
Genetic disorders of calcium, phosphorus, and bone homeostasis
Abstract
Calcium and phosphorus [in the form of phosphate (H2PO4-/HPO4–2)] are the most abundant elements in the body where they subserve many functions – the most prominent of which is the formation of hydroxyapatite [Ca10(PO4)10(OH)2] – the mineral portion of bone [1, 2]. Calcium and phosphate are present in every cell in the body and are essential for normal cellular function. Calcium is indispensable for transmission of neural signals, muscle contractility, intracellular signal transduction, and secretion of cellular products. Phosphate is required for formation of nuclear and mitochondrial DNA and RNA, phospholipids for cell membrane formation, glycolysis and generation of high energy bonds (ATP), and intracellular signaling by guanosine triphosphate (GTP)-bearing proteins (G-proteins) and cyclic adenosine monophosphate (AMP). Phosphorylation of networks of intracellular proteins by many different kinases propagate the transmission of signals from the cell’s plasma membrane into the nucleus to regulate gene expression and cellular function. Genetic disorders of calcium, phosphate, and skeletal homeostasis lead to hypercalcemia, hypocalcemia, rickets or osteomalacia, osteopenia with marked skeletal fragility, and excessively dense bones.
Abbreviations
AD | Autosomal dominant |
ADHR | Autosomal dominant hypophosphatemic rickets |
AHO | Albright’s hereditary osteodystrophy |
APECED | Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy |
AR | Autosomal recessive |
ASARM | Acidic serine- and aspartate-rich motif |
BMP | Bone morphogenetic protein |
CASR | Calcium sensing receptor |
DMP1 | Dentin matrix protein-1 |
FGF | Fibroblast growth factor |
FGFR | FGF receptor |
GPCR | G-protein coupled receptor |
MEPE | Matrix extracellular phosphoglycoprotein |
NFκB | Nuclear factor kappa B |
NPT | Sodium/phosphate cotransporter |
OI | Osteogenesis imperfecta |
PHP | Pseudohypoparathyroidism |
PKA | Protein kinase A |
PPHP | Pseudopseudohypoparathyroidism |
PTG | Parathyroid gland |
PTH | Parathyroid hormone |
PTHR | PTH receptor |
PTHrP | PTH related protein |
RANK | Receptor activator of nuclear factor κB |
RANKL | Rank ligand |
RXR | Retinoid X receptor |
sFRP-4 | Secreted frizzled-related protein-4 |
SIBLINGS | Small integrin-binding ligand, N-linked glycoprotein |
TF | Transcription factor |
TGFβ | Transforming growth factor β |
TIO | Tumor induced osteomalacia |
VDR | Vitamin D receptor |
VDRE | VDR response element |
WSTF | Williams syndrome transcription factor |
XHR | X-linked hypophosphatemic rickets |
25OHD3 | 25-Hydroxyvitamin D3 (calcidiol) |
1,25(OH)2D3 | 1,25-Dihydroxyvitamin D3 (calcitriol) |
1Normal calcium, phosphate, and skeletal homeostasis
1.1Calcium and phosphate
Ninety-nine percent of body calcium is deposited in bone as hydroxyapatite. Circulating, extracellular and intracellular calcium, and surface bone account for approximately one percent of total body calcium. Within bone calcium is both deeply deposited where turnover rate is relatively slow (several weeks) and on the bone surface where it is immediately accessible for homoeostatic needs. In blood, calcium circulates in the free or ionized state (Ca2+), bound to proteins (primarily albumin), and complexed to citrate, bicarbonate, or lactate. It is the Ca2+ concentration that is the biologically active form of plasma calcium and hence its concentration is closely controlled by the interaction of parathyroid hormone (PTH), calcitriol (1,25-dihydroxyvitamin D3), and calcitonin, a product of the thyroid parafollicular (C) cell, upon the kidney, bone, and intestinal tract [1, 3]. The circulating concentration of Ca2+ is monitored by the calcium sensing receptor (CaSR), a 7 transmembrane G-protein coupled receptor (GPCR) situated on the cell membranes of the parathyroid gland chief cell and renal tubular cells [4]. Binding of Ca2+ to the CaSR leads to activation of the Gαq subunit of the G-protein followed by increase in phospholipase C activity with ensuing enzymatic conversion of membrane-bound phosphatidylinositol 4,5-bisphosphate to cytosolic diacylglycerol and inositol trisphosphate; the latter then increases intracellular levels of Ca2+ by releasing it from storage sites in the endoplasmic reticulum. In the parathyroid chief cell, increases in intracellular concentrations of Ca2+ depress expression of PTH and decrease synthesis and secretion of its product. In the renal tubule, increased cytoplasmic Ca2+ levels depress reabsorption of filtered calcium and increase its urinary excretion.
PTH is synthesized as a preprohormone of 115 amino acids that is subsequently metabolized to a prohormone (90 amino acids) and then to the mature product (84 amino acids) by the enzyme furin. PTH is further metabolized to smaller carboxyl and amino terminal fragments either within the parathyroid glands (PTGs) themselves prior to secretion, a process dependent on the cytosolic Ca2+ concentration, or in the periphery (primarily the kidney) after secretion. The first 34 amino acids of PTH are essential for bioactivity as they contain the sites of binding to the PTH receptor (PTHR1). PTHR1 is a G-protein coupled receptor (GPCR) that mediates the effects of PTH and PTH related protein (PTHrP). PTHR1 is structurally related to the GPCRs for growth hormone releasing hormone, corticotropin releasing hormone, secretin, glucagon, and vasoactive intestinal polypeptide. PTH inhibits renal tubular reabsorption of phosphate. It enhances renal tubular reabsorption of calcium from glomerular filtrate and bone (Fig. 1). PTH also amplifies absorption of intestinal calcium by increasing renal tubular synthesis of calcitriol. In bone, PTH stimulates the stromal cell/osteoblast to synthesize agents that regulate osteoclastogenesis (Receptor Activator of Nuclear Factor κB ligand = RANKL and its antagonist Osteoprotegerin). PTHrP (MIM 168470) is a 144 amino acid peptide with sequence homology to PTH in its first 13 amino acids; it binds to PTHR1 with affinity equal to that of PTH. PTHrP is active primarily in the fetus as a regulator of endochondral bone development and the formation of teeth and breasts; it is frequently a cause of humoral hypercalcemia of malignancy due to its ectopic secretion by neoplastic tumors.
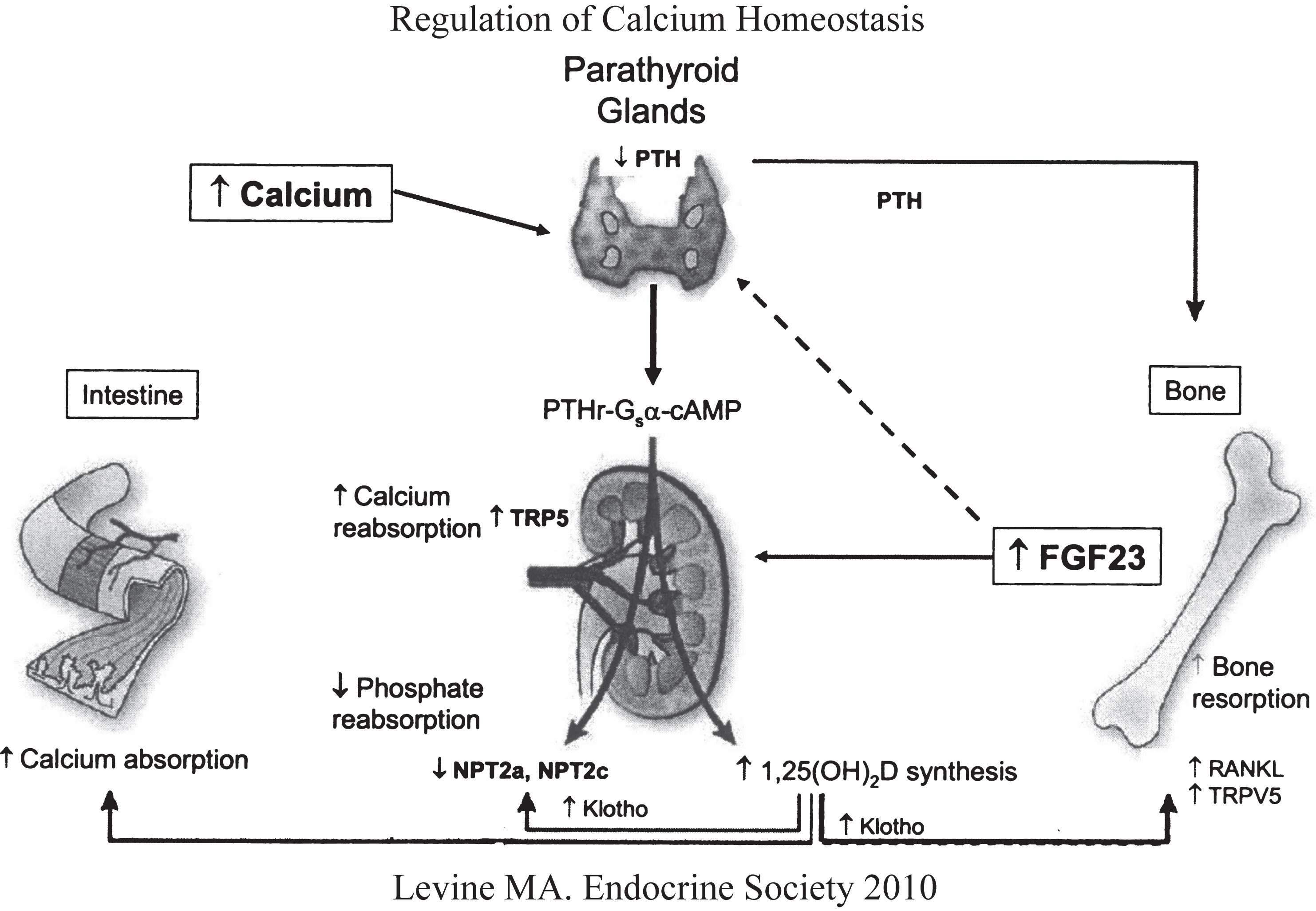
Fig.1
Regulation of calcium and phosphate homeostasis by the parathyroid glands, bone, kidney, and intestinal tract. Increase in serum Ca2+ stimulates secretion of PTH that acts upon the renal tubule (increases calcium reabsorption, decreases phosphate reabsorption, increases synthesis of calcitriol) and bone (stimulates osteoclastogenesis and reabsorption of skeletal calcium and phosphate. Increases in serum phosphate levels stimulates skeletal production of FGF23 (depresses renal tubular reabsorption of phosphate and synthesis of calcitriol). (Reproduced with permission from Levine MA. Investigation and management of hypocalcemia. Meet-the-Professor. Meeting of the Endocrine Society, 2010, p 81-86.)
The bulk of vitamin D3 (cholecalciferol) is synthesized in skin by exposure to ultraviolet light and heat [5, 6] (Fig. 2). In the liver, cholecalciferol is hydroxylated at carbon-25 to form calcidiol (25-hydroxyvitamin D3 = 25OHD3); in the kidney, calcidiol is hydroxylated at carbon-1 to form calcitriol [1,25-dihydroxyvitamin D3 = 1,25(OH)2D3], the biologically most active metabolite of vitamin D. Intracellularly, calcitriol binds to a nuclear transcription factor – the vitamin D receptor (VDR) – that then links as a heterodimer with the retinoid-X receptor (RXR) to the vitamin D response element (VDRE) in the 5’ promoter region of target genes to either stimulate or inhibit their transcription. Calcitriol is catabolized by renal 1,25(OH2)D3-24 hydroxylase to calcitroic acid and then excreted [7]. Calcitriol stimulates intestinal absorption of calcium and phosphate and increases reabsorption of calcium from bone and renal glomerular filtrate.
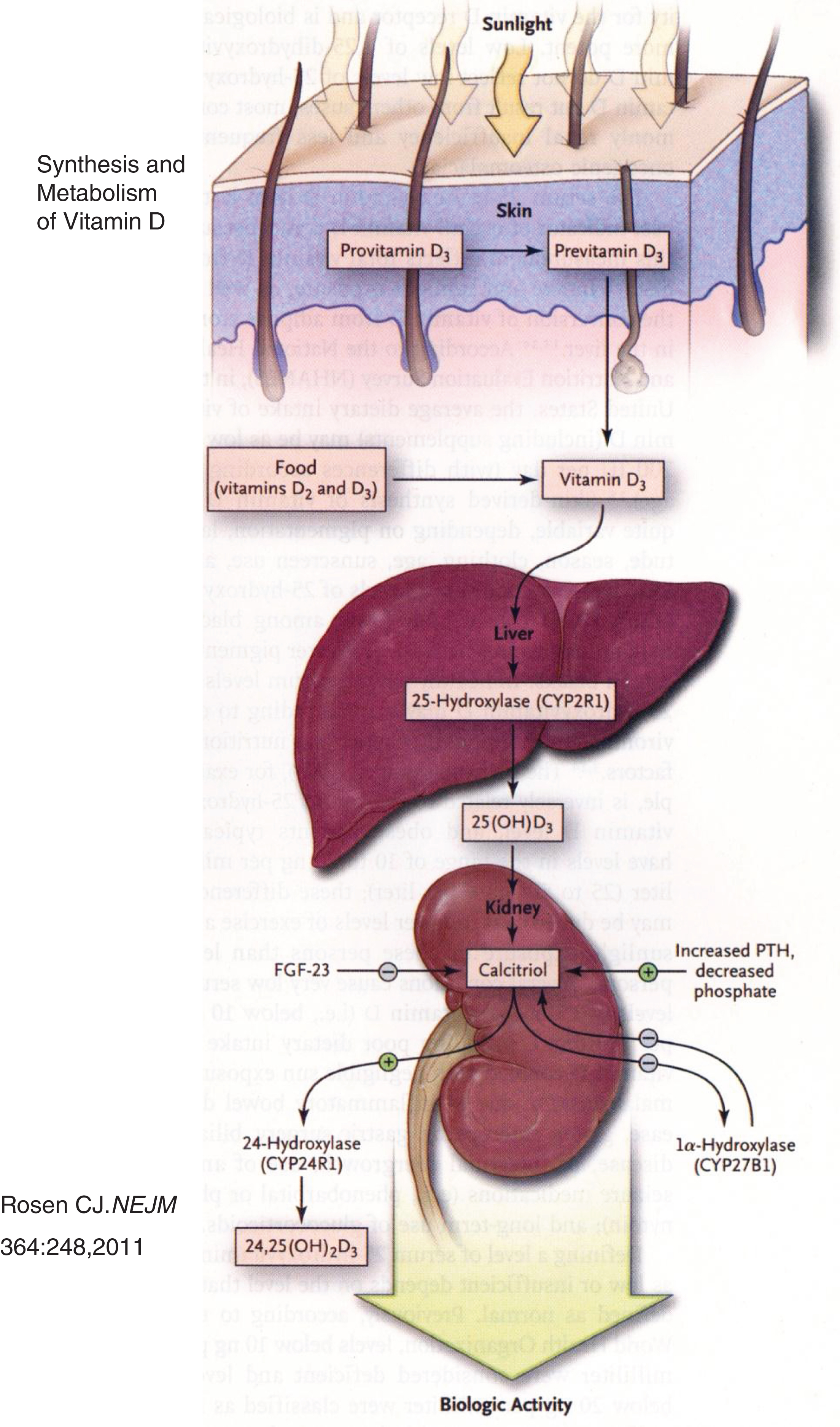
Fig.2
Synthesis and metabolism of vitamin D. Precursors of vitamin D synthesized in skin are metabolized to cholecalciferol in response to ultraviolet light and heat. Cholecalciferol is then successively hydroxylated in the liver and the kidney to form calcitriol. (Reproduced with permission from Rosen CJ. Clinical practice. Vitamin D insufficiency. N Engl J Med 364:248-254,2011.)
Calcitonin is a 32 amino acid hypocalcemic peptide encoded by CALCA (MIM 114130, chromosome 11p15.2-15.1) that is synthesized by the parafollicular C cells of the thyroid gland in response to rising concentrations of Ca2+. It antagonizes the effects of PTH and calcitriol on bone mineral reabsorption and intestinal calcium absorption, respectively. In the kidney, calcitonin binding to the renal tubule inhibits reabsorption of phosphate and calcium. CALCA also encodes calcitonin gene-related protein, a neuromodulatory peptide involved in regulation of the autonomic nervous system, and katacalcin, a second hypocalcemic factor. Acting through its GPCR – CALCR (CALCR, MIM 114131, chromosome 7q21.3) expressed on the plasma membrane of osteoclasts and other responsive cells, calcitonin stimulates signal transduction through stimulation of adenylyl cyclase activity. Serum concentrations of calcitonin serve as a marker of C cell hyperplasia and of medullary carcinoma of the thyroid.
Phosphate is present primarily in bone bound to calcium in the hydroxyapatite crystal. In the circulation it is complexed to various cations. Intracellularly, phosphate is a component of the G-proteins, cyclic AMP, and numerous intermediates in all of the many signal transduction systems that culminate in gene expression and cellular function. Phosphate is an irreplaceable part of nucleic acids linking individual nucleic acids to form chains of DNA and RNA. Phosphate is essential for cellular glucose processing [e.g., glucose-6-phosphate is the first compound formed during intracellular generation of high energy bonds (i.e., ATP)] and hence for energy utilization and cell metabolism [8]. Approximately 70 percent of phosphate filtered through the renal glomerulus is actively reabsorbed in the proximal renal tubule through apical membrane situated sodium-phosphate cotransporters – NPT2a and NPT2c (Fig. 3) [9]. PTH inhibits renal tubular reabsorption of phosphate by stimulating the rapid internalization and degradation of these cotransporters; PTH also down-regulates the expression of the genes encoding these transporters, mechanisms also promoted by the hypophosphatemic phosphatonins.
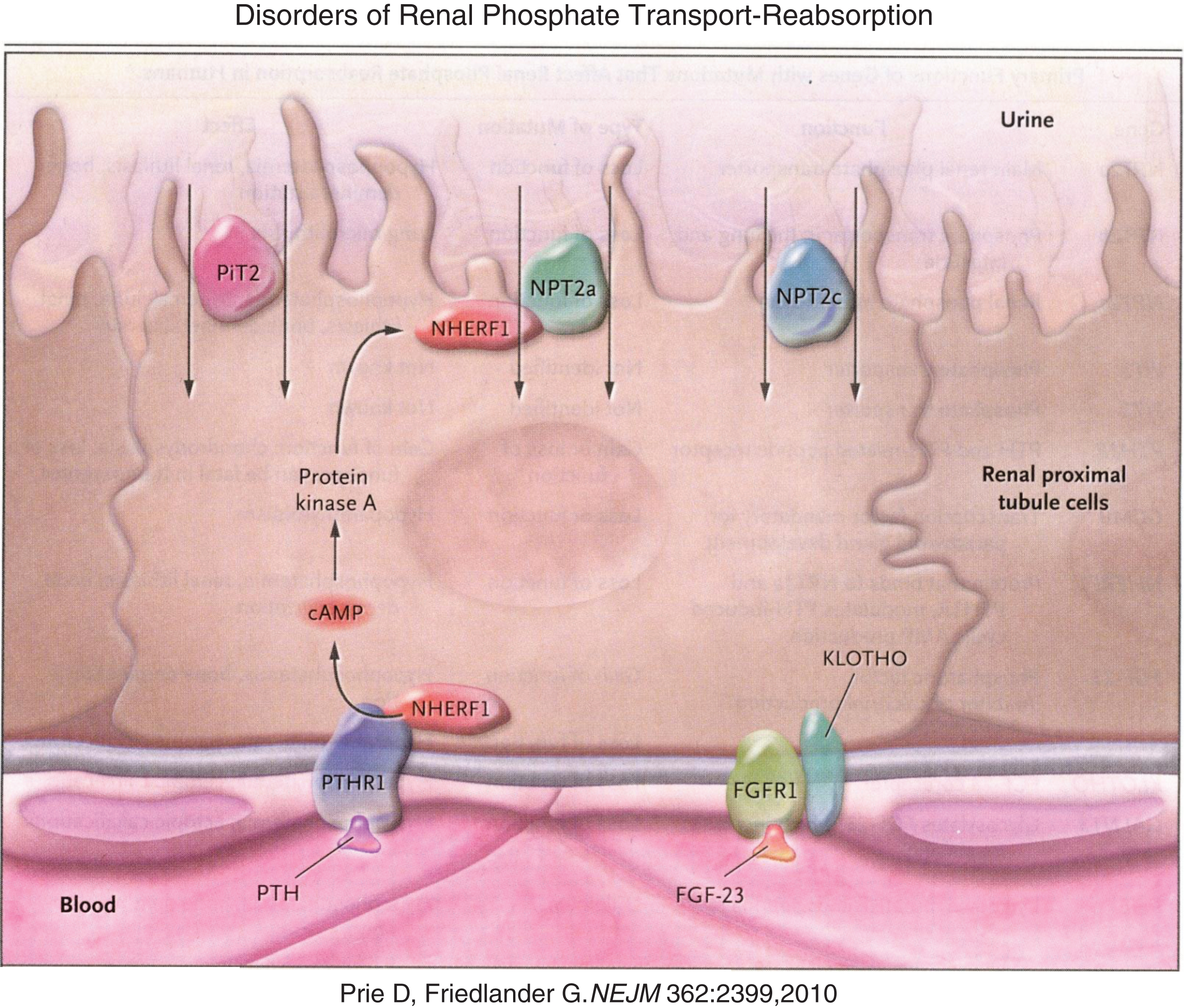
Fig.3
Reabsorption of phosphate from the renal tubule is blocked by PTH and FGF23 acting upon their respective receptors to down regulate the number of sodium phosphate luminal transmembrane channels through which phosphate is reabsorbed from the glomerular filtrate. (Reproduced with permission from Prie D, Friedlander G. Genetic disorders of renal phosphate transport. N Engl J Med 362:2399-2409,2010.)
Phosphatonins are substances that increase renal tubular excretion of phosphate and depress renal tubular synthesis of calcitriol – the latter by down regulating expression of the gene encoding 25-hydroxyvitamin D-1α-hydroxylase. Four substances with phosphatonin-like activity have been identified – primarily because they have been produced by mesenchymal tumors associated with hypophosphatemic osteomalacia: Fibroblast growth factor 23 (FGF23), Matrix extracellular phosphoglycoprotein (MEPE), Secreted frizzled-related protein-4 (sFRP-4), and Fibroblast growth factor 7. FGF23 is a 251 amino acid peptide whose full length is required for optimal bioactivity. Its amino and carboxyl terminal domains are linked at an enzymatic cleavage site located within amino acids 176–180; mutations at this site delay degradation of FGF23 and prolong its biological half-life leading to autosomal dominant hypophosphatemic rickets (ADHD) [10]. FGF23 is synthesized and secreted by osteoblasts and osteocytes in response to rising serum concentrations of phosphate and calcitriol. There is a direct positive relationship between serum levels of phosphate and those of FGF23. In addition to its inhibitory effects on phosphate reabsorption in the proximal renal tubule and upon synthesis of calcitriol, FGF23 also impairs expression of PTH in the PTGs thereby decreasing the synthesis and secretion of PTH [11]. FGF23 is produced in excess in patients with X-linked dominant hypophosphatemic rickets (XHR) and autosomal dominant and recessive forms of hypophosphatemic rickets. In addition, FGF23 is secreted ectopically by neoplasms (tumor-induced osteomalacia = TIO) and non-ossifying fibrous dysplastic tissue (McCune-Albright syndrome) and by patients with osteoglophonic dysplasia, Jansen type metaphyseal chondrodysplasia, and linear nevus sebaceous syndrome; concentrations of FGF23 are also increased by the intravenous administration of ferric oxide-maltose – a drug employed in the treatment of iron deficiency anemia [12]. Loss of function mutations in FGF23 leading to decreased FGF23 synthesis result in autosomal recessive hyperphosphatemic familial tumoral calcinosis [13]. UDP-N-acetyl-alpha-D-galactosamine: polypeptide N-acetylgalactosaminyltransferase 3 (GALNT3) is an enzyme that glycosylates FGF23 amino acid number 178 (threonine) in the ortho position; O-glycosylation at Thr178 is essential for normal translation of FGF23 and its subsequent cleavage between amino acids Arg179-Ser180. Loss of function mutations of GALNT3 lead to disordered post-translational processing of FGF23, decreased secretion of FGF23, and thence also to autosomal recessive hyperphosphatemic familial tumoral calcinosis with large amounts of calcium being deposited in skin and subcutaneous tissues, particularly the eyelids. Hyperostosis of the long bones may also occur in some patients with this syndrome [14].
FGF23 transmits its message through binding to cell membrane fibroblast growth factor receptor 1 (FGFR1). The extracellular domain of the FGFRs contains three immunoglobulin-like sequences, the third of which may be differentially incorporated into the FGFR protein by alternative splicing of messenger RNA during translation. A tyrosine kinase domain is present within the intracellular portion of the FGFRs. FGF23 is recognized specifically by the isoform – FGFR1(IIIc) – in which part of the third immunoglobulin-like domain is lacking. αKlotho (encoded by KL) is a 1012 amino acid transmembrane protein that also binds FGF23. (Klotho is the name of the Fate in Greek mythology who spun the thread of life, so named because αKlotho also plays a role in the aging process.) Binding of αKlotho to FGF23 enables FGFR1(IIIc) to recognize FGF23; αKlotho and FGFR1(IIIc) acting in concert constitute the FGF23 receptor. Loss of function mutations in KL lead also to autosomal recessive hyperphosphatemic familial tumoral calcinosis [15]. Thus, inactivating mutations in three different but functionally interrelated genes (FGF23, GALNT3, KL) result in the same clinical disease phenotype of autosomal recessive hyperphosphatemic familial tumoral calcinosis.
1.2Bone
Bone is composed of a protein matrix of collagen type I [an intertwined heterotrimeric structure of two molecules of collagen type I subunit α1(I) and one molecule of collagen type I subunit α2(I) bridged by disulfide bonds] that provides an extracellular platform upon which is deposited the mineral portion of bone in the form of the hydroxyapatite lattice [Ca8(PO4)10(OH)2]. Approximately 10% of bone matrix consists of non-collagenous proteins such as osteocalcin and osteonectin. Bone is composed of a cortex of compact bone braced by internal trabecular (spongy or cancellous) bone [16]. Bone formation is regulated by osteoblast function, while bone reabsorption is mediated by osteoclasts. Osteoblasts are derived from multipotential mesenchymal cell under the guidance of many transcription factors including bone morphogenetic proteins (BMP2, BMP7), WNT1, RUNX2, and SP7 [17, 18]. Osteoblasts produce collagen type I – subunits 1 and 2, as they express both COL1A1 and COL1A2. Each gene encodes a core structure of 1014 amino acids at both the amino and carboxyl terminals by propeptides that are removed during post-translational processing. Glycine comprises every third amino acid in collagen; lysine and proline are other commonly present amino acids. In the endoplasmic reticulum of the osteoblast, procollagen type I subunits 1 and 2 are linked to form type I procollagen that, in turn, undergoes complex post-translational modifications including 4-prolyl hydroxylation, lysyl hydroxylation, and glycosylation of hydroxylysine before assuming a stable three-dimensional configuration. Hydroxylation at carbon-3 of proline residue 986 of collagen type I subunit α1(I) is essential for optimal stability of the helical form of intact collagen type I [16]. Prolyl-3 hydroxylation is accomplished by a complex of three proteins; prolyl-3-hydroxylase-1 (LEPRE1), cartilage-associated protein (CRTAP), and peptidyl-prolyl isomerase B (PPIB). Chaperones (e.g., HSP47, FKBP65) guide the post-translational modifications of procollagen type I and the folding process that results in the three dimensional configuration of the protein. After exiting the endoplasmic reticulum, procollagen type I transits the Golgi apparatus, is entrapped in secretory vesicles, secreted into the extracellular matrix, and further processed by removal of amino and carboxyl terminal propeptide amino acid sequences forming mature collagen type I. When embedded within formed bone, osteoblasts become terminally differentiated osteocytes that are interconnected by cytoplasmic strands within canaliculi [19]. Osteocytes function as a network sensing mechanical loading and alterations in bone strength; this network modulates bone remodeling by sending signals to both osteoclasts and osteoblasts; they also respond to PTH and PTHrP.
In addition to collagen type I, osteoblasts secrete into bone matrix a number of non-collagenous proteins that aid and abet bone mineralization. SIBLINGS (Small integrin-binding ligand, N-linked glycoprotein) are matrix proteins such as bone sialoprotein, matrix extracellular glycoprotein (MEPE), dentin matrix protein-1 (DMP1), and osteopontin that bind avidly to and coat hydroxyapatite. They share a sequence or motif of acidic serine- and aspartate-rich amino acids (ASARM). ASARM may be released into the circulation by catalytic proteolysis of its parent protein; phosphorylated ASARM inhibits bone mineral deposition. Bone contains 99 percent of the total body calcium content in two functional compartments – newly deposited surface bone that is part of the readily exchangeable and rapidly available calcium pool as is the calcium in serum and interstitial fluid, and more deeply deposited bone that although not inert as it turns over with a half life of approximately six weeks is not immediately available for maintenance of normal extra- and intracellular calcium levels; deeply deposited bone preserves the firm structural integrity of the skeleton.
Bone is remodeled by osteoclasts that reabsorb the mineral and protein components of bone. Osteoclasts are mutinucleated giant cells derived from mesenchymal mononuclear cells that differentiate and fuse under the guidance of a number of osteoclast activating and differentiating factors (e.g., PTH, calcitriol, prostaglandins, interleukins, macrophage-colony stimulating factor) [20, 21] (Fig. 4). Osteoclast activating factors stimulate the stromal cell/osteoblast to express on its cell surface and to release into the circulation RANKL, a protein that enhances osteoclastogenesis. RANKL is recognized by RANK expressed on the plasma membrane of an osteoclast precursor cell derived from hematopoietic stem cells. Binding of RANKL to RANK promotes further differentiation of committed pre-osteoclasts and their fusion to form the mature, multinucleated osteoclast (osteoclastogenesis). Osteoblasts also secrete osteoprotegerin – a protein that binds to RANKL and prevents its interaction with RANK, thereby inhibiting osteoclastogenesis (i.e., osteoprotegerin serves as a “decoy” receptor). Remodeling of bone begins with the attraction of an osteoclast to a site of bone injury summoned by a signal from the osteocyte network. The osteoclast adheres or seals to the bone surface at the site of the injured bone. On its undersurface, the osteoclast forms an irregular (ruffled) border through which it secretes hydrochloric acid into a sub-osteoclastic lacuna to dissolve the mineral portion of bone and enzymes (cathepsin K, matrix metalloproteins 9 and 13) to break down collagen and other bone matrix proteins. Intracellularly, hydrochloric acid is generated by conversion of carbon dioxide to carbonic acid, a process catalyzed by carbonic anhydrase II (encoded by CA2). Protons (H+) are “pumped” into the sub-osteoclastic lacuna through a vacuolar H+ATPase-regulated 6-transmembrane protein linked to a chloride channel (encoded by CLCN7) and chloride-bicarbonate exchanger. From the sub-osteoclast lacuna, catabolized bone products are absorbed into and transported through the osteoclast and then released into the circulation. As bone is dissolved, osteoblasts are attracted to the site and begin to form new bone in a process of continual remodeling. Errors in osteoblast function lead to impaired bone formation and various forms of osteogenesis imperfecta, while errors in osteoclast differentiation and function compromise bone reabsorption resulting in high bone mass, osteopetrosis, and osteosclerosis.
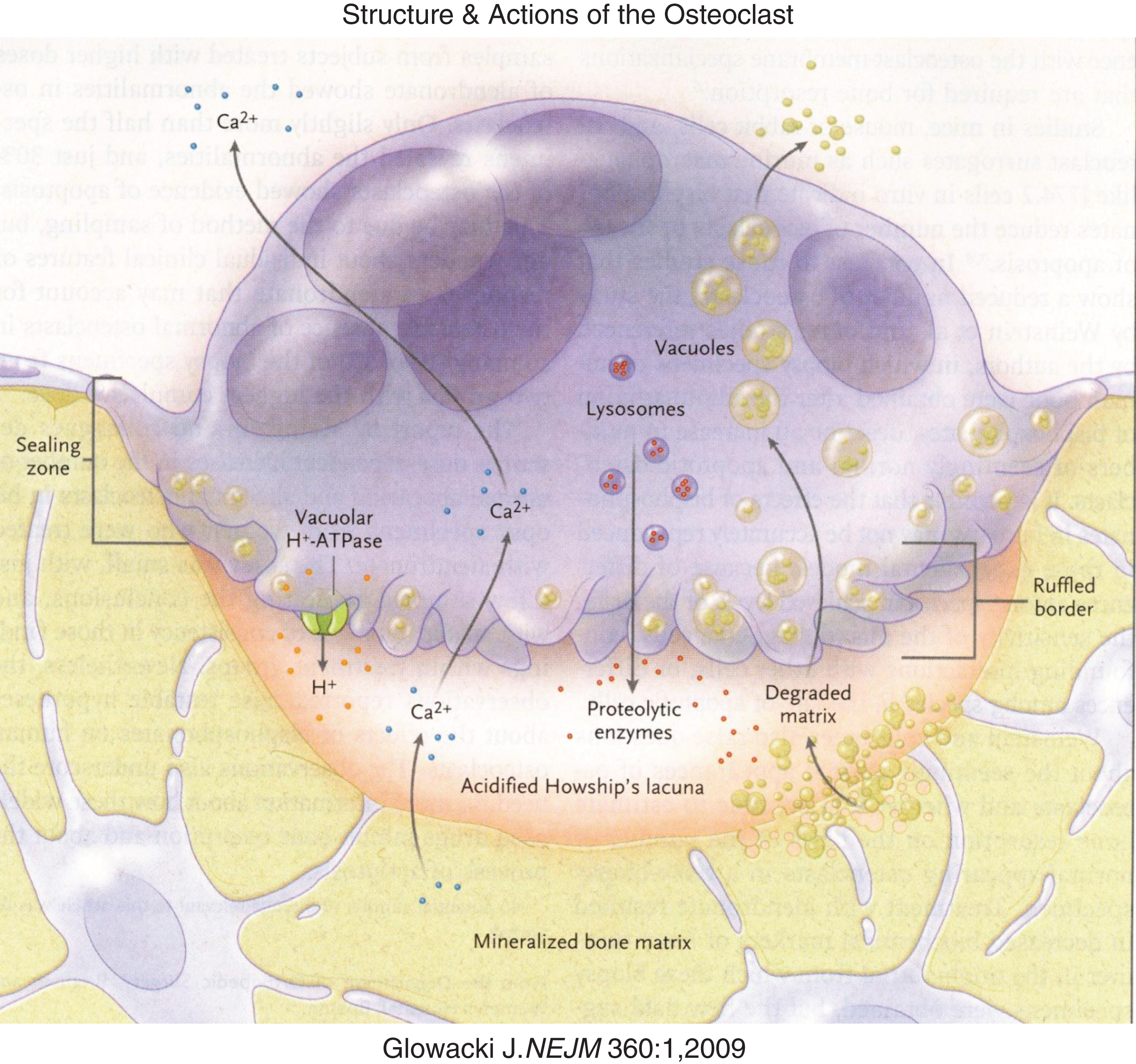
Fig.4
Diagrammatic representation of osteoclastic bone reabsorption depicting the osteoclast adherent to bone, subosteoclastic lacuna, secretion of acid and proteolytic enzymes into the lacuna and transport of digested products of reabsorption through the osteoclast. (Reproduced with permission from Glowacki J The deceiving appearances of osteoclasts. N Engl J. Med 360:80-82,2009.)
Bone mineralization is under extensive genetic regulation – an estimated 60 to 90 percent of individual variation in bone mineralization is related to genetic factors with a plethora of involved genes each accounting for only a small fraction (perhaps 1 percent) of the total genetic variability in bone mass [22]. Genome wide association studies have identified genes related to bone mineralization that are essential for bone formation. These include SP7 (MIM 606633) encoding Osterix, a transcription factor that is indispensable for osteoblast differentiation and WNT1 (MIM 164820) encoding a circulating protein that signals through the 7-transmembrane domain-Frizzled receptor (MIM 603408) with intracellular signal transduction by stabilization of β-catenin (MIM 116806) that leads to differentiation and proliferation of osteoblasts and to transcription of COL1A1 and COL1A2 and formation of collagen type I [23]. Lipoprotein receptor-related protein 5 (LRP5 – MIM 603506) is a cell surface protein that is expressed on the plasma membrane of the osteoblast and osteocyte. LRP5 has three domains: the extracellular domain of LRP5 contains four amino acid sequences resembling those of epidermal growth factor (EGF repeats); there are also single transmembrane and cytoplasmic domains. Gain of function mutations in LRP5 are associated with high bone mass, and loss of function mutations result in low bone mass. In vitro and in vivo studies indicate that LRP5 functions as a co-receptor (with Frizzled) for WNT enabling signal transduction through β-catenin. Both Dickkopf-related protein 1 (MIM 605189) and sclerostin (MIM 605740) antagonize LRP5-mediated WNT1 signaling in the osteoblast by binding to the extracellular domain of LRP5 and impairing formation of the LRP5-WNT1-Frizzled complex. However, it has also been reported that rather than acting through the WNT1-Frizzled-β-catenin pathway in osteoblasts, LRP5 affects bone mineral accrual through its expression in intestinal enterochromaffin cells. There, LRP5 inhibits expression of TPH1 (MIM 191060) encoding tryptophan hydroxylase and consequently the synthesis of serotonin [24–26]. In bone, serotonin inhibits osteoblast proliferation and mineralization. Thus, through this suggested pathway, upregulated LRP5-mediated inhibition of intestinal serotonin production results in enhanced osteoblast function and high bone mass and vice versa. Experimentally, support for the enteral-serotonin mechanism of LRP5 function has been challenged [27]. However, it may be that depending on extant conditions LRP5 may exert its effects through both mechanisms. Many genes that normally regulate bone formation have had deleterious effects on mineralization when rendered non-functional by noxious mutations – e.g., SP7, SOST, TNFSF11A (RANKL), and TNFRSF11A (RANK) (Tables 3–5) [22].
Alkaline phosphatase (encoded by ALPL – MIM 171760) is an ectoenzyme produced by and expressed on the surface of osteoblasts that degrades endogenous inhibitors of mineralization (e.g., pyrophosphate, pASARM) while increasing local concentrations of phosphate (natural substrates pyridoxal-5’-phosphate and phosphoethanolamine) to values that exceed the calcium-phosphate solubility product allowing deposition of calcium-phosphate as the hydroxyapatite crystal upon collagen type 1. Inactivating mutations in ALPL lead to hypophosphatasia of variable severity depending on the site of mutation.
2Hypocalcemia
Hypocalcemia in children and adolescents is most commonly an acquired abnormality. In preterm and/or highly stressed neonates, hypocalcemia is the cumulative result of limited secretion of PTH, hypercalcitonemia, vitamin D deficiency, hypomagnesemia, glucocorticoid inhibition of intestinal calcium absorption, and limited calcium intake [2]. In older patients, hypocalcemia may be the result of vitamin D deficiency or an autoimmune destructive process (autoimmune polyglandular syndrome type 1) or surgical insult to the parathyroid glands. Genetic disorders that cause hypocalcemia include those due to mutations in genes that control development of the PTGs, PTH synthesis or biologic effectiveness, vitamin D synthesis or functionality, and the CaSR (Table 1).
Table 1
Genetic causes of hypocalcemia
| Gene Chromosome MIM | Pathophysiology | Mutation – Clinical manifestations |
| Hypoparathyroidism | ||
| GCM2 – Glial cells missing, Drosphila, homolog of, 2 6p24.2 603716 | TF essential for normal development of PTGs | Loss of function mutations lead to familial isolated hypoparathyroidism; AR, AD |
| TBX1 – T-Box 1 22q11.2 602054 | TF that also interacts with transcriptional co-regulators; regulates expression of GCM2 – TF essential for differentiation of the PTGs | Hemizygous deletion of chromosome 22q11.2 (DiGeorge &velocardiofacial syndromes) – Hypoparathyroidism, immune dysfunction, cleft palate. conotruncal congenital heart disease, AD; monoallelic inactivating mutations of TBX1 lead to several of the manifestations of the chromosome del22q11.2 syndrome |
| SOX3 – SRY-Box 3 Xq26.3 313430 | TF expressed in the developing PTG | Abnormal expression of SOX3 may be related to pathogenesis of X-linked hypoparathyroidism |
| GATA3 – GATA-binding protein3 10p15 131320 | TF regulating expression of GCM2 – a TF requisite for differentiation of the PTGs; GATA3 also essential for development of auditory structures &kidneys &immune regulation | Monoallelic loss of function mutations result in Barakat (HDR) syndrome: Hypoparathyroidism, sensorineural deafness, renal disease, AD |
| TBCE – Tubulin specific chaperone E 1q42-43 604934 | Encodes a chaperone protein necessary for proper formation of the cytoskeleton, protein transport, &secretion | Inactivating mutations lead to – Sanjad-Sakati (HRD) syndrome: Hypoparathyroidism-retardation-dysmorphism syndrome, AR; Kenny-Caffey syndrome, type 1: Hypoparathyroidism, medullary stenosis, osteosclerosis, recurrent bacterial infections, AR |
| PTH – Parathyroid hormone 11p15.3-p15.1 168450 | Peptide that mobilizes calcium from bone by stimulating osteoclastogenesis and increases its reabsorption from glomerular filtrate; enhances intestinal absorption of calcium by stimulating synthesis of calcitriol | Loss of function mutations result in familial isolated hypoparathyroidism; AR, AD |
| CASR – Calcium sensing receptor 3q13.3-q21 601199 | GPCR that monitors ambient Ca2+ concentrations | Gain of function mutations results in autosomal dominant hypoparathyroidism, AD |
| TRPM6 – Transient receptor potential cation channel, subfamily M, member 6 9q21 607009 | One member of a TRPM6/TRPM7 complex that is essential for epithelial transport of Mg2+ in kidney &intestine | Loss of function mutations lead to familial hypomagnesemia with secondary hypocalcemia, AR |
| Resistance to PTH | ||
| PTHR1 – PTH receptor 1 3p22-p21.1 156400 | GPCR for PTH &PTHrP | Loss of function mutations result in Blomstrand chondrodysplasia &hypocalcemia despite elevated serum levels of PTH, AR |
| GNAS – GNAS complex locus 20q13.2 139320 | Stimulatory alpha subunit of G-protein (Gαs) that responds to ligand binding of GPCR; activates adenylyl cyclase generating cyclic AMP resulting in activation of PKA | Monoallelic inactivating mutations result in pseudohypoparathyroidism type 1a; methylation defects in maternal allele lead to type 1 |
| PRKAR1A – Protein kinase, cAMP-dependent regulatory, type 1, alpha 17q23-q24 188830 | Component of PKA response to cyclic AMP that leads to cascade of intracellular signal transduction signals in response to Gαs that regulate cell division, differentiation, metabolism, apoptosis | Gain of function mutation leads to acrodysostosis &peripheral resistance to the biologic effects of PTHrP, PTH &TSH; (de novo) |
| Other | ||
| AIRE1 – Autoimmune regulator 21q22.3 607358 | TF expressed in medulla of thymus that enables this organ to differentiate self-antigens from foreign antigens | Loss of function mutations result in autoimmune polyendocrine syndrome type 1 with autoimmune hypoparathyroidism &other endocrinopathies, mucocutaneous candidiasis, ectodermal dystrophy, AR |
Hypoparathyroidism may be transmitted as an autosomal recessive or dominant characteristic or as an X-linked trait or it may be part of more complex congenital disorders such as the deletion of chromosome 22q11.2 syndrome [28]. Autosomal recessive familial isolated hypoparathyroidism due to agenesis of the PTGs has been attributed to loss of function mutations in GCM2, a nuclear transcription factor expressed only in and essential for formation of these structures. Expression of GCM2 is regulated by both GATA3 and TBX1; loss of function mutations in these genes result in hypoparathyroidism in association with other malformations (vide infra) [29]. Mutations in GCM2 may also be transmitted as an autosomal dominant trait when the mutation exerts a dominant-negative effect on the product of the wt allele [30]. Interestingly, GCM2 mutations transmitted as autosomal recessive traits occur in the DNA binding domains of GCM2 nearer to the amino terminal of the protein, while those transmitted as dominant characteristics occur in the second transactivation domain near the carboxyl terminus of GCM2.
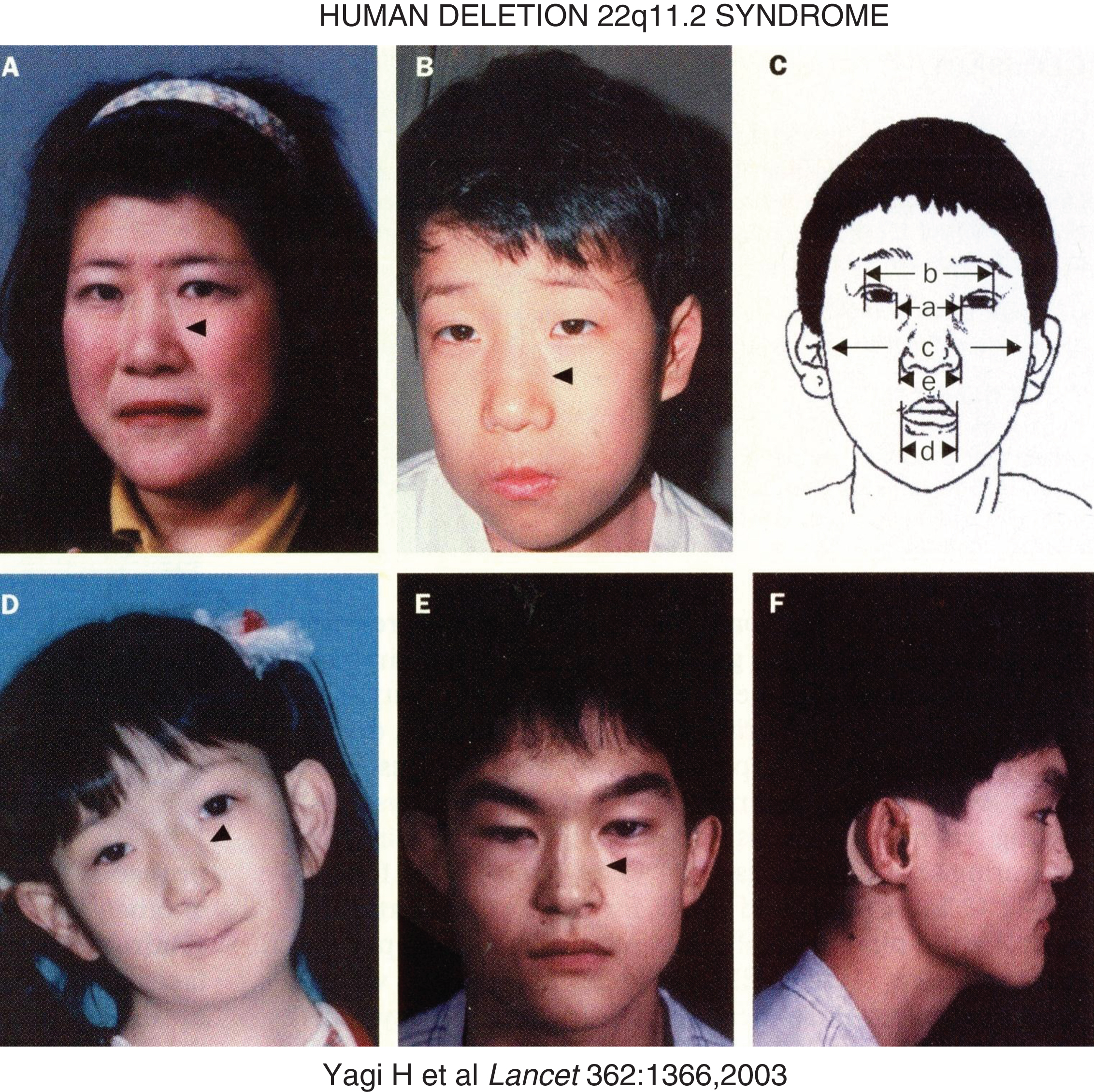
Hemizygous deletions of chromosome 22q11.2 of variable lengths occur in approximate 1 in 4000 births [31]. They are present in patients with the DiGeorge syndrome (DGS – MIM 188400) of hypoplasia of the thymus and PTGs (leading to mild to moderately severe immune deficiencies and hypoparathyroidism, respectively) and anatomic malformations of the outflow tract of the heart and in patients with the velocardiofacial syndrome (MIM 192430) comprised of cleft palate, velopharyngeal insufficiency, conotruncal cardiac anomalies, atypical face (hypertelorism, short palpebral fissures, micrognathia), and learning disabilities (III and IV pharyngeal arch syndromes) (Fig. 5) [32–34]. Isolated hypoparathyroidism may also be observed in patients with chromosome 22delq11. Chromosome 22q11 segment is susceptible to deletion because it is bracketed by low copy repeat numbers that make this region intrinsically unstable during meiotic exchange leading to asynchronous replication [31]. One of the genes lost in deletion 22q11.2 is TBX1, a transcription factor that also interacts with a histone methyltransferase complex to regulate gene expression [35]. TBX1 is essential for normal cardiac and PTG differentiation. Monoallelic inactivating mutations in TBX1 alone (haploinsufficiency) lead to five major abnormalities that occur in patients with the chromosome deletion 22q11 syndrome: conotruncal face (nasal segmentation, hypertelorism, short palpebral fissures, small mouth); velopharyngeal insufficiency with cleft palate; cardiac defects (tetrology of Fallot, atrial and ventricular septal defects, interrupted aortic arch); thymic hypoplasia; hypoparathyroidism [34]. Manifestations of the chromosome deletion 22q11.2 syndromes may be influenced by modifier genes (e.g., VEGF polymorphism).
Fig.5
Facial characteristics of the deletion chromosome 22.q11 syndrome and/or mutations in TBX1. Panels A and B are mother and son – arrowheads point to the divide into upper and lower segments. Panel C schematically depicts a – ocular hypertelorism; b – short palpebral fissures; d – small mouth (a – inner canthal distance; b – outer canthal distance; c – transverse facial width; d – oral width; e – nasal width). Panel D depicts the face of a patient with conotruncal anomaly face syndrome, intact chromosome 22q11, mutation in TBX1. Panels E and F display frontal and lateral views of a patient with the DiGeorge syndrome, intact chromosome 22q11, and mutation in TBX1. (Reproduced with permission from Yagi H, Furutani Y, Hamada H, et al. Role of TBX1 in human del22q11.2 syndrome. Lancet 362:1366-1373,2003.)
The presence of an interstitial deletion-insertion involving chromosomes Xq27.1 near SOX3 and 2p25.3 and the known expression of SOX3 in the developing PTG have suggested that abnormal expression of this gene may be of etiopathogenic significance in patients with X-linked hypoparathyroidism associated with agenesis of the PTGs [36]. The autosomal dominant Barakat syndrome (MIM 146255) of hypoparathyroidism, sensorineural deafness, and renal dysfunction is due to loss of function mutations in GATA3, a transcription factor that regulates expression of GCM2 and is also involved in differentiation of the auditory system and kidneys. The Sanjad-Sakati syndrome (MIM 241410) of hypoparathyroidism, intrauterine and postnatal growth retardation, developmental delay, seizures, dysmorphic facial features (microcephaly, prominent forehead, deeply set eyes, beaked nose, thin upper lip, micrognathia, large and droopy external ears), and increased susceptibility to infections is an autosomal recessive disorder due to inactivating mutations in TBCE encoding tubulin specific chaperone E; this disorder may be genetically heterogeneous, however [37–39]. The intact product of TBCE facilitates proper folding of proteins – specifically alpha and beta tubulin heterodimers that interconnect microtubules (structures essential for normal architecture of the cytoskeleton) and endocytic vesicles and facilitate the intracellular transport and secretion of proteins. Inactivating mutations in TBCE are also present in patients with the Kenny-Caffey syndrome type 1 (MIM 244460), an autosomal recessive disorder with many findings similar to those in the Sanjad-Sakati syndrome but with the additional manifestations of medullary stenosis of the long bones and cranial osteosclerosis. (Kenny-Caffey syndrome type 2 is transmitted as an autosomal dominant characteristic due to variants of FAMILIA.)
Inactivating mutations in PTH that obstruct the processing of preproPTH to its bioactive product (PTH) result in functional hypoparathyroidism that may be transmitted as an autosomal dominant or recessive characteristic. Depending on the specificity of the immunoassay for PTH, serum levels of PTH may be low, normal or even high in these patients. Autosomal dominant hypoparathyroidism is the result of gain of function mutations in CASR. In this disorder very low levels of Ca2+ activate the CaSR and repress parathyroid chief cell transcription of PTH as well as renal tubular reabsorption of calcium leading to paradoxical hypercalciuria – the mirror image of familial hypocalciuric hypercalcemia (vide infra). Magnesium is essential for release of PTH from the PTGs and for normal peripheral responsiveness to PTH. Familial hypomagnesemia type 1 with secondary hypocalcemia (MIM 602014) is an autosomal recessive disorder due to inactivating mutations in TRPM6 encoding an essential subunit of a TRPM6/TRPM7 renal and intestinal epithelial co-transporter for this cation. Mutations in FXYD2 (MIM 601814), CLDN16 (MIM 603959), EGF (MIM 131530), and CLDN19 (MIM 610036) have been identified in patients with renal hypomagnesemia types 2–5, respectively. In addition, many patients with Bartter (MIM 241200) or Gitelman (MIM 263800) syndromes may be hypomagnesemic.
Patients with inactivating mutations in PTHR1, encoding the GPCR for PTH and PTHrP, have functional hypoparathyroidism (hypocalcemia, hyperphosphatemia) although serum concentrations of PTH are elevated. Additionally, these subjects have an osteochondrodystrophy – Blomstrand chondrodysplasia (MIM 215045) – characterized by advanced skeletal and dental maturation and very short extremities – abnormalities detectable in utero by fetal ultrasonography. Histological examination of the growth plates of long bones of these patients reveals that loss of function mutations in PTHR1 are typified by a narrow zone of proliferating epiphyseal cartilage, erratically distributed and vacuolated chondrocytes, and few osteoclasts. The disorder is due to defective in utero response to PTHrP, an essential factor for normal fetal cartilage differentiation and maturation. Blomstrand chondrodysplasia is often lethal in early infancy.
Patients with loss of function mutations in GNAS encoding the stimulatory alpha subunit (Gαs) of G-protein also develop hormone resistance – primarily pseudohypoparathyroidism (PHP) with or without the phenotype of Albright’s hereditary osteodystrophy (AHO). The clinical manifestations of AHO include short stature, brachydactyly, obesity, round face, and heterotopic subcutaneous calcifications. Whether the AHO phenotype is expressed in the patient with PHP depends on which parent has donated the inactive GNAS allele, because the tissue expression of GNAS in renal tissue is imprinted and determined by the parent of origin allele. Both GNAS alleles are expressed in most tissues including bone; however, in the kidney only maternal GNAS is expressed. Hence, the clinical manifestations of PHP in an individual patient depend on whether the inherited maternal or paternal GNAS allele is defective. Thus, loss of function mutations of the maternal GNAS allele result in PHP type Ia with end-organ resistance to many protein hormones including PTH (resulting in hypocalcemia and hyperphosphatemia) as well as the skeletal defects of AHO. Inactivating mutations of paternal GNAS result in pseudopseudohypoparathyroidism (PPHP) with intact hormonal responsiveness (including renal reactivity to PTH) but abnormal skeletal phenotype due to haploinsufficiency of GNAS in bone. In PHP type Ib, lack of maternal GNAS expression is confined to the kidney resulting in selective renal resistance to PTH; skeletal expression of maternal GNAS and hence skeletal formation are normal. PHP type Ib is due to a methylation defect in the imprinting region that regulates maternal GNAS expression in the kidney. Patients with PHP type II have a normal skeletal phenotype but are partially resistant to the renal effects of PTH; its cause is unknown as yet. [Although not yet described clinically, it may be anticipated that a loss of function mutation in LRP6 (encoding lipoprotein receptor-related protein 6 – MIM 603507) will be associated with resistance to the biologic effect of PTH. In addition to its role as a cell surface protein necessary for receptor-mediated endocytosis of lipoproteins, LRP6 is essential for the movement of Gαs to the plasma membrane and for its coupling to PTHR1 [40]. Experimentally, in vitro “knock down” or inactivating mutation of Lrp6 decreases the cellular response to PTH.]
The next step in the signal transduction pathway initiated by activation of Gαs entails cAMP-mediated activation of protein kinase A (PKA). A patient with a de novo germline gain-of-function mutation (Arg368Stop) in PRKAR1A, encoding the cyclic AMP-dependent regulatory subunit of PKA leading to hormone resistance (specifically PTH and TSH) and to acrodysostosis, a chondrodysplasia with many features of AHO (MIM 101800 – short stature, obesity, brachydactyly, abnormal face with nasal and maxillary hypoplasia, advanced skeletal maturation) has been identified [41]. Translation of the mutant messenger RNA of PRKAR1A resulted in a shortened protein product with enhanced function, because the mutated regulatory subunit lacked one of its two cyclic AMP binding domains and was catabolized very slowly. Consequently, the mutated regulatory subunit was unresponsive to normal levels of cyclic AMP; it bound tightly to the catalytic subunit of PKA preventing its release and thus maintaining the inactive state as evidenced experimentally by the reduced bioactivity of the resultant PKA in response to cyclic AMP in vitro.
Children and adolescents with inactivating mutations in AIRE develop hypoparathyroidism (usually between infancy and 10 years of age) together with other autoimmune endocrinopathies (adrenalitis, primary hypogonadism, insulin-dependent diabetes mellitus, thyroiditis), mucocutaneous candidiasis, and ectodermal dystrophy (APECED) resulting in autoimmune polyendocrine syndrome type 1 (APS1 – MIM 240300) [42, 43]. The cytoplasmic PTG autoantigen against which many APS1 patients with hypoparathyroidism develop antibodies is NLRP5 (Nacht domain-, leucine-rich repeat-, and Pyd-containing protein 5, MIM 609658), a peptide that is one component of a multi-protein inflammasome [44]. Other autoimmune disorders associated with APS1 are chronic active hepatitis, juvenile-onset pernicious anemia, vitiligo, and alopecia. AIRE encodes a zinc-finger transcription activating factor that regulates thymic function enabling it to differentiate between self and foreign proteins (antigens). In order to do so, AIRE interacts with thymic proteins involved with nuclear transport, chromatin binding, transcription, and pre-mRNA processing [45].
Hypocalcemia occurs in infants with inactivating mutations in CYP27B1, the gene that encodes 25-hydroxyvitamin D-1α-hydroxylase, the enzyme essential for synthesis of calcitriol, and in VDR encoding the vitamin D receptor (Table 3). Hypocalcemia may also be found in patients with mitochondrial fatty acid oxidation disorders due to mutations in mitochondrial DNA in association with other distinguishing characteristics [Kearns-Sayre (MIM 530000 – ophthalmoplegia, pigmentary degeneration of the retina, cardiomyopathy), MELAS (MIM 540000 – myopathy, encephalopathy, lactic acidosis, diabetes mellitus)].
3Hypercalcemia
In children and adolescents, hypercalcemia is most commonly an acquired abnormality due to immobilization, excessive vitamin D or vitamin A intake, increased synthesis of calcitriol by inflammatory monocytes (e.g., subcutaneous fat necrosis, sarcoidosis), or medication ingestion (e.g., thiazide diuretics, lithium, alkali-containing antacids) [46, 47]. Familial isolated primary hyperparathyroidism may be due to hyperplasia, adenoma, or carcinoma of the PTGs. Familial isolated hyperparathyroidism types 1 (MIM 145000) and 2 (MIM 145001 – associated with functional cystic adenomas and carcinomas of the PTGs as well as ossifying fibromas of the maxilla and/or mandible) are due to heterozygous germline activating mutations in CDC73 (Table 2). This gene encodes parafibromin, a member of a protein complex that associates with both a RNA polymerase II and a histone methyltransferase complex and plays roles in both gene transcription and translation. Parafibromin is also a component of the WNT/β-catenin intracellular signal transduction pathway. Familial isolated primary hyperparathyroidism type 3 (MIM 610071) has been linked to chromosome 2p14-p13.3, but no specific gene has as yet been identified in this region as its cause. In addition to the syndrome of multiple endocrine neoplasia type I, mutations in MEN1 have been associated with familial isolated primary hyperparathyroidism. Monoallelic inactivating germline mutations in CDKN1B, encoding a tumor suppressor gene, are of etiopathogenic significance in patients with MEN type 4 (MIM 610755) which is associated with adenomas of the parathyroid and pituitary glands and tumors of the kidneys and testes.
Table 2
Genetic causes of hypercalcemia
| Gene Chromosome MIM | Pathophysiology | Mutation – Clinical manifestations |
| Hyperparathyroidism | ||
| MEN1 – Menin 11q13 131100 | Menin is a nuclear protein that inhibits transcription activated by JunD, SMAD3 &other transcription factors &serves as a tumor suppressor | Germline loss of function mutations in MEN1 and somatic loss of MEN1 sum to produce multiple endocrine neoplasia type 1 or familial isolated hyperparathyroidism; AD |
| CDKN1B – Cyclin dependent kinase inhibitor 1B 12p13.1 600778 | Inhibitor of cyclin-mediated cell proliferation; tumor suppressor | Monoallelic loss of function mutations lead to parathyroid, pituitary, renal, testicular tumors (MEN type 4), AD |
| CDC73 – Cell division cycle protein 73, S. cerevisiae, homolog of 1q31.2 607393 | Also designated HRPT2; encodes parafibromin, a protein involved in gene transcription &translation | Germline gain of function mutations result in hyperparathyroidism type 2 – hyperparathyroidism-jaw tumor syndrome (HRPT2) &familial isolated hyperparathyroidism type 1; AD |
| PTHR1 – PTH receptor 1 3p22-p21.1 156400 | GPCR for PTH &PTHrP | Monoallelic activating mutations result in Murk-Jansen metaphyseal chondrodysplasia &hypercalcemia with low serum levels of PTH |
| CASR – Calcium sensing receptor 3q13.3-q21 601199 | GPCR for Ca2+ | Monoallelic loss of function mutations result in clinically occult familial benign hypocalciuric hypercalcemia; biallelic mutations lead to neonatal severe hyperparathyroidism; AD, AR |
| ELN – Elastin 7q11.23 130160 | One of the genes encoded on chromosome 7q11.23 – hemizygous loss likely account for the vascular malformations associated with WBS | Williams-Beuren syndrome – infantile hypercalcemia, supravalvular aortic stenosis, characteristic face – contiguous gene deletion syndrome at chromosome 7q11.23; AD |
| BAZ1B – Bromodomain adjacent to zinc finger domain, 1B 7q11.2 605681 | Encodes Williams syndrome transcription factor (WSTF)- haploinsufficiency interferes with calcitriol-VDR mediated inhibition of transcription of CYP27B1 &synthesis of calcitriol | Williams-Beuren syndrome – infantile hypercalcemia, supravalvular aortic stenosis, characteristic face – contiguous gene deletion syndrome at chromosome 7q11.23; AD |
| CYP24A1 – Cytochrome P450, family 24, subfamily A, polypeptide 1 20q13.2-13.3 126065 | Encodes renal 1,25(OH2)D3-24 hydroxylase enzyme that initiates inactivation of 1,25(OH2)D3 and its excretion as calcitroic acid | Loss of function mutations result in idiopathic infantile hypercalcemia, AR |
Among the more frequent (albeit still unusual) genetic causes of hypercalcemia due to hyperparathyroidism are loss of function mutations in CASR, encoding the calcium sensing receptor, that result in decreased sensitivity to the PTH-suppressing effects of Ca2+. Consequently, higher serum concentrations of Ca2+ are required to suppress synthesis and secretion of PTH. In most instances, familial benign hypocalciuric hypercalcemia (type I) (MIM 145980) is an autosomal dominant trait due to monoallelic loss of function mutations in CASR. These patients have asymptomatic hypercalcemia (total calcium level 11-12 mg/dL), relative hypocalciuria (calcium/creatinine ratio <0.1), and normal or slightly elevated serum concentrations of PTH and are usually identified during routine chemical screening or during family surveys when another family member is found to be hypercalcemic. Biallelic inactivating mutations in CASR are associated with neonatal severe hyperparathyroidism (NSHPT) (MIM 239200), at times a life threatening disorder as serum calcium concentrations are markedly elevated (total calcium >15 mg/dL) as are levels of PTH. Newborns bearing a monoallelic mutation in CASR may develop NSHPT if the offspring is born to a normal mother and affected father because of the fetal need to increase secretion of PTH in utero in order to maintain the high levels of calcium present in the fetus. Occasionally, NSHPT is transmitted as an autosomal recessive disorder by parents whose clinical and biochemical phenotypes are normal [49, 50]. Other forms of familial benign hypocalciuric hypercalcemia have been linked to chromosome 19p13.3 (type II) and to chromosome 19q13 (type III), but no gene mutations at these loci have as yet been identified.
Activating mutations of PTHR1 result in Murk-Jansen metaphyseal chondrodysplasia in association with hypercalcemia (MIM 156400). Patients with this disorder are very short (adult height 100 cm); the metaphyses of the long bones are long and extremely disorganized and skeletal maturation is quite delayed; in adults the cranial bones are sclerotic. Biochemically, serum and urine analyte values are comparable to those in patients with primary hyperparathyroidism except that PTH concentrations are extremely low (hypercalcemia and hypercalciuria, hypophosphatemia but hyperphosphaturia, increased urinary excretion of cyclic AMP, elevated serum concentrations of calcitriol). The chondrodysplasias of Blomstrand due to inactivating mutations in PTHR1 (vide supra) and Murk-Jansen are mirror images of one another. [Eiken syndrome (MIM 60002) is associated with multiple epiphyseal dysplasias and markedly delayed skeletal maturation but normal serum concentrations of calcium, phosphate, and PTH. It has ben associated with biallelic mutations in that portion of PTHR1 encoding the carboxyl terminal cytoplasmic tail of the receptor [51].]
Williams-Beuren syndrome (WBS – MIM 194050) is a hemizygous contiguous gene deletion syndrome (involving 26 to 28 genes on chromosome 7q11.23) affecting multiple systems that occurs in approximately 1:10,000 individuals and is transmitted as an autosomal dominant disorder [52]. WBS is characterized by a distinctive pixie-like face (flat nasal bridge, upturned nose, full lips), congenital heart disease (supravalvular aortic stenosis, peripheral pulmonary arterial stenoses, mitral valve prolapse), developmental delay (mean IQ 58, range 20–106) and weak visuospatial skills but enhanced language skills, vocabulary, and musical ability (Fig. 6) [47, 52]. When present, infantile hypercalcemia usually resolves by two years of age but occasionally persists into childhood and adulthood. The pathogenesis of hypercalcemia in WBS subjects may relate to loss of the Williams syndrome transcription factor (WSTF) encoded by BAZ1B. This nuclear protein is part of a very large multimeric, ATP-dependent, chromatin remodeling complex termed WINAC (WSTF including nucleoside assembly complex) [53]. Independently of ligand-binding, the VDR interacts with WINAC; normally, when calcitriol binds to the VDR-WINAC complex, it represses renal tubular transcription of CYP27B1, the gene encoding 25-hydroxyvitamin D-1α-hydroxylase, and stimulates transcription of CYP24A1, the gene encoding 25-hydroxyvitamin D-24-hydroxylase. Hence, haploinsufficiency of WSTF permits increased and relatively unregulated synthesis of calcitriol and at the same time slows its rate of degradation; consequently, the intestinal absorption of calcium increases and hypercalcemia ensues. Other findings in patients with WBS include short stature, hypertension, narrowing of the thoracic and abdominal aortae and renal and cerebral arteries, hyperacusis in childhood leading to nerve deafness, and somewhat early onset of puberty in girls. Other endocrinopathies associated with WBS include glucose intolerance and hypothyroidism. Deletion chromosome 7q11.23 is the result of unequal crossover of genes between chromosome 7 homologs during meiosis (unequal meiotic recombination) and occurs with equal frequency in the gametes of both parents. Among the 26 to 28 genes deleted in chromosome 7q11.23, hemizygous loss of ELN encoding elastin is the likely cause of the cardiovascular malformations and hypertension observed in WBS. Hemizygosity for LIMK1 (MIM 601329) may be a partial cause of visuospatial and cognitive difficulties in WBS. Hemizygous loss of STX1A (MIM 186590) may be pathogenetically related to development of glucose intolerance in WBS subjects [52].
Fig.6
Facial characteristics (pixie-like face with flat nasal bridge, upturned nose, full lips) of a patient with the Williams-Beuren syndrome from infancy to elderly. (Reproduced with permission from Pober BR. Williams-Beuren syndrome. N Engl J Med 362:239-252,2010.)

Idiopathic infantile hypercalcemia (MIM 143880) is manifested by failure to thrive, hypercalcemia, hypercalciuria, and nephrocalcinosis. It has been attributed to ingestion of excessive amounts of vitamin D in fortified formulas and milk, although the amounts of vitamin D (500 to 4000 IU/day) consumed by affected infants are far less than those known to be associated with toxicity in the general population [5]. Although hypercalcemic, these patients usually have normal serum concentrations of calcidiol and normal or slightly increased values of serum calcitriol, factors that differentiate these patients from those with vitamin D poisoning. Thus, patients with idiopathic infantile hypercalcemia have been considered to be extremely “sensitive” to vitamin D. Biallelic loss of function mutations in CYP24A1, encoding the mitochondrial enzyme that inactivates 1,25(OH2)D3 by converting it to 1,24,25(OH)3D3 and then to calcitroic acid and by converting 25OHD3 to 24,25(OH)2D3, have been identified in patients with this disorder [7]. Thus, hypercalcemia in these subjects is likely due to ingestion of relatively high amounts of vitamin D3 and the associated prolonged increases in serum levels of calcitriol due to its slow rate of catabolism. Patients with hypophosphatasia (MIM 171760) are also “hypersensitive” to calcitriol and may develop hypercalcemia when consuming usual amounts of cholecalciferol, although the mechanism of this phenomenon is as yet unidentified.
Hypercalcemia, hypercalciuria, and nephrocalcinosis of undetermined pathogenesis are found in patients with the blue diaper syndrome (MIM 211000) associated with a defect in intestinal transport of tryptophan, lactase deficiency, and disaccharide intolerance. Bartter syndrome (MIM 601678) is the designation of a group of diseases that collectively is due to attenuated reabsorption of sodium chloride in the renal tubular thick ascending loop of Henle. These autosomal recessive disorders are characterized by hypokalemic alkalosis, salt wasting, and hypercalciuria. Hypercalcemia may occur in some infants with antenatal Bartter syndrome due to loss of function mutations in SCL12A1 (type 1 – MIM 600839) encoding the furosemide-sensitive NaK-2Cl-cotransporter or KCNJ1 (type 2 – MIM 600359) encoding the renal outer-medullary potassium channel (ROMK) – an inwardly rectifying potassium channel [47].
4Disorders of bone mineralization
4.1Rickets
Clinically, rickets is a disorder of bone formation and remodeling in the growing infant, child, and adolescent in which defective bone mineralization due to impairment of hydroxyapatite deposition onto collagen type I in bone matrix results in skeletal deformations (genu valgum, genu varum, metaphyseal flaring). Hypocalcemia may be a presenting manifestation of vitamin D deficiency in the neonate or infant. In the adult, osteomalacia leading to increased fracture risk is the outcome of a similar process. Rickets is due to inadequate supplies of calcium or phosphate (dietary deficiencies, lack of vitamin D, malabsorption syndromes) or to excessive urinary loss of calcium or phosphate or to abnormalities in the synthesis or function of alkaline phosphatase [2]. Osteoporosis is the result of decreased synthesis or excessively rapid degradation of bone matrix proteins, particularly collagen type I, that leads to osteopenia because of decreased collagen scaffolding upon which to deposit hydroxyapatite. In children and adolescents, decreased bone mineralization has been defined as bone mineral content or concentration that is two standard deviations below the mean normal for similar gender and chronologic age by the method employed for its determination. Osteopenia has been defined as bone mineralization between –1 and –2 standard deviations for similar gender and age. Bone mineralization is considered increased if it is more than two standard deviations above the mean normal for similar gender and chronologic age. However, when evaluating bone mineralization data it is imperative to consider not only the gender and chronologic age of the patient but also his/her race, ethnicity, height, weight, and stage(s) of sexual maturation.
Deficiency of vitamin D due to either limited dietary intake or insufficient exposure to sunlight impairing endogenous synthesis of cholecalciferol is the most common cause of rickets. Genetically restricted synthesis of endogenous calcitriol or functional responsiveness to calcitriol are uncommon causes of rickets (Table 3). Defective hepatic 25-hydroxylation of vitamin D3 (cholecalciferol) due to an inactivating mutation in CYP2R1 results in vitamin D hydroxylation-deficient rickets type 1 B (MIM 600081). Loss of function mutations in CYPB27B1 encoding renal 25-hydroxyvitamin D3-1α-hydroxylase result in decreased synthesis of calcitriol and vitamin D dependent rickets type 1A (MIM 264700). Inactivating mutations in VDR impair tissue responsiveness to calcitriol and lead to vitamin D dependent rickets type 2A (MIM 277440). In the latter patients, alopecia and hypocalcemia are often present. HNRNPC encodes a ribonucleoprotein that transiently occupies hormone response elements within the 5’ upstream promoter region of target genes; when overexpressed, HNRNPC interferes with interaction of the VDR/RXR heterodimer with the VDRE resulting in a secondary form of vitamin D resistance termed vitamin D dependent rickets type 2B (MIM 600785) in which clinical manifestations are similar to those recorded in patients with mutations in VDR [54]. The mechanism by which HNRNPC is overexpressed in patients with vitamin D dependent rickets type 2B is unknown as yet.
Table 3
Disorders of mineralization: Rickets and osteomalacia
| Gene Chromosome MIM | Pathophysiology | Mutation – Clinical manifestations |
| Disorders of vitamin D metabolism | ||
| CYP2R1 – Cytochrome P450, subfamily IIR, polypeptide 1 11p15.2 608713 | Hepatic 25-Hydroxylase – enzyme that converts vitamin D3 to 25OHD3 (calcidiol) | Biallelic loss of function mutation leads to vitamin D hydroxylation-deficient rickets type 1B (also termed vitamin D dependent rickets type 1B), AR |
| CYP27B1 – Cytochrome P450, subfamily XXVII, polypeptide 1 12q13.1-q13.3 609506 | 25OHD3-1α hydroxylase – enzyme that converts 25OHD3 to 1,25(OH)2D3 (calcitriol) | Biallelic inactivating mutations result in vitamin D dependent rickets type 1A, AR |
| VDR – Vitamin D receptor 12q12-q14 601769 | Vitamin D receptor – transcription factor that transduces the effects of calcitriol on gene activation or repression | Biallelic loss of function mutations lead to resistance to calcitriol and vitamin D dependent rickets type 2A, AR |
| HNRNPC – Heterogeneous nuclear ribonucleoprotein C 164020 | Encodes a ribonucleoprotein that regulates gene transcription by reciprocally and transiently occupying the VDRE in the upstream promoter region of vitamin D-responsive target genes | Overexpression in vitamin D-responsive tissues leads to prolonged occupancy of the VDRE that interferes with interaction of the VDR/RXR with the VDRE resulting in vitamin D dependent rickets type 2B |
| Disorders of phosphate metabolism | ||
| SLC34A1 – (Solute carrier family 34 (sodium phosphate cotransporter), member 1 5q35 182309 | Encodes NPT2a – sodium/phosphate cotransporter expressed on the apical membrane of the proximal renal tubule; under the inhibitory control of PTH | Loss of function mutations result in hypophosphatemia with urolithiasis and osteopenia; AD |
| SLC34A2 – (Solute carrier family 34 (sodium phosphate cotransporter), member 2 2p15.31-p15.2 604217 | Encodes NPT2b – sodium/phosphate cotransporter expressed in the small intestine, lung, and testes | Loss of function mutations associated with pulmonary alveolar microlithiasis and testicular microlithiasis. AR |
| SLC34A3 – (Solute carrier family 34 (sodium phosphate cotransporter), member 3 9q34 609826 | Encodes NPT2c – sodium/phosphate cotransporter expressed on the apical membrane of the proximal renal tubule | Loss of function mutation results in hereditary hypophosphatemic rickets with hypercalciuria, AR |
| SLC9A3R1 – Solute carrier family 9, member 3, regulator 1 17q25.1 604990 | Encodes NHERF1 – a renal tubular sodium/hydrogen exchange regulatory factor that binds NPT2a anchoring it to the luminal membrane of the proximal renal tubule; phosphorylation by PTH leads to its dissociation from &endocytosis of NPT2a | Loss of function mutations result in autosomal recessive hypophosphatemic rickets &nephrolithiasis, AR |
| CLCN5 – Chloride channel 5 Xp11.23-p11.22 300008 | Encodes a proximal renal tubular exchanger of chloride &hydrogen ions | Loss of function mutations result in X-linked recessive hypophosphatemic rickets, hypercalciuria, nephrocalcinosis, XLR |
| PHEX – Phosphate-regulating endopeptidase homolog, X-linked Xp22.2-p22.1 300550 | Ectoenzyme expressed on the cell membrane of osteoblasts; its physiological substrate may be MEPE &pASARM, its phosphorylated degradation product that coats hydroxyapatite &hinders mineral deposition | Loss of function mutation leads to X-linked hypophosphatemic rickets; associated with increased expression of FGF23; X-linked dominant |
| DMP1 – Dentin matric acidic phosphoprotein 1 4q21 600980 | Non-collagenous, serine-rich, bone matrix protein; a small integrin-binding ligand, N-linked glycoprotein (SIBLING) expressed in osteocytes | Loss of function mutations lead to increased osteocyte synthesis of FGF23, hyperphosphaturia, &autosomal recessive hypophosphatemic rickets, AR |
| ENPP1 – Ectonucleotide pyrophosphatase/ phosphodiesterase 1 6q22-q23 173335 | Ectoenzyme expressed by chondrocytes, bone, &plasma cells that hydrolyzes ATP to pyrophosphate, an inhibitor of bone mineralization | Loss of function mutations result in autosomal recessive hypophosphatemic rickets with increased expression of FGF23, AR |
| ANKH – ANK, mouse, homolog of 5p15.2-141 605145 | Transmembrane-spanning cell surface protein that regulates pyrophosphate secretion | Inactivating mutations result in mild hypophosphatemia &joint ankylosis, developmental delay, deafness, &dentinogenesis imperfecta; AR |
| FGF23 – Fibroblast growth factor 23 12p13.3 605380 | Product of osteoblast &osteocyte that depresses renal tubular reabsorption of phosphate &inhibits synthesis of calcitriol | Gain of function mutation that decreases the rate of degradation of FGF23 results in autosomal dominant hypophosphatemic rickets, AD; excessive ectopic synthesis by neoplasms leads to hypophosphatemic rickets; loss of function mutation leading to decreased FGF23 synthesis results in autosomal recessive hyperphosphatemic familial tumoral calcinosis, AR |
| GALNT3 – UDP-N-Acetyl-alpha-D-Galactosamine: polypeptide N-acetylglactosaminyl transferase 3 1q24-q31 601756 | Encodes an enzyme that is essential for O-glycosylation of Thr178 of FGF23 during post-translational processing; failure of this step leads to degradation of FGF23 prior to its secretion | Loss of function mutation leads to autosomal recessive hyperphosphatemic familial tumoral calcinosis, AR |
| KL – Klotho 13q12 604824 | Co-receptor with FGFR1(IIIc) for FGF23 that converts FGFR1(IIIc) into the specific FGF23 receptor enabling signal transduction | Loss of function mutation leads to autosomal recessive hyperphosphatemic familial tumoral calcinosis, AR |
| Hypophosphatasia | ||
| ALPL – Alkaline phosphatase, liver 1p36.12 171760 | Tissue non-specific alkaline phosphatase – ectoenzyme expressed on the cell membrane of osteoblasts, removes organically bound phosphate; major substrates are pyrophosphate, phosphoethanolamine, pyridoxal-5’-phosphate | Loss of function mutations lead to hypophosphatasia &rickets of variable severity &onset: perinatal, infancy, childhood, adult, odontohypophosphatasia; AR, AD |
Adapted from Farrow EG, White KE. Recent advances in renal phosphate handling. Nature Rev/Nephrol 6:207-217,2010.
Hypophosphatemic rickets may be due to abnormalities of intestinal phosphate absorption or its reabsorption in the proximal renal tubule (Table 3). SLC34A1 encodes a sodium/phosphate cotransporter (NPT) expressed in the proximal renal tubule; loss of function mutations in SLC34A1 result in hypophosphatemia due to hyperphosphaturia, renal calculi, and osteopenia. Inactivating mutations in SLC9A3R1 encoding NHERF1, a renal tubular protein that binds NPT2a thereby anchoring it to the luminal membrane of the proximal renal tubule, lead to autosomal recessive hypophosphatemic rickets with urolithiasis; mutations in SLC9A3R1 potentiate PTH-induced generation of cyclic AMP and inhibition of proximal renal tubular reabsorption of phosphate. Inactivating mutations of SLC34A3, a gene encoding a second proximal renal tubular sodium/phosphate cotransporter, lead to autosomal recessive hereditary hypophosphatemic rickets with hypercalciuria, the latter due to excessive synthesis of calcitriol in response to systemic hypophosphatemia [55].
The most common form of hypophosphatemic rickets encountered is X-linked (XHR) due to inactivating mutations in PHEX. This gene encodes a zinc-metalloendopeptidase situated on the surface of osteoblasts and osteocytes whose physiologic substrate has proven difficult to identify with certainty. Unexpectedly, FGF23 is not a physiologic substrate of PHEX. Rather, current data indicate that a phosphorylated peptide derived from MEPE is the primary substrate for PHEX action [56–58]. MEPE is a 525 amino acid non-collagenous protein secreted by osteoblasts that is present in extracellular bone matrix; it is also found in teeth and in brain. MEPE inhibits renal tubular phosphate uptake and, thus, is a phosphatonin. It is also one of the SIBLING group of non-collagenous matrix proteins. MEPE contains an ASARM in its carboxyl terminal region that can be released by cathepsin B. Normally, PHEX and MEPE bind to one another in a non-proteolytic manner as a result of which MEPE is not degraded by cathepsin B and ASARM is not released. In the absence of normal PHEX activity, ASARM is released from MEPE by cathepsin B. Indeed, serum ASARM levels are increased in patients with XHR. Serine-phosphorylated ASARM binds directly to hydroxyapatite by interaction with calcium atoms and prevents further deposition of calcium and phosphate thereby inhibiting mineralization. Additionally, PHEX proteolytically cleaves pASARM between serine-glutamate and serine-aspartate residues, thereby destroying pASARM and further preventing pASARM-mediated inhibition of bone mineralization. The role of non-phosphorylated ASARM in the mineralization process is as yet incompletely understood; ASARM can also bind to hydroxyapatite, albeit weakly, and inhibit mineralization; ASARM too is enzymatically cleaved by PHEX. Thus, in XHR the primary pathophysiologic abnormalities appear to be failure of mutated and bioinactive PHEX 1) to prevent proteolytic cleavage of MEPE and release of ASARM and 2) to degrade pASARM derived from MEPE thereby extending pASARM-mediated inhibition of mineralization. It is of interest that in XHR, two phosphatonins – FGF23 and MEPE – are present in excess further exacerbating the mineralization defect by increasing renal phosphate excretion and lowering still more the supply of phosphate ions necessary for construction of hydroxyapatite. Interestingly, pASARM increases expression of FGF23 in bone cells [58]. pASARM catalytically released from osteopontin also binds to hydroxyapatite and inhibits mineralization; osteopontin-derived pASARM is a PHEX substrate but non-phosphorylated ASARM is not [57]. The pathophysiologic role of osteopontin and its pASARM in XHR is uncertain at present. Mutations in DMP1, ENPP1, and ANKH result in autosomal recessive forms of hypophosphatemic rickets. X-linked recessive hypophosphatemic rickets (MIM 300554) is due to inactivating mutations in CLCN5 (MIM 30008) encoding a proximal renal tubular exchanger of chloride and hydrogen ions [59]. This disorder is characterized clinically by failure of proximal renal tubular reabsorptive function, hypophosphatemic rickets, hypercalciuria, nephrocalcinosis, and proteinuria progressing to renal failure (Dent disease).
Fibroblast growth factor 23 is synthesized by osteoblasts and osteocytes; it depresses renal tubular reabsorption of phosphate and inhibits synthesis of calcitriol. Gain of function mutations in FGF23 decrease the rate of degradation of FGF23 and result in autosomal dominant hypophosphatemic rickets. Excessive ectopic synthesis of FGF23 by neoplasms and by the fibrodysplastic lesions of the McCune-Albright syndrome and neurofibromatosis type 1 also leads to hypophosphatemic rickets. Loss of function mutations in FGF23 lead to decreased FGF23 synthesis resulting in autosomal recessive hyperphosphatemic familial tumoral calcinosis. This disorder that may also be due to inactivating mutations in GALNT3 (MIM 601756) encoding an enzyme that is essential for O-glycosylation of FGF23 during post-translational processing and whose failure leads to degradation of FGF23 prior to its secretion and KL (MIM 604824), encoding Klotho – a co-receptor with FGFR1(IIIc) for FGF23 – whose loss results in renal tubular unresponsiveness to FGF23.
The clinical and biochemical manifestations of rickets due to hypophosphatasia are due to loss of function in ALPL (MIM 171760) encoding alkaline phosphatase, the osteoblast enzyme essential for degradation of inhibitors of bone mineralization such as pyrophosphate and pASARM and for increasing local concentrations of phosphate enabling deposition of hydroxyapatite onto collagen type I. Perinatal/infantile (MIM 241500), childhood (MIM 241510), and adult/odontohypophosphatasia (premature shedding of deciduous and/or secondary teeth, dental caries) (MIM 146300) forms of this disorder have been described. In all subjects with hypophosphatasia, serum bone alkaline phosphatase activity is inappropriately low. The severity of the disorder depends on the site of the mutation and residual enzymatic activity and whether the affected subject has a biallelic or a monoallelic mutation in ALPL.
4.2Low bone mass
Decreased nutritional intake, limited physical activity, chronic diseases, and hormonal deficiencies are the most common factors leading to low bone mass in children and adolescents. In addition, several deleterious gene mutations lead to osteopenia in children (Table 4). Mutations in genes controlling normal collagen synthesis are the major causes of osteogenesis imperfecta or “brittle bone disease,” a disorder characterized by low bone mass and increased osseous fragility (Table 5). Loss of function mutations in LRP5 result in the osteoporosis-pseudoglioma syndrome (MIM 259770), an autosomal recessive disorder associated with marked osteopenia leading to skeletal fractures, deformities, and short stature and pseudotumors of the retina – at times mistaken for retinoblastoma; monoallelic carriers of these mutations may have reduced bone mass of variable severity. Marfan syndrome (MIM 154700) is a connective tissue disorder characterized by abnormalities of the skeleton (tall stature, long limbs, arachnodactyly, joint laxity, osteopenia, scoliosis/lordosis, highly arched palate, dental crowding), eye (myopia, lenticular subluxation), and cardiovascular system (mitral valve prolapse and regurgitation, dilatation of the aortic root, aortic valve regurgitation, aortic aneurysm and dissection). Marfan syndrome is due to inactivating mutations in fibrillin-1 encoded by FBN1, a glycosylated component of the microfibrillar system of extracellular matrix elastic fibers to which transforming growth factor (TGF)β binds and within which it is sequestered. Loss of function mutations in FBN1 permit TGFβ to mount an inflammatory state that leads to degradation of elastin fibers and weakening of connective tissue [60]. Patients with homocystinuria (MIM 236200) have a phenotype similar to that of Marfan syndrome and are also osteopenic. The Ehlers-Danlos syndromes are a group of connective tissue disorders whose clinical manifestations include hyperextensibility of the skin, hypermobility of the joints, tissue fragility, and in many patients substantially decreased bone mineralization; in some subjects, this disorder is related to monoallelic inactivating mutations in COL1A1. Mutations in COL3A1, COL5A1, COL5A2, and PLOD1 have also resulted in clinical Ehlers-Danlos syndromes.
Table 4
Disorders of mineralization: Low bone mass (Osteogenesis imperfecta Types I XI – see Table 5)
| Gene Chromosome MIM | Pathophysiology | Mutation – Clinical manifestations |
| LRP5 – Low density lipoprotein receptor-related protein 5 11q13.4 603506 | Transmembrane receptor expressed on plasma membrane of osteoblasts, osteocytes &enterochromaffin cells; in osteoblasts &osteocytes, LRP5 &Frizzled are co-receptors for WNT &the complex stimulates signal transduction by β-catenin; in enterochromaffin cells activation of LRP5 inhibits synthesis of tryptophan hydroxylase &serotonin decreasing serotonin-mediated inhibition of osteoblast proliferation &function | Inactivating mutations result in osteoporosis-pseudoglioma, AR |
| FBN1 – Fibrillin 1 15q21.1 134797 | Glycosylated component of the microfibrillar system of extracellular matrix elastic fibers that are essential components of elastic and non-elastic connective tissue | Inactivating mutations lead to Marfan syndrome (MIM 154700), AD, AR |
| CBS – Cystathionine beta synthase 21.q22.3 613381 | Encodes enzyme that conjugates homocysteine and serine to form cystathionine | Inactivating mutations lead to homocystinuria (MIM 236200) – phenotype similar to that of Marfan syndrome |
| TNFRSF11B – Tumor necrosis factor receptor superfamily, member 11B 8q24 602643 | Encodes osteoprotegerin, decoy receptor for RANKL | Juvenile Paget disease |
| COL1A1 – Collagen type I, alpha-1 17q21.3-q22 120150 | Encodes a protein that forms collagen type I | Inactivating mutations result in osteogenesis imperfecta types I–IV (see Table 5), Ehlers-Danlos syndrome type I (MIM 130000) |
Table 5
Disorders of mineralization: Osteogenesis imperfecta
| Type – MIM: Disease – Gene Chromosome | Severity | Clinical features | Growth impairment | Blue sclera | Inheritance | Functional defect |
| I – 166200 – COL1A1 – Collagen type I, alpha-1 17q21.3-q22 1120150 | Mild | Few fractures, little deformity, hearing loss in 50%; rarely dentinogenesis imperfecta | Minimal | Present – intense | AD | Nonsense &frameshift mutations resulting in premature STOP codons result in decreased production of collagen type I |
| IIA – 166210 – COL1A1 or COL1A2 – Collagen type I, alpha-2 7q22.1 120160 | Perinatal lethal – congenital | Many rib &long bone fractures in utero &at birth, severe long bone deformities, unmineralized calvarium | Severe | Present | AD, parental mosaicism | A – Glycine substitutions in COL1A1 or COL1A2 result in structurally abnormal collagen type I |
| IIB – 610854 – CRTAP – Cartilage associated protein 3p22 605497 | Perinatal lethal – congenital | Many rib &long bone fractures at birth, severe long bone deformities, unmineralized calvarium | Severe | Present | AR | B – Inactivating mutations of CRTAP impair 3-prolyl (986) hydroxylation of procollagen α1(I) |
| III – 259420 – COL1A1 or COL1A2 | Severe, progressive, deforming | Moderate to severe bowing, multiple fractures, dentinogenesis imperfecta, hearing loss | Severe | Present but lighten with age | AD | Glycine substitutions in COL1A1 or COL1A2 result in structurally abnormal collagen type I |
| IV -166220 COL1A1 or COL1A2 | Moderately deforming | Mild to moderate bowing, fractures | Moderate, variable | Greyish or absent | AD | Glycine substitutions in COL1A1 or COL1A2 result in structurally abnormal collagen type I |
| V – 610967 IFITMS-Interferon-induced transmembrane protein V IIP15.5614757 | Moderately deforming, clinically similar to Type IV | Mild to moderate bone fragility, ossification of interosseous membranes of forearm, hypertrophic callus formation at fracture sites | Mild to moderate | Absent | AD | Unknown |
| VI – 610968 FKBP65 – FK506-binding protein 10 17q21.2 607062 | Moderately to severely deforming, clinically similar to Type III | Onset of fractures in infancy, increased alkaline phosphatase activity &osteoid, “fish-scale” pattern of lamellation | Moderate to severe | Absent or faint | AR | Loss of function mutations in a chaperone protein -FKBP65 – essential for post-translational processing of procollagen type I; may co-segregate with epidermolysis bullosa; also mutated in Bruck syndrome type 1 |
| VII – 610682 *CRTAP – Cartilage associated protein 3p22 605497 | Moderately deforming | Fractures present at birth, rhizomelia, limb deformities | Moderate | Absent or faint | AR | Inactivating mutations (duplication) of CRTAP impair hydroxyation of 3-prolyl (986) of procollagen α1(I) |
| VIII- 610915 *LEPRE1 – Leucine- and proline-enriched proteoglycan 1 1p34 610339 | Severely deforming, overlaps type II &III | Phenotype overlaps those of types II and III | Severe | Absent | AR | Inactivating mutations of LEPRE encoding prolyl-3-hydroxylase (leprecan) impair hydroxylation of 3-prolyl (986) of procollagen α1(I) |
| IX – 259440 – *PPIB- Peptidyl-prolyl isomerase B 15q21-q22 123841 | Lethal to severe | Shortened, bowed, &fractured long bones in mid-gestation | Severe | Grey | AR | Inactivating mutations of PPIB encoding peptidyl-prolyl isomerase B impair hydroxylation of 3-prolyl (986) of procollagen α1(I) |
| X – 613848 – SERPINH1 – Serpin peptidase inhibitor, clade H, member 1 11q13.5 600943 | Severe to lethal | Short bowed femora in utero, multiple fractures in first month of life | Severe | Present | AR | Loss of function mutations in a chaperone protein – HSP47 – essential for post-translational processing of procollagen type I |
| XI – 613849 – SP7 – Transcription factor specificity factor (Sp)7 12q13.13 606633 | Severe, resembles OI type IV | Multiple fractures in early infancy; bowing of long bones | Severe | Absent | AR | Inactivating mutations in Osterix, a transcription factor essential for osteoblast differentiation |
| PLOD2 – Procollagen-lysine, 2-oxoglutarate 5-dioxygenase 2 3q24 601865 | Severe, OI with congenital joint contractures – Bruck syndrome 2 – (MIM 609222) | Fractures in infancy, pterygia, scoliosis | Severe | Absent | AR | Encodes a lysyl hydroxylase necessary for formation of collagen type 1 teleopeptide |
(Modified from 2 Root AW, Diamond FB Jr. Disorders of bone mineral metabolism in the newborn, infant, child, and adolescent. in Sperling MA (ed). Pediatric Endocrinology, 3rd ed, Saunders/Elsevier, Philadelphia, 2008, p 686-769, with permission.). *Form a complex essential for hydroxylation of carbon 3 of proline at position 986 of procollagen type α1(I).
4.3Osteogenesis imperfecta
The predominant clinical characteristic of osteogenesis imperfecta (OI) is bone fragility due to low bone mass leading to bone deformation and fracture [16]. Loss-of function mutations in the genes encoding the two subunits of collagen type I (COL1A1, COL1A2) predominate in children with OI, although there is substantial genetic heterogeneity of this disease as we learn more about the factors that regulate and assist normal collagen formation (Table 5) [61, 62]. Traditionally, the forms (types) of OI have been classified by their clinical manifestations as initially described by Sillence. Depending on the site, mutations in COLIA1 or COL1A2 impair either synthesis or structure of the subunits of collagen type I, may vary from lethal to mild in severity, and may be transmitted as either autosomal dominant or autosomal recessive traits (OI types I–IV). A number of harmful sequence variations have also been identified in genes that control the post-translational processing of both subunits of collagen type I and whose disease phenotypes often overlap those of other types of OI, making a molecular diagnosis increasingly important in the evaluation and management of children with OI. The mildest clinical form of OI (type I) is that due to mutations in COL1A1 that reduce its rate of production. Mutations in COL1A1 or COL1A2 that alter the structure of collagen type I and interfere with the function of the collagen fiber result in more severe clinical disorders (types II–IV).
OI type V due to variants of IFITMS is characterized by radiographically dense metaphyseal bands adjacent to the cartilage growth plate, calcification of the interosseous membrane between the radius and ulna, exuberant callus formation at the site of fractures, and distinct and irregular “mesh-like” appearance of the iliac lamellae microscopically. In OI type VI, the microscopic appearance of bone is that of an array of “fish scales.” OI type VI is due to inactivating mutations in FKBP65, a gene that encodes a chaperone that is essential for normal post-translational processing of procollagen type I. In several Turkish families, OI type VI co-segregated with epidermolysis bullosa due to an inactivating mutation in KRT14 encoding keratin 14, also situated on chromosome 17q21 [61]. Osteogenesis imperfecta type X is a severe form of OI due to a loss of function mutation in SERPINH1, encoding collagen binding protein 2 (CPB2) or heat shock protein 47 (HSP47), another chaperone [63]. HSP47 is essential for normal localization of procollagen type I within the osteoblast; in its absence procollagen type I accumulates in the Golgi apparatus. HSP47 is also necessary for formation and stabilization of the collagen triple helix configuration.
In the endoplasmic reticulum, a proline residue at position 986 within procollagen type IA must be specifically hydroxylated by a tripartite complex of prolyl 3-hydroxylase 1 (encoded by LEPRE), cartilage associated protein (encoded by CRTAP), and peptidyl-prolyl isomerase B (encoded by PPIB). The third member of the prolyl 3-hydroxylase complex, peptidyl-prolyl isomerase B, is also termed cyclophilin B. Loss of function mutations in CRTAP, LEPRE, or PPIB result in moderate to severe and often lethal forms of osteogenesis imperfecta (designated types IIB, VII, VIII, and IX, respectively). One child has been described with autosomal recessive OI (type XI) due to a loss-of-function mutation in SP7 encoding osterix, a transcription factor essential for osteoblast differentiation and activation of COL1A1 [64].
Increased bone fragility is found in children with Bruck syndromes 1 and 2 (MIM 259450; 609220) characterized by congenital joint contractures, pterygia, fractures beginning in infancy or early childhood, short stature, and scoliosis. In patients with Bruck syndrome type 1, mutations in FKBP10, (also known as FKBP65 – see osteogenesis imperfecta type VI) encoding a collagen chaperone, have been identified [65]. In Bruck syndrome type 2, biallelic mutations in PLOD2 (MIM 601865) encoding a lysyl hydroxylase necessary for normal formation of collagen type 1 teleopeptide have been found [66, 67]. The Cole-Carpenter syndrome (MIM 112240) is manifested by multiple fractures developing in the first year of life, craniosynostosis, progressive ocular proptosis, hydrocephalus, and an unusual face (frontal bossing, mid-face hypoplasia, micrognathia), it is due to variants of P4HB or SEC24D [68].
4.4Increased bone mass/Osteopetrosis
Increased bone mineralization may be the result of an excessive rate of bone formation or of a decreased rate of reabsorption of already formed bone (Table 6). Sequence variations (e.g., Gly171Val, Arg214Val) in LRP5 are associated with high bone mass and resistance to fractures in otherwise normal subjects. These clinically benign sequence variants impair the binding to and thus the inhibitory effects of Dickkopf-1 (encoded by DKK1) and sclerostin (encoded by SOST) upon LRP5. More significant activating mutations in LRP5 lead to autosomal dominant osteopetrosis type 1 (607634) whose phenotype closely resembles that of high bone mass patients with endosteal osteosclerosis (MIM 144750) or van Buchem disease type 2 (MIM 607636). Inactivating mutations in SOST (expressed in osteoblasts and osteocytes and secreted by osteoclasts) that are unable to modulate LRP5-WNT-Frizzled stimulation of bone formation result in pathologic forms of high bone mass including sclerosteosis (cortical hyperostosis with cutaneous syndactyly – MIM 269500) and van Buchem disease (osteosclerosis of skull, long bones, clavicle, ribs – MIM 239100).
Disorders of bone dissolution result in several forms of osteopetrosis that are characterized histologically by either abundant or scarce osteoclasts (Table 6). Osteopetrosis in which bone biopsy reveals a paucity of osteoclasts include those associated with loss of function mutations in TNFSF11, TNFRSF11A, and IKBKG. Type 2 autosomal recessive osteopetrosis (MIM 259710) is a disorder of variable severity characterized by progressive blindness, anemia, thrombocytopenia, hepatosplenomegaly, skeletal and dental deformities, mandibular osteomyelitis, and increased fracture risk; it is due to loss of function mutations in TNFSF11 encoding RANKL, a member of the tumor necrosis factor ligand superfamily, thereby decreasing the stimulus to osteoclastogenesis [69]. Loss of function mutations in TNFRSF11A encoding RANK result in impaired osteoclastogenesis and immune dysfunction (autosomal recessive osteopetrosis type 7 – MIM 612301). Some mutations in TNFSF11 reduce binding of RANKL to RANK [70]. Inactivation of the NFκB essential modulator (NEMO – the cytosolic regulatory subunit of IkappaB kinase essential for NFκB signaling that is encoded by IKBKG) impairs NFκB-mediated immune responsiveness and osteoclastogenesis resulting in a complex of immunodeficiency, lymphedema, anhidrotic ectodermal dysplasia, and osteopetrosis transmitted as an X-linked trait (MIM 300301) [71].
Table 6
Disorders of mineralization: Increased bone mass
| Gene Chromosome MIM | Pathophysiology | Mutation – Clinical manifestations |
| LRP5 – Low density lipoprotein receptor-related protein 5 11q13.4 603506 | Transmembrane receptor expressed on plasma membrane of osteoblasts, osteocytes &enterochromaffin cells; in osteoblasts &osteocytes, LRP5 &Frizzled are co-receptors for WNT &the complex stimulates signal transduction by β-catenin; in enterochromaffin cells activation of LRP5 inhibits synthesis of tryptophan hydroxylase &serotonin decreasing serotonin-mediated inhibition of osteoblast proliferation &function | Gain of function mutations lead to autosomal dominant osteopetrosis type I, endosteal hyperostosis, osteosclerosis, van Buchem disease type 2, AD |
| SOST – Sclerostin 17q11.2 605740 | Antagonist of WNT signaling of osteoblast function, inhibitor of bone morphogenetic protein-stimulated differentiation of osteoblasts – secreted by osteoclasts | Sclerosteosis, van Buchem disease, AR |
| TNFSF11 – Tumor necrosis factor ligand superfamily, member 11 13q14 602642 | Encodes RANKL – activates RANK on the membrane of the pre-osteoclast enhancing osteoclastogenesis through signaling to NFκB | Osteopetrosis, autosomal recessive type 2 – relatively less severe form of osteopetrosis than type 1 |
| TNFRSF11A – Tumor necrosis factor receptor superfamily, member 11A 18q21.1 603499 | Encodes RANK – activation results in NFκB-mediated osteoclastogenesis &osteoclast function | Osteopetrosis, autosomal recessive type 7 – with hypogammaglobulinemia |
| IKBKG – Inhibitor of kappa light polypeptide gene enhancer in B cells, kinase of, gamma Xq28 300248 | Also termed NEMO (NFκB essential modulator) – component of IkappaB kinase complex that is essential for NFκB signaling | Inactivating mutations lead to a syndrome complex of immunodeficiency, lymphedema, anhidrotic ectodermal dysplasia, osteopetrosis; X-linked |
| PLEKHM1 – Pleckstrin homology domain-containing protein, family M, member 1 17q21.31 611466 | Encodes a protein that may affect vesicular transport within the osteoclast &the attachment of the osteoclast to bone. | Osteopetrosis, autosomal recessive type 6 – intermediate phenotype, Erlenmeyer flask deformity of long bone |
| CA2 – Carbonic anhydrase II 8q22 611492 | Carbonic anhydrase – osteoclast enzyme that generates protons (H+) from carbonic acid | Osteopetrosis, autosomal recessive type 3 – associated with renal tubular acidosis |
| TCIRG1 – T cell immune regulator type 1 11q13.2 604592 | Subunit of vacuolar proton pump that transports H+ into the sub-osteoclastic lacunae to dissolve hydroxyapatite | Osteopetrosis, autosomal recessive type 1 – infantile malignant osteopetrosis |
| CLCN7 – Chloride channel 7 16p13 602727 | Chloride channel that transports Cl– into the sub-osteoclastic lacunae to dissolve hydroxyapatite | Osteopetrosis, autosomal recessive type 4; monoallelic mutations lead to autosomal dominant osteopetrosis type II – of intermediate severity |
| OSTM1 – Osteopetrosis-associated transmembrane protein 1 6q21 607649 | CLCN7 &OSTM1 form a molecular complex that cooperatively enable the transport of Cl– from the osteoclast into the sub-osteoclastic lacuna permitting dissolution of hydroxyapatite | Osteopetrosis, autosomal recessive type 5 – associated with retinal and neural dysplasia |
| CTSK – Cathepsin K 1q21 601105 | Cysteine endoproteinase essential for degradation of bone extracellular organic matrix | Pycnodysostosis, AR |
| LEMD3 – LEM domain-containing protein 3 12q14 607844 | Nuclear membrane protein that modulates signal transduction by bone morphogenetic proteins | Osteopoikilosis, AD |
| COL1A1 – Collagen type I, alpha-1 17q21.3-q22 1120150 | COL1A1 &COL1A2 form the triple helix of collagen type 1 | Infantile cortical hyperostosis (Caffey disease), AD with variable penetrance |
| SAMD9 – Sterile alpha motif domain-containing protein 9 7q21.2 610455 | Encodes a protein expressed in skin endothelial cells and fibroblasts | Homozygous mutation results in familial normophosphatemic tumoral calcinosis, AR |
| ACVR1 – Activin A receptor, type I 2q24.1 102576 | Transmembrane receptor for BMP type 1 (&activin), members of the TGFβ super family | Heterozygous gain of function mutations lead to fibrodysplasia ossificans progressiva, AD |
Forms of osteopetrosis in which bone biopsy reveals abundant but poorly or non-functional osteoclasts are those associated with loss of function mutations in CA2, TCIRG1, CLCN7, OSTM1, and PLEKHM1. Autosomal recessive osteopetrosis type 6 (MIM 611497) is a relatively mild form of osteopetrosis characterized by high bone mass and slight deformities of long bones that is due to loss of function mutations in PLEKHM1. This gene encodes a protein that enables the osteoclast to form ruffled borders; thus its loss prevents sealing of osteoclasts to bone and initiation of the reabsorption process. PLEKHM1 may also assist the transport of vesicles. Inability of the osteoclast to synthesize carbonic acid prevents formation of H+ and dissolution of bone mineral resulting in autosomal recessive osteopetrosis type 3 (MIM 259730); it is due to inactivating mutations in CA2 encoding carbonic anhydrase II. Patients with this moderately severe form of osteopetrosis are characterized by fractures in early childhood, short stature, visual impairment, and developmental challenges. Loss of function mutations in TCIRG1 impair transport of H+ into the subosteoclastic lacuna preventing dissolution of hydroxyapatite resulting in autosomal recessive infantile malignant/lethal osteopetrosis type 1 (MIM 259700). This disorder has also been termed Albers-Schonberg or marble bone disease. Beginning in utero and progressing rapidly postnatally, affected patients manifest macrocephaly, progressive blindness and deafness, severe anemia, and hepatosplenomegaly. The clinically severe infantile form of osteopetrosis may also be due to mutations in CLCN7 (autosomal recessive osteopetrosis type 4 – MIM 611490) and OSTM1 (autosomal recessive osteopetrosis type 5 – MIM 259720) (vide infra). The phenotypic spectrum of osteopetrosis due to inactivating mutations in CLCN7 is wide [72]. Clinically, biallelic mutations in CLCN7 encoding the osteoclast chloride channel are associated with autosomal recessive infantile malignant osteopetrosis, while monoallelic mutations lead to a much less severe form of autosomal dominant osteopetrosis type II (MIM 166600). Loss of function mutations in OSTM1 encoding a protein that forms a complex with CLCN7 that enables the transport of Cl- into the sub-osteoclastic lacuna and the reabsorption of hydroxyapatite also result in early onset form of often lethal osteopetrosis paired with neuroaxonal and retinal dystrophies [73, 74].
Clinically, pycnodysostosis (MIM 265800) is manifested by short stature, osteosclerosis, cranial deformities, and osseous fragility. It is due to biallelic inactivating mutations in CTSK, encoding a cysteine endoproteinase necessary for degradation of components of skeletal extracellular organic matrix. The process of bone demineralization is normal in patients with pycnodysotosis. LEMD3 encodes a nuclear membrane protein that antagonizes intracellular signal transduction by bone morphogenetic proteins. Heterozygous loss of function mutations in LEMD3 result in osteopoikilosis (MIM 166700) characterized by bone “spots” composed of discrete sclerotic areas present in epiphyses and metaphyses of long bones, pelvis, and scapulae, often in association with distinct skin lesions; it is a relatively benign disorder. Infantile cortical hyperostosis (Caffey disease – MIM 114000) is manifested by acute onset of tenderness and warmth of the skull or ribs that is self limited; it has been associated with a specific heterozygous mutation (Arg836Cys) in COL1A1.
Hyperphosphatemic familial tumoral calcinosis is an autosomal recessive disorder characterized by subcutaneous calcifications that may be due to inactivating mutations in FGF23, GALNT3, or KL as discussed in section IV.A (Table 3). Normophosphatemic familial tumoral calcinosis (MIM 610455) is an autosomal recessive disease in which subcutaneous calcified nodules develop over the limbs during infancy and early childhood. Calcification is preceded by cutaneous inflammation. It is due to biallelic loss of function mutations in SAMD9, a protein that is regulated by interferon-γ and of pathogenic importance in suppression of inflammation [76]. Fibrous dysplasia ossificans progressiva (also termed myositis ossificans progressiva – MIM 135100) is characterized by heterotopic development of mature endochondral bone within extraskeletal sites (muscle, tendons, ligaments) and is usually associated with congenital anomalies of the big toes (monophalangic) and fingers (short thumbs, clinodactyly) [77, 78]. This disorder is due to gain of function mutations in ACVR1 encoding a receptor for both activin (a stimulus to the secretion of pituitary follicle stimulating hormone) and BMP type 1, members of the transforming growth factor β (TGFβ) super family [79]. One heterozygous mutation (Arg206His) has been identified most frequently (but not exclusively) in patients with familial disease and in sporadically affected subjects.
4.5Osteochondrodysplasia
Dysplasias of cartilage and bone are manifested by widely variable clinically characteristics and heterogeneous genetic mutations involving the vertebrae and epiphyses and metaphyses of the long bone [80]. Selected dysplasias of bone and cartilage development, many of them with a metabolic basis, are listed in Table 7.
Table 7
Osteochondrodystrophies
| Gene Chromosome MIM | Pathophysiology | Clinical disorder (MIM) |
| TRIP11 – Thyroid hormone interactor 11 14q31-q32 604505 | Co-activator of the nuclear triiodothyronine receptor; interacts with microtubules &Golgi apparatus | Achondrogenesis, type IA (200600), AR |
| SLC26A2 – Solute carrier family 26 (sulfate transporter) 5q31-q34 606718 | Encodes sulfate transporter essential for normal collagen synthesis | Achondrogenesis, type IB (600972), AR; Atelosteogenesis II (256050), AR; Diastrophic dysplasia (222600), AR |
| COL2A1 – Collagen, type II, alpha 1 12q13.11 120140 | Subunit of collagen type II, the major collagen of cartilage comprised of three 3-alpha 1(II) chains | Achondrogenesis, type II, (200610), AD; Spondyloepiphyseal dysplasia congenita (183900), AD; Spondylometaphyseal dysplasia (184252), AD |
| FLNB – Filamin B 3p14.3 603381 | Protein that influences vertebral segmentation, endochondral ossification, joint formation | Atelosteogenesis I (108720), AR; Atelosteogenesis III (108721), AR |
| FGFR1 – Fibroblast growth factor receptor 1 8p11.2-p11.1 136350 | Transmembrane tyrosine kinase receptor for FGFs | Pfeiffer (101600), AD |
| FGFR2 – Fibroblast growth factor receptor 2 10q25.3-q26 176943 | Transmembrane tyrosine kinase receptor for FGFs | Apert (101200), AD Crouzon (123500), AD Jackson-Weiss (123150), AD; Antley-Bixler with normal steroidogenesis(207410), AD |
| FGFR3 – Fibroblast growth factor receptor 3 4p16.3 134934 | Transmembrane tyrosine kinase receptor for FGFs | Achondroplasia (100800), AD Hypochondroplasia (146100), AD Thanatophoric dysplasia types I (187600) &II (187601), AD |
| COMP – Cartilage oligomeric matrix protein 19p13.1 600310 | Chondrocyte protein that binds calcium &collagen types I, II, &IX | Pseudoachondroplasia (177170), AD Multiple epiphyseal dysplasia (132400), AD |
| SHOX – Short stature homeobox Xpter-p22.32 312865 | Homeobox gene transcription factor located on the pseudoautosomal region of Yp | Leri-Weill dyschondrosteosis (127300), X-linked dominant; Langer dysplasia (249700), biallelic |
| SOX9 – SRY-box 9 17q23 608160 | Transcription factor essential for chondrogenesis &testicular differentiation | Campomelic dysplasia (114290), AD |
| RUNX2 – Runt-related transcription factor 2 6p21 600211 | Osteoblast specific transcription factor | Cleidocranial dysostosis (119600), AD |
| PTHR1 – PTH receptor 1 3p22-p21.1 168468 | GPCR recognizing PTH &PTHrP with equal affinity | Inactivating mutations lead to Blomstrand osteochondrodysplasia &hypocalcemia (215045), AR; activating mutations result in hypercalcemia &Murk-Jansen metaphyseal chondrodysplasia (156400); Eiken syndrome (600002); AD |
| PRKAR1A – Protein kinase, cAMP-dependent regulatory, type 1, alpha 17q23-q24 188830 | Component of PKA response to cyclic AMP that leads to cascade of intracellular signal transduction signals in response to Gαs that regulate cell division, differentiation, metabolism, apoptosis | Gain of function mutation leads to Acrodysostosis &peripheral resistance to the biologic effects of PTHrP, PTH &TSH; (de novo) |
| HSPG2 – Heparan sulfate proteoglycan of basement membrane 1p36.1-p34 142461 | Stabilizes basement membranes &influences permeability; co-receptor for FGFR2 | Inactivating mutations result in Schwartz-Jampel type 1 myotonic chondrodystrophy (255800), AR |
| POR – Cytochrome P450 oxidoreductase 7q11.2 124015 | Flavoprotein co-factor that donates electrons to microsomal 17α-hydroxylase, 21-hydroxylase, &aromatase | Inactivating mutations lead to congenital adrenal hyperplasia usually with genital ambiguity in both 46XX &46XY subjects &at times in association with skeletal abnormalities (Antley-Bixler syndrome – craniosynostosis, mid-face hypoplasia, choanal stenosis, femoral bowing, radioulnar synostosis – 201750); AR |
| PAPSS2 – 3-Prime-phosphoadenosine 5-prime-phosphosulfate synthase2 10q24 603005 | Enzyme that synthesizes the sulfate donor (3’-phosphoadenosine 5’-phosphosulfate) from ATP &sulfate that is the co-factor for adrenocortical sulfotransferase | Spondyloepimetaphyseal dysplasia (Pakistani type) (612847) |
| PTPN11 – Protein tyrosine phosphatase, non-receptor, type 11 12q24.1 176876 | Encodes SHP2 – a non-receptor protein tyrosine phosphatase that acts upstream of RAS | Monoallelic loss of function mutations lead to metachondromatosis (MIM 156250) or Noonan syndrome with multiple lentigenes; monoallelic gain of function mutations result in Noonan syndrome; cranio-facio-cutaneous syndrome |
References
[1] | Root A.W. , Disorders of bone mineral metabolism: Normal homeostasis. In Sperling M.A. (ed). Pediatric Endocrinology, 3rd ed, Saunders/Elsevier. Philadelphia, (2008) , pp. 74–126. |
[2] | Root A.W. , Diamond F.B. Jr , Disorders of mineral homeostasis in the newborn, infant, child, and adolescent. In Sperling M.A. (ed). Pediatric Endocrinology, 3rd ed, Saunders/Elsevier, Philadelphia, (2008) , pp. 686–769. |
[3] | Levine M.A. , Investigation and management of hypocalcemia. Meet-the-Professor, Meeting of the Endocrine Society ((2010) ), 81–86. |
[4] | Magno A.L. , Ward B.K. and Ratajczak T. , The calcium-sensing receptor: A molecular perspective, Endocrine Reviews 32: ((2011) ), 3–30. |
[5] | Holick M.F. and Vitamin D. , Evolutionary, physiological and health perspectives, Curr Drug Targets 12: ((2011) ), 4–18. |
[6] | Rosen C.J. , Clinical practice. Vitamin D insufficiency, N Engl J Med 364: ((2011) ), 248–254. |
[7] | Schlingmann K.P. , Kaufmann M. , Weber S. , et al., Mutations in CYP24A1 and idiopathic infantile hypercalcemia, N Engl J Med 365: ((2011) ), 410–425. |
[8] | Farrow E.G. and White K.E. , Recent advances in renal phosphate handling, Nature Rev/Nephrol 6: ((2010) ), 207–217. |
[9] | Prie D. and Friedlander G. , Genetic disorders of renal phosphate transport, N Engl J Med 362: ((2010) ), 2399–2409. |
[10] | Shimada T. , Muto T. , Urakawa I. , et al., Mutant FGF-23 responsible for autosomal dominant hypophosphatemic rickets is resistant to proteolytic cleavage and causes hypophosphatemia in vivo, Endocrinology 143: ((2002) ), 3179–3182. |
[11] | Galitzer H. , Ben-Dov I. , Lavi-Moshayoff V. , et al., Fibroblast growth factor 23 acts on the parathyroid to decrease parathyroid hormone secretion, Curr Opin Nephrol Hypertens 17: ((2008) ), 363–367. |
[12] | Hori M. , Shimuzu Y. and Fukumoto S. , Minireview: Fibroblast growth factor 23 in phosphate homeostasis and bone metabolism, Endocrinology 152: ((2011) ), 4–10. |
[13] | Chefetz I. , Heller R. , Galli-Tsinopoulou A. , et al., A novel homozygous mutation in FGF23 causes familial tumoral calcinosis associated with disseminated visceral calcification, Hum Genet 118: ((2005) ), 261–266. |
[14] | Ichikawa S. , Baujat G. , Seyahi A. , et al., Clinical variability of familial tumoral calcinosis caused by novel GALNT3 mutations, Am J Med Genet 152A: ((2010) ), 896–903. |
[15] | Ichikawa S. , Imel E.A. , Kreiter M.L. , et al., A homozygous mutation in human KLOTHO causes severe tumoral calcinosis, J Clin Invest ((2007) ), 117-2684–2691. |
[16] | Bishop N. , Characterizing and treating osteogenesis imperfecta, Early Hum Devel 86: ((2010) ), 743–746. |
[17] | Krause C. , de Gorter D.J.J. , Karperien M. and ten Dijke P., Signal transduction cascades controlling osteoblast differentiation. In Rosen C.J. (ed). Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism, 7th ed, American Society for Bone and Mineral Research, (2008) , pp. 10–16. |
[18] | Zhou X. , Zhang Z. , Feng J.Q. , et al., Multiple functions of Osterix are required for bone growth and homeostasis in postnatal mice, PNAS 107: ((2010) ), 12919–12924. |
[19] | Bonewald L. , The holy grail of high bone mass, Nature Med 17: ((2011) ), 657–658. |
[20] | Glowacki J. , The deceiving appearances of osteoclasts, N Engl J Med 360: ((2009) ), 80–82. |
[21] | Ross F.P. , Osteoclast biology and bone resorption. In Rosen C.J. (ed). Primer on the Metabolic Bone Diseasesand Disorders of Mineral Metabolism, 7th ed, American Society for Bone and Mineral Research, (2008) , pp. 16–22. |
[22] | Duncan E.L. and Brown M.A. , Genetic determinants of bone density and fracture risk – State of the art and future directions, J Clin Endocrinol Metab 95: ((2010) ), 2576–2587. |
[23] | Kubota T. , Michigami T. and Ozono K. , Wnt signaling in bone metabolism, J Bone Miner Metab 27: ((2009) ), 265–271. |
[24] | Bliziotes M. , Update in serotonin and bone, J Clin Endocrinol Metab 95: ((2010) ), 4124–4132. |
[25] | Karsenty G. and Yadav V.K. , Regulation of bone mass by serotonin: Molecular biology and theraeutic implications, Ann Rev Med 62: ((2011) ), 323–331 . |
[26] | Yadav V.K. , Ryu J.H. , Suda N. , et al., Lrp5 controls bone formation by inhibiting serotonin synthesis in the duodenum, Cell 135: ((2008) ), 825–837. |
[27] | Cui Y. , Niziolek P.J. , MacDonald B.T. , et al., Lrp5 functions in bone to regulate bone mass, Nature Med 17: ((2011) ), 684–691. |
[28] | Shoback D. , Hypoparathyroidism, N Engl J Med 359: ((2008) ), 391–403. |
[29] | Grigorieva I.V. , Mirczuk S. , Gaynor K.U. , et al.,Gata3-deficient mice develop parathyroid abnormalities due to dysregulation of the parathyroid-specific transcription factor Gcm2, J Clin Invest 120: ((2010) ), 2144–2155. |
[30] | Mirczuk S.M. , Bowl M.R. , Nesbit M.A. , et al., A missense Glial Cells Missing Homolog B (GCMB) mutation, Asn502His, causes autosomal dominant hypoparathyroidism, J Clin Endocrinol Metab 95: ((2010) ), 3512–3516. |
[31] | McDonald-McGinn D.M. and Sullivan K.E. , Chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/Velocardiofacial syndrome), Medicine 90: ((2011) ), 1–18. |
[32] | Kobrynski L.J. and Sullivan K.E. , Velocardiofacial syndrome, DiGeorge syndrome: The chromosome 22q11.2 deletion syndromes, Lancet 370: ((2007) ), 1443–1452. |
[33] | Robin N.H. and Shprintzen R.J. , Defining the clinical spectrum of deletion 22q11.2, J Pediatr 147: ((2005) ), 90–96. |
[34] | Yagi H. , Furutani Y. , Hamada H. , et al., Role of TBX1 in human del22q11.2 syndrome, Lancet 362: ((2003) ), 1366–1373. |
[35] | Stoller J.Z. , Huang L. , Tan C.C. , et al., Ash21 interacts with Tbx1 and is required during early embryogenesis, Exper Biol Med 235: ((2010) ), 569–576. |
[36] | Bowl M.R. , Nesbit M.A. and Harding B. , et al. An interstitial deletion-insertion involving chromosome 2p25.3 and Xq27.1 near SOX3, causes X-linked recessive hypoparathyroidism, J Clin Invest 115: ((2005) ), 2822–2831. |
[37] | Courtens W. , Wuyts W. , Poot M. , et al., Hypoparathyroidism-retardation-dysmorphism in a girl: A new variant not caused by a TBCE mutation – clinical report and review, Am J Med Genet 140: ((2006) ), 611–617. |
[38] | Hershkovitz E. , Parvari R. , Diaz G.A. and Gorodischer R. , Hypoparathyroidism, retardation-dysmorphism (HRD) syndrome – A review, J Pediatr Endocrinol Metab 17: ((2005) ), 1583–1590. |
[39] | Naguib K.K. , Gouda S.A. , Elshafey A. , et al., Sanjad-Sakati syndrome/Kenny-Caffey syndrome type 1: A study of 21 cases in Kuwait, Eastern Mediter Health J 15: ((2009) ), 345–352. |
[40] | Wan M. , Li J. , Herbst K. , et al., LRP6 mediates cAMP generation by G-protein coupled receptors through regulating the membrane targeting of Gαs, Sci Signal 4: (2011), ra15. |
[41] | Linglart A. , Menguy C. , Couvineau A. , et al., Recurrent PRKAR1A mutation in acrodysostosis with hormone resistance, N Engl J Med 364: ((2011) ), 2218–1126. |
[42] | Faiyez-Ul-Haque M. , Bin-Abbas B. , Al-Abdullatif A. , et al., Novel and recurrent mutations in the AIRE gene of autoimmune polyendocrinopathy syndrome type 1, Clin Genet 76: ((2009) ), 431–440. |
[43] | Husebye E.S. and Anderson M.S. , Autoimmune polyendocrine syndromes: Clues to type 1 diabetes pathogenesis, Immunity 32: ((2010) ), 479–487. |
[44] | Alimohammadi M. , Bjorklund P. , Hallgren A. , et al., Autoimmune polyendocrine syndrome type 1 and NALP5, a parathyroid autoantigen, N Engl J Med 358: ((2008) ), 1018–1028. |
[45] | Abramson J. , Giraud M. , Benoist C. and Mathis D. , Aire’s partners in the molecular control of immunological tolerance, Cell 140: ((2010) ), 123–135. |
[46] | Benjamin R.W. , Moats-Staats B.M. , Calikoglu’s A. , et al., Hypercalcemia in children, Pediatr Endocrinol Rev 5: ((2008) ), 778–784. |
[47] | Lietman S.A. , Germain-Lee E.L. and Levine M.A. , Hypercalcemia in children and adolescents, Curr Opin Pediatr 22: ((2010) ), 508–515. |
[48] | Egbuna O.I. and Brown E.M. , Hypercalcaemic and hypocalcaemic conditions due to calcium sensing receptor mutations, Best Pract Res Clin Rheumatol 22: ((2008) ), 129–148. |
[49] | Diaz-Thomas A. , Iyer P. , Root A. and Cannon J. , Functional characterization of calcium sensing receptors (CaR) bearing loss-of-function or gain-of-function missense mutations. (Abstract presented at the annual meeting of the Endocrine Society, Boston, MA, June (2011) ). |
[50] | Lietman S.A. , Tenenbaum-Rakover Y. , Jap T.S. , et al., A novel loss-of-function mutation, Gln459Arg, of the calcium-sensing receptor gene associated with apparent autosomal recessive inheritance of familial hypocalciuric hypercalcemia, J Clin Endocrinol Metab 94: ((2009) ), 4372–4379. |
[51] | Duchatelet S. , Ostergaard E. , Cortes D. , et al., Recessive mutations in PTHR cause contrasting skeletal dysplasia in Eiken and Blomstrand syndromes, Hum Molec Genet 14: ((2005) ), 1–5. |
[52] | Pober B.R. , Williams-Beuren syndrome, N Engl J Med 362: ((2010) ), 239–252. |
[53] | Barnett C. and Krebs J.E. , WSTF does it all: A multifunctional protein in transcription, repair, and replication, Biochem Cell Biol 89: ((2011) ), 12–23. |
[54] | Chen H. , Hewison M. and Adams J.S. , Functional characterization of heterogeneous nuclear ribonuclear protein C1/C2 in vitamin D resistance: A novel response-element binding protein, J Biol Chem 281: ((2006) ), 39114–39120. |
[55] | Bergwitz C. , Roslin N.M. , Tieder M. , et al., SLC34A3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria predict a key role for sodium-phosphate cotransporter Nap(i)-IIc in maintaining phosphate homeostasis, Am J Hum Genet 78: ((2006) ), 179–192. |
[56] | Addison W.N. , Nakano Y. , Loisel T. , et al., MEPE-ASARM peptides control extracellular matrix mineralization by binding to hydroxyapatite: An inhibition regulated by PHEX cleavage of ASARM, J Bone Miner Res 23: ((2008) ), 1638–1649. |
[57] | Addison W.N. , Masica D.L. , Gray J.J. and McKee M.D. , Phosphorylation dependent inhibition of mineralization by osteopontin ASARM peptides in regulated by PHEX cleavage, J Bone Miner Res 25: ((2010) ), 695–705. |
[58] | Martin A. , David V. , Laurence J.S. , et al., Degradation of MEPE, DMP1, and release of SIBLING ASARM-peptides (minhibins): ASARM-peptide(s) are directly responsible for defective mineralization in HYP, Endocrinology 149: ((2008) ), 1757–1772. |
[59] | Novarino G. , Weinert S. , Rickheit G. and Jentsch T.J. , Endosomal chloride-proton exchange rather than chloride conductance is crucial for renal endocytosis, Science 32: ((2010) ), 1398–1401. |
[60] | Holm T.M. , Habashi J.P. , Doyle J.J. , et al., Noncanonical TGFβ signaling contributes to aortic aneurysm progression in Marfan syndrome mice, Science 332: ((2011) ), 358–361. |
[61] | Alanay Avaygan Y. , Camacho N., et al., Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesis imperfecta, Am J Hum Genet 86: ((2010) ), 551–559. |
[62] | Van Dijk F.S. , Pals G., Van Rijn R.R., et al., Classification of osteogenesis imperfecta revisited, Eur J Med Genet 53: ((2010) ), 1–5. |
[63] | Christiansen H.E. , Schwarze U. , Pyott S.M. , et al., Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta, Am J Hum Genet 86: ((2010) ), 389–398. |
[64] | Lapunzina P. , Aglan M. , Temtany S. , et al., Identification of a frameshift mutation in Osterix in a patient with recessive osteogenesis imperfecta, Am J Hum Genet 87: ((2010) ), 110–114. |
[65] | Kelley B.P. , Malfait F. , Bonafe L. , et al., Mutations in FKBP10 cause recessive osteogenesis imperfecta and Bruck syndrome, J Bone Miner Res 26: ((2011) ), 666–672. |
[66] | Ha-Vinh R. , Alanay Y. , Banks R.A. , et al., Phenotypic and molecular characterization of Bruck syndrome (osteogenesis imperfecta with contractures of the large joints) caused by a recessive mutation in PLOD2, Am J Med Genet A 131: ((2004) ), 115–120. |
[67] | Hvry M. , Lantto J. and Myllyharju J. , Missense mutations that cause Bruck syndrome affect enzymatic activity, folding, and oligomerization of lysyl hydroxylase 2, J Biol Chem 284: ((2009) ), 30917–30924. |
[68] | Amor D.J. , Savarirayan R. , Schneider A.S. and Bankier A. , New case of Cole-Carpenter syndrome, Am J Hum Genet 92: ((2000) ), 273–274. |
[69] | Sobacchi C. , Frattini A. , Guerrini M.M. , et al., Osteoclast-poor human osteopetrosis due to mutations in the gene encoding RANKL, Nature Genet 38: ((2007) ), 960–962. |
[70] | Ta H.M. , Nguyen G.T.T. , Jun N.M. , et al., Structure-based development of a receptor activator of nuclear factor-κB ligand (RANKL) inhibitor peptide and molecular basis for osteopetrosis, Proc Natl Acad Sci 107: ((2010) ), 20281–20286. |
[71] | Doffinger R. , Smahi A. , Bessia C. , et al., X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling, Nature Genet 27: ((2001) ), 277–285. |
[72] | Pangrazio A. , Pusch M. , Caldana E. , et al., Molecular and clinical heterogeneity in CLCN7-dependent osteopetrosis: Report of 20 novel mutations, Human Mut 31: ((2010) ), E071–E1080. |
[73] | Maranda B. , Chabot G. , Decarie J.C. , et al., Clinical and cellular manifestations of OSTM1-related infantile osteopetrosis, J Bone Miner Res 23: ((2008) ), 296–300. |
[74] | Pangrazio A. , Poliani P.L. , Megarbane A. , et al., Mutations in OSTM1 (grey-lethal) define a particularly severe form of autosomal recessive osteopetrosis with neural involvement, J Bone Miner Res 21: ((2006) ), 1098–1105. |
[75] | Gensure R.C. , Makitie O. , Barclay C. , et al., A novel COL1A1 mutation in infantile cortical hyperostosis (Caffey disease) expands the spectrum of collagen-related disorders, J Clin Invest 115: ((2005) ), 1250–1257. |
[76] | Hershkovitz D. , Gross Y. , Nahum S. , et al., Functional characterization of SAMD9, a protein deficient in normophosphatemic familial calcinosis, J Invest Dermatol 131: ((2011) ), 662–669. |
[77] | Mavrogenis A.F. and Soucacos P.N. , Papagelopoulos PJ. Heterotopic ossification revisted, Orthopedics 34: ((2011) ), 177. |
[78] | Trigui M. , Acadi K. , Zribi M. , et al., Fibrodysplasia ossificans progressiva: Diagnosis and surgical management, Acta Orthopaed Belg 77: ((2011) ), 139–144. |
[79] | Kaplan F.S. , Seemann P. , Haupt J. , et al., Investigations of activated ACVR1/ALK2, a bone morphogenetic type 1 receptor, that causes fibrodysplasia ossificans progressiva, Methods Enzymol 484: ((2010) ), 357–373. |
[80] | Alanay Y. , Rimoin D.L. , Chondrodysplasias. In Rosen C.J. , (ed). Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism, 7th ed, American Society for Bone and Mineral Research, (2008) , pp. 428–429. |

