
Impact of RARα and miR-138 on retinoblastoma etoposide resistance
Abstract
BACKGROUND:
Retinoblastoma (RB) is the most common childhood eye cancer. Chemotherapeutic drugs such as etoposide used in RB treatment often cause massive side effects and acquired drug resistances. Dysregulated genes and miRNAs have a large impact on cancer progression and development of chemotherapy resistances.
OBJECTIVE:
This study was designed to investigate the involvement of retinoic acid receptor alpha (RARα) in RB progression and chemoresistance as well as the impact of miR-138, a potential RARα regulating miRNA.
METHODS:
RARα and miR-138 expression in etoposide resistant RB cell lines and chemotherapy treated patient tumors compared to non-treated tumors was revealed by Real-Time PCR. Overexpression approaches were performed to analyze the effects of RARα on RB cell viability, apoptosis, proliferation and tumorigenesis. Besides, we addressed the effect of miR-138 overexpression on RB cell chemotherapy resistance.
RESULTS:
A binding between miR-138 and RARα was shown by dual luciferase reporter gene assay. The study presented revealed that RARα is downregulated in etoposide resistant RB cells, while miR-138 is endogenously upregulated. Opposing RARα and miR-138 expression levels were detectable in chemotherapy pre-treated compared to non-treated RB tumor specimen. Overexpression of RARα increases apoptosis levels and reduces tumor cell growth of aggressive etoposide resistant RB cells in vitro and in vivo. Overexpression of miR-138 in chemo-sensitive RB cell lines partly enhances cell viability after etoposide treatment.
CONCLUSIONS:
Our findings show that RARα acts as a tumor suppressor in retinoblastoma and is downregulated upon etoposide resistance in RB cells. Thus, RARα may contribute to the development and progression of RB chemo-resistance.
1Introduction
Retinoblastoma is the most common primary intraocular tumor in childhood with an incidence of 1:16,000 livebirth, accounting for 2–4% of all childhood malignancies [1]. The mean age at diagnosis is 27 month for unilateral and 15 month for bilateral cases [2]. There are different mechanisms and several mutations involved in RB development which has been defined as a multi-step process from normal retinal tissue towards RB tumor [3]. Beside several potential target genes, RB is primarily initiated by the inactivation of the RB1 gene. In addition MYCN amplifications [4] and dysregulated microRNAs (miRs) [5, 6] play a role in RB development. By now, RB is highly curable in the developed countries, but delayed diagnosis may increase the mortality of RB by 70% [1] and untreated RBs often lead to extraocular spread of RB tumor cells into regional lymph nodes, bone and the CNS [7]. Treatment of RB is often accompanied by massive side effects induced by chemotherapeutics like vincristine, etoposide and carboplatin, used in the combined RB VEC-therapy. Therefore, the search of clinically reliable biomarkers for early RB diagnosis/detection and the development of new strategies to improve RB therapies are main challenges.
Retinoic acid receptors alpha (RARα) plays a role in human tumors such as acute promyelocytic leukemia (APL), esophageal carcinoma [8] and retinoblastoma [9]. A PML-RARα fusion protein can be found in most APL cases [10], in which a successful beneficial retinoid based therapy has been achieved in clinical evaluations [11]. Moreover, RARs are involved in RA/BMP4 mediated induction of apoptosis in the human retinoblastoma cell line WERI-Rb1 [9].
Retinoids, including all trans retinoic acids (RA) are signaling molecules binding to retinoic acid receptors (RARs), which are composed of three subtypes: RARα (NR1B1), RARβ (NR1B2), and RARγ (NR1B3), all belonging to the nuclear hormone receptor superfamily [11, 12]. RA signaling contributes to crucial roles in a wide range of biological processes and communication networks. These events comprise development of chordate animals [13], proliferation, differentiation and apoptosis and are mediated through RAR transcription factors predominantly acting as heterodimers with nuclear rexinoid receptors (RXR) [11]. These RXR-RAR heterodimers regulate gene expression by binding to specific RA-responsive elements (RAREs) in the promotor region [14] leading to repression or activation of their target genes [15].
In this context several studies revealed the involvement of miRs and their target genes in RB development and progression [7].
MiRs are 18–24 nucleotides long noncoding RNA molecules, which negatively regulate post-transcriptional processes by repressing mRNA translation [16]. These molecules play key roles in mostly all biological processes, representing conserved binding sites for at least one miRNA in sixty percent of all human genes [17]. Dysregulated miRs have been described in all types of cancer with great impact on tumor progression [16, 18]. They can be classified as onco-miRs or tumor suppressor miRs depending on their function on the targeted mRNA [6]. A large amount of dysregulated miRs were connected to different functions in RB progression like proliferation, apoptosis, metastasis, hypoxia and chemoresistance [6]. Among others, RARα is a predicted target gene of miR-138 in Alzheimer’s disease [19]. MiR-138 was also described as a tumor suppressor in human RB by targeting pyruvate dehydrogenase kinase 1 (PDK1) resulting in increased apoptosis and decreased growth kinetics of RB cells [20]. Moreover, a dysregulation of miR-138 has been shown to contribute to drug resistance in multiple myeloma [21].
In this study, we examined the involvement of RARα and miR-138 in retinoblastoma chemotherapy resistance. We could show that RARα is downregulated in etoposide resistant RB cells, while its potentially regulating miR-138 is endogenously upregulated. Besides, the overexpression of RARα significantly inhibits proliferation and tumor formation of aggressive etoposide resistant RB cells in vitro and in vivo. The upregulation of miR-138 partly increases cell viability of chemo-sensitive RB cells treated with etoposide. Therefore, RARα is a promising candidate for RB tumor therapy.
2Materials and methods
2.1Human retina and retinoblastoma samples
Post mortem human retina samples from cornea donors and retinoblastoma samples were used for comparative expression studies. The Ethics Committee of the Medical Faculty of the University of Duisburg-Essen approved the use of human retina (approval # 06-30214) and retinoblastoma samples (approval # 14-5836-BO) for research conducted in the course of the study presented and written informed consent has been obtained from patients’ relatives or parents.
2.2Cell culture
The human RB cell line RB355, established and first described by Griegel et al. (1990) [22], and formerly donated by K. Heise, was kindly provided by Dr. H. Stephan. Y79 [23] and WERI-Rb1 [24], originally purchased from the Leibniz Institute DSMZ (German Collection of Microorganisms and Cell Cultures). The cell lines were cultivated as described previously [25].
The corresponding etoposide resistant RB cell lines RB355-Etop, Y79-Etop and WERI-Etop, were established and kindly provided by Dr H. Stephan. The cultivation of the etoposide resistant RB cell lines and Human embryonic kidney cells (HEK293-T) were described previously [25, 26].
2.3Plasmids and lentiviral expression vectors
To generate a lentiviral RARα expression vector (pLenti_CMV_RARa), the pDONR223_RARA_WT_V5 vector (#83018, Addgene, Cambridge, USA) [27] was used as gateway entry vector. RARα was finally cloned into the pLenti CMV Puro Dest vector (Addgene, Cambridge, USA) by the use of a Gateway LR Clonase II Enzyme Mix (Invitrogen, Karlsruhe, Germany), following the manufacturer‘s protocol. The empty pLenti CMV Puro Dest vector was used as control in all transduction experiments.
To generate a miR-138 expression vector (pSG5-miR-138), the human miR-138 sequence was amplified from cDNA of HEK293T cells by RT-PCR using the forward primer 5′-CGGAATTCTGGCTGTTCTCTGTCCTCTC-3′ and the reverse primer 5′-CGGGATCCAGTGGAGCATTTGTGTTGGG-3′ containing EcoRI and BamHI restriction sites. The PCR product was cloned into the pCRII-TOPO vector (Invitrogen, Karlsruhe, Germany), excised by digestion with EcoRI and BamHI and ligated into the pSG5 vector (#216201, Stratagene, La Jolla, USA) digested with the same restriction enzymes. The empty pSG5 vector was used as control in all transfection experiments.
To generate the wildtype and mutant miR-138 binding sites of the 3′-UTR of RARα, the human sequences were amplified from cDNA of HEK293T cells by RT-PCR and cloned into the pMIR-TK-RNL [28] vector. The following forward and reverse primers containing SpeI and SacI restriction sites (underlined; Table 1) were used. In order to generate the mutant binding site forward and reverse primers containing a SalI restriction site (bold; Table 1) were used in combination with the mentioned primers for the wildtype construct.
Table 1
Primer sequences for cloning
| RARα wildtype binding site 1 (RARα_WT_BSI) | 5′-CGACTAGTTGAGTGAGGCCCCTGGTC-3′ |
| 5′-CGGAGCTCGGAGGTTGAGGTTCCCCAG-3′ | |
| RARα wildtype binding site 2 (RARα_WT_BSII) | 5′-CGACTAGTGTCAGGGCCCACATCATCTA-3′ |
| 5′-CGGAGCTCGTGTGTGTTTGTGTGTGTGC-3′ | |
| RARα mutant binding site 1 (RARα_Mut_BSI) | 5′-TCACCACATCTTCATGTCGACAACGCCAGGACTTGG-3′ |
| 5′-CCAAGTCCTGGCGTTGTCGACATGAAGATGTGGTGA-3′ | |
| RARα mutant binding site 2 (RARα_Mut_BSII) | 5′-ACCCCCGGCCTCAGCGTCGACCCCCCATAGGGCCCC-3′ |
| 5′-GGGGCCCTATGGGGGGTCGACGCTGAGGCCGGGGGT-3′ |
The final PCR products were cloned into the pCRII-TOPO vector, excised by digestion with SpeI and SacI and ligated into the pMIR-TK-RNL vector digested with the same restriction enzymes. All vector constructs were verified by sequencing.
2.4Luciferase assay
The binding of miR-138 to the predicted binding sites in the 3′-UTR region of RARα was measured by luciferase activity. HEK293T cells were transiently co-transfected with the pSG5-miR-138 or the control vector (pSG5) in combination with the pMIR-TK-RNL vector including wildtype or mutant RARα binding sites. Cells were lyzed after 48 h in 1x Passive-Lyse Buffer (Promega, Mannheim, Germany) and the luciferase-activity was measured with the “Dual Luciferase reporter assay system” (E1910; Promega, Mannheim, Germany and Glomax 20/20 Luminometer) as described by the manufacturer. The relative luciferase-activity was determined as the quotient of firefly-luciferase and renilla-luciferase-activity. The analysis was performed in triplicates.
2.5Treatment with chemotherapeutics after transient transfection of miR-138
For transient miR-138 overexpression, Y79, WERI-Rb1 and RB355 cells were transfected as described previously by our group [29]. The vectors pSG5-miR-138 and the empty vector control (pSG5) were used for the transfection. 24 h before transient transfection 0.6×106 RB cells/ml were seeded in growth medium without antibiotics. 48 h after transient transfection miRNA was isolated and 72 h after transient transfection RB cells were treated with different concentrations of etoposide (0,01 –5μM) for 72 h. After 72 h of treatment with etoposide cell viability was analyzed by WST-1 assay and normalized to untreated controls.
2.6RARα overexpression in WERI-Etop and RB355-Etop retinoblastoma cells
For virus production, HEK293T cells were transfected as described previously [25] with each of the following plasmid DNAs: packaging vector pczVSV-G [30], pCD NL-BH [30] and either pLenti CMV_RARα or empty pLenti CMV. Experimental conditions for the lentiviral transductions were the same as described previously [29].
2.7RNA extraction and quantitative real-time PCR
RNA isolations from RB cells were performed using the the miRNeasy Kit (Qiagen, Hilden, Germany) and the miRNeasy FFPE Kit (Qiagen, Hilden, Germany), respectively.
For quantitative Real-time PCR analyses cDNA was synthesized with the QuantiTect Reverse Transcription Kit (Qiagen, Hilden, Germany) following the manufacturer’s protocol and the following human Taqman Gene Expression Assays (Applied Biosystems, Dreieich, Germany) were used: RARα (Hs00940446_m1) and 18S (Hs99999901_s1). The latter was used as an endogenous control. Using the TaqMan Universal PCR Master Mix (Applied Biosystems, Dreieich, Germany), Real-time PCR reactions were performed in duplicate and a total volume of 20μl was applied to the following program: 2 min at 50°C, 10 min at 95°C, 40 cycles of 15 s at 95°C and 60 s at 60°C.
For miRNA analyses cDNA was synthesized and quantitative Real-Time PCR analyses were performed using the miScript PCR Starter Kit (# 2181193; Qiagen, Hilden, Germany) as described by the manufacturer. The “miScript HiSpec Buffer” for quantification of mature miRNA was used with specific primers for miR-138 (5’-AGCTGGTGTTGTGAATCAGGCCG-3‘) and 5.8S RNA (5’-CTACGCCTGTCT GAGCGTCGCTT-3’) as endogenous control. The reactions were performed in duplicates using a 7300 Real-Time PCR System (Applied Biosystems, Dreieich, Germany).
2.8Western blotting
After washing in PBS, total protein was lyzed for 60 min at 4°C in RIPA buffer [31] plus additives (see: [32]) and cleared by a 10 min centrifugation step at 14,000 rpm (4°C). Equal amounts of protein extracts were separated on a 12% SDS-PAGE and transferred onto nitrocellulose membranes. Primary antibodies used (incubated overnight at 4°C): RARα (1:1000): #2554, (Cell Signalling, Leiden, The Netherlands); ß-actin (1:1,000): #4967, (Cell Signaling, Leiden, The Netherlands). Secondary antibodies used: HRP-conjugated goat-anti-rabbit (1:10,000): P0448; (DAKO, Düsseldorf, Germany). Signals were developed by the WesternBright Chemiluminescence Reagent (Advansta, San Jose, USA).
2.9Cell viability
To determine cell viability, 1× 105 cells in 100μl medium were seeded in two triplicates into a 96 well plate. Afterwards, 10μl of a water soluble tetrazolium-1 (WST-1) solution was added to each well and the cells were incubated at 37°C for different time points. Quantification of the formazan dye produced by viable cells was performed by measuring the absorbance at 450 nm in a microplate reader.
2.10Cell proliferation and apoptosis detection
Cell proliferation was determined by 5-Bromo-2′-deoxyuridine (BrdU; Sigma, Hamburg, Germany) incorporation. In order to determine changes in apoptosis levels, cells were stained with 4’,6-Diamidino-2-phenylindole (DAPI; Sigma, Hamburg, Germany). For each experiment six coverslips were stained and the percentages of proliferating or apoptotic cells were calculated as described previously by our group [33].
2.11CAM assay
In order to test for changes in tumor formation and migration capacity, RARα overexpressing WERI-Etop and RB355-Etop cells and control cells were grafted on the chorioallantoic membrane (CAM) [25] mainly following Zijlstra and Palmers protocols [34, 35]. Six to ten eggs were grafted in at least 3 independent experiments. Seven days after grafting (E10-17) tumors which formed from the grafted cells were excised, measured and photographed as described in Große-Kreul et al. 2016 [29]. All experiments have been conducted according to relevant national guidelines of the State Office for Nature, Environment and Consumer Protection (LANUV). According to the German Animal Experiment and Welfare Guidelines, ethic approval is only required if animals are intended to live beyond hatching and might suffer from the experimental treatment, which was not the case in our study as we only used chicken embryos until day E17 of development and thus, prior to hatching.
2.12Statistical analysis
All assays were performed at least in triplicates. Statistical analyses were performed using GraphPad Prism 6. Data represent means±SEM of three independent experiments from independent RB cell cultures. Results were analyzed by a Student‘s t-test or one-way ANOVA and Newman-Keuls Post test and considered significantly different if *P-value < 0.05, **P-value < 0.01 or ***P-value < 0.001.
3Results
3.1Reduced RARα and increased miR-138 expression in etoposide resistant RB cell lines
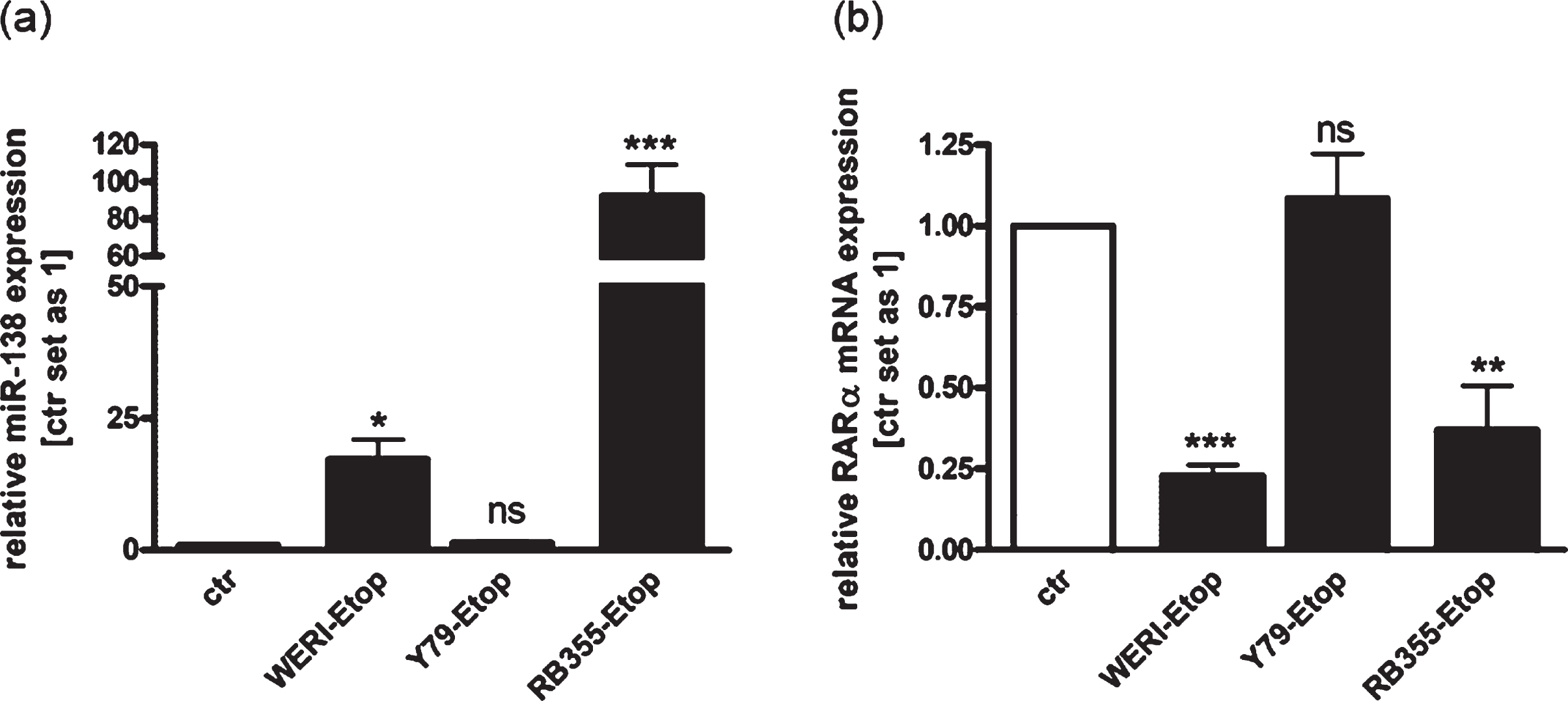
In order to investigate RB chemo-resistance mechanisms, we analyzed the expression of RARα and miR-138 in three etoposide resistant RB cell lines in comparison to the chemo-sensitive cells of origin. We could show increased miR-138 expression in the etoposide resistant RB cell lines WERI-Rb1 and RB355 (Fig. 1(a)), simultaneously displaying reduced RARα expression levels compared to the control cells (Fig. 1(b)). The expression of miR-138 and RARα was not significantly altered in etoposide resistant Y79 cells compared to their sensitive counterparts, but an opposing regulation of both was likewise seen. These results suggest that miR-138 may regulate endogenous expression of RARα in etoposide resistant RB cell lines.
Fig. 1
Expression analyses revealed increased endogenous expression of miR-138 and decreased expression of RARα in etoposide resistant RB-cells compared to chemo-sensitive control cells. (a) Quantitative Real-time PCR analysis of miR-138 expression in etoposide resistant WERI-Rb1 (WERI), Y79 and RB355 RB-cells compared to chemo-sensitive control cells (ctr). (b) Quantitative Real-time PCR analysis of RARα expression in etoposide resistant WERI-Rb1, Y79 and RB355 RB-cells compared to chemo-sensitive control cells (ctr). Values are means from at least 3 independent experiments±SEM. *P-value < 0.05; **P-value < 0.01; ***P-value < 0.001; ns: not significant; statistical differences compared to the control group calculated by Student’s t-test.
3.2Regulation of RARα and miR-138 in primary RB tumor specimens
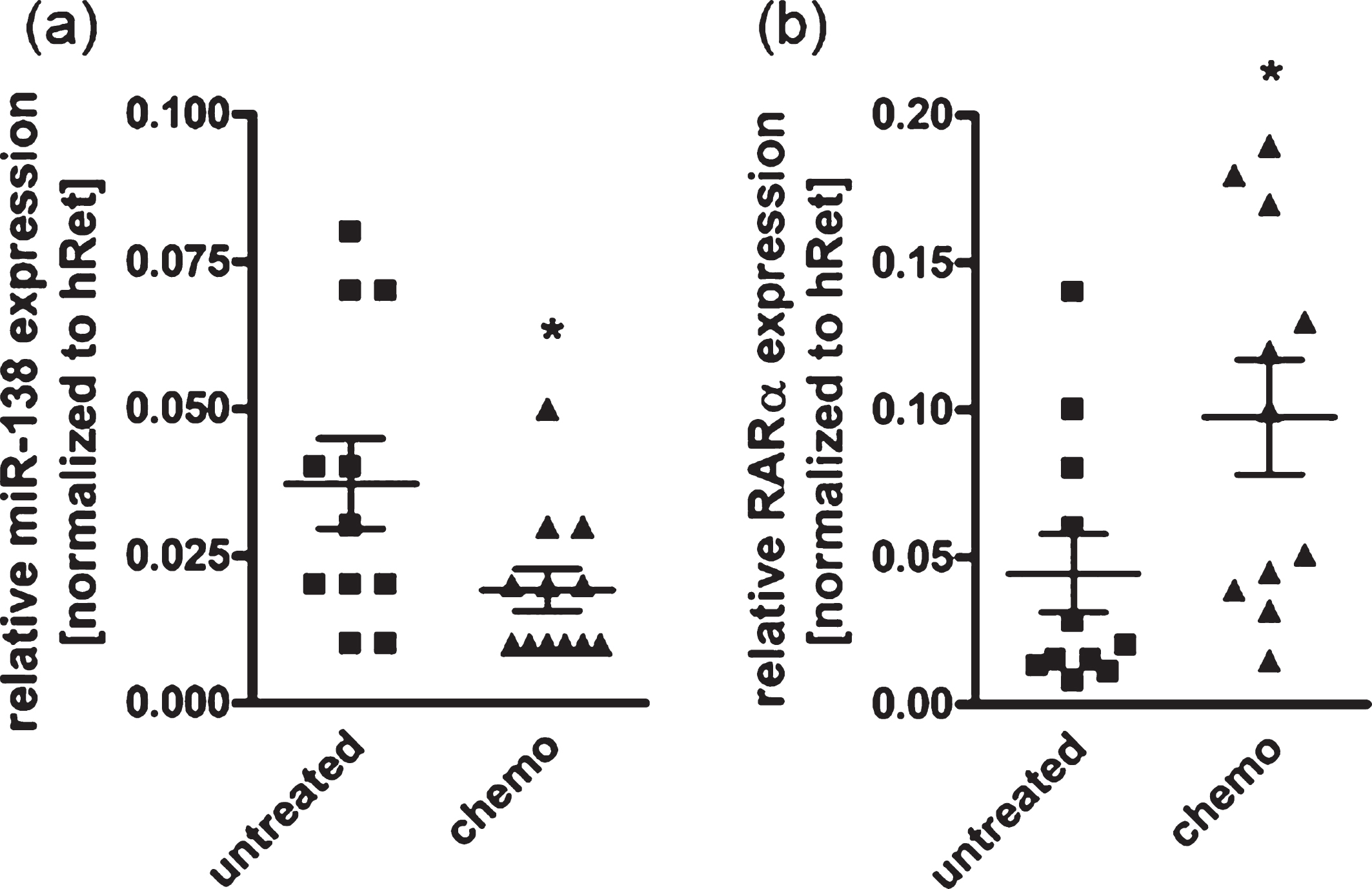
In order to investigate if chemotherapeutic treatment might affect RARα and miR-138 expression levels in RB patients, we analyzed and compared tumor specimens from patients with and without chemotherapy prior to enucleation of the effected eye. We could show a significantly reduced expression of miR-138 (Fig. 2(a)) in tumor specimens of patients with preoperative chemotherapeutic treatment compared to untreated tumors. RARα expression was concomitantly upregulated in drug treated primary RB samples (Fig. 2(b)). These results suggest that chemotherapeutic treatment of RB tumors simultaneously influences the expression of miR-138 and RARα in an opposing manner.
Fig. 2
Expression analysis of miR-138 and RARα revealed a chemotherapy-dependent regulation in patient RB tumor specimen. Quantitative Real-time PCR analyses of miR-138 (a) and RARα expression (b) of RB tumor specimen with (chemo) and without (untreated) chemotherapy prior to enucleation of the affected eye. Values are means from 11 individual tumor samples±SEM. *P-value < 0.05; statistical differences compared to the control group calculated by Student’s t-test.
3.3RARα overexpression inhibits viability and induces RB cell apoptosis
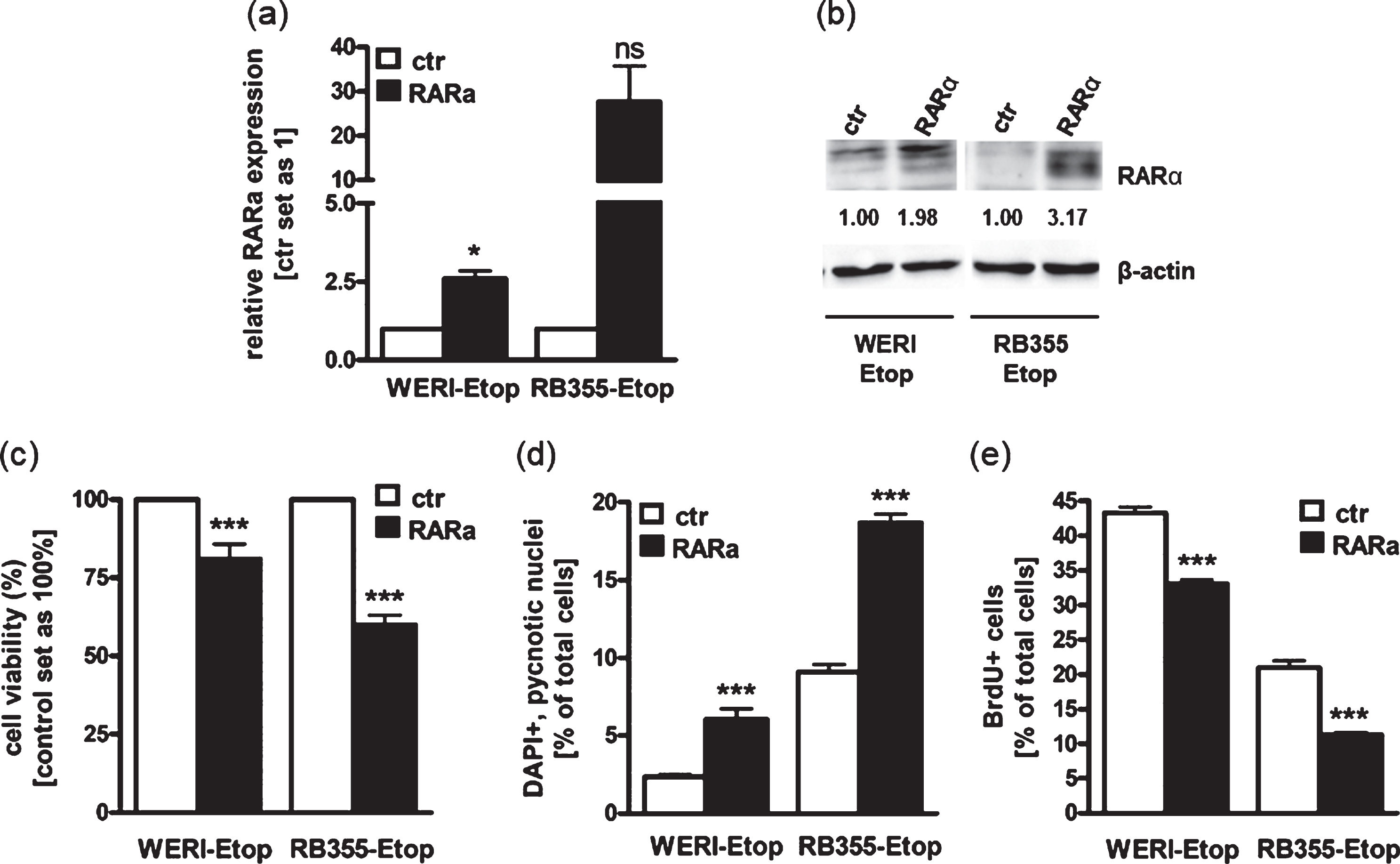
Against the background that RARα levels are downregulated in etoposide resistant RB cell lines in vitro and RARα is differentially expressed in RB patients‘ tumor specimens after chemotherapy treatment, we next addressed effects induced in etoposide resistant RB cells after re-expressing RARα to mimic the status prior to resistance. For this purpose, we stably overexpressed RARα in the two etoposide resistant RB cell lines WERI-Rb1-Etop and RB355-Etop (Fig. 3(a) + (b)), displaying significantly reduced RARα expression levels after developing resistances against etoposide (Fig. 1(b)). RARα overexpression was confirmed by Real-time PCR (Fig. 3(a)) and Western Blot analyses (Fig. 3(b)). We could show that both RARα overexpressing RB cell lines exhibit a significant reduction in cell viability (Fig. 3(c)) with a concurrent significant increase in apoptosis levels (Fig. 3(d)) and considerably reduced proliferation levels (Fig. 3(e)). These results suggest that RARα overexpression counteracts induced chemotherapy resistance by reducing growth and inducing apoptosis of etoposide resistant RB cells.
Fig. 3
Stable RARα overexpression induces RB apoptosis and lowers proliferation leading to reduced cell viability. (a) Quantitative Real-time PCR analysis of RARα expression levels after stable RARα overexpression in etoposide resistant WERI-Rb1 (WERI-Etop) and RB355 (RB355-Etop) cells compared to the control cells (ctr). (b) Western blot confirmation of RARα overexpression using total cell lysates from etoposide resistant WERI-Rb1 (WERI-Etop) and RB355 (RB355-Etop) cells or the control cells (ctr). Beta-Actin (β-actin) was used as loading control. (c)-(e) Stably RARα overexpressing WERI-Etop and RB355-Etop RB cells (RARα) displayed significantly reduced cell viability, significantly induced apoptosis and reduced proliferation rates compared to control cells (ctr) as revealed by WST-1 assay (c), DAPI cell counts (d) and BrdU stains (e). Values are means from at least 3 independent experiments±SEM. *P-value < 0.05; ***P-value < 0.001; statistical differences compared to the control group calculated by Student’s t-test.
3.4RARα overexpression decreases tumor growth in vivo
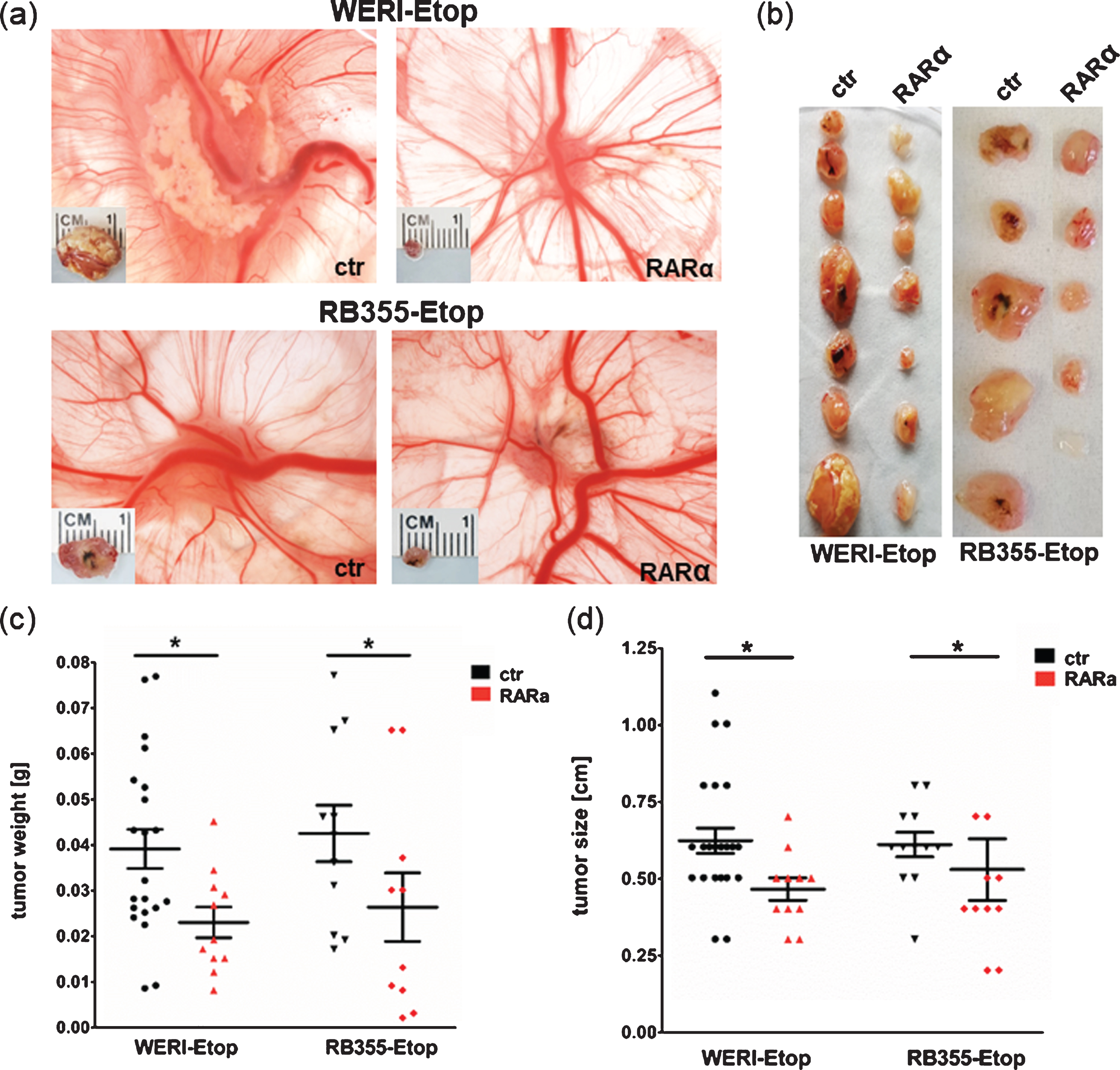
Our in vitro results indicated that RARα overexpression reduces growth kinetics of etoposide resistant RB cells. The involvement of RARα in etoposide resistance was further confirmed in vivo examining the effect of RARα overexpression on tumor growth in a chicken chorioallantoic membrane (CAM) model (Fig. 4). Compared to control cells RARα overexpressing etoposide resistant WERI-Rb1-Etop and RB-355-Etop cells developed strikingly smaller CAM tumors (Fig. 4(a) + (b)). Quantification revealed that CAM tumors forming from grafted RARα overexpressing RB cells display significant lower weights and sizes compared to tumors developing from control cells (Fig. 4(c) + (d)), indicating that RARα overexpression reduces tumor growth capacity of etoposide resistant RB cell lines in vivo.
Fig. 4
Stable lentiviral RARα overexpression reduces tumor growth of etoposide resistant RB cells in vivo. (a) Photographs of CAM tumors in situ and ruler measurements of excised tumors after grafting RARα overexpressing WERI-Rb1 (WERI)-Etop and RB355-Etop (RARa) or control cells (ctr) onto the CAM membrane. (b) Depiction of excised tumors revealing that tumors forming on the upper CAM after grafting RARα overexpression of WERI-Etop and RB355-Etop (RARα) are significantly smaller compared to those developing from control cells (ctr). (c) Quantification of tumor weight and (d) tumor size. Values are means from at least 3 independent experiments±SEM. *P-value < 0.05; statistical differences compared to the control group calculated by Student’s t-test.
3.5RARα overexpression does not re-sensitize etoposide resistant RB cell lines
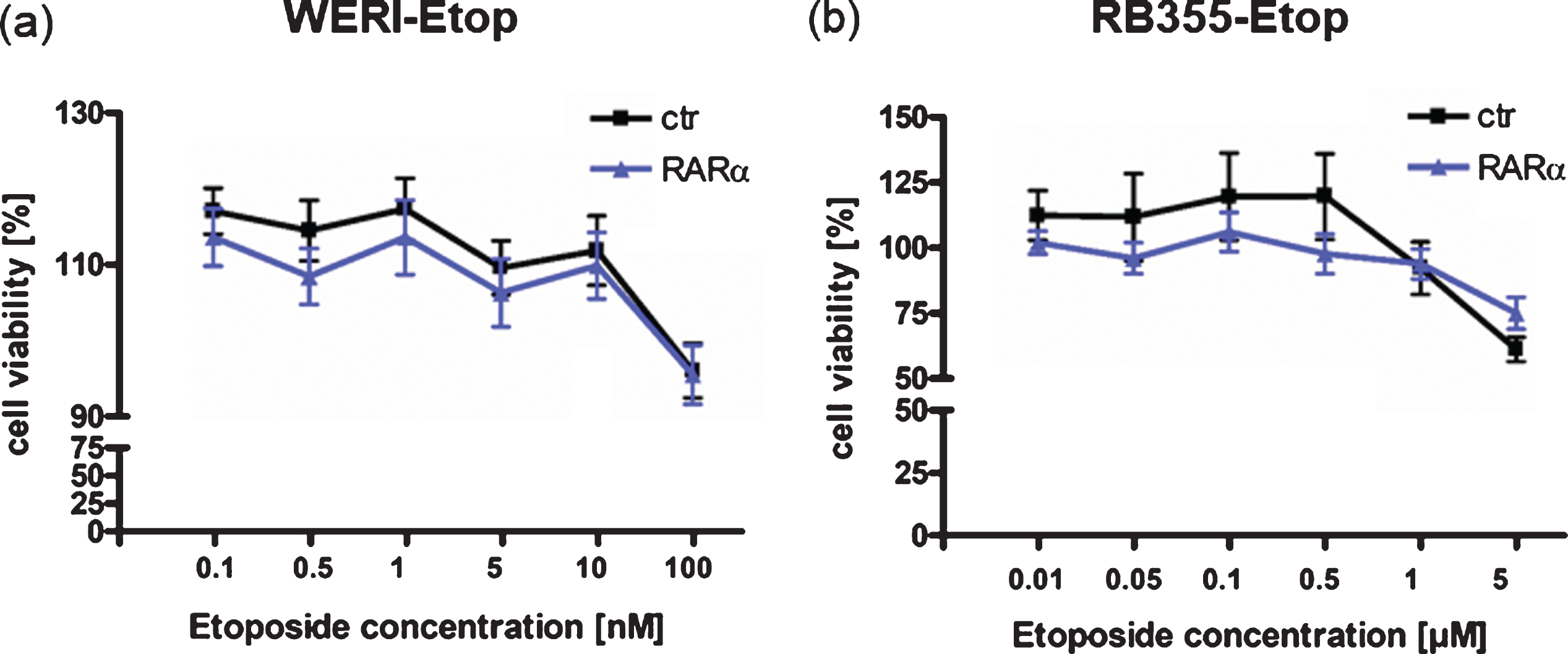
To determine whether RARα additionally contributes to a re-sensitization of etoposide resistant RB cells, we again overexpressed RARα in etoposide resistant WERI-Rb1-Etop and RB355-Etop cell lines. Treatment of RARα overexpressing etoposide resistant RB cell lines with etoposide, however, did not result in decreased cell viability (Fig. 5(a) + (b)) indicating that re-expression of RARα does not re-sensitize etoposide resistant RB cells for this chemotherapeutic drug.
Fig. 5
Effect of RARα overexpression on etoposide re-sensitization of etoposide resistant RB cell lines. (a) Compared to control cells (ctr), RARα overexpression in etoposide resistant WERI-Rb1 cells (WERI-Etop) cells did not induce significant changes in cell viability after etoposide treatment. (b) Compared to control cells (ctr) RARα overexpression did not significantly change cell viability of drug treated etoposide resistant RB355 cells (RB355-Etop) cells. Etoposide resistant RB cell lines were treated with different etoposide concentrations (0.01 –5μM) for 72 h and WST-1 assays were performed. Values are means from 3 independent experiments ±SEM and were normalized against untreated controls. No statistical differences compared to the control group could be calculated by Student‘s t-test.
3.6miR-138 overexpression leads to increased cell viability after etoposide treatment in some RB cells
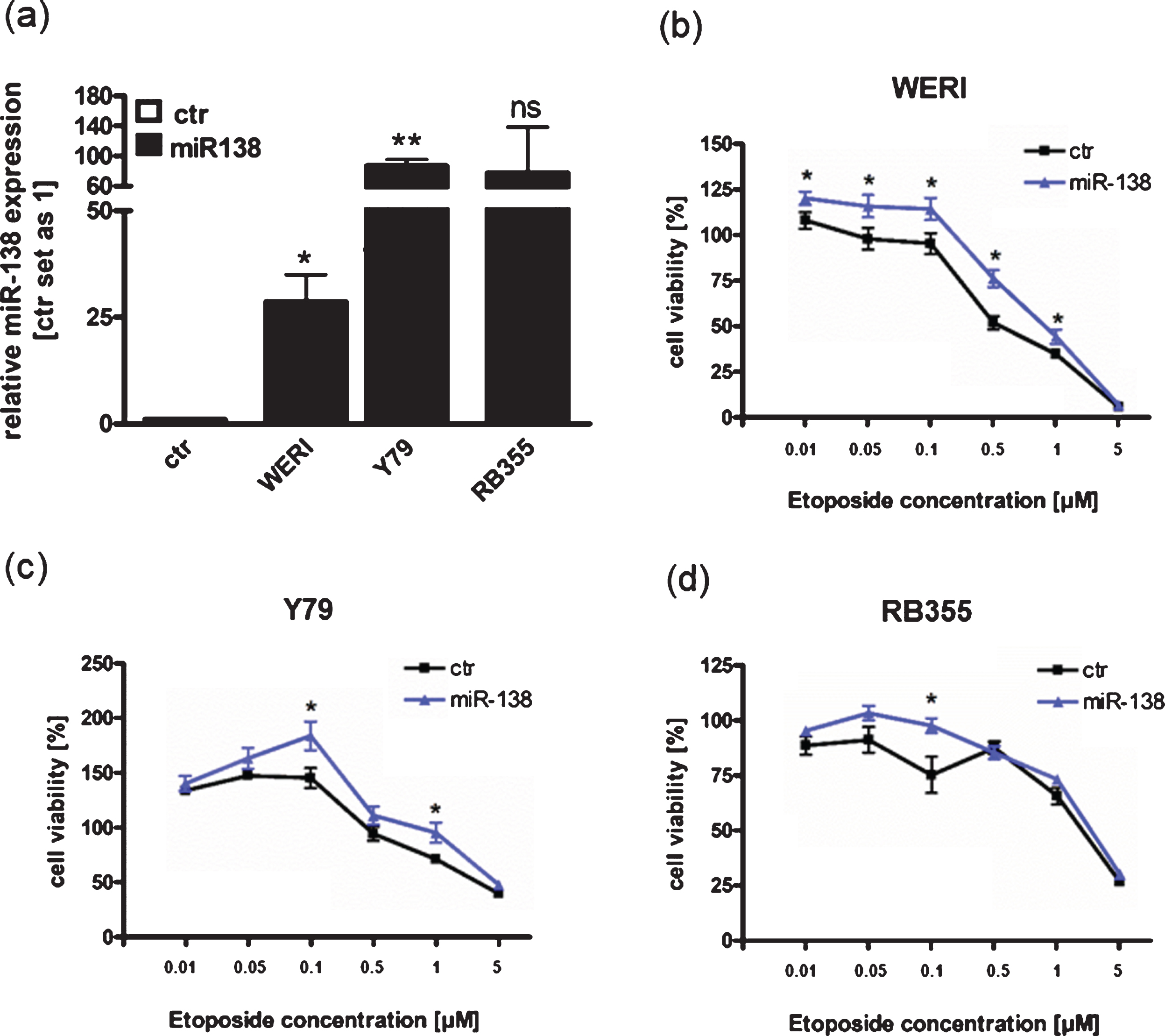
Since we could show that miR-138 is endogenously upregulated in etoposide resistant RB cell lines compared to the sensitive counterparts (Fig. 1(a)), we hypothesized that miR-138 expression may influence chemotherapy resistance in RB cells. We therefore overexpressed miR-138 in the three different chemotherapy sensitive cell lines Y79, WERI-Rb1 and RB355 RB. Transient overexpression of miR-138 was verified by Real-time PCR for all three RB cells lines investigated (Fig. 6(a)). Next, miR-138 overexpressing cell lines were treated with different concentrations of etoposide and cell viability was analyzed (Fig. 6(b)-(d)). MiR-138 overexpressing WERI-Rb1 cells displayed significantly increased cell viability upon etoposide treatment, except for the highest etoposide concentration applied (Fig. 6(b)). MiR-138 overexpression in Y79 and RB355 RB cells led to slightly higher cell viability, which, however, did only reach significance for individual etoposide concentrations (Fig. 6(c) + (d)). MiR-138 overexpressing WERI-Rb1 cells exhibited a significant increased IC50 value following etoposide treatment (p = 0.019), but no significant IC50 changes could be detected for the other RB cell lines investigated (data not shown). These results indicate that in some RB cells miR-138 overexpression may lead to an increase in cell viability after etoposide treatment, partially reflecting the intrinsic regulation seen in etoposide resistant RB cell lines.
Fig. 6
MiR-138 overexpression partially enhances cell viability of RB cell lines after treatment with etoposide. (a) Quantitative Real-time PCR analysis of miR-138 expression in WERI-Rb1 (WERI), Y79 and RB355 RB-cells compared to control cells (ctr) after transient transfection of miR-138 vector. (b) MiR-138 overexpression in WERI-Rb1 RB cells leads to significantly increased cell viability after treatment with etoposide compared to control cells (ctr). (c) Compared to control cells (ctr), miR-138 overexpression in Y79 cells partially increased cell viability after treatment with individual concentrations of etoposide. (d) MiR-138 overexpression in RB355 RB cells does not increase cell viability after etoposide treatment compared to control cells (ctr). RB cells were treated with different concentrations of etoposide (0.01 –5μM) for 72 h and WST-1 assays were performed. Values are means from at least 3 independent experiments±SEM and were normalized against untreated controls. **P-value < 0.01; *P-value < 0.05; statistical differences compared to the control group calculated by Student’s t-test.
3.7Binding of miR-138 to the 3′-UTR region of RARα
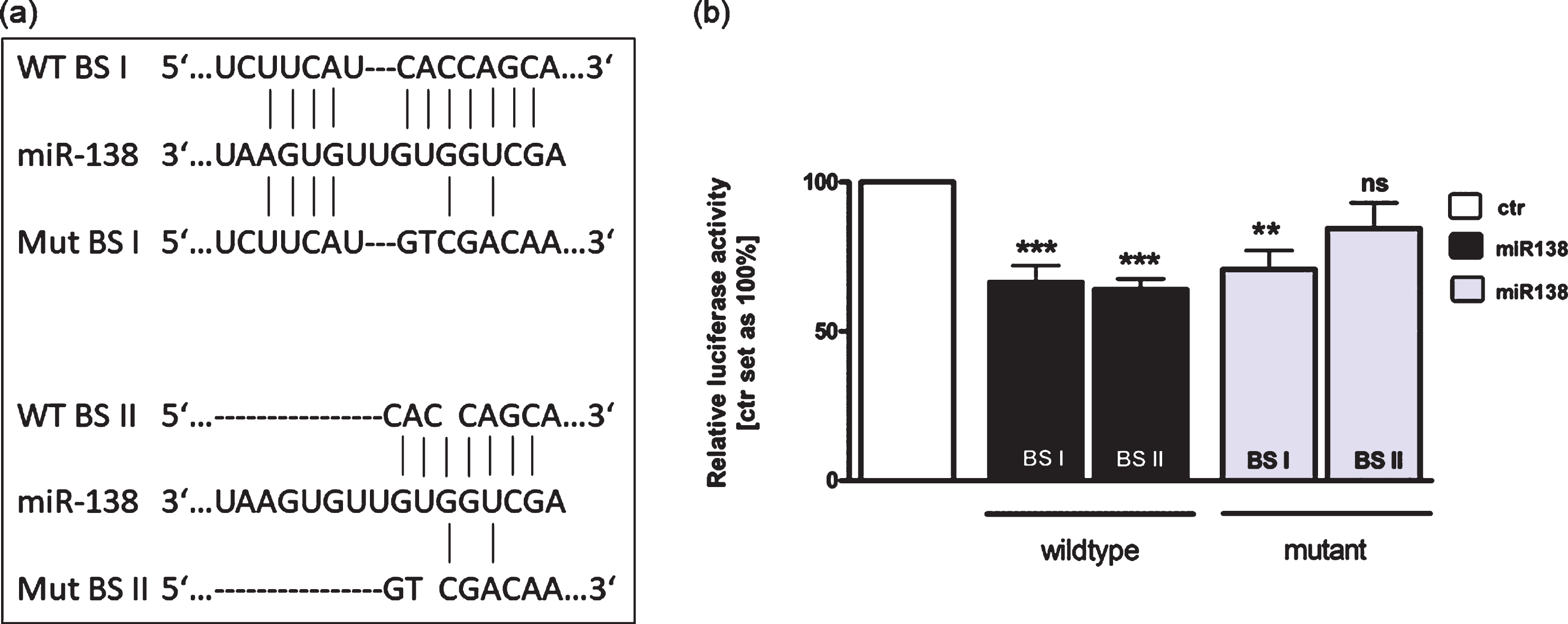
Using TargetScan 6.2 and miRanda tools, we identified two binding sites for miR-138 in the 3′-UTR region of RARα (Fig. 7(a)). In order to investigate the binding capacity of miR-138 to the 3′-UTR region of RARα we used luciferase activity assays. These assays revealed that miR-138 significantly inhibits luciferase activity by interacting with both binding sites (BSI and BSII) of the wild type RARα 3′-UTR (Fig. 7(b)). After complete mutation of BSII luciferase activity was not significantly reduced (Fig. 7(b)). Partial mutation of BSI, however, resulted in a sustained significant reduction of the luciferase signal most likely due to binding to the short (four nucleotide) wild type sequence (Fig. 7(b)).
Fig. 7
Binding studies of miR-138 to the 3’UTR region of RARα. (a) The RARα 3’-UTR region contains two potential binding sites for miR-138. Wild type binding site I and II (WT BS I and II), mutant binding site I and II (Mut BS I and II). (b) HEK293T cells were co-transfected with a miR-138 vector in addition to an empty vector (ctr) or the wild type I or II (WT BS I or II) or the mutant I or II (Mut BS I or II) vector containing the binding sequence of the RARα 3′-UTR. After 48 hours cells were harvested and dual luciferase activity were determined. Values are means of at least 3 independent experiments ±SEM. **P-value < 0.01; ***P-value < 0.001 statistical differences compared to the control group calculated by one-way ANOVA and Newman-Keuls Post test.
In order to verify the regulation of RARα by miR-138 in RB cells we overexpressed miR-138 in three RB cell lines and analyzed the expression levels of RARα. We could show that miR-138 overexpression only slightly reduces RARα mRNA expression levels without changing RARα protein levels in all three RB cell lines investigated (data not shown). These results indicate that although miR-138 is generally capable to bind to and regulate RARα, it does not influence RARα expression in chemo-sensitive RB cell lines.
4Discussion
Etoposide is one of the commonly used chemotherapeutic agents in vincristine, etoposide, carboplatin (VEC)-therapy RB treatment regimen. This therapy targets the DNA topoisomerase II enzyme, finally leading to induction of apoptosis. Our group recently demonstrated that etoposide resistant RB cells become more aggressive compared to the chemo-sensitive cells of origin. Resistant RB cells display significantly higher proliferation rates and an increased tumor formation capacity [26]. Thus, the development of chemotherapy resistance poses difficulties in cancer therapy as resistant cancer cells not only no longer respond to the anticancer drug initially used, but also potentially acquire a non-desirable, more tumorigenic phenotype. Therefore, understanding the mechanisms of chemotherapy resistances are main challenges in developing new treatment strategies for resistant tumor cells.
In the study presented, we could show that the retinoid receptor RARα is significantly downregulated in two out of three etoposide resistant RB cell lines compared to the chemo-sensitive cells of origin. In the absence of ligands (retinoic acid, RA) the ligand-activated nuclear receptor RARα acts as a transcriptional repressor by binding to co-repressor complexes and recruiting histone deacetylases (HDACs) [36]. Several other groups demonstrated that the expression of retinoid receptors is downregulated in several tumor entities and that the commonly induced signaling pathways regulate crucial biological functions, such as development, differentiation, proliferation and apoptosis [37–41]. In line with these observations, we identified RARα to be significantly downregulated in two out of three highly proliferative, tumorigenic etoposide resistant RB cell lines compared to the chemo-sensitive cells of origin. In order to proof the hypothesized tumor suppressive function of RARα in retinoblastoma, we overexpressed RARα in etoposide resistant RB cell lines. and could indeed demonstrate a significant apoptosis induction, proliferation reduction, and significantly reduced tumor growth in vivo. Fittingly, repression of RARα leads to enhanced cell proliferation in rhabdomyosarcoma [42]. Moreover, poor response to first-line chemotherapy in gastric cancer [43] probably depends on RARα regulation. In the study presented, we could show a strong tumor-suppressive effect of RARα re-expression in etoposide resistant RB cells in vivo. RARα, however, did not re-sensitize etoposide resistant RB cells towards this drug in vitro.
The expression of retinoid receptors in cancers can be altered by different mechanisms including 1) mutation or deletion of the gene, 2) transcriptional repression or 3) post-transcriptional repression [44]. We were interested in RARα regulation at the post-transcriptional level by miRs, resulting in either destabilization of the target mRNA or translational repression [45]. Among other miRs deregulated in RB, we identified miR-138 as a miR potentially targeting RARα. Interestingly, we identified upregulated miR-138 expression levels in etoposide resistant RB cells compared to the chemo-sensitive counterparts potentially leading to RARα downregulation observed in etoposide resistant RB355 and WERI-Rb1 cells. An opposite expression pattern was observed in the etoposide resistant cell line Y79 displaying reduced expression of miR-138 and concordantly higher RARα expression. These results led us to the assumption that in contrast to the other RB cell lines investigated etoposide resistant Y79 cells use a different, yet unknown mechanism to become chemo-resistant. Since we observed that chemotherapeutic treatment of RB patients’ tumors simultaneously downregulates expression patterns of miR-138 and upregulates RARα levels, we hypothesized that RARα expression is regulated by miR-138 in the development and/or progression of etoposide resistance in RB cells. Along this line, we could show a cell line dependent increase in cell viability after miR-138 overexpression in chemo-sensitive RB cell lines following etoposide treatment. In addition to previous findings [19] and in silico binding predictions we could show that miR-138 in general binds to the 3′-UTR region of RARα. A residue of four nucleotides upstream of the intrinsic binding site might allow for a binding of miR-138. Nevertheless, overexpression of miR-138 in chemo-sensitive RB cells did not influence endogenous RARα expression levels. RARα overexpression in turn did not induce re-sensitization for etoposide. Hence, the increase in cell viability seen after miR-138 overexpression and etoposide treatment of sensitive RB cells is apparently not mediated by RARα. Therefore, miR-138 triggered induction of cell viability after etoposide treatment most likely involves other signaling molecules adjacent to the regulation of RARα. The primary finding of elevated miR-138 expression levels in etoposide resistant RB tumor cells contradicts findings of other groups identifying miR-138 as a tumor suppressor miRNA in multiple malignancies [42, 46, 47]. These studies suggest that miR-138 most likely downregulates target genes belonging to the family of oncogenes as their downregulation sensitized cells for chemotherapeutic drugs. In line with our results, another report describes miR-138 as an oncogenic miR in brain tumors promoting self-renewal of glioma stem cells [48]. It could also been shown that transcriptional activation of miR-138 in glioma is mediated by C/EBPβ [49]. Moreover, miR-138 promotes acquired resistance to temozolomide (TMZ) by targeting the apoptosis regulator BIM [50]. In the end, the regulatory mechanisms and signaling pathways of miR-138 and its functions are not fully understood in many cancers and the understanding of the complex network of involved genes needs further investigation. However, besides its target gene regulation, miRNAs especially those changing their expression patterns upon chemotherapeutic treatment like miR-138, are promising RB biomarkers in cancer therapy. In particular, circulating miRNAs [51] are of special interest upon availability in liquid biopsies of the eye [6].
In summary, the study presented could show that the expression of RARα and miR-138 are altered in etoposide resistant RB cell lines and in RB tumor specimens after chemotherapeutic treatment. Thus, despite the fact that miR-138 does not regulate RARα expression in RB cells this miRNA might serve as a potential future RB biomarker. RARα induces strong tumor suppressive effects in retinoblastoma by decreasing the tumorigenic potential of highly aggressive etoposide resistant RB cells in vivo rendering this gene a highly interesting target in future RB therapy approaches.
Acknowledgments
The authors would like to thank Dr. H. Hanenberg for the lentiviral vectors. We also would like to thank U. Gerster for excellent technical assistance and D. Gioè for valuable help with the cell counts.
This research was funded by Else Kröner-Fresenius-Stiftung (grant number 2018_A35).
Author contributions
All the authors have accepted responsibility for the entire content of this submitted manuscript and have approved its submission.
Conflict of interest
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical considerations
The Ethics Committee of the Medical Faculty of the University of Duisburg-Essen approved the use of human retina (approval # 06-30214) and retinoblastoma samples (approval # 14-5836-BO) for research conducted in the course of the study presented and written informed consent has been obtained from patients’ relatives or parents.
References
[1] | Dimaras H , Kimani K , Dimba EAO , Gronsdahl P , White A , Chan HSL , et al. Retinoblastoma. The Lancet.. (2012) ;379: :1436–46. doi: 10.1016/S0140-6736(11)61137-9 |
[2] | Soliman SE , ElManhaly M , Dimaras H . Knowledge of genetics in familial retinoblastoma. Ophthalmic Genet.. (2017) ;38: :226–32. doi: 10.1080/13816810.2016.1195846 |
[3] | Corson TW , Gallie BL . One hit, two hits, three hits, more? Genomic changes in the development of retinoblastoma. Genes, Chromosomes Cancer. (2007) ;46: :617–34. doi: 10.1002/gcc.20457 |
[4] | Rushlow DE , Mol BM , Kennett JY , Yee S , Pajovic S , Thériault BL , et al. Characterisation of retinoblastomas without RB1 mutations: Genomic, gene expression, and clinical studies: Genomic, gene expression, and clinical studies. The Lancet Oncology. (2013) ;14: :327–34. doi: 10.1016/S1470-2045(13)70045-7 |
[5] | Singh U , Malik MA , Goswami S , Shukla S , Kaur J . Epigenetic regulation of human retinoblastoma. Tumour Biol. (2016) ;37: :14427–41. doi: 10.1007/s13277-016-5308-3 |
[6] | Delsin LEA , Salomao KB , Pezuk JA , Brassesco MS . Expression profiles and prognostic value of miRNAs in retinoblastoma. J Cancer Res Clin Oncol. (2019) ;145: :1–10. doi: 10.1007/s00432-018-2773-7 |
[7] | Dimaras H , Corson TW . Retinoblastoma, the visible CNS tumor: A review. J Neurosci Res. (2019) ;97: :29–44. doi: 10.1002/jnr.24213 |
[8] | Mao X-M , Li H , Zhang X-Y , Zhou P , Fu Q-R , Chen Q-E , et al. Retinoic Acid Receptor α Knockdown Suppresses the Tumorigenicity of Esophageal Carcinoma via Wnt/β-catenin Pathway. Dig Dis Sci. (2018) ;63: :3348–58.doi: 10.1007/s10620-018-5254-6 |
[9] | Müller P , Doliva R , Busch M , Philippeit C , Stephan H , Dünker N . Additive Effects of Retinoic Acid (RA) and Bone Morphogenetic Protein 4 (BMP-4) Apoptosis Signaling in Retinoblastoma Cell Lines. PLoS ONE. (2015) ;10: :e0131467. doi: 10.1371/journal.pone.0131467 |
[10] | Redner RL , Rush EA , Faas S , Rudert WA , Corey SJ . The t(5;17) variant of acute promyelocytic leukemia expresses a nucleophosmin-retinoic acid receptor fusion. Blood. (1996) ;87: :882–86. doi: 10.1182/blood.V87.3.882.bloodjournal873882 |
[11] | Altucci L , Gronemeyer H . The promise of retinoids to fight against cancer. Nature Reviews Cancer. 2001:181-193. doi: 10.1038/35106036 |
[12] | Le Maire A , Teyssier C , Balaguer P , Bourguet W , Germain P . Regulation of RXR-RAR Heterodimers by RXR- and RAR-Specific Ligands and Their Combinations. Cells. (2019) ;8: :1392. doi: 10.3390/cells8111392 |
[13] | Ghyselinck NB , Duester G . Retinoic acid signaling pathways. Development. (2019) ;146: :dev167502. doi: 10.1242/dev.167502 |
[14] | Rochette-Egly C , Germain P . Dynamic and combinatorial control of gene expression by nuclear retinoic acid receptors (RARs). Nucl Recept Signal. (2009) ;7: (1):e005. doi: 10.1621/nrs.07005 |
[15] | Chatagnon A , Veber P , Morin V , Bedo J , Triqueneaux G , Sémon M , et al. RAR/RXR binding dynamics distinguish pluripotency from differentiation associated cis-regulatory elements. Nucleic Acids Res. (2015) ;43: :4833–54. doi: 10.1093/nar/gkv370 |
[16] | Peng Y , Croce CM . The role of MicroRNAs in human cancer. Signal Transduct Target Ther. (2016) ;1: :15004. doi: 10.1038/sigtrans.2015.4 |
[17] | Friedman RC , Farh KK-H , Burge CB , Bartel DP . Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. (2009) ;19: :92–105. doi: 10.1101/gr.082701.108 |
[18] | Berindan-Neagoe I , Monroig PdC , Pasculli B , Calin GA . MicroRNAome genome: a treasure for cancer diagnosis and therapy. CA Cancer J Clin. (2014) ;64: :311–36. doi: 10.3322/caac.21244 |
[19] | Wang X , Tan L , Lu Y , Peng J , Zhu Y , Zhang Y , et al. MicroRNA-138 promotes tau phosphorylation by targeting retinoic acid receptor alpha. FEBS Letters. (2015) ;589: :726–29. doi: 10.1016/j.febslet.2015.02.001 |
[20] | Wang Z , Yao Y-J , Zheng F , Guan Z , Zhang L , Dong N , et al. Mir-138-5p acts as a tumor suppressor by targeting pyruvate dehydrogenase kinase 1 in human retinoblastoma. Eur Rev Med Pharmacol Sci. (2017) ;21: :5624–29. |
[21] | Rastgoo N , Pourabdollah M , Abdi J , Reece D , Chang H . Dysregulation of EZH2/miR-138 axis contributes to drug resistance in multiple myeloma by downregulating RBPMS. Leukemia. (2018) ;32: :2471–82. doi: 10.1038/s41375-018-0140-y |
[22] | Griegel S , Hong C , Frötschl R , Hülser DF , Greger V , Horsthemke B , et al. Newly established human retinoblastoma cell lines exhibit an “immortalized” but not an invasive phenotype in vitro. Int J Cancer. (1990) ;46: :125–32. doi: 10.1002/ijc.2910460123 |
[23] | Reid TW , Albert DM , Rabson AS , Russell P , Craft J , Chu EW , Tralka TS , Wilcox JL . Characteristics of an established cell line of retinoblastoma. J Nat Cancer Inst. (1974) ;53: (2):347–60. doi: 10.1093/jnci/53.2.347 |
[24] | McFall RC , Sery TW , Makadon M . Characterization of a new continuous cell line derived from a human retinoblastoma. Cancer Res. (1977) ;37: :1003–10. |
[25] | Busch M , Philippeit C , Weise A , Dünker N . Re-characterization of established human retinoblastoma cell lines. Histochem Cell Biol. (2015) ;143: :325–38. doi: 10.1007/s00418-014-1285-z |
[26] | Busch M , Papior D , Stephan H , Dïnker N . Characterization of etoposide- and cisplatin-chemoresistant retinoblastoma cell lines. Oncol Rep. (2018) ;39: :160–172. doi: 10.3892/or.2017.6100 |
[27] | Berger AH , Brooks AN , Wu X , Shrestha Y , Chouinard C , Piccioni F , et al. High-throughput Phenotyping of Lung Cancer Somatic Mutations. Cancer Cell. (2016) ;30: (2):214–28. doi: 10.1016/j.ccell.2016.06.022 |
[28] | Imig J , Motsch N , Zhu JY , Barth S , Okoniewski M , Reineke T , et al. microRNA profiling in Epstein-Barr virus-associated B-cell lymphoma. Nucleic Acids Res. (2011) ;39: (5):1880–93. doi: 10.1093/nar/gkq1043 |
[29] | Große-Kreul J , Busch M , Winter C , Pikos S , Stephan H , Dünker N . Forced Trefoil Factor Family Peptide 3 (TFF3) Expression Reduces Growth, Viability, and Tumorigenicity of Human Retinoblastoma Cell Lines. PLoS ONE. (2016) ;11: :e0163025. doi: 10.1371/journal.pone.0163025 |
[30] | Hartmann L , Neveling K , Borkens S , Schneider H , Freund M , Grassman E , et al. Correct mRNA processing at a mutant TT splice donor in FANCC ameliorates the clinical phenotype in patients and is enhanced by delivery of suppressor U1 snRNAs. Am J Hum Genet. (2010) ;87: :480–93. doi: 10.1016/j.ajhg.2010.08.016 |
[31] | Spruck C , Sun D , Fiegl H , Marth C , Mueller-Holzner E , Goebel G , et al. Detection of low molecular weight derivatives of cyclin E1 is a function of cyclin E1 protein levels in breast cancer. Cancer Res. (2006) ;66: (14):7355–60. doi: 10.1158/0008-5472.CAN-05-3240 |
[32] | Weise A , Dünker N . High trefoil factor 1 (TFF1) expression in human retinoblastoma cells correlates with low growth kinetics, increased cyclin-dependent kinase (CDK) inhibitor levels and a selective down-regulation of CDK6. Histochem Cell Biol. (2013) ;139: :323–38. doi: 10.1007/s00418-012-1028-y |
[33] | Haubold M , Weise A , Stephan H , Dünker N . Bone morphogenetic protein 4 (BMP4) signaling in retinoblastoma cells. Int J Biol Sci. (2010) ;6: :700–15. doi: 10.7150/ijbs.6.700 |
[34] | Zijlstra A , Mellor R , Panzarella G , Aimes RT , Hooper JD , Marchenko ND , et al. A quantitative analysis of rate-limiting steps in the metastatic cascade using human-specific real-time polymerase chain reaction. Cancer Res. (2002) ;62: :7083–92. |
[35] | Palmer TD , Lewis J , Zijlstra A . Quantitative analysis of cancer metastasis using an avian embryo model. J Vis Exp. (2011) ;51: :e2815. doi:10.3791/2815 |
[36] | Cordeiro TN , Sibille N , Germain P , Barthe P , Boulahtouf A , Allemand F , et al. Interplay of Protein Disorder in Retinoic Acid Receptor Heterodimer and Its Corepressor Regulates Gene Expression. Structure. (2019) ;27: :1270–1285.e6. doi: 10.1016/j.str.2019.05.001 |
[37] | Bushue N , Wan Y-JY . Retinoid pathway and cancer therapeutics. Adv Drug Deliv Rev. (2010) ;62: :1285–98. doi: 10.1016/j.addr.2010.07.003 |
[38] | Guo X , Ruiz A , Rando RR , Bok D , Gudas LJ . Esterification of all-trans-retinol in normal human epithelial cell strains and carcinoma lines from oral cavity, skin and breast: reduced expression of lecithin:retinol acyltransferase in carcinoma lines. Carcinogenesis. (2000) ;21: :1925–33. doi: 10.1093/carcin/21.11.1925 |
[39] | Ablain J , Thé H de . Retinoic acid signaling in cancer: The parable of acute promyelocytic leukemia. Int J Cancer. (2014) ;135: :2262–72. doi: 10.1002/ijc.29081 |
[40] | Esteller M , Guo M , Moreno V , Peinado MA , Capella G , Galm O , et al. Hypermethylation-associated Inactivation of the Cellular Retinol-Binding-Protein 1 Gene in Human Cancer. Cancer Res. (2002) ;62: :5902–05. |
[41] | Di Martino O , Welch JS . Retinoic Acid Receptors in Acute Myeloid Leukemia Therapy. Cancers (Basel). (1915) ;11: :1915. doi: 10.3390/cancers11121915 |
[42] | Tombolan L , Zampini M , Casara S , Boldrin E , Zin A , Bisogno G , et al. MicroRNA-27a Contributes to Rhabdomyosarcoma Cell Proliferation by Suppressing RARA and RXRA. PLoS ONE. (2015) ;10: :e0125171. doi: 10.1371/journal.pone.0125171 |
[43] | Huang D , Wang H , Liu R , Li H , Ge S , Bai M , et al. miRNA27a is a biomarker for predicting chemosensitivity and prognosis in metastatic or recurrent gastric cancer. J Cell Biochem. (2014) ;115: :549–56. doi: 10.1002/jcb.24689 |
[44] | Yin S , Bleul T , Zhu Y , Isayev O , Werner J , Bazhin AV . MiRNAs are Unlikely to be Involved in Retinoid Receptor Gene Regulation in Pancreatic Cancer Cells. Cell Physiol Biochem. (2017) ;44: :644–56. doi: 10.1159/000485276 |
[45] | Huntzinger E , Izaurralde E . Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet. (2011) ;12: :99–110. doi: 10.1038/nrg2936 |
[46] | Jin Z , Guan L , Song Y , Xiang G-M , Chen S-X , Gao B . MicroRNA-138 regulates chemoresistance in human non-small cell lung cancer via epithelial mesenchymal transition. Eur Rev Med Pharmacol Sci. (2016) ;20: :1080–86. |
[47] | Yeh M , Oh CS , Yoo JY , Kaur B , Lee TJ . Pivotal role of microRNA-138 in human cancers. Am J Cancer Res.. (2019) ;9: :1118–26. |
[48] | Chan XHD , Nama S , Gopal F , Rizk P , Ramasamy S , Sundaram G , et al. Targeting glioma stem cells by functional inhibition of a prosurvival oncomiR-138 in malignant gliomas. Cell Rep. (2012) ;2: (3):591–602. doi: 10.1016/j.celrep.2012.07.012 |
[49] | Di Pascale F , Nama S , Muhuri M , Quah S , Ismail HM , Chan XHD , et al. C/EBPβ mediates RNA polymerase III-driven transcription of oncomiR-138 in malignant gliomas. Nucleic Acids Res. (2018) ;46: :336–49. doi: 10.1093/nar/gkx1105 |
[50] | Stojcheva N , Schechtmann G , Sass S , Roth P , Florea A-M , Stefanski A , et al. MicroRNA-138 promotes acquired alkylator resistance in glioblastoma by targeting the Bcl-2-interacting mediator BIM. Oncotarget. (2016) ;7: :12937–50. doi: 10.18632/oncotarget.7346 |
[51] | Beta M , Venkatesan N , Vasudevan M , Vetrivel U , Khetan V , Krishnakumar S . Identification and Insilico Analysis of Retinoblastoma Serum microRNA Profile and Gene Targets Towards Prediction of Novel Serum Biomarkers. Bioinform Biol Insights. (2013) ;7: :21–34. doi: 10.4137/BBI.S10501 |


