
Practical management of organic acidemias
Abstract
Organic acidemias or acidurias (OAs) are a group of metabolic disorders which result in organic acids detected in the urine (or plasma). They include the systemic OAs, the cerebral OAs, and the ketogenic/ketolytic OAs. In general, all types of OA can be treated using a similar conceptual organization. The goals of therapy (acute and chronic) include reversal/prevention of catabolism, limitation of non-tolerated precursors, scavenging of toxic intermediates, replacement of the deficient product, use of cofactors when able and treatment of complications using standard methods. In the future, this framework will also include replacement of dysfunctional enzyme with a functional one. Here we review how this conceptional framework applies to the systemic OAs (propionic acidemia, methylmalonic aciduria without homocystinuria, and isovaleric acidemia), the cerebral OA (glutaric aciduria 1) and the ketogenic/ketolytic OA (HMG-CoA lyase deficiency). We review the current recommendations and use examples of how to use this conceptional framework.
1Introduction
Organic acidemias and acidurias (OAs) are, by definition inherited enzyme disorders in which excessive organic acids (e.g. dicarboxylic acids) are accumulated in the urine or blood [1]. There are 3 categories of organic acidemias: 1) systemic organic acidemias, 2) cerebral organic acidemias, and 3) ketolytic/ketogenic organic acidemias (Table 1). For each of these, there are different approaches to treatment, but all have diagnostic organic acids found in urine (and blood).
Table 1
List of organic acidemias, their dysfunctional enzymes and their OMIM number
| Type | Disorder | Genes | OMIM(s) |
| Systemic | Propionic acidemia | PCCA | 606054 |
| PCCB | |||
| Methylmalonic aciduria (without homocystinuria) | MMUT | 251000 | |
| MMAA | 251100 | ||
| MMAB | 251110 | ||
| MCEE | |||
| Isovaleric aciduria | IVDH | 243500 | |
| 3-methylcrotonylglycinuria | MCCC1 | 210200 | |
| MCCC2 | |||
| 3-methylglutaconic acidurias | HTRA2 | 617248 | |
| DNAJC19 | 610198 | ||
| SERAC1 | 614739 | ||
| AUH | 250950 | ||
| CLPB | 616271 | ||
| TIMM50 | 617698 | ||
| OPA3 | 258501 | ||
| TAZ | 302060 | ||
| MGCA1 | 250951 | ||
| D-2-hydroxyglutaric aciduria | D2HGDH | 600721 | |
| Combined malonic and methylmalonic aciduria | ACF3 | 61426524 | |
| Cerebral | Glutaric aciduria I | GCDH | 231670 |
| L-2-hydroxyglutaric aciduria | L2HGDH | 236792 | |
| 4-hydroxybutyric aciduria | ALDH5A1 | 271980 | |
| Malonic aciduria | MLYCD | 248360 | |
| Canavan Disease | ASPA | 271900 | |
| Ketolytic/Ketogenic | 3-hydroxy-3-methylglutaryl-CoA Lyase | HMGCL | 246450 |
| 3-hydroxy-3-methylglutaryl-CoA synthase | HMGCS2 | 605911 | |
| Succinyl-CoA:3-oxoacid-CoA transferase | SCOT | 245050 | |
| 3-oxothiolase/betaketothiolase | ACAT1 | 203750 |
The systemic OAs (e.g. propionic acidemia (PA), methylmalonic aciduria (MMA), or isovaleric aciduria (IVA)) often result in metabolic acidosis. On the other hand, the cerebral organic acidemias (e.g. glutaric aciduria I (GA1)) may not have systemic acidemia, but rather only organic acids identified in urine [2]. Ketolytic/ketogenic OAs result in organic acids identifiable in the urine, but they are more likely to have hypoglycemia due to abnormal ketone production as a presenting finding.
Recognition of OAs may be via newborn screening or symptoms. Several OAs are detectable by newborn screening, but different locales may have different disorders screened. Diagnosis is based on recognizing the diagnostic organic acid in the urine (or sometimes blood) and acylcarnitines. The diagnosis is often confirmed by genetic testing looking for the mutation(s) in the gene encoding the enzyme which is dysfunctional.
In general, systemic OAs are also numbered among the intoxication-type metabolic disorders and can be classified into those which impact the entire individual (thus called systemic) [2]. For the systemic OAs, neonates (and older children) can progress through symptoms that resemble sepsis (i.e. lethargy, confusion, decreased intake, vomiting, coma and if untreated, death) [2–4]. However, they can also present in a myriad of other manners especially as they get older (e.g. cardiomyopathy, cardiac arrhythmia, renal failure, insulin-requiring hyperglycemia, etc.), and if a condition is nonresponsive to typical treatment, this category of disorders should be considered. One of the major contributors to the presentation is a secondary inhibition of the urea cycle which results in hyperammonemia. The presence of metabolic acidosis and organic acids in the urine/blood help differentiate this disorder from urea cycle disorders.
This review includes discussion of the three most common systemic OAs, IVA, PA, and MMA, but there are many other systemic OAs. These three were chosen because they are more common and can be complicated by life-threatening hyperammonemia. The systemic organic OAs include propionic acidemia (PA, OMIM #606054), methylmalonic acidurias without homocystinuria (MMUT, #25100; MMAA, #251100; MMAB, #251110), isovaleric aciduria (IVA, #243500), 3- methylcrotonylglycinuria (3MCCC, #210200), 3-methylglutaconic acidurias (#250950, #250951, #302060, #610198, #614739, #616271, #617248, and #617698), D-2 hydroxyglutaric aciduria (D2HGDH, #6007121) and several others.
Some also consider maple syrup urine disease (MSUD, #248600) to be a systemic OA due to the presence of dicarboxylic acids in urine in this disorder, but others do not. In our opinion, MSUD is closer to an aminoacidopathy and so the identification and treatment of MSUD is beyond the scope of this review.
Similar to the above systemic OAs in terms of whole body impact, there are several ketogenic/ketolytic systemic OAs which have abnormal ketone metabolism resulting in hypoglycemia and abnormal protein metabolism resulting in organic acids detectable in urine. The ketogenic/ketolytic organic acidemias include 3-hydoxy-3-methylglutaryl-CoA lyase deficiency (HMGCoA, #246450), 3-oxothiolase (beta-ketothiolase, #203750) deficiency and several others. They can present with hypoglycemia and occasionally hyperammonemia.
Cerebral organic acidemias include glutaric aciduria 1 (GA1, #231670), L-2-hydroxyglutaric aciduria (#236792), and several others which are rarer. They do not affect organs other than the brain, but they continue to have detectable organic acids in the urine and blood.
Each type of organic acidemia shares some of the same treatment approaches but there are also some differences.
Table 1: list of the different types of OAs
1.1General management of OAs
In general, all OAs are treated by 1) reversal or prevention of catabolism, 2) decreased exposure to the compounds which cannot be metabolized, 3) scavenging of the “toxins” produced, 4) replacement of the non-manufactured down-stream intermediates, 5) use of cofactors and supplements to make that which is disturbed, work better, and 6) treatment of the complications using standard methods (Table 2). This list of steps can be used conceptualizing acute and long-term management.
Table 2
The Conceptional framework for treating individuals with OAs and some common management approaches for acute presentations and chronic management
| Concept | Acute Management | Chronic Management |
| Reversal or prevention of catabolism | 10% dextrose with electrolytes (GIR 6–10 mg/kg/day) | Avoid fasting |
| Decrease exposure | May hold protein for no more than 48 hours | Diet to avoid the non-tolerated amino acids |
| Scavenge the “toxins” produced | *Specific for disorder | *Specific for disorders |
| Replace the non-manufactured product | # e.g. glucose | Not yet available for anything other than glucose |
| Cofactors and supplements to make increase enzyme work betteractivity | **e.g. Biotin, hydroxocobalamin | **e.g. Biotin, hydroxocobalamin |
| Treat other organ complications | Standard therapies | Standard therapies |
| Fix the broken enzyme | Research on-going | Research on-going |
*Specific for disorder includes nitrogen scavengers if hyperammonemia or levocarnitine. #ketolytic/ketogenic need additional glucose, but typically research. **PCC is a biotin requiring enzyme, MMUT is a hydroxocobalamin requiring enzyme.
Table 2: The Conceptional framework for treating individuals with OAs and some common management approaches for acute presentations and chronic management
1.2Reversal and avoidance of catabolism and decreased exposure
In the acute decompensated state, therapies for OAs include reversal of catabolism and a 12- to 48-hour hold on protein intake. Oftentimes, catabolism can be reversed by using 10% dextrose with electrolytes at a glucose delivery rate of 6 to 10 mg/kg/min (about 1.5 times maintenance) [2, 5–7]. Insulin can be used to help deliver glucose to the cells and so in many cases a continuous infusion (with continued dextrose delivery) is used. Glucose delivery is particularly important in the ketolytic/ketogenic OAs since they do not have appropriate ketone production.
Intralipids may or may not be used in the systemic and cerebral OAs. Evidence for intralipids use is limited and controversial. Some studies have been advocates [8], however, intralipids are not universally used [3] due to the fact that at these glucose delivery rates, insulin should be released and thus, fatty acid oxidation inhibited. Most ketolytic/ketogenic disorders have disturbed fat metabolism and intralipids should be avoided.
In the chronic state (and sometimes for acute management), the diet focuses on adequate calories [9]. For the systemic OAs, PA and MMA, WHO requirements for natural proteins is necessary and precursor-free amino acid mixtures can help to prevent catabolism from inadequate calories [10]. Studies in MMA have shown that too much precursor-free amino acid mixture [11] may be problematic and so adequate natural protein is important even though hyperammonemia can be a complication. For IVA, WHO adequate calories +/- precursor-free amino acid mixtures are used depending on the severity of the disease (EIMD website).
For the ketogenic/ketolytic OAs, there are no current recommendations available, however, again, adequate calories for age (according to WHO guidelines) is important. Protein restriction has been discussed in the field but does not appear to have significant role in chronic management, especially outside the infancy period.
Individuals with the cerebral OAs should avoid catabolism in the same way as the other OAs. Prevention of complications often requires 6–10 mg/kg/min glucose delivery during illness like the systemic and ketolytic/ketogenic OAs. Dietary recommendations are discussed below.
1.3Scavenge the toxins
There are two types of toxins seen in the OAs. The first is due to the accumulation of the intermediate proximal to the enzyme block, which may result in other precursors and this is seen in all the OAs to some extent. The second is ammonium which is the result from secondary inhibition of the urea cycle, presumably by compounds that accumulate in the first type and is predominately seen in the systemic and ketolytic/ketogenic OAs. Scavenging of both types (or removal of each by binding to another compounds) is important to decrease these causes of complications.
Scavenging the accumulated intermediates proximal to the enzyme block is often accomplished by levocarnitine. There may be additional compounds used to scavenge these intermediates, but when used they will be addressed later in this review.
Elevations in ammonia must be treated rapidly, and this can be done using intravenous scavengers (such as sodium phenylacetate with sodium benzoate), oral scavengers (e.g. sodium phenylbutarate or carbaglumic acid) or through hemodialysis, hemofiltration, or extracorporeal membrane oxidation (ECMO). In the case of ketolytic/ketogenic, scavengers may not be necessary if adequate dextrose calories are provided, but most will receive at least some scavenger treatment in the early phases of disease.
Metabolic acidosis from the OAs can be controlled by using calories in the form of 10% dextrose containing fluids, precursor-free amino acid mixtures, as well as hemodialysis, hemofiltration, or ECMO.
Chronic management includes treatment with levocarnitine to scavenge proximal toxins and in some patients’ nitrogen scavengers. There is some discussion whether glycine can be used as a scavenger in IVA, but its use is not universal.
1.4Cofactors
The carboxylases (which include the enzymes which are disrupted in PA and 3MCCC) are biotin requiring enzymes. Rarely will an individual require supplementation with biotin to regain function. A short trial can be attempted, but rarely is helpful.
Methylmalonyl-CoA mutase (MMUT) is a cobalamin requiring enzyme. Some individuals respond to intramuscular hydroxocobalamin. A trial should be attempted for every patient. Multiple approaches to this trial have been used but often with a dose of 1 mg intramuscular per day for 3 days a change of 50% in urine and plasma methylmalonic acid per MMA/PA guidelines denotes responsiveness [3, 12]. There can be variable responses from no response to a complete correction of enzyme function.
1.5Treatment of underlying complications
Complications from residual damage from metabolic decompensations are treated by standard protocols.
Long term organ damage, especially seen in the systemic OAs are treated by standard clinical interventions. For example, in PA patient with cardiomyopathy, there is no current therapy that is specific for PA, but rather usual medications and interventions for cardiomyopathy are used. Screening recommendations for long term organ complications are listed in the next section which highlights specific interventions for selected representative OAs.
2Disease specific interventions
2.1Approach to propionate pathway (PA and MMA)
PA and MMA are systemic OAs of the intoxication-type that can present in the typical intoxication-type manner, as a sepsis mimic, and as a disorder diagnosed via NBS.
PA is caused by the inability of propionyl-CoA carboxylase to convert propionyl-CoA (produced from catabolism of cholesterol, valine, odd-chain fatty acids, methionine, isoleucine and threonine, also known as c-VOMIT) to methylmalonyl-CoA. Patients can present with hyperammonemia, metabolic acidosis, or both during episodes of increased catabolism (Fig. 1) [3].
Fig.1
The propionate pathway with the toxins, organic acid detectable and enzyme impacted. Abbreviations: C-VOMIT: cholesterol, valine, odd chain fatty acids, methionine, isoleucine and threonine.
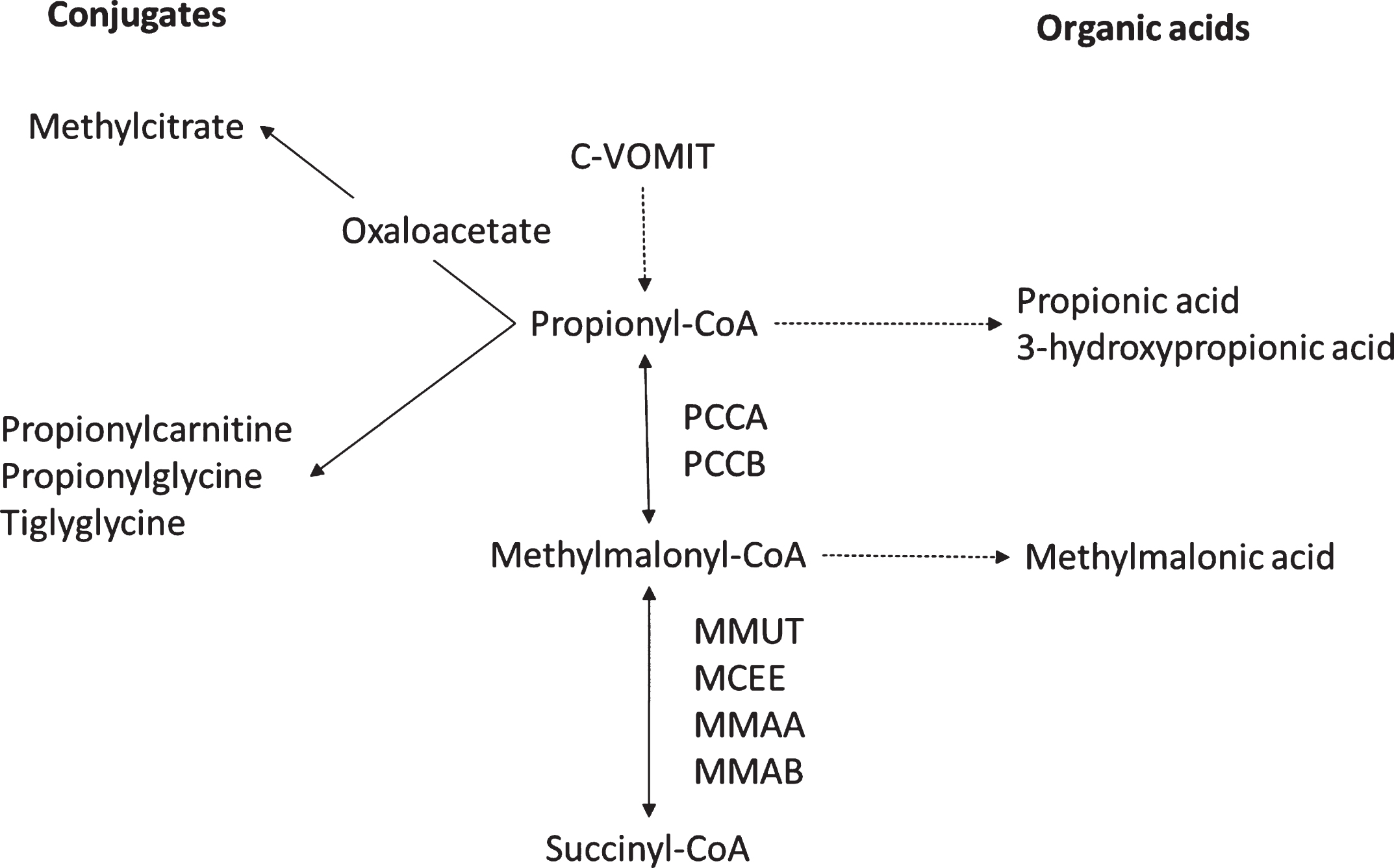
MMA is caused by the inhibition of the next step in the propionate pathway since it is caused by the inability of methylmalonyl-CoA to be converted to succinyl-CoA (intermediate in Krebs cycle) due to dysfunction of methylmalonyl-CoA mutase (a vitamin B12–responsive enzyme), cobalamin A deficiency (gene mutations in MMAA), or cobalamin B deficiency (with gene mutations in MMAB). MMA presents in a manner similar to PA but also has elevations of methylmalonic acid in urine and blood [3] (Fig. 1).
If available, newborn screening can identify both PA and MMA, but individuals may present prior to the return of results.
Treatment for PA and MMA are similar for both the short-term metabolic decompensations and long term. The few differences in management will be addressed below. Day to day management for PA and MMA focuses on avoiding extra propionate precursors in the diet and decreasing production by gut flora. Usually decreased exposure in the diet requires a precursor-free amino acid mix added to natural protein source. The goal is to provide at least WHO protein requirements with at least 2/3 of protein needs from natural protein. Complicating therapy is the estimate that about 30% of propionate in the body is produced by the gut flora. Current therapy includes aggressive prevention of constipation and potentially antibiotic use including metronidazole courses [3]. Levocarnitine is used as a scavenger for organic acids both during an acute metabolic decompensation event and as part of long-term management.
During metabolic decompensations, 10% dextrose with electrolytes, with or without intralipids, is often used [3, 6]. The goal is to provide 6–10 mg/kg/min glucose delivery [3, 6, 7]. Insulin infusions (with continued dextrose delivery) may be useful in sites where this can be closely monitored to prevent hypoglycemia.
Dialysis or ECMO may be necessary in severe metabolic acidosis or hyperammonemia during decompensations.
MMA may be responsive to administration of intramuscular or subcutaneous vitamin B12. Typical dosing in those responsive is 1 mg intramuscularly daily; the approach to determine responsivity is listed above (under 1.4).
The enzyme that is dysfunctional in PA is biotin dependent, and very rarely biotin may help.
Long term complications include cardiac, ophthalmological, neurological, and renal difficulties or dysfunction, and possibly increased insulin requirement.
Cardiac complications are more common in PA than in MMA and these include cardiac arrhythmias and cardiomyopathy. Long QTc syndrome usually is related to age [13] (unpublished communication EIMD). Cardiomyopathy is also more likely with older ages. Both arrhythmia and cardiomyopathy usually occur gradually, but acute events are noted. Screening annually and treatment when identified is recommended [3, 14]
Neurological complications including developmental delay, intellectual disability, autistic features and stroke-like episodes are common [15] energy strokes especially of the basal ganglia can lead to movement disorders; unfortunately the exact pathophysiology of the stroke-like episodes or energy strokes is unknown, but they are suspected to be related to secondary energy difficulties [16].
Renal failure is more common MMA, but dysfunction is seen in PA especially with electrolyte wasting during metabolic decompensations.
Hyperammonemia can occur not only during metabolic decompensations, but also at maximal baseline therapy (more in PA than MMA). Medications such as sodium benzoate, sodium phenylbutyrate, and carbaglumic acid have been used. The exact choice of medication is somewhat dependent on the availability of each. Sodium benzoate has the benefit of also impacting glycine levels. Carbaglumic acid is thought to overcome the secondary urea cycle block [17–19].
With the renal failure seen in MMA, renal transplant has been used. It has been observed that renal transplant must be repeated in MMA and this is thought to be a consequence of exposure to toxins. Thus, liver-renal transplants are increasing in number especially in MMA. With improved outcomes for all liver transplants in the last 10 years, liver-only transplants are also used in PA and MMA to ameliorate the disease and there is growing evidence of its efficacy [20, 21].
2.2Approach to isovaleric acidemia
IVA can have a milder phenotype, but in certain settings it can have devastating complications [22–24]. IVA is a disorder of the leucine catabolic pathway in which the intermediate isovaleryl-CoA cannot be metabolized by isovaleryl-CoA dehydrogenase (Fig. 2), resulting in metabolic acidosis and in some cases hyperammonemia. Therapy focuses on adding the scavengers, levocarnitine and glycine as medications. In times of illness or stress, patients should receive increased calories and fluids (intravenous fluids with 10% dextrose and electrolytes may be used). Hyperammonemia, if found, should be treated aggressively. Dietary therapy is individualized in that not everyone will require a leucine restriction. Genotype can be of help when trying to predict the milder patient.
Fig.2
Metabolism of Leucine with its intermediates and enzymes.
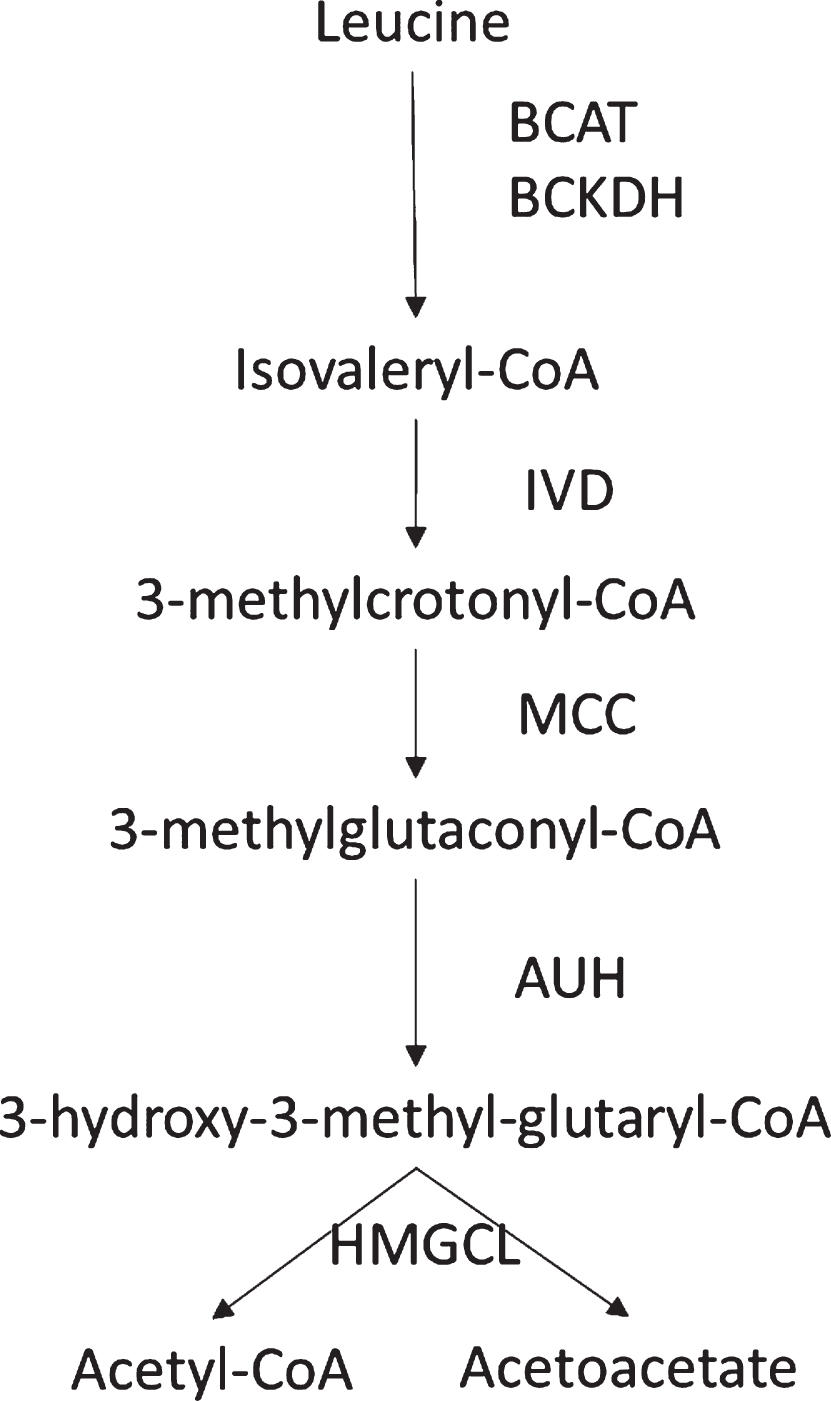
If newborn screening is available, IVA can be identified. Because of the possible milder phenotype, infants whose mothers have IVA may have a positive newborn screen.
2.3Approaches for ketolytic/ketogenic OAs
Ketolytic/ketogenic OAs are a group of disorders caused by enzymes which impact both protein and ketone metabolism.
Individuals with these disorders have a predominant difficulty with hypoglycemia related to fasting. Some of these disorders may also have elevated ammonia in extremis. For example, HMG-CoA lyase often will have hypoglycemia and mildly elevated ammonia in its neonatal presentation.
There are limited management recommendations published for these disorders; however,similar principles as in other OAs treatment are applicable. Management includes providing adequate calories (with adequate dextrose) to prevent the need for ketones. Usually making sure that at least 6–10 mg/kg/min glucose delivery is used is adequate in the acute phase, but some infants will require additional calories. Elevated ammonia usually responds to dextrose-containing calories.
Long term complications are not typically observed unless initial presentation had long lasting hypoglycemia or hyperammonemia.
Whether any dietary protein restriction is needed is unclear [5]. We try to place our patients on vegetarian diets, with goal towards limiting dietary fat and protein to healthy levels. Moreover, we recommend limited fasting. Overnight fasting (while in the healthy state) is limited to no longer than 4 hours of fasting for 1–4 month old, 5 hours for 5 month old, 6 hours for 6 month old, 7 hours for 7 month old, 8 hours for 8 month old, 9 hours for 9 month old, 10 hours for 10 month old, 11 hours for 11 month old up to 12 hours in all equal to older than 12 months. Using these dietary recommendations, the only decompensation after the neonatal period we have seen in our patient population was in a patient with HMG-CoA lyase deficiency who was receiving high dose steroids as part of a treatment for a very rare metastatic overgrowth of lymph channels, kaposiform lymphangiomatosis. Her kaposiform lymphangiomatosis case is described in Fernandes et al. [25].
2.4Approaches to treatment of GA1
GA1 is considered the quintessential cerebral OA. It is caused by dysfunction of glutaryl-CoA dehydrogenase (GCDH, #231670) and inability to convert glutaryl-CoA to crotonyl-CoA in the catabolism of lysine and tryptophan [26]. Blockage at GCDH leads to accumulation of glutaric acid and 3-hydroxyglutaric acid in blood and urine. Patients have macrocephaly and can develop striatal injury usually between 6 months and 6 years which is usually related to illness or catabolic stress and can lead to a movement disorder.
Aggressive and early treatment of illness or fever (and aggressive prevention during surgery and around immunizations is important). Use of antipyretics during fever is particularly important. Aggressive therapy during risk periods focuses on using dextrose with electrolyte containing fluids at 6–10 mg/kg/min glucose delivery and making sure adequate calories are provided. Diet alterations include a low lysine diet with low lysine (+/–) low tryptophan metabolic foods during the risk years [26]. There is some discussion about whether metabolic foods with extra arginine may be helpful, but currently no consensus [27]. Levocarnitine is used as a scavenger. There do not appear to be any cofactor supplements which appear to be useful.
Complications are usually related to striatal injury resulting in dystonia. This dystonia is treated using standard therapies. The goal is to prevent these episodes.
2.5Other important interventions for all the OAs
Every patient (and their caregivers) with an OA should know the name of their disorder and have access to a metabolic specialist to help personalize their therapy. They should have contact information for their metabolic provider and a plan for contact during travel when they are on holiday. It is important for each to have an Emergency protocol which can be provided to caregivers in an emergency room setting. This protocol should at the very least, list the disorder and common complications, provide instructions for initial therapy and provide the metabolic provider’s contact information. If the local hospital is not a metabolic center, the local hospital should have a plan in place, including knowledge of the patient with an OA, availability of adequate dextrose containing fluids and plan for transporting the patient to a metabolic center for more specific care. Patients (and their caregivers) should also have instructions about when to contact the metabolic provider (including what temperature is a fever, how much vomiting is acceptable, when surgery is planned, etc).
Individuals with OAs do not have a contraindication to regular childhood immunizations based on their OA. There is no data supporting the concern for increased metabolic decompensations. In a cohort of a similar patient population—individuals with urea cycle disorders—there was no increase in hospitalizations the month following vaccination [28]. Individuals with OAs should be vaccinated using the standard vaccine schedule. Antipyretics should be given immediately prior as all as the 24 hours following vaccine as a precaution.
2.6Research and enzyme function recovery
Several therapeutic approaches are on the horizon for these disorders. Some look at replacing down-stream intermediates; others look to scavenge toxins; and still others look to replace the dysfunctional enzyme by either enzyme replacement or nucleotide-based approaches. Most of the recent research has focused on the propionate pathway disorders, PA and MMA, but the approaches may work for other OAs.
Since more common in other disorders, the initial therapeutic approach section includes replacement of deficient down-stream intermediates, but this is not currently standard in the OA management. Some work has been done to examine whether replacement of these intermediates could be helpful in the propionate pathway OAs. Recent studies have shown that citrate supplements can replace some missing downstream intermediates in individuals with PA or MMA, but not necessarily other compounds [29].
Looking at scavengers, the propionate pathway disorders seem to have elevations in reactive oxygen species. More recently studies have looked at scavenging these damaging molecules, but these therapies have not yet reached the clinic [30–32].
2.7Enzyme replacement or nucleotide production of new enzyme
Enzyme replacement has been designed for PA and initial studies have looked promising in mice [33] but these have not progressed to human studies and have been predominately proof-of-principle studies.
On the other hand, nucleotide therapy to provide either RNA or DNA replacement of the abnormal PCCA/PCCB/MMUT gene have all been described and appear to be farther along the development pathway [34–36]. Several approaches have been tested in mice, but no active studies were open for patients at the time of this writing. As with other disorders treated with gene therapy approaches, the delivery method of the therapy is important and no perfect single vector, which can deliver to all the needed tissues, has been identified. As a result, currently the question of whether several different vectors are needed or whether correction in only certain organs would be sufficient is still yet to be answered.
All these studies look promising to help improve management and although predominately focused on the propionate pathway OAs, their approaches may be attempted in other OAs.
Summary
The treatment of OAs can be conceptualized into 6 important steps. First is reversal or prevention of catabolism. The second is avoidance of non-metabolized precursors (especially through diet). The third is scavenge the “toxins”. Fourth is replace that which is not made. Fifth is try cofactors with the goal of making the enzyme work better. Finally, the sixth step is to screen and treat the known complications of the disorder. The future will bring a seventh step which is replace the dysfunctional enzyme with a functional one. Keeping these steps in mind, using the resources available to you, everyone should be able to take care of OA (or at least get started, allowing for time to ask and get an answer from an expert).
Acknowledgments
I would like to thank the NIDDK for their gracious support through a K08DK105233 (KAC), as well as my partners and colleagues. I would also like to thank all the patients and families of the Children’s National Rare Disease Institute who teach us about their problems or disorders every day.
References
[1] | Fenton W.A. , Gravel R. , Rosenblatt D. . Disorders of Propionate and Methylmalonate Metabolism, in: Scriver C.R., Beaudet A.L., Valle D., Sly W. (Eds.), The Metabolic & Molecular Bases of Inherited Disease, McGraw-Hill, New York, (2006) , pp. 2165–2204. |
[2] | Saudubray J.M. , Sedel F. and Walter J.H. , Clinical approach to treatable inborn metabolic diseases: An introduction, J Inherit Metab Dis 29: ((2006) ), 261–274. |
[3] | Baumgartner M.R. , Horster F. , Dionisi-Vici C. , Haliloglu G. , Karall D. , Chapman K.A. , Huemer M. , Hochuli M. , Assoun M. , Ballhausen D. , Burlina A. , Fowler B. , Grunert S.C. , Grunewald S. , Honzik T. , Merinero B. , Perez-Cerda C. , Scholl-Burgi S. , Skovby F. , Wijburg F. , MacDonald A. , Martinelli D. , Sass J.O. , Valayannopoulos V. and Chakrapani A. , Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia, Orphanet Journal of Rare Diseases 9: ((2014) ), 130. |
[4] | Prietsch V. , Lindner M. , Zschocke J. , Nyhan W.L. and Hoffmann G.F. , Emergency management of inherited metabolic diseases, Journal of Inherited Metabolic Disease 25: ((2002) ), 531–546. |
[5] | Vademecum Metabolicum: Diagnosis and Treatment of Inborn Errors of Metabolism, Milupa, Friedrichsdorf, Germany, (2011) . |
[6] | Chapman K.A. , Gropman A. , MacLeod E. , Stagni K. , Summar M.L. , Ueda K. , Ah M.N. , Franks J. , Island E. , Matern D. , Pena L. , Smith B. , Sutton V.R. , Urv T. , Venditti C. and Chakrapani A. , Acute management of propionic acidemia, Mol Genet Metab 105: ((2012) ), 16–25. |
[7] | Tsalikian E. , Simmons P. , Gerich J.E. , Howard C. and Haymond M.W. , Glucose production and utilization in children with glycogen storage disease type I, Am J Physiol 247: ((1984) ), E513–E519. |
[8] | Fraser J.L. and Venditti C.P. , Methylmalonic and propionic acidemias: Clinical management update Current opinion in pediatrics, 28: ((2016) ), 682–693. |
[9] | Evans M. , Truby H. and Boneh A. , The Relationship between Dietary Intake, Growth, and Body Composition in Inborn Errors of Intermediary Protein Metabolism, The Journal of Pediatrics 188: ((2017) ), 163–172. |
[10] | Jurecki E. , Ueda K. , Frazier D. , Rohr F. , Thompson A. , Hussa C. , Obernolte L. , Reineking B. , Roberts A.M. , Yannicelli S. , Osara Y. , Stembridge A. , Splett P. and Singh R.H. , Nutrition management guideline for propionic acidemia: An evidence- and consensus-based approach Molecular genetics and metabolism, ((2019) ). |
[11] | Manoli I. , Myles J.G. , Sloan J.L. , Carrillo-Carrasco N. , Morava E. , Strauss K.A. , Morton H. and Venditti C.P. , A critical reappraisal of dietary practices in methylmalonic acidemia raises concerns about the safety of medical foods. Part 2: Cobalamin C deficiency Genetics in medicine: Official journal of the American College of Medical Genetics, ((2015) ). |
[12] | Fowler B. , Leonard J.V. and Baumgartner M.R. , Causes of and diagnostic approach to methylmalonic acidurias, Journal of Inherited Metabolic Disease 31: ((2008) ), 350–360. |
[13] | Baumgartner D. , Scholl-Burgi S. , Sass J.O. , Sperl W. , Schweigmann U. , Stein J.I. and Karall D. , Prolonged QTc intervals and decreased left ventricular contractility in patients with propionic acidemia, J Pediatr 150: ((2007) ), 192–197. |
[14] | Sutton V.R. , Chapman K.A. , Gropman A.L. , MacLeod E. , Stagni K. , Summar M.L. , Ueda K. , Ah Mew N. , Franks J., Island E., Matern D., Pena L., Smith B., Urv T., Venditti C. and Chakarapani A., Chronic management and health supervision of individuals with propionic acidemia, Mol Genet Metab 105: ((2012) ), 26–33. |
[15] | Pena L. and Burton B.K. , Survey of health status and complications among propionic acidemia patients, Am J Med Genet A 158A: ((2012) ), 1641–1646. |
[16] | Schreiber J. , Chapman K.A. , Summar M.L. , Ah M.N. , Sutton V.R. , MacLeod E. , Stagni K. , Ueda K. , Franks J. , Island E. , Matern D. , Pena L. , Smith B. , Urv T. , Venditti C. , Chakarapani A. and Gropman A.L. , Neurologic considerations in propionic acidemia, Mol Genet Metab 105: ((2012) ), 10–15. |
[17] | Ah Mew N. , McCarter R., Daikhin Y., Nissim I., Yudkoff M. and Tuchman M., N-carbamylglutamate augments ureagenesis and reduces ammonia and glutamine in propionic acidemia, Pediatrics 126: ((2010) ), e208–e214. |
[18] | Filippi L. , Gozzini E. , Fiorini P. , Malvagia S. , la M.G. and Donati M.A. , N-carbamylglutamate in emergency management of hyperammonemia in neonatal acute onset propionic and methylmalonic aciduria, Neonatology 97: ((2010) ), 286–290. |
[19] | Schwahn B.C. , Pieterse L. , Bisset W.M. , Galloway P.G. and Robinson P.H. , Biochemical efficacy of N-carbamylglutamate in neonatal severe hyperammonaemia due to propionic acidaemia, Eur J Pediatr 169: ((2010) ), 133–134. |
[20] | Charbit-Henrion F. , Lacaille F. , McKiernan P. , Girard M. , de Lonlay P. , Valayannopoulos V., Ottolenghi C., Chakrapani A., Preece M., Sharif K., Chardot C., Hubert P. and Dupic L., Early and late complications after liver transplantation for propionic acidemia in children: A two centers study American journal of transplantation: Official journal of the American Society of Transplantation and the American Society of Transplant Surgeons, 15: ((2015) ), 786–791. |
[21] | Li M. , Dick A. , Montenovo M. , Horslen S. and Hansen R. , Cost-effectiveness of liver transplantation in methylmalonic and propionic acidemias Liver transplantation: Official publication of the American Association for the Study of Liver Diseases and the International Liver Transplantation Society, 21: ((2015) ), 1208–1218. |
[22] | Ensenauer R. , Vockley J. , Willard J.M. , Huey J.C. , Sass J.O. , Edland S.D. , Burton B.K. , Berry S.A. , Santer R. , Grunert S. , Koch H.G. , Marquardt I. , Rinaldo P. , Hahn S. and Matern D. , A common mutation is associated with a mild, potentially asymptomatic phenotype in patients with isovaleric acidemia diagnosed by newborn screening, Am J Hum Genet 75: ((2004) ), 1136–1142. |
[23] | Vockley J. and Ensenauer R. , Isovaleric acidemia: New aspects of genetic and phenotypic heterogeneity, Am J Med Genet C Semin Med Genet 142C: ((2006) ), 95–103. |
[24] | Dionisi-Vici C. , Deodato F. , Roschinger W. , Rhead W. and Wilcken B. , ‘Classical’ organic acidurias, propionic aciduria, methylmalonic aciduria and isovaleric aciduria: Long-term outcome and effects of expanded newborn screening using tandem mass spectrometry, J Inherit Metab Dis 29: ((2006) ), 383–389. |
[25] | Fernandes V.M. , Fargo J.H. , Saini S. , Guerrera M.F. , Marcus L. , Luchtman-Jones L. , Adams D. and Meier E.R. , Kaposiform lymphangiomatosis: Unifying features of a heterogeneous disorder, Pediatric Blood & Cancer 62: ((2015) ), 901–904. |
[26] | Kolker S. , Christensen E. , Leonard J.V. , Greenberg C.R. , Boneh A. , Burlina A.B. , Burlina A.P. , Dixon M. , Duran M. , Garcia C.A. , Goodman S.I. , Koeller D.M. , Kyllerman M. , Muhlhausen C. , Muller E. , Okun J.G. , Wilcken B. , Hoffmann G.F. and Burgard P. , Diagnosis and management of glutaric aciduria type I - revised recommendations, J Inherit Metab Dis 34: ((2011) ), 677–694. |
[27] | Kolker S. , Boy S.P. , Heringer J. , Muller E. , Maier E.M. , Ensenauer R. , Muhlhausen C. , Schlune A. , Greenberg C.R. , Koeller D.M. , Hoffmann G.F. , Haege G. and Burgard P. , Complementary dietary treatment using lysine-free, arginine-fortified amino acid supplements in glutaric aciduria type I - A decade of experience Molecular genetics and metabolism, 107: ((2012) ), 72–80. |
[28] | Morgan T.M. , Schlegel C. , Edwards K.M. , Welch-Burke T. , Zhu Y. , Sparks R. and Summar M. , Vaccines are not associated with metabolic events in children with urea cycle disorders Pediatrics, 127: ((2011) ), e1147–e1153. |
[29] | Longo N. , Price L.B. , Gappmaier E. , Cantor N.L. , Ernst S.L. , Bailey C. and Pasquali M. , Anaplerotic therapy in propionic acidemia, Molecular Genetics and Metabolism 122: ((2017) ), 51–59. |
[30] | Gallego-Villar L. , Perez B. , Ugarte M. , Desviat L.R. and Richard E. , Antioxidants successfully reduce ROS production in propionic acidemia fibroblasts, Biochemical and Biophysical Research Communications 452: ((2014) ), 457–461. |
[31] | Gallego-Villar L. , Perez-Cerda C. , Perez B. , Abia D. , Ugarte M. , Richard E. and Desviat L.R. , Functional characterization of novel genotypes and cellular oxidative stress studies in propionic acidemia, Journal of Inherited Metabolic Disease 36: ((2013) ), 731–740. |
[32] | Gallego-Villar L. , Rivera-Barahona A. , Cuevas-Martin C. , Guenzel A. , Perez B. , Barry M.A. , Murphy M.P. , Logan A. , Gonzalez-Quintana A. , Martin M.A. , Medina S. , Gil-Izquierdo A. , Cuezva J.M. , Richard E. and Desviat L.R. , In vivo evidence of mitochondrial dysfunction and altered redox homeostasis in a genetic mouse model of propionic acidemia: Implications for the pathophysiology of this disorder Free radical biology & medicine, 96: ((2016) ), 1–12. |
[33] | Guenzel A.J. , Collard R. , Kraus J.P. , Matern D. and Barry M.A. , Long-term sex-biased correction of circulating propionic acidemia disease markers by adeno-associated virus vectors, Human Gene Therapy 26: ((2015) ), 153–160. |
[34] | An D. , Schneller J.L. , Frassetto A. , Liang S. , Zhu X. , Park J.S. , Theisen M. , Hong S.J. , Zhou J. , Rajendran R. , Levy B. , Howell R. , Besin G. , Presnyak V. , Sabnis S. , Murphy-Benenato K.E. , Kumarasinghe E.S. , Salerno T. , Mihai C. , Lukacs C.M. , Chandler R.J. , Guey L.T. , Venditti C.P. and Martini P.G.V. , Systemic Messenger RNA Therapy as a Treatment for Methylmalonic Acidemia Cell reports, 21: ((2017) ), 3548–3558. |
[35] | Chandler R.J. , Chandrasekaran S. , Carrillo-Carrasco N. , Senac J.S. , Hofherr S. , Barry M.A. and Venditti C.P. , Adeno-associated virus serotype 8 (AAV8) Gene Transfer Rescues a Neonatal Lethal Murine Model of Propionic Acidemia, Hum Gene Ther ((2010) ). |
[36] | Chandler R.J. , Chandrasekaran S. , Carrillo-Carrasco N. , Senac J.S. , Hofherr S.E. , Barry M.A. and Venditti C.P. , Adeno-associated virus serotype 8 gene transfer rescues a neonatal lethal murine model of propionic acidemia, Human Gene Therapy 22: ((2011) ), 477–481. |


