates the involvement of Akt1 in the regulation of ΔNp63α expression in TNBC cells harboring PIK3CA mutations. Furthermore, the decreased levels of AKT and p-AKT observed in p110αH1047L cells suggest that the p110αH1047L mutation may inhibit Akt1 expression and activity, thereby contributing to the downregulation of ΔNp63α and the enhanced metastatic potential of TNBC cells. The p110αH1047L partially inhibited Akt1 expression, suggesting the importance of Akt1 in p110αH1047L-induced ΔNp63α downregulation and cell migration and invasion. In p110αH1047L, the levels of AKT and p-AKT decreased, while ERK1/2 remained unaltered and had more phosphorylation than in the WT. The study emphasizes the importance of the PI3K/Akt signaling pathway and of Akt1 in TNBC metastasis. The p110αH1047R/L mutations in the p110α/PIK3CA complex affect ΔNp63α expression and activity.
Fig. 4
Inhibition of ΔNp63α expression by p110αH1047L via Akt1-mediated inhibition and tyrosine kinase receptor signaling. The BT-549 cells were transduced with recombinant retroviruses encoding wild-type p110α (WT) and mutant derivatives (H1047L or H1047R). (A) qRT-PCR data and (B) representative western blot analysis of p110α, ΔNp63α, and Akt1-mediated signaling pathways in various groups. (C) Structure and Domain Organization of the p110α/ΔNp63α Complex. Schematic representation of the ΔNp63α and P110α protein isoforms. (D) The tertiary structure of ΔNp63α. The DBD domain contains three important helices, H1 (residues 234–236), H2 (residues 245–249), and H3 (residues 348–355), which play a crucial role in the interaction with PIK3. (E) The p110α/ΔNp63α complex’s 3D structure highlights its contacts with the kinase, helical, ABD, RBD, and C2 domains of PIK3CA. The different domains of PIK3CA are colored for visual differentiation, with the ABD (residues 16–105) in green, the RBD (residues 187–289) in yellow, the C2 domain (residues 330–487) in blue, the helical domain (residues 517–694) in red, the kinase domain (residues 797–1068) in purple, and the ΔNp63α DBD in pink. Western blotting was performed on whole-cell lysates (50–70μg) using GAPDH as a control. Values are presented as mean±SEM, n = 3, *P < 0.05 and **P < 0.001 compared to the V group and #P < 0.05, and ##P < 0.01 versus the WT group.
3.3Conformational changes in the WT and mutant structure of the p110α/ΔNp63α complex
The study analyzed the PIK3CA gene’s WT structure and two mutations (p.H1047R and p.H1047L) in the p110α kinase domain using computational methods. Our results show that the three-helix regions of the ΔNp63α protein’s DNA-binding domain (DBD) were located at the p110α/ΔNp63α interface and maintained stable interactions with p110α’s kinase domain (Fig. 4D). The results suggested that the mutations in the linker motif 797–1068 could potentially affect the interdomain allosteric interactions in the complex. The three-helix regions (H1, H2, and H3) of the DNA-binding domain (DBD) of ΔNp63α were found to be at the interface with p110α, and the hydrophobic cleft could also be affected by the mutations (Fig. 4E). A molecular dynamics simulation was then performed to assess the stability of the WT and mutant structures over time. The results showed a continuous loss in compactness and conformational change in the mutant structures, with the most severe change observed in the p.H1047L mutant. Further studies are required to confirm these findings.
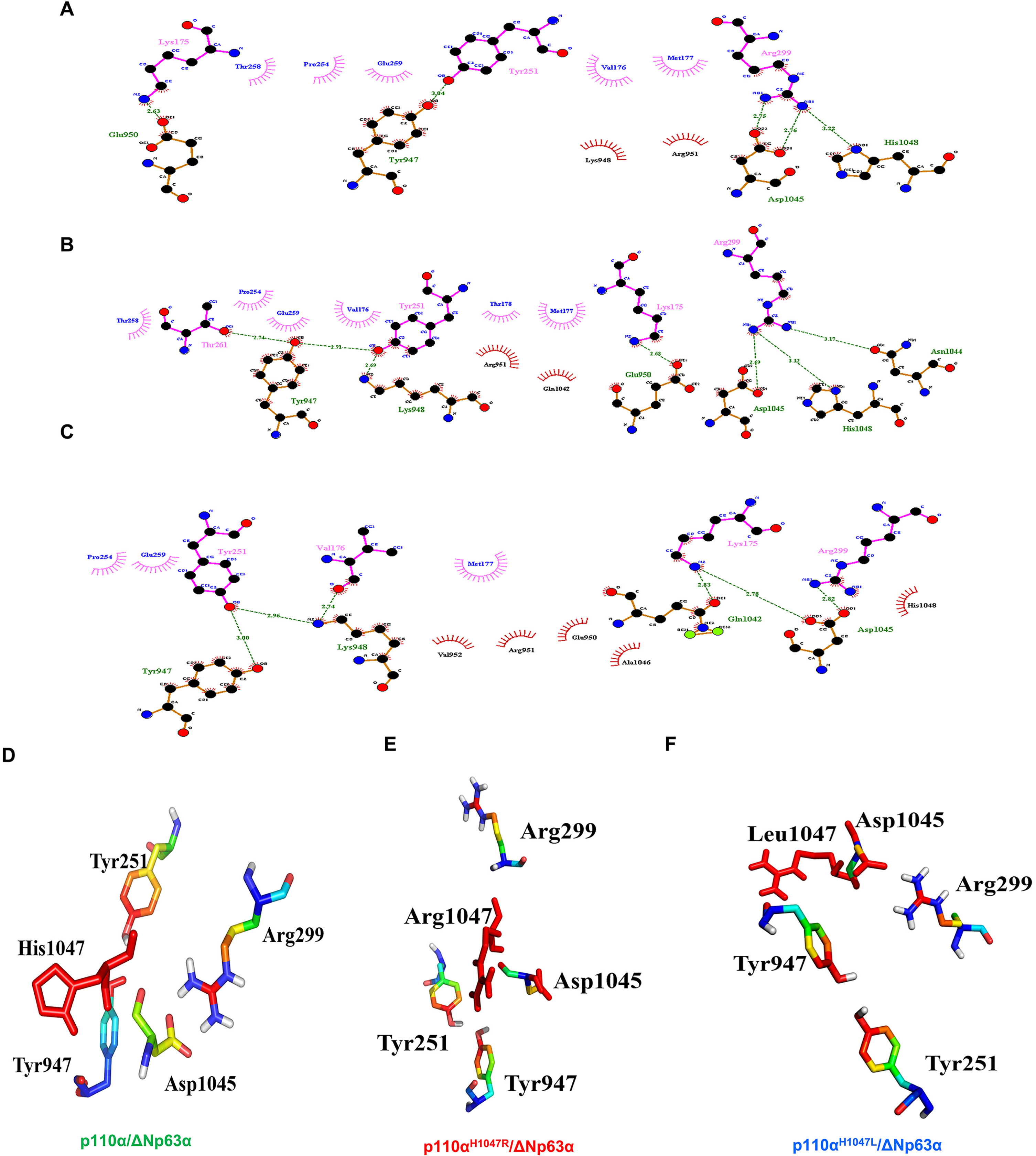
3.4Hydrogen bonding in the p110α/ΔNp63α complex
Figure 5 shows the H-bonds in the p110α/ΔNp63α complex for the WT and both mutant systems. All residues involved in the binding between p110α and ΔNp63α are present in the WT (Fig. 5A), as well as in the p.H1047R (Fig. 5B) and p.H1047L (Fig. 5C) mutant systems. In the WT, Arg299 interacts with Asp1045, while Tyr947 bonds with Tyr261. In the p.H1047R mutant, Arg299 forms a bond with Asp1045, while Tyr947 bonds with Thr261. In the p.H1047L mutant, Asp1045 interacts with Arg299, and Tyr947 interacts with Tyr251. The residues Tyr947, Lys948, Glu950, Asn1044, Asp1045, and His1048 of PIK3CA are involved in H-bonding with the DBD of ΔNp63α. The common residues in the H-bond between the p110α/ΔNp63α complex in the WT and both mutant models are Tyr947-Tyr251 and Asp1045-Arg299. These findings suggest that mutations in the linker motif 797–1068 could impair interdomain allostery in the p110α/ΔNp63α complex through the hydrophobic cleft and H-bonding. Validation through further computational studies is necessary.
Fig. 5
A model of the p110α/ΔNp63α complex interaction. The residues involved in the binding interaction between p110α and ΔNp63α are shown in wild-type (WT) (A), the p.H1047R mutant system (B), and the p.H1047L mutant system (C). Tyr947, Lys948, Glu950, Asn1044, Asp1045, and His1048 of PIK3CA participate in the H-bonding with the ΔNp63α DBD domain. The residues present in the H-bond between the p110α/ΔNp63α complex in WT and both hotspot mutant models are Tyr947-Tyr251 and Asp1045-Arg299 (D-F).
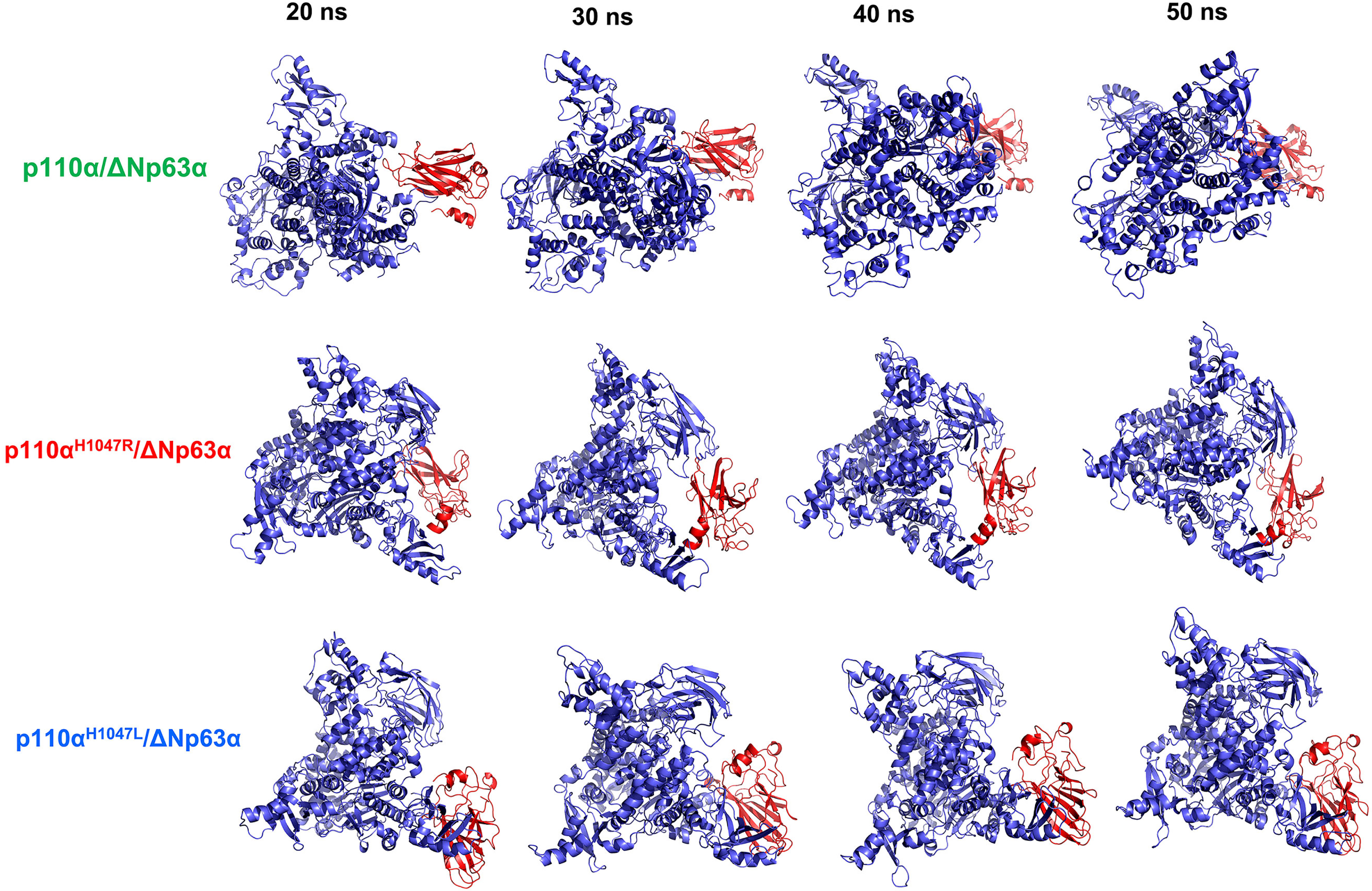
3.5Molecular dynamics simulation analysis of the WT and hotspot mutants of the p110α/δnp63α complex
A molecular dynamics simulation was conducted to evaluate the structural stability of the WT and the two hotspot mutant models of the p110α/ΔNp63α complex. Four snapshots were selected to examine the effect of mutations on the structure and function of the complex. The tertiary structural stability of the three models at 20, 30, 40, and 50 ns is shown in Fig. 6. The results indicate a continuous decline in compactness and conformational changes in the mutant structures until the end of the simulation, which is supported by the DSSP analysis. The p110αH1047R/L structures both exhibited an increased β-sheet content in specific regions of PIK3CA residues over the 20–50 ns time frame. The study findings suggest that the suppression effects between the ΔNp63α domain and the kinase domain of PIK3CA become closer at different times, with a visible shift in the orientation of ΔNp63α, an increase in its compactness, and motion towards the core binding site. Among all the models examined, the p.H1047L mutant exhibited the most significant conformational change.
Fig. 6
Stability of the tertiary structure of the wild-type (WT) and hotspot mutant forms of the p110α/ΔNp63α complex. The structures of the p110α (blue color) and ΔNp63α (red color) domains were derived from simulations of 20, 30, 40, and 50 ns. The ΔNp63α domain of the mutant forms showed increased interaction over various time intervals but showed differences in interaction sites compared to the WT.
4Discussion
Here, for the first time, we suggest that the expression of either p110αH1047R or p110αH1047L gradually increases during the development of MBC tumors. The findings indicate that both p110αH1047R and p110αH1047L inhibit the expression of ΔNp63α, which could significantly contribute to the oncogenic Akt1-mediated cell motility and cancer metastasis. The p110αH1047L-activated PI3K/Akt and tyrosine kinase receptors signaling pathway results in a greater increase in cell migration, scattered cell growth, and invasion in MBC compared to the p110αH1047R-activated pathways. The functional effect of p110αH1047R/L was dependent on human breast tumor cells and was able to suppress several malignancy parameters [11, 42]. In our study, we found that the p.H1047L mutation in the p110α protein caused a structural change in the helix motif, altering the activation-loop conformation and disrupting the interaction site between p110α and ΔNp63α in an AKT-dependent manner. Our MD simulation analysis showed that both p110αH1047R and p110αH1047L mutations resulted in the loss of H-bonds in the protein core and disturbed proper folding due to their location on the protein surface. This is the first report that sheds light on the structural changes resulting from these disease-causing mutations in the context of the p110α/ΔNp63α complex [43]. The mutation at that residue could disturb the interactions with ΔNp63α or the other parts of the protein [44, 45]. Additionally, the mutations at p110αH1047R/L may result in alterations in the hydrophobic interactions between the p110α protein and other molecules due to their differing hydrophobicity. This finding was further supported by the 3D model results, which showed the mutations’ placement on the active site of the p110α protein [46]. Thus, it is crucial to consider their impact on downstream interactions, such as the PI3K/ERK/MAPK signaling pathways. The results of this study also suggest that the tyrosine kinase receptor is affected by these mutations. Further investigation is needed to determine which specific tyrosine kinase receptor is more likely to be involved in the p110α/ΔNp63α complex. These findings highlight the potential for the development of new treatments for TNBC by targeting both the PI3K downstream signaling pathway and the tyrosine kinase receptor.
The p110αH1047R/L mutations are considered common, and impact the lipid membrane composition and the activity of the mutated systems, particularly p.H1047L, by altering the substrate interactions with the kinase-bound PI3Ks compared to the WT PIK3CA [11, 21, 28, 47]. The hotspot mutations offer a promising target for precision therapeutic approaches and improved patient management for the MBC treatment [48]. Studies have revealed the role of ΔNp63α proteins as metastasis inhibitors in a different type of BC [10, 14, 49]. There is evidence suggesting that the reduced expression of ΔNp63α is a result of alternative splicing and is linked to elevated cellular motility and cancer metastasis. This phenomenon is observed in the presence of induced oncogenic signaling pathways, such as PI3K, Ras, Her2, and Akt [10, 50–52]. Downregulation of the tumor-suppressor protein p63 is caused by the activation of oncogenic hotspot mutations in the PI3KCA gene, which activate the PI3K signaling and PI3K/Akt and tyrosine kinase receptors signaling pathways, and trigger tumorigenicity through various pathways such as Akt-FOXO, Akt-mTOR, Akt-IKK, and Akt-GSK3β [53, 54].
According to this study, the ERK/p63 signaling pathways induced by the p110αH1047L mutation transactivate genes involved in the development of MBC. The ERK checkpoint targets molecules involved in regulating cell migration, and its decrease has been associated with a poor prognosis in patients with urothelial cancer [55, 56]. The study shows that a decrease in p-Erk1/2 (not Erk1/2) effectively reversed the cell migration caused by the p63 ablation [55, 57]. Phospho-ERK1/2 is activated through the AKT signaling pathway and further evaluations are needed to determine if p110αH1047L acts as a downstream molecule in the AKT pathway [58–61]. Negative modulation of these epigenetic regulators may provide a promising approach for the ERK-targeted therapy [62].
This pathway clinical trials are assessing the efficacy of tyrosine kinase inhibitors in cancer treatments [63, 64]. In the TNBC, the dysregulation of the tyrosine kinase receptor pathway often leads to the promotion of tumor growth and oncogenic transformation through the activation of downstream signaling pathways, such as Akt-FOXO, Akt-mTOR, Akt-IKK, and Akt-GSK3β [65–68]. Mutations in the PI3KCA gene, particularly the p110αH1047R/L mutations, activate the PI3K/Akt and tyrosine kinase receptors signaling pathway and trigger oncogenic transformation in TNBC cells [11, 19, 21]. These mutations also decrease the inhibitory effect of the tumor-suppressor protein ΔNp63α on the p110α kinase domain, enhancing the downstream signaling activity and promoting the tumorigenicity [69]. Overall, a deeper understanding of the tyrosine kinase receptors signaling pathway and its role in cancer could lead to the development of more effective and targeted therapies for cancer patients [64].
The activation of MBC signaling pathways, such as PI3K and mTOR, was shown to increase cancer cell motility and promote tumor metastasis through the repression of ΔNp63α expression in an in vitro assay [70]. The results of the present study exhibited the importance of the dysregulation of the ΔNp63α-p110α pathway for TNBC metastasis. However, in vivo models are necessary to decipher the specific contribution of each p63 isoform, as well as isoform interactions with the other members of the p110α and p53 family, to develop strategies for more effective treatments and prevention of BC metastasis [19]. Considering the resistance of FOXO3a-ΔNp63α to Akt phosphorylation, and consequently its role as a constitutive activator of p110αH1047R/L in the MBC, further research is essential to confirm triggering the contribution of the ΔNp63α-p110α to susceptibility to MBC in Akt/FOXO signaling pathways [19, 71]. The FOXO3a could be analyzed in ΔNp63α expression through the p63 knockdown in the MBC [11, 19].
Further research is necessary to investigate the impact of p110αH1047R/L on angiogenesis, as well as their effects on FOXO3a, AKT, and ERK1/2 expression and phosphorylation in MBC cell lines and mouse xenograft models. The role of p110α in cancer and its impact on tumor growth and metastasis could then be better understood. Clinical studies are necessary to confirm the prognostic value of p110α mutation levels in MBC and determine if more aggressive therapy is required for certain patients. The correlation between PI3KCA and p63 expression with ΔNp63α downregulation in human breast cancers highlights the importance of the ΔNp63α-p110α signaling pathway in cancer development [72]. To comprehensively relate p110α expression to the metastatic potential and advanced disease, it is recommended to incorporate a non-metastatic PIK3CA model in future research endeavors. This approach will allow for a more thorough exploration of the role of p110α in cancer progression and enhance our understanding of its clinical implications. In addition, in vivo model research is needed to determine the effect of HER2 activation on cell motility and tumor metastasis by suppressing ΔNp63α expression. The formation of the p110α/ΔNp63α complex in BT-549 cells can be confirmed by conducting in vitro co-immunoprecipitation assays using WT or over-expressed p110αH1047L/R hotspot mutations.
In conclusion, this study sheds light on the significance of the ΔNp63α-p110α pathway in the development of TNBC and the possible involvement of p110αH1047R/L in reducing ΔNp63α’s inhibitory effect on p110α. The results also underscore the significance of downstream signaling pathways of tyrosine kinase receptors in promoting tumorigenesis. Further investigations are needed to deepen our understanding of the complex interplay between p110α PIK3CA/ΔNp63α and their functional specificity in cancer, and to devise new therapeutic approaches for TNBC.
Acknowledgments
The authors would like to extend their appreciation and acknowledge the support provided by the Department of Oncology and the Department of Pathology at the Affiliated Hospital of Southwest Medical University in Luzhou, China, during the conduct of this study.
Author contributions
All authors contributed to the study’s conception and design. Material preparation, data collection, and analysis were performed by WM, XH, YZ, and YZ. MDS, HH, and MM performed the molecular dynamic and bioinformatics analysis. The first draft of the manuscript was written by WH, XH, and SI. QLW, QW, and QJW edited and revised the paper. All authors have read and agreed to the published version of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Data availability
The raw data and codes utilized for the MD simulations of p110αH1047R/L mutation’s conformational diversity on three-dimensional models of the p110α/ΔNp63α complex are available on zenodo ( https://zenodo.org/deposit?page=1&size=20). Our previous publications provide comprehensive information regarding the clinical predictive value of targeted genes and their evaluation.
Ethical considerations
This study was approved by the Ethics Review Board of the Affiliated Hospital of Southwest Medical University (Approval No. KY2019041) following the Declaration of Helsinki (1983 Revision). Furthermore, all laboratory assessments were conducted by the local guidelines of the Ethics Committee of the Oncology Department at the Affiliated Hospital of Southwest Medical University, Sichuan, China.
Funding
This research received partial support from the Southwest Medical University Grant and the Research Foundation of the Science and Technology Department of Sichuan Province (No, 18080), as well as the Talent Scientific Research Project of Zhejiang Shuren University (No, KXJ1723104). The funders did not play a role in the study design, data collection, analysis, decision to publish, or preparation of the manuscript. No additional external funding was received.
References
[1] | Lukasiewicz S , Czeczelewski M , Forma A , Baj J , Sitarz R , Stanislawek A Breast cancer-epidemiology, risk factors, classification, prognostic markers, and current treatment strategies-an updated review, Cancers (Basel) (2021) ;13: (17). DOI: 10.3390/cancers13174287. |
[2] | Howard FM , Olopade OI Epidemiology of triple-negative breast cancer: A review, Cancer J (2021) ;27: (1):8–16. DOI: 10.1097/PPO.0000000000000500. |
[3] | Radenkovic S , Konjevic G , Isakovic A , Stevanovic P , Gopcevic K , Jurisic V HER2-positive breast cancer patients: Correlation between mammographic and pathological findings, Radiat Prot Dosimetry (2014) ;162: (1-2):125–8. DOI: 10.1093/rpd/ncu243. |
[4] | Baselga J Targeting the phosphoinositide-3 (PI3) kinase pathway in breast cancer, Oncologist (2011) ;16: (Suppl 1):12–9. DOI: 10.1634/theoncologist.2011-S1-12. |
[5] | Zardavas D , Fumagalli D , Loi S Phosphatidylinositol 3-kinase/AKT/mammalian target of rapamycin pathway inhibition: A breakthrough in the management of luminal (ER+/HER2-) breast cancers? Curr Opin Oncol (2012) ;24: (6):623–34. DOI: 10.1097/CCO.0b013e328358a2b5. |
[6] | Coughlin CM , Johnston DS , Strahs A , Burczynski ME , Bacus S , Hill J , et al. Approaches and limitations of phosphatidylinositol-3-kinase pathway activation status as a predictive biomarker in the clinical development of targeted therapy, Breast Cancer Res Treat (2010) ;124: (1):1–11. DOI: 10.1007/s10549-010-1108-4. |
[7] | Bartlett JM Biomarkers and patient selection for PI3K/Akt/mTOR targeted therapies: Current status and future directions, Clin Breast Cancer (2010) ;10: (Suppl 3):S86–95. DOI: 10.3816/CBC.2010.s.017. |
[8] | Gatti V , Bongiorno-Borbone L , Fierro C , Annicchiarico-Petruzzelli M , Melino G , Peschiaroli A p63 at the Crossroads between Stemness and Metastasis in Breast Cancer, Int J Mol Sci (2019) ;20: (11). DOI: 10.3390/ijms20112683. |
[9] | Nekulova M , Holcakova J , Gu X , Hrabal V , Galtsidis S , Orzol P , et al. DeltaNp63alpha expression induces loss of cell adhesion in triple-negative breast cancer cells, BMC Cancer (2016) ;16: (1):782. DOI: 10.1186/s12885-016-2808-x. |
[10] | Niu M , He Y , Xu J , Ding L , He T , Yi Y , et al. Noncanonical TGF-beta signaling leads to FBXO3-mediated degradation of DeltaNp63alpha promoting breast cancer metastasis and poor clinical prognosis, PLoS Biol (2021) ;19: (2):e3001113. DOI: 10.1371/journal.pbio.3001113. |
[11] | Hu L , Liang S , Chen H , Lv T , Wu J , Chen D , et al. DeltaNp63alpha is a common inhibitory target in oncogenic PI3K/Ras/Her2-induced cell motility and tumor metastasis, Proc Natl Acad Sci U S A (2017) ;114: (20):E3964–73. DOI: 10.1073/pnas.1617816114. |
[12] | Di Giacomo V , Tian TV , Mas A , Pecoraro M , Batlle-Morera L , Noya L , et al. DeltaNp63alpha promotes adhesion of metastatic prostate cancer cells to the bone through regulation of CD82, Oncogene (2017) ;36: (31):4381–92. DOI: 10.1038/onc.2017.42. |
[13] | Kumar S , Wilkes DW , Samuel N , Blanco MA , Nayak A , Alicea-Torres K , et al. DeltaNp63-driven recruitment of myeloid-derived suppressor cells promotes metastasis in triple-negative breast cancer, J Clin Invest (2018) ;128: (11):5095–109. DOI: 10.1172/JCI99673. |
[14] | Bergholz J , Zhang Y , Wu J , Meng L , Walsh EM , Rai A , et al. DeltaNp63alpha regulates Erk signaling via MKP3 to inhibit cancer metastasis, Oncogene (2014) ;33: (2):212–24. DOI: 10.1038/onc.2012.564. |
[15] | Barbieri CE , Barton CE , Pietenpol JA Delta Np63 alpha expression is regulated by the phosphoinositide 3-kinase pathway, The Journal of biological chemistry (2003) ;278: (51):51408–14. DOI: 10.1074/jbc.M309943200. |
[16] | Holcakova J , Nekulova M , Orzol P , Nenutil R , Podhorec J , Svoboda M , et al. DeltaNp63 activates EGFR signaling to induce loss of adhesion in triple-negative basal-like breast cancer cells, Breast Cancer Res Treat (2017) ;163: (3):475–84. DOI: 10.1007/s10549-017-4216-6. |
[17] | Dirican E , Kaya Z , Gullu G , Peker I , Ozmen T , Gulluoglu BM , et al. Detection of PIK3CA gene mutations with HRM analysis and association with IGFBP-5 expression levels in breast cancer, Asian Pacific Journal of Cancer Prevention: APJCP (2014) ;15: (21):9327–33. DOI: 10.7314/apjc2014.15.21.9327. |
[18] | Dornan GL , Burke JE Molecular mechanisms of human disease mediated by oncogenic and primary immunodeficiency mutations in class IA phosphoinositide 3-kinases, Front Immunol (2018) ;9: :575.. DOI: 10.3389/fimmu.2018.00575. |
[19] | Wang Y , Li J , Gao Y , Luo Y , Luo H , Wang L , et al. Hippo kinases regulate cell junctions to inhibit tumor metastasis in response to oxidative stress, Redox Biol (2019) ;26: :1012–33. DOI: 10.1016/j.redox.2019.101233. |
[20] | Wang Q , Liu P , Spangle JM , Von T , Roberts TM , Lin NU , et al. PI3K-p110alpha mediates resistance to HER2-targeted therapy in HER2+, PTEN-deficient breast cancers, Oncogene (2016) ;35: (27):3607–12. DOI: 10.1038/onc.2015.406. |
[21] | Cheng H , Liu P , Ohlson C , Xu E , Symonds L , Isabella A , et al. PIK3CA(HR)- and Her2-initiated mammary tumors escape PI3K dependency by compensatory activation of MEK-ERK signaling, Oncogene (2016) ;35: (23):2961–70. DOI: 10.1038/onc.2015.377. |
[22] | Fritsch R , de Krijger I , Fritsch K , George R , Reason B , Kumar MS , et al. RAS and RHO families of GTPases directly regulate distinct phosphoinositide 3-kinase isoforms, Cell (2013) ;153: (5):1050–63. DOI: 10.1016/j.cell.2013.04.031. |
[23] | Wang R , Peng C , Song J , Hua Y , Wu Q , Deng L , et al. Downregulated RRS1 inhibits invasion and metastasis of BT549 through RPL11-c-Myc-SNAIL axis, Int J Oncol (2022) ;60: (3). DOI: 10.3892/ijo.2022.5323. |
[24] | Bilir B , Kucuk O , Moreno CS Wnt signaling blockage inhibits cell proliferation and migration, and induces apoptosis in triple-negative breast cancer cells, J Transl Med (2013) ;11: :280. DOI: 10.1186/1479-5876-11-280. |
[25] | Ho SN , Hunt HD , Horton RM , Pullen JK , Pease LR Site-directed mutagenesis by overlap extension using the polymerase chain reaction, Gene (1989) ;77: (1):51–9. DOI: 10.1016/0378-1119(89)90358-2. |
[26] | Yang Q , Ni L , Imani S , Xiang Z , Hai R , Ding R , et al. Anlotinib suppresses colorectal cancer proliferation and angiogenesis via inhibition of AKT/ERK signaling cascade, Cancer Manag Res (2020) ;12: :4937–48. DOI: 10.2147/CMAR.S252181. |
[27] | Wei C , Yang L , Cheng J , Imani S , Fu S , Lv H , et al. A novel homozygous variant of GPR98 causes usher syndrome type IIC in a consanguineous Chinese family by next generation sequencing, BMC Med Genet (2018) ;19: (1):99. DOI: 10.1186/s12881-018-0602-0. |
[28] | Cheng J , Fu S , Wei C , Tania M , Khan MA , Imani S , et al. Evaluation of PIK3CA mutations as a biomarker in Chinese breast carcinomas from Western China, Cancer Biomarkers: Section A of Disease Markers (2017) ;19: (1):85–92. DOI: 10.3233/cbm-160380. |
[29] | Imani S , Hosseinifard H , Cheng J , Wei C , Fu J Prognostic value of EMT-inducing Transcription Factors (EMT-TFs) in metastatic breast cancer: A systematic review and meta-analysis, Sci Rep (2016) ;6: :28587. DOI: 10.1038/srep28587. |
[30] | Khan MA , Tania M , Wei C , Mei Z , Fu S , Cheng J , et al. Thymoquinone inhibits cancer metastasis by downregulating TWIST1 expression to reduce epithelial to mesenchymal transition, Oncotarget (2015) ;6: (23):19580–91. DOI: 10.18632/oncotarget.3973. |
[31] | Yi ZH , Luther Y , Xiong GH , Ni YL , Yun F , Chen J , et al. Association between diabetes mellitus and lung cancer: Meta-analysis, Eur J Clin Invest. (2020) ;50: (10):e13332. DOI: 10.1111/eci.13332. |
[32] | Xie F , Ding R-L , He W-F , Liu Z-J-L , Fu S-Z , Wu J-B , et al. In vivo antitumor effect of endostatin-loaded chitosan nanoparticles combined with paclitaxel on Lewis lung carcinoma, Drug Delivery (2017) ;24: (1):1410–8. |
[33] | He W , Yang G , Liu S , Maghsoudloo M , Shasaltaneh MD , Kaboli PJ , et al. Comparative mRNA/micro-RNA co-expression network drives melanomagenesis by promoting epithelial-mesenchymal transition and vasculogenic mimicry signaling, Transl Oncol. (2021) ;14: (12):101237. DOI: 10.1016/j.tranon.2021.101237. |
[34] | Imani S , Ijaz I , Shasaltaneh MD , Fu S , Cheng J , Fu J In silico data analyses of the hotspot mutations of CHM gene in choroideremia disease, Data Brief (2018) ;18: :1217–23. 10.1016/j.dib.2018.04.023. |
[35] | Dominguez C , Boelens R , Bonvin AM HADDOCK: A protein-protein docking approach based on biochemical or biophysical information, J Am Chem Soc (2003) ;125: (7):1731–7. DOI: 10.1021/ja026939x. |
[36] | Wallace AC , Laskowski RA , Thornton JM LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions, Protein Eng (1995) ;8: (2):127–34. DOI: 10.1093/protein/8.2.127. |
[37] | Imani S , Cheng J , Shasaltaneh MD , Wei C , Yang L , Fu S , et al. Genetic identification and molecular modeling characterization reveal a novel PROM1 mutation in Stargardt4-like Macular Dystrophy. Oncotarget. 2017;Profreading. |
[38] | Imani S , Ijaz I , Shasaltaneh MD , Fu S , Cheng J , Fu J Molecular genetics characterization, homology modeling and molecular dynamics of the mutation of CHM Gene: Association studies with choroideremia. Human Mutation. 2017;Under review.. |
[39] | Imani S , Ijaz I , Shasaltaneh MD , Fu S , Cheng J , Fu J Molecular genetics characterization and homology modeling of the CHM gene mutation: A study on its association with choroideremia, Mutat Res Rev Mutat Res (2018) ;775: :39–50. 10.1016/j.mrrev.2018.02.001. |
[40] | Zhou J , Imani S , Shasaltaneh MD , Liu S , Lu T , Fu J PIK3CA hotspot mutations HR and HL sensitize breast cancer cells to thymoquinone treatment by regulating the PI3K/Akt1 pathway, Mol Biol Rep (2022) ;49: (3):1799–816. DOI: 10.1007/s11033-021-06990-x. |
[41] | Dunn S , Lombardi O , Lukoszek R , Cowling VH Oncogenic PIK3CA mutations increase dependency on the mRNA cap methyltransferase, RNMT, in breast cancer cells, Open Biol (2019) ;9: (4):190052. DOI: 10.1098/rsob.190052. |
[42] | Wan G , Pehlke C , Pepermans R , Cannon JL , Lidke D , Rajput A The HR point mutation in p110 alpha changes the morphology of human colon HCT116 cancer cells, Cell Death Discov (2015) ;1: :15044. DOI: 10.1038/cddiscovery.2015.44. |
[43] | Sharma J , Bhardwaj V , Purohit R Structural perturbations due to mutation (HR) in phosphoinositide-3-kinase (PI3Kalpha) and its involvement in oncogenesis: An in Silico Insight, ACS Omega (2019) ;4: (14):15815–23. DOI: 10.1021/acsomega.9b01439. |
[44] | Gkeka P , Evangelidis T , Pavlaki M , Lazani V , Christoforidis S , Agianian B , et al. Investigating the structure and dynamics of the PIK3CA wild-type and HR oncogenic mutant, PLoS Comput Biol. (2014) ;10: (10):e1003895. DOI: 10.1371/journal.pcbi.1003895. |
[45] | Lee KH , Hwang HJ , Noh HJ , Shin TJ , Cho JY Somatic mutation of PIK3CA (HR) is a common driver mutation hotspot in canine mammary tumors as well as human breast cancers, Cancers (Basel) (2019) ;11: (12). DOI: 10.3390/cancers11122006. |
[46] | Spangle JM , Von T , Pavlick DC , Khotimsky A , Zhao JJ , Roberts TM PIK3CA C-terminal frameshift mutations are novel oncogenic events that sensitize tumors to PI3K-alpha inhibition, Proc Natl Acad Sci U S A (2020) ;117: (39):24427–33. DOI: 10.1073/pnas.2000060117. |
[47] | Rasti AR , Guimaraes-Young A , Datko F , Borges VF , Aisner DL , Shagisultanova E PIK3CA mutations drive therapeutic resistance in human epidermal growth factor receptor 2-positive breast cancer, JCO Precis Oncol (2022) ;6: :e2100370. DOI: 10.1200/PO.21.00370. |
[48] | Garay JP , Smith R , Devlin K , Hollern DP , Liby T , Liu M , et al. Sensitivity to targeted therapy differs between HER2-amplified breast cancer cells harboring kinase and helical domain mutations in PIK3CA, Breast Cancer Res (2021) ;23: (1):81. DOI: 10.1186/s13058-021-01457-0. |
[49] | Gao G , Chen J , Wang D , Li Q , Yang X , Wang J , et al. TGF-beta1 facilitates TAp63alpha protein lysosomal degradation to promote pancreatic cancer cell migration, Biology (Basel) (2021) ;10: (7). DOI: 10.3390/biology10070597. |
[50] | Cai J , Chen S , Yi M , Tan Y , Peng Q , Ban Y , et al. DeltaNp63alpha is a super enhancer-enriched master factor controlling the basal-to-luminal differentiation transcriptional program and gene regulatory networks in nasopharyngeal carcinoma. Carcinogenesis. 2019. DOI:10.1093/carcin/bgz203. |
[51] | Stacy AJ , Zhang J , Craig MP , Hira A , Dole N , Kadakia MP TIP60 up-regulates DeltaNp63alpha to promote cellular proliferation, The Journal of biological chemistry (2019) ;294: (45):17007–16. DOI: 10.1074/jbc.RA119.010388. |
[52] | Rekhtman N , Paik PK , Arcila ME , Tafe LJ , Oxnard GR , Moreira AL , et al. Clarifying the spectrum of driver oncogene mutations in biomarker-verified squamous carcinoma of lung: Lack of EGFR/KRAS and presence of PIK3CA/AKT1 mutations, Clinical Cancer Research: An Official Journal of the American Association for Cancer Research (2012) ;18: (4):1167–76. DOI: 10.1158/1078-0432.ccr-11-2109. |
[53] | Wu G , Nomoto S , Hoque MO , Dracheva T , Osada M , Lee CC , et al. DeltaNp63alpha and TAp63alpha regulate transcription of genes with distinct biological functions in cancer and development, Cancer Research (2003) ;63: (10):2351–7. |
[54] | Goyal B , Duncavage EJ , Martinez D , Lewis JS Jr , Chernock RD Next-generation sequencing of salivary high-grade neuroendocrine carcinomas identifies alterations in RB1 and the mTOR pathway, Exp Mol Pathol (2014) ;97: (3):572–8. DOI: 10.1016/j.yexm2014.10.011. |
[55] | Cook SJ , Lochhead PA ERK5 signalling and resistance to ERK1/2 pathway therapeutics: The path less travelled? Front Cell Dev Biol97 (2022) ;10: :839997. DOI: 10.3389/fcell.2022.839997. |
[56] | Sale MJ , Balmanno K , Cook SJ Resistance to ERK1/2 pathway inhibitors; sweet spots, fitness deficits and drug addiction, Cancer Drug Resist (2019) ;2: (2):365–80. DOI: 10.20517/cdr.2019.14. |
[57] | Roskoski R Jr Targeting oncogenic Raf protein-serine/threonine kinases in human cancers, Pharmacol Res (2018) ;135: :239–58. DOI: 10.1016/j.phrs.2018.08.013. |
[58] | Bos JL Ras oncogenes in human cancer: A review, Cancer Research (1989) ;49: (17):4682–9. |
[59] | Li J , Wang R , Hu X , Gao Y , Wang Z , Li J , et al. Activated MEK/ERK pathway drives widespread and coordinated overexpression of UHRF1 and DNMT1 in cancer cells, Sci Rep (2019) ;9: (1):907. DOI: 10.1038/s41598-018-37258-3. |
[60] | Jones PA , Issa JP , Baylin S Targeting the cancer epigenome for therapy, Nat Rev Genet (2016) ;17: (10):630–41. DOI: 10.1038/nrg.2016.93. |
[61] | Steelman LS , Pohnert SC , Shelton JG , Franklin RA , Bertrand FE , McCubrey JA JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis, Leukemia (2004) ;18: (2):189–218. DOI: 10.1038/sj.leu.2403241. |
[62] | Yuan Z , Yu X , Chen W , Chen D , Cai J , Jiang Y , et al. Epigenetic silencing and tumor suppressor gene of HAND2 by targeting ERK signaling in colorectal cancer, Cell Commun Signal (2022) ;20: (1):111. DOI: 10.1186/s12964-022-00878-4. |
[63] | Ediriweera MK , Tennekoon KH , Samarakoon SR Role of the PI3K/AKT/mTOR signaling pathway in ovarian cancer: Biological and therapeutic significance. Semin Cancer Biol (2019) ;59: :147–60. DOI: 10.1016/j.semcancer.2019.05.012. |
[64] | Alzahrani AS PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside, Semin Cancer Biol (2019) ;59: :125–32. DOI: 10.1016/j.semcancer.2019.07.009. |
[65] | Schuchardt S , Borlak J Quantitative mass spectrometry to investigate epidermal growth factor receptor phosphorylation dynamics, Mass Spectrom Rev (2008) ;27: (1):51–65. DOI: 10.1002/mas.20155. |
[66] | Butti R , Das S , Gunasekaran VP , Yadav AS , Kumar D , Kundu GC Receptor tyrosine kinases (RTKs) in breast cancer: Signaling, therapeutic implications and challenges, Mol Cancer. (2018) ;17: (1):34. DOI: 10.1186/s12943-018-0797-x. |
[67] | Gujral TS , Karp RL , Finski A , Chan M , Schwartz PE , MacBeath G , et al. Profiling phospho-signaling networks in breast cancer using reverse-phase protein arrays, Oncogene (2013) ;32: (29):3470–6. DOI: 10.1038/onc.2012.378. |
[68] | Duda P , Akula SM , Abrams SL , Steelman LS , Martelli AM , Cocco L , et al. Targeting GSK3 and associated signaling pathways involved in cancer, Cells (2020) :9: (5). DOI: 10.3390/cells9051110. |
[69] | He Y , Sun MM , Zhang GG , Yang J , Chen KS , Xu WW , et al. Targeting PI3K/Akt signal transduction for cancer therapy, Signal Transduct Target Ther (2021) ;6: (1):425. DOI: 10.1038/s41392-021-00828-5. |
[70] | Karami Fath M , Akhavan Masouleh R , Afifi N , Loghmani S , Tamimi P , Fazeli A , et al. PI3K/AKT/mTOR signaling pathway modulation by circular RNAs in breast cancer progression, Pathol Res Pract. (2022) ;241: :154279. DOI: 10.1016/j.pr2022.154279. |
[71] | Nasrollahzadeh A , Momeny M , Fasehee H , Yaghmaie M , Bashash D , Hassani S , et al. Anti-proliferative activity of disulfiram through regulation of the AKT-FOXO axis: A proteomic study of molecular targets, Biochim Biophys Acta Mol Cell Res (2021) ;1868: (10):119087. DOI: 10.1016/j.bbamcr.2021.119087. |
[72] | Chong X , Chen J , Zheng N , Zhou Z , Hai Y , Chen S , et al. PIK3CA mutations-mediated downregulation of circLHFPL2 inhibits colorectal cancer progression via upregulating PTEN, Mol Cancer (2022) ;21: (1):118. DOI: 10.1186/s12943-022-01531-x. |


