
Ligand-induced structural changes analysis of ribose-binding protein as studied by molecular dynamics simulations
Abstract
BACKGROUND:
The ribose-binding protein (RBP) from Escherichia coli is one of the representative structures of periplasmic binding proteins. Binding of ribose at the cleft between two domains causes a conformational change corresponding to a closure of two domains around the ligand. The RBP has been crystallized in the open and closed conformations.
OBJECTIVE:
With the complex trajectory as a control, our goal was to study the conformation changes induced by the detachment of the ligand, and the results have been revealed from two computational tools, MD simulations and elastic network models.
METHODS:
Molecular dynamics (MD) simulations were performed to study the conformation changes of RBP starting from the open-apo, closed-holo and closed-apo conformations.
RESULTS:
The evolution of the domain opening angle
CONCLUSIONS:
The motions showed a strong one-to-one correspondence with the slowest modes from our previous study of RBP with the anisotropic network model (ANM). The results obtained for RBP contribute to the generalization of robustness for protein domain motion studies using either the ANM or PCA for trajectories obtained from MD.
1.Introduction
Bacterial periplasmic binding proteins (PBPs) are well-known receptors that undergo significant changes in conformation during ligand binding. These proteins convey a variety of basic processes such as ligand transport, chemotax and quorum sensing. These changes allow the entry and exit of ligands and allow ligand bound proteins to interact properly with the membrane components per system. PBP have been identified as a variety of ligands, including ions, vitamins, amino acid sand carbohydrates. PBPs include two
RBP is one of the representative structures of the bacterial periplasmic binding proteins. RBP consists of open and closed forms in the solution. Without ligands, the open form is dominant and they adopt a number of conformations that are demonstrated by different open structures. The ribose binding to the gap between the two domains can cause conformation changes, which corresponds to the closure of the two domains surrounding the ligand. The open and the closed conformations play an important role in chemotaxis and transport, which have different protein binding surfaces. The membrane-bound receptors can only recognize the closed conformation. The connection therefore functions as a switch between the inactive open form and the active closed form.
RBP has been widely investigated by structural analysis and computational studies. RBP was chosen to study function of the large-scale site-directed mutagenesis for protein stability by Vercillo et al. [16]. Small-angle X-ray scattering (SAXS) experiments indicated that RBP from gram negative bacteria and two related maltose and glucose binding proteins were monomers with reduced radius of gyration (Rg) to ligand binding [17]. Matthew et al. reported the crystal structure of a Thermoanaerobacter tengcongensis ribose-binding protein (tteRBP) with 1.9 Å resolution by X-ray experiments and they found that the tteRBP is significantly more stable than the ecRBP [19]. Mowbray et al. compared the structures and conformational changes of the RBP and two repressors, PurR and LacI. The opening and closing mechanism of RBP was examined by the method of umbrella sampling molecular dynamics [4].
Various forms of RBP have been found, and the conforming pathways connecting these forms are known when ligands are recovered and released into the membrane-bound permease system. In this study, we performed MD simulations to investigate the conformation changes of RBP starting from the open-apo, closed-holo and closed-apo conformations, respectively. As a control from the complex trajectory, our goal was to investigate the conformation changes induced by detachment of the ligand and the results have been revealed from two computational methods, MD simulations and elastic network models.
2.Materials and methods
The crystal structures of RBP with 271 residues (open apo structure PDB ID:1URP; closed holo structure PDB ID: 2DRI) were selected from the Protein Data Bank (see Fig. 1). By removing the binding ribose from the ligand binding structure (2DRI), a closed apo state structure was achieved. Three simulations have been carried out. Two simulations have been started from the open-apo and closed-holo conformations, respectively. One simulation started from the closed apo state structure. The RBP structures were solvated in a rectangular box with the minimum solute-box boundary distance being set to 1.2 nm. The water box is the same size in all simulations. The water keep equilibrated and one sodium ions were added to produce a neutral simulation system. By reducing the restraining force on all protein atoms (1000, 800, 600, 400, 200, 0 kJ/mol Nm
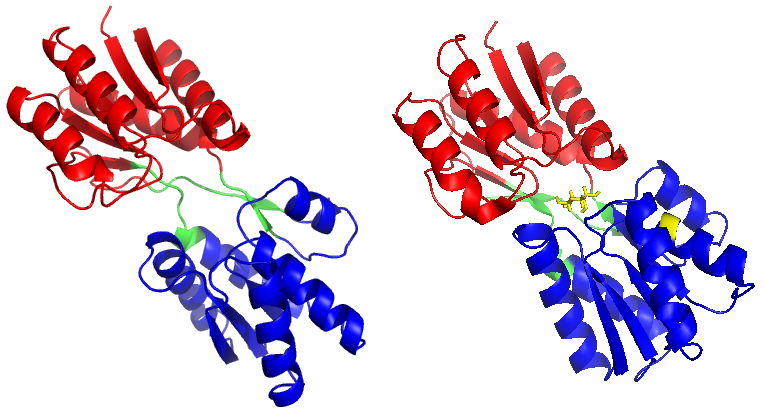
Figure 1.
(a) The X-ray crystal structure of RBP in the open-apo state (PDB code: 1URP) and (b) the closed-holostate (PDB code: 2DRI). Ribose is marked in stick representation and coloured in yellow. The C-domain of RBP is marked in red, the N-domain of RBP is marked in blue, and the hinge segments is marked in green.
All simulations were performed with GROMACS v4.5.5 [20]. The protein and the ribose were performed using the CHARMM27 force field, with TIP3P water. The simulations were represented in an NPT ensemble with a Berendsen thermostat [21] and a coupling constant of 0.1 ps at 300 K. Protein and water/ions were coupled independently. Pressure coupling used the Berendsen barostat with a coupling constant of 0.5 ps and a compressibility of 4.510-5bar-1. The long-range electrostatic interaction was treated with the particle-mesh Ewald method [22] with a 0.9 nm cut-off. The time step for the MD integrator was set to 2 fs and LINCS [23] was applied to constrain all bond lengths. Each system was subjected to three independent 50 ns simulations and the coordinates were recorded every 2 ps.
3.Results and discussion
3.1Molecular dynamics
RBPs have been established by X-ray crystallography using the data in PDB: 1URP of the open form and 2DIR of the closed form. The structures is different according to the 43
Figure 2.
Distribution of domain opening
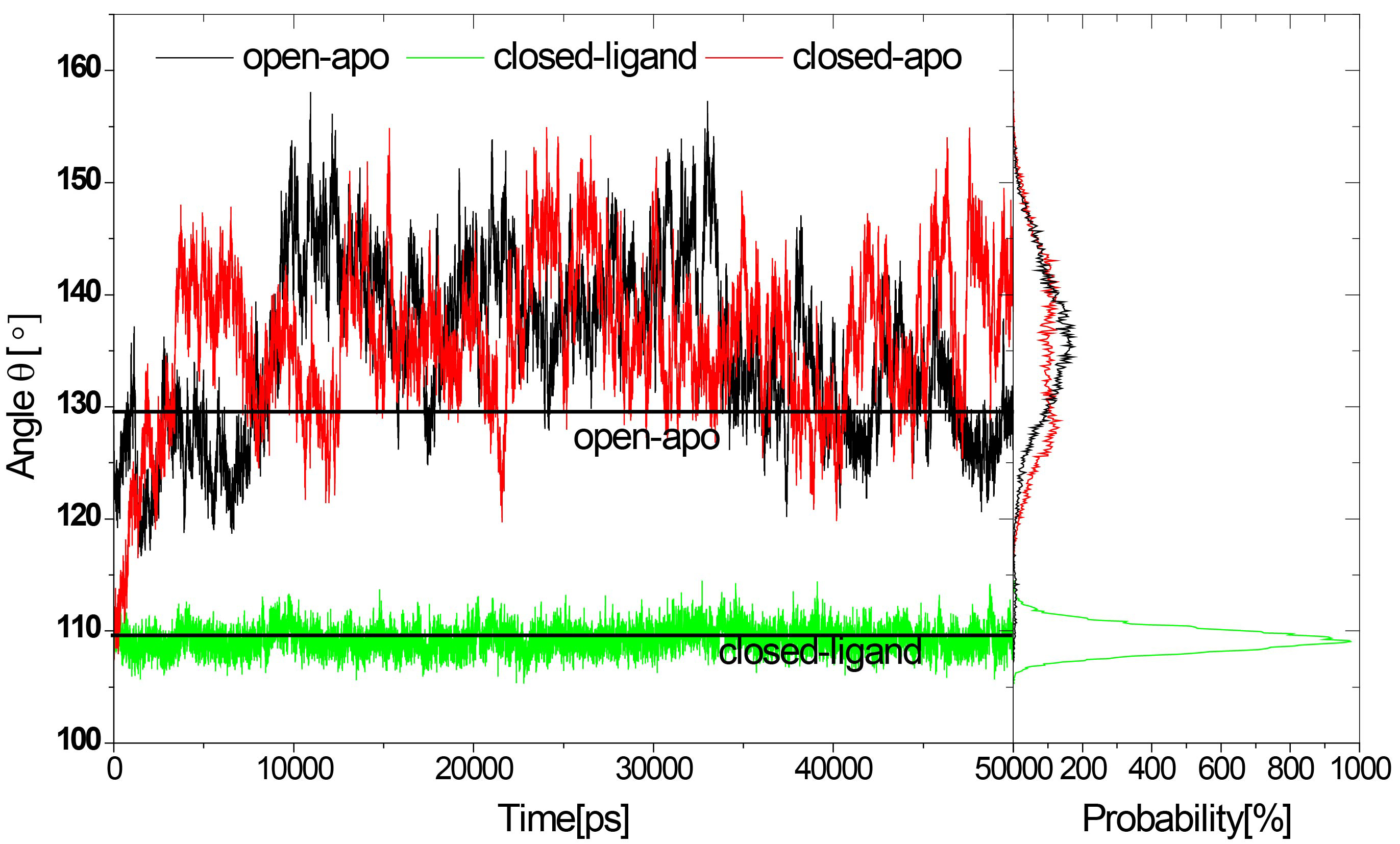
Small-angle X-ray scattering experiments (SAXS) showed that Rg of RBP decreased in solution by 1.4 (
In our previous study on the RBP with elastic network model, we found that the change in RBP configuration can be described as an inflexible rotation between two domains, but the internal structure of these two domains is basically unchanged. We investigated the root mean-square deviation (RMSD) of the N-domain (residues 108–231, and residues 269–271) and the C-domain (residues 1–100, and residues 236–259) respectively and analyzed their stability (see Figs 3 and 4).
Figure 3.
RMSD of the Ca atoms from the crystal structures for the three systems as a function of time.
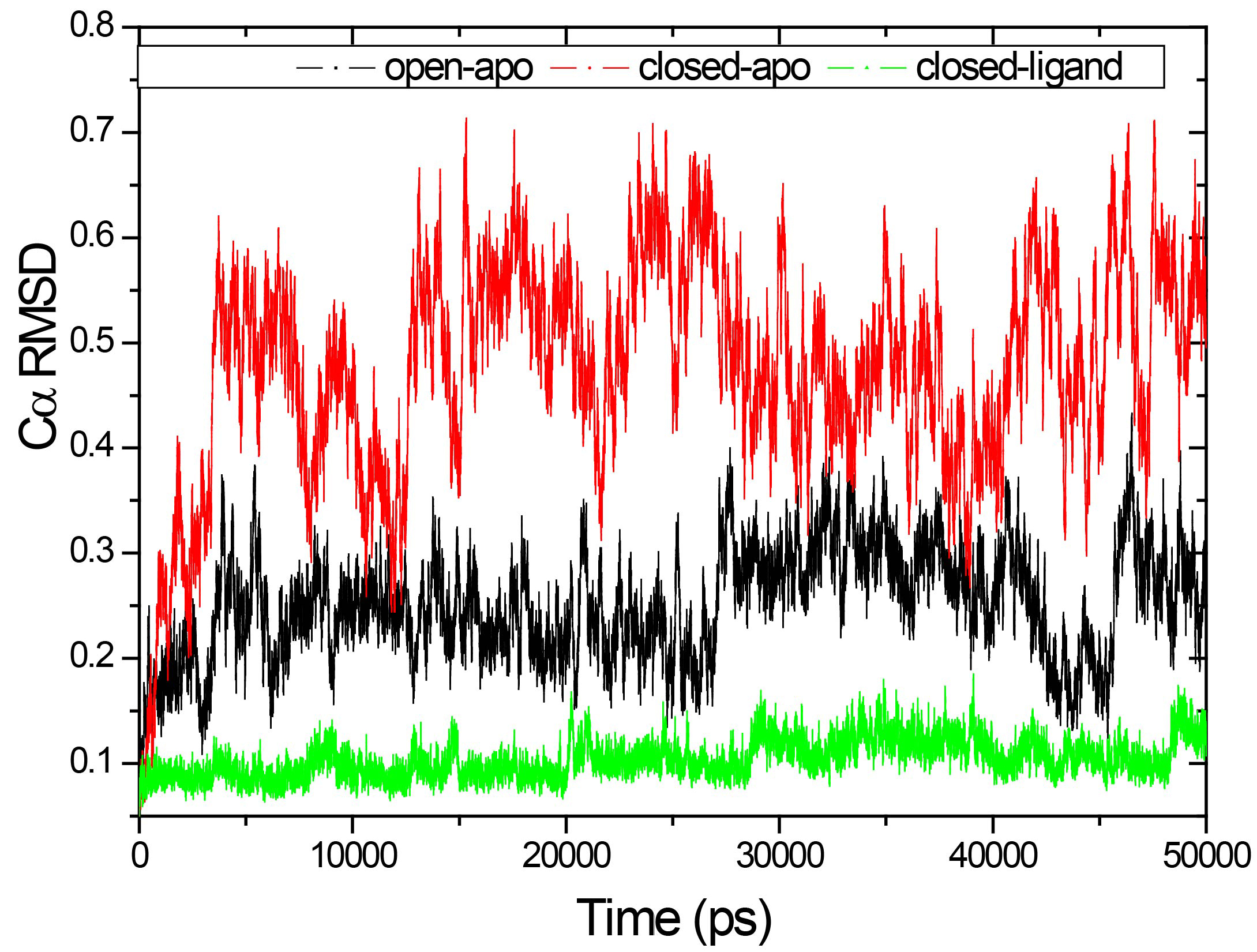
Figure 4.
Structural stability of each domain individually: the RMSD of the Ca atoms from the starting structure of the N-terminal domain (a) and C-terminal domain (b).
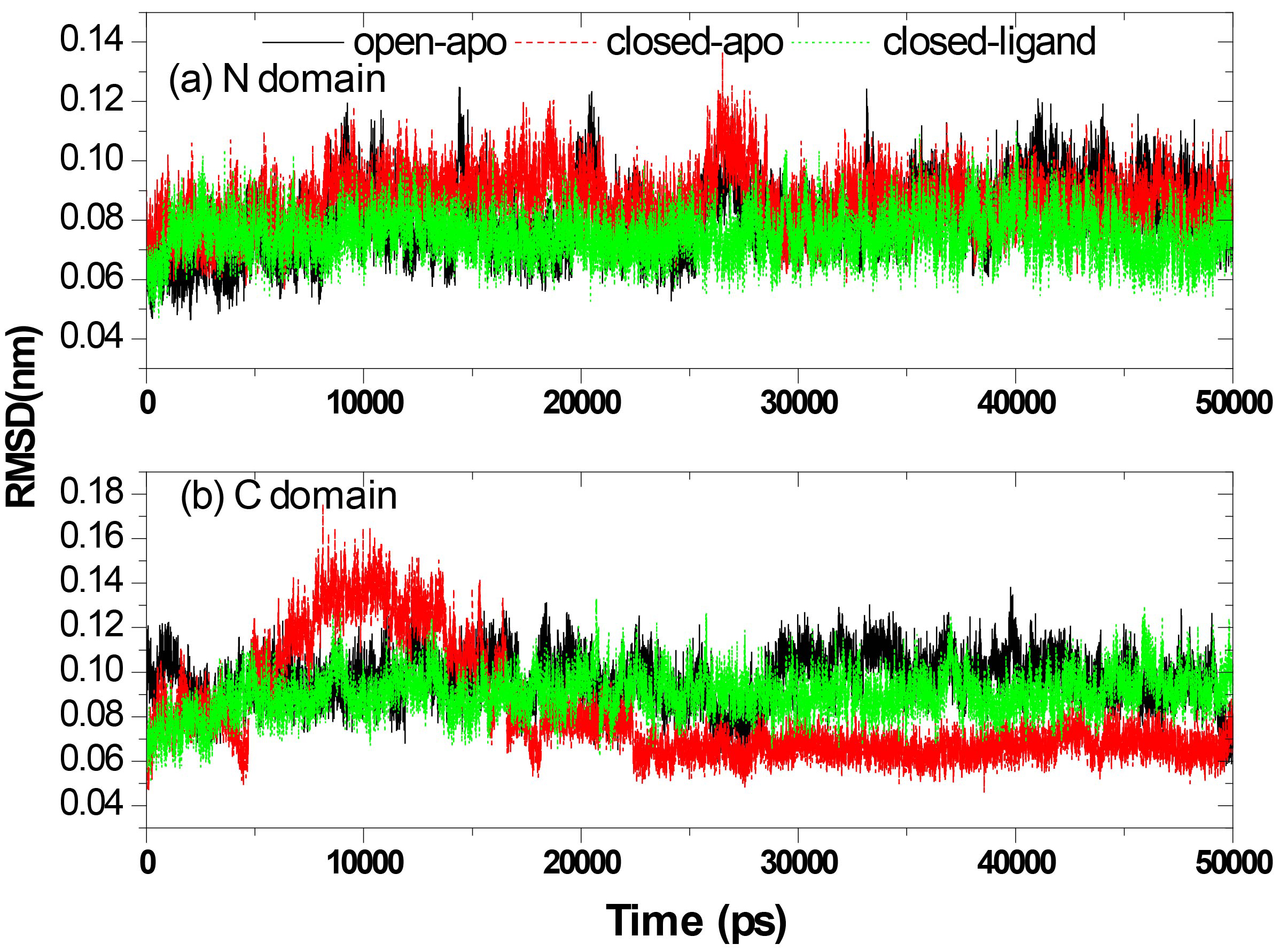
Figure 3 shows the RMSD of the Ca atoms from the crystal structures for the three systems as a function of time. The results correspond to the results of the above calculation of gyration radius. The RMSD values are fairly stable in the closed-ligand and open-apo simulations (the average values and the standard deviation of the former are1.039 and 0.1795 Å and those of the latter are 2.508 and 0.517 Å). Since the beginning of the closed-apo conformation, there have been major changes in the simulation process (mean and SD were 4.760 and 1.022 Å respectively), and the RMSD-value quickly reached a relative equilibrium after approximately 4 ns of the simulation period. In Fig. 4, the RMSD values of both the C domain (Fig. 4A, between 0.459 to 1.756 Å in three different simulations) and the N-domain (Fig. 4B, between 0.47 to 1.365 Å in three different simulations) are smaller than that of the whole structure. This means that both C and N domains are more stable than the entire conformation. The results are consistent with our previous study by elastic network model. Further comparing the two domains of in the same simulation, it can be found that RMSD values of the N domain are small and relatively stable than the C domain. Especially in the closed-ligand simulations, in the C domain a clear fluctuation appears beginning from 5 ns to 18 ns, and the fluctuation range reaches 1.756 Å. This means that the movement between domains can mainly come from the flexibility of C domains.
The total mobility of a single amino acid at each trajectory is characterized by means of the root mean-square fluctuation (RMSF) curve of the alpha-carbon atoms, as shown in Fig. 5. Due to the binding of ligand, most residues in the simulation of closed ligand fluctuate slightly, while there is the same fluctuation trend in the three simulations starting from different conformations. The results of the RMSF analysis indicate that there were significant fluctuations around residues 44–56, 68–80, 163–179, 192–203 and 218–226 in both the open-apo and closed-apo conformations. Residues 44–56 and 68–80 in the C-domain and residues 163–179, 192–203 and 218–226 in the N-domain form several helices at the top position in each domain, respectively. The other regions around residue 12, 93 and 134 also have large fluctuations both in open-apo and the closed-apo simulations. The wild type x-ray structure showed that these regions are involved in important interdomain contacts. These regions include known major transport sites (residues 11, 12, 45, 52, 67, 72, 165 and 166) as well as locations of chemotax and transport (residues 44, 70, 73 and 134). In the closed-apo simulation, the residue 134 with high mobility was located in one
Figure 5.
Root mean square fluctuation (RMSF) profile of the Ca atoms obtained from the three simulations.
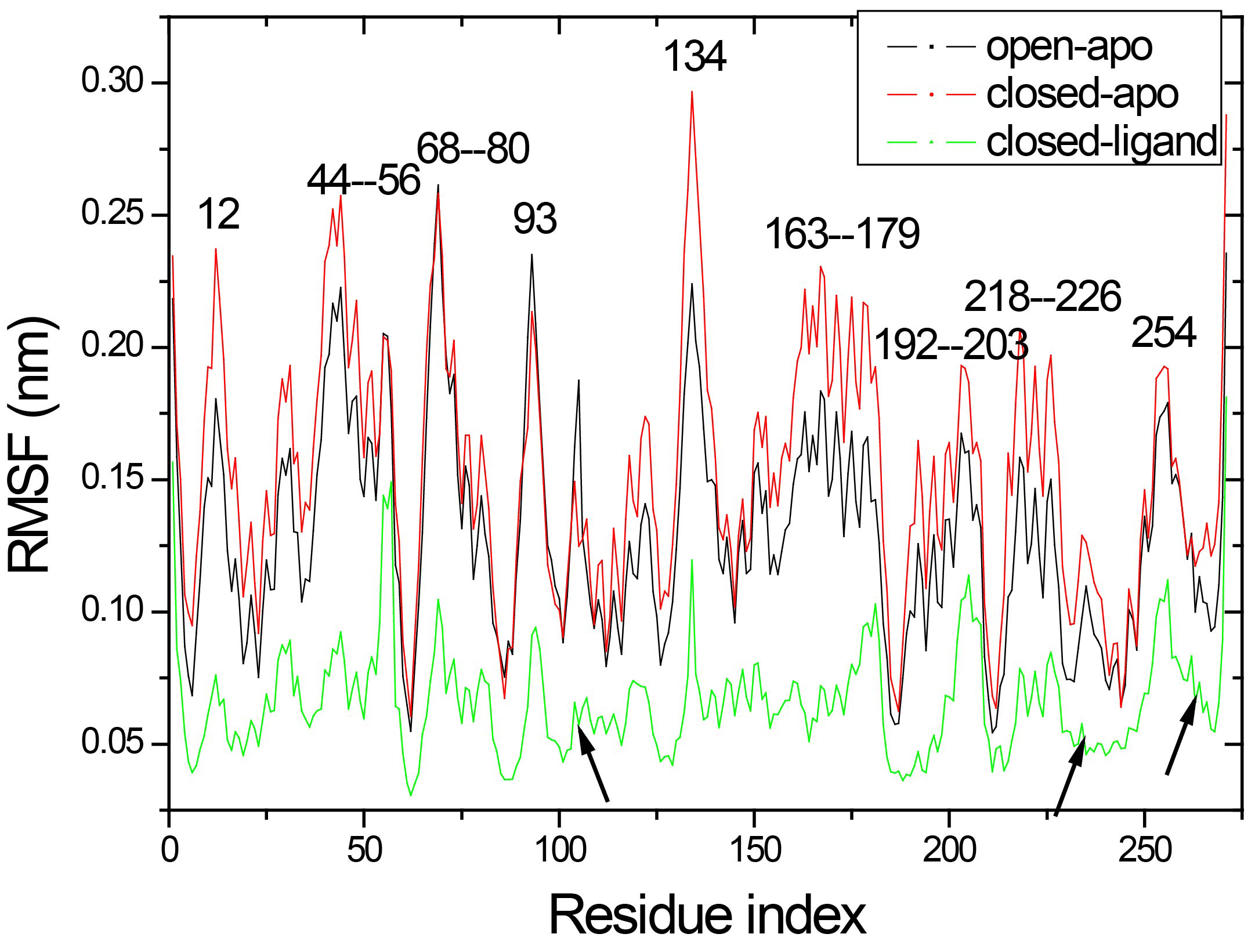
It is interesting to note that the hinge segment (residues 101–107, 232–235 and 260–268 indicate by arrow in Fig. 5) are showing a relatively rigidity in three simulations. While, comparing the fluctuation in the hinge region of three simulations, the open-apo simulation and the closed-apo simulation show greater fluctuation than the closed-ligand simulation. Obviously, the hinge region of monomer has exhibit much more flexibility than the complex. This endows the monomer to have a larger flexibility and provides the necessary conditions to make the conformational change from open to close. It is found that nineteen hydrogen bonds (analysis in Section 3.2) are formed between ribose and RBP, which are considered as stiffness of the hinge area. It also indicates that the ligand binding in the closed structure enable the binding pocket turn into more stable than in the open-apo form and this directly decreases the flexibility of the hinge region.
3.2Hydrogen bonds and hydrophobic contacts
In order to describe more comprehensively the difference in the consistent movement, we investigated the time-dependent hydrogen bonding patterns between RBP domains and between Ribose and RBP. Here, the geometry criterion of H-bonds is that the donor-H-acceptor angle is more than 120
Table 1
Interdomain H-bonds in the three simulations
| Closed-ligand | Occurrence probability | Closed-apo | Occurrence probability | Open-apo | Occurrence probability |
|---|---|---|---|---|---|
| O | 15.2% | O | 3% | N | 3.5% |
| N Ser68-O Gly134 | 44.9% | N Ser136-O | 1.5% | ||
| NGln91-O | 98.7% |
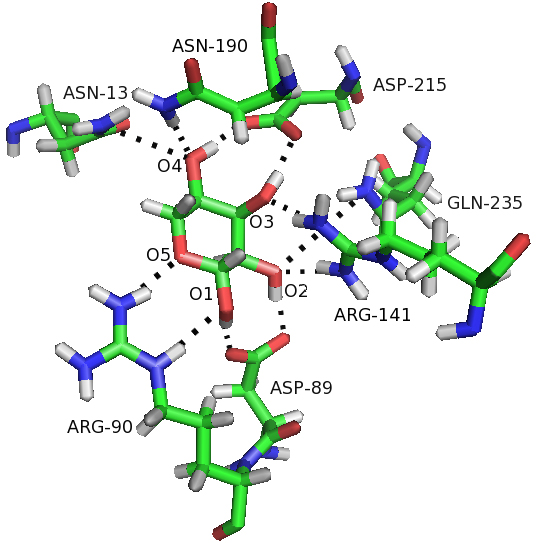
Figure 6.
Hbonds were formed between ribose and residues of RBP in the closed-ligand simulation. Hbonds are represented as broken lines.
We also examined the behavior of the interdomain H-bonds in the three simulations. The three inter-domain H-bonds (O
Figure 7.
The three interdomain H-bonds (O
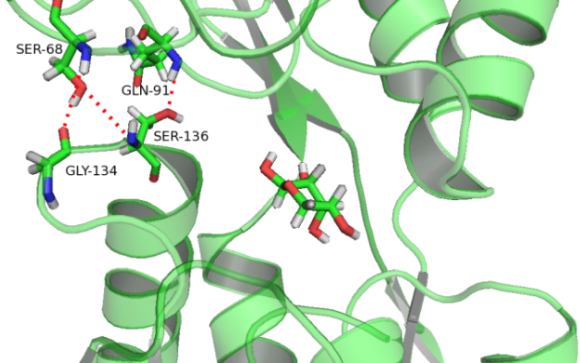
Figure 8.
The hydrophobic contact distances between the four residues and ribose all remain constant in the closed-ligand simulation.
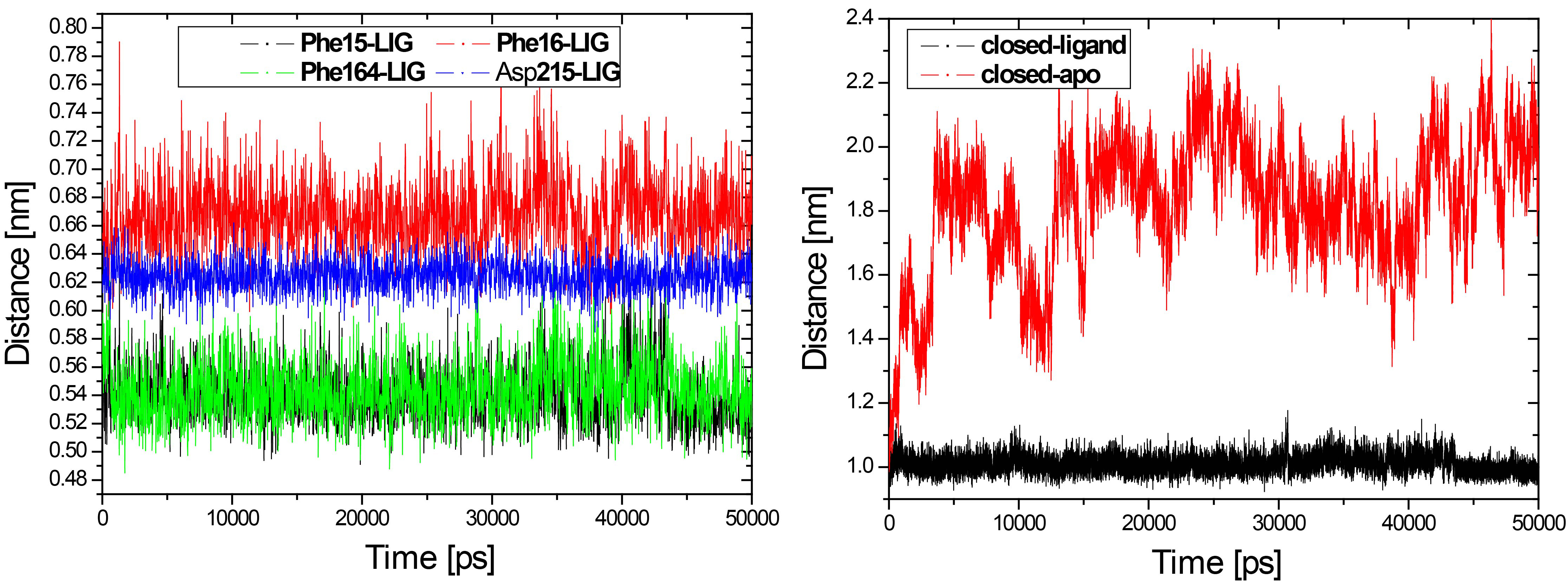
Figure 9.
The snapshots are taken from the closed-ligand simulation (left) and closed-apo simulation (right) at 30 ns.
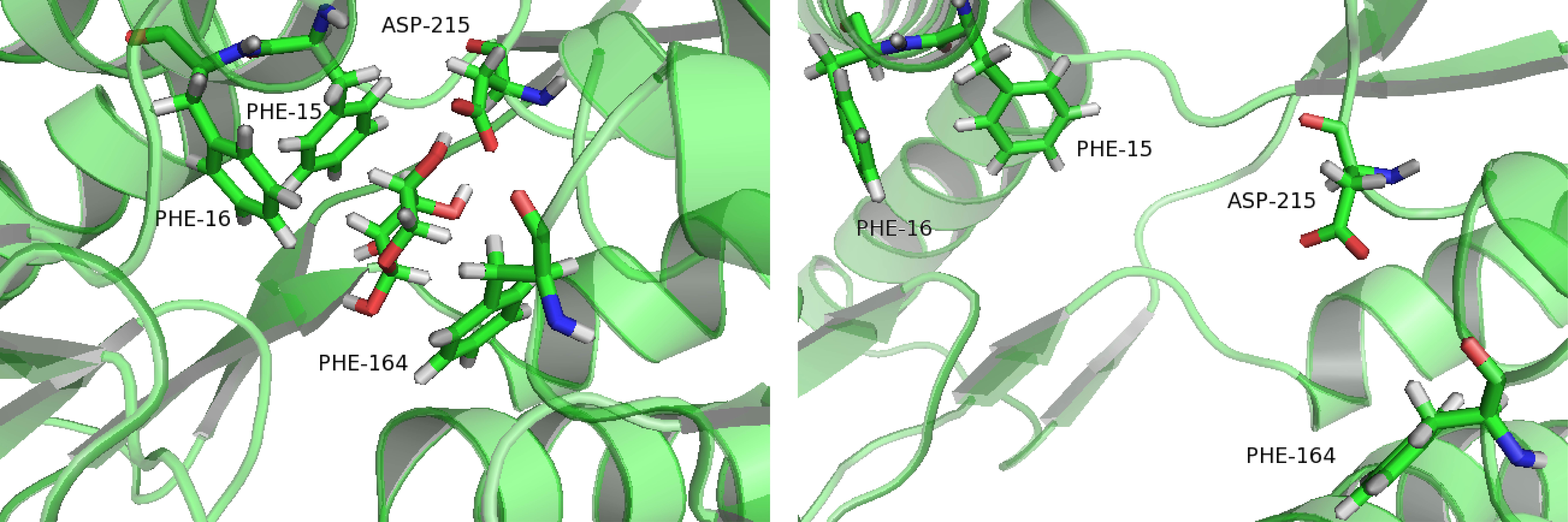
Another important interaction that stabilizes the closed conformation is the hydrophobic contacts between RBP and ribose. Four hydrophobic residues Phe15, Phe16, Phe164 and Asp215 had hydrophobic contact with ribose in the closed form of RBP. We have calculated the distance between the four residues and ribose respectively, as shown in Fig. 8. The hydrophobic contacts distance between the four residues and ribose all remain constant in the closed-ligand simulation (see Fig. 8). Especially, the distance between residue Phe15 and ribose is about 5 Å and the similar distance is found between residue Phe164 and ribose. In contrast, we have calculated the distance between residue Phe15 and Phe164 from the closed-ligand and the closed-apo simulations, respectively. It is found that the distance between the two residues in the closed-ligand simulation is very stable with a mean value of 10 Å. While, this distance increased quickly and reached at 22 Å in the closed-apo simulation.
The residues Phe15, Phe164 and ribose operates as a stable sandwich pattern (Fig. 9) that maintains the ligand-bound RBP in the closed form. This snapshot is taken from the closed-ligand simulation at 30 ns. Compare the result with the closed-apo simulation at 30 ns, it is found that the sandwich pattern was destroyed and the residues Phe15 and Phe164 were going further away with each other. It indicated that binding to ribose stabilized the closed form of RBP. There are more H-bonds and more hydrophobic contacts formed between ribose and RBP in the closed state.
3.3Essential dynamics
The essential dynamics analysis can be used to identify important molecular inner motions in a trajectory in order to understand more about large correlated protein motions and how they are critical to biological function [24]. The most important motions can be described by the largest eigenvectors associated eigenvalues. The three simulation trajectories of each structure are connected and the covariance matrixes are calculated. As shown in Fig. 10, projecting all three simulations in the same image, we estimate the similarity between our three trajectories and the overlap of sampling phase space, as the first two eigenvectors of the apo-transition trajectory.
Figure 10.
Projections of all three simulations of the apo-transition trajectory onto the first two eigenvectors.
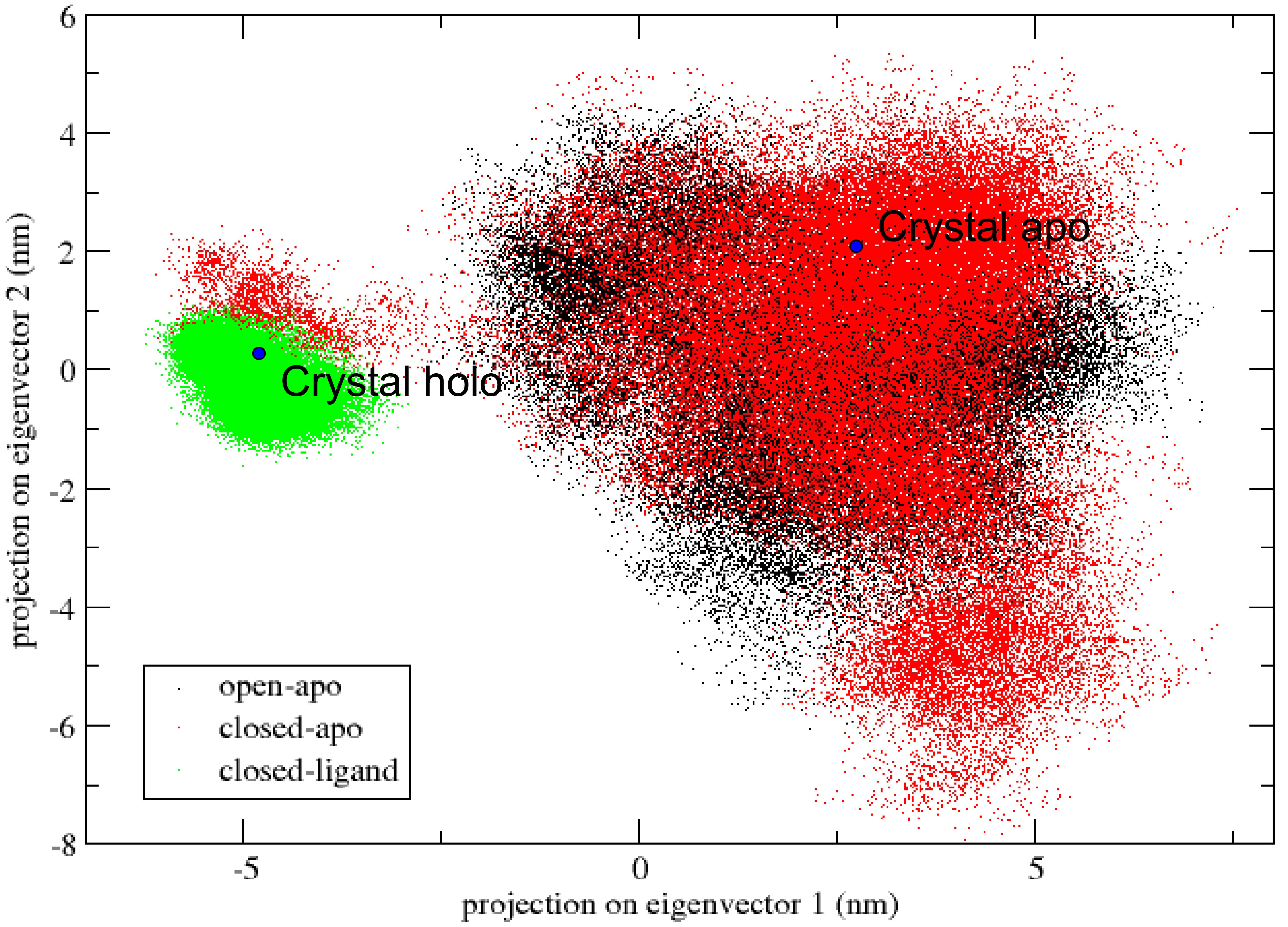
The simulated open-apo (black) and closed-ligand (green) took out the non-overlapping areas of the configuration space and remained close to their each initial structure. The transition from one state to another is obvious in the closed-apo simulation. Closed-apo simulation (red) first samples the region of closed RBP conformation. The conformation then changes during the transition to the open state and fills the overlapping region with the open-apo simulation process. The simulations indicate that the closed states in the absence of ribose are inclined to transition to the open states and the conformations of ribose-free RBP exist in a wide distribution. The results are in accord with the related study of RBP via umbrella sampling molecular dynamics [2]. As shown in the Fig. 10 is visible, and the movement associated with the first eigenvector can be described as an open movement.
Figure 11.
The movements along the first three eigenvectors for the closed-apo simulations (a) and the slowest modes of RBP by ANM (b).
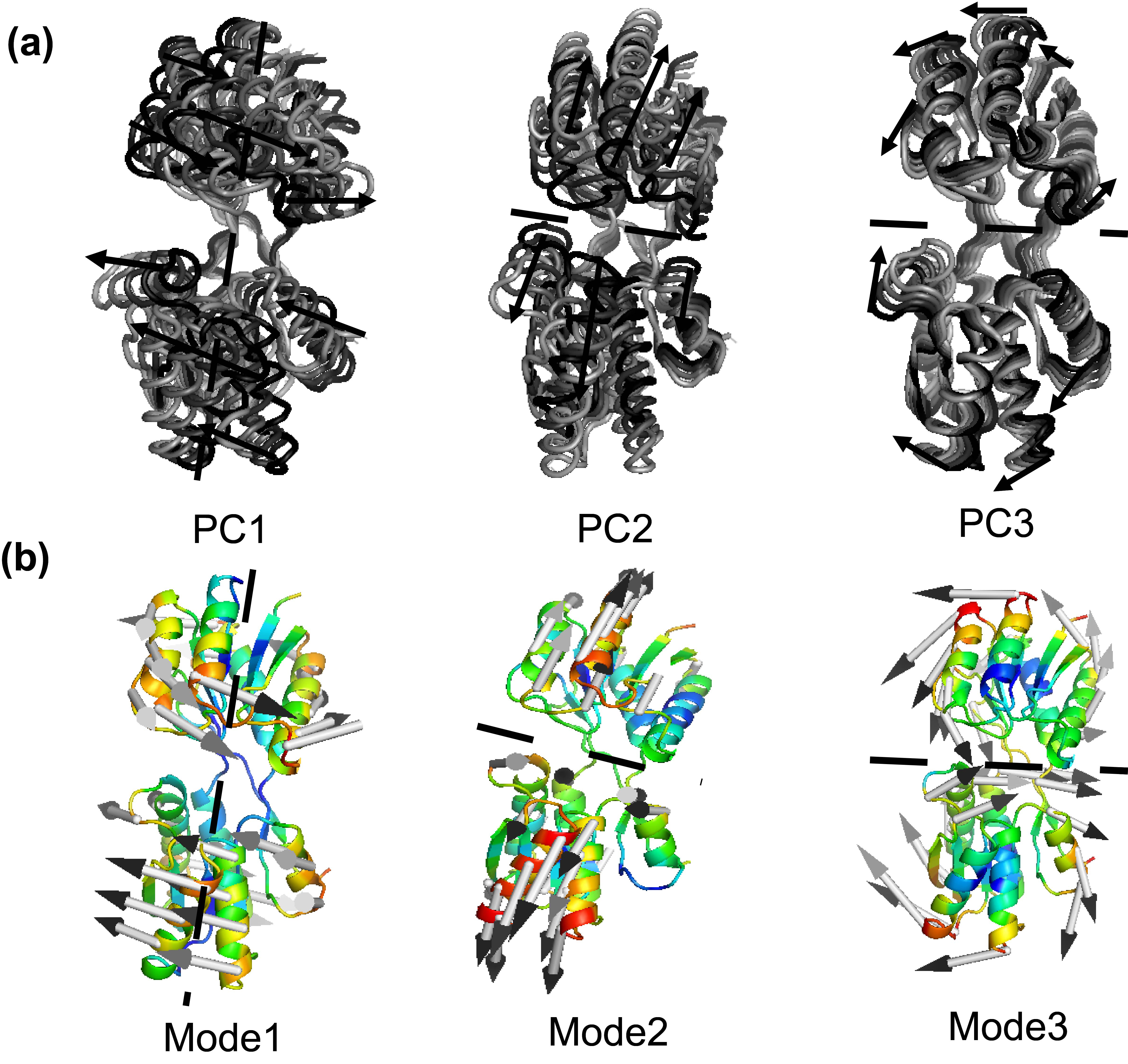
For a detailed analysis, motions belong to the first three eigenvectors for the closed-apo simulations are displayed in Fig. 11a. Significantly, it is found that the motions associated with the first three eigenvectors showed a strongly one-to-one correspondence with the slowest modes in our previous study of RBP by ANM [13, 25], as shown in Fig. 11b. PC1 of two domains (mode 1 of ANM) drives a domain continuously to rotate a roughly fixed rotation axis relative to the other. This movement can be important for the self-regulation of molecules, so that all immutable residues at the intersection and the cracking interfaces of the two domains come close and interact with the ligands. PC2 (mode 2 of ANM) drives the hinge connection and the cleft opening/closure movement. This Movement can be responsible for the binding or release of ligand ribose. These results are in accord with the previous studies of Bjorkman and Mowbray [2]. They described the open movement of RBP with rotating axis (PC1 or Mode 1) and rotating angle (PC2 or Mode 2) necessary to align two corresponding domains of closed and open configurations, and their alignment overlaps the other two domains. In PC3 (mode 3 of ANM) two domains make a twisting motion with each other. One domain moves left/right, the other one moves right/left, as shown in Fig. 11 (PC3 or Mode 3). It is speculated that the ligand binding protein will first interact with the membrane-bound permease complex. The opening of the binding protein coupled with ligand is transferred to the membrane components. The first three major movements that emerge from the closed-apo trajectories are rotation, bending and torsion, which explain the large-scale redesign of domains from ligand-bound (closed) to ligand-free (open) conformations. The results we obtain from the RBP contribute to the generalization of robust for the study of protein domain motions, using either ANM or PCA of trajectories from the MD.
4.Conclusions
Ligand-induced hinge bending movement is an important feature of the periplasmic binding protein superfamily. In this work, the ribose-binding protein (RBP) belong to Escherichia coli was subjected to an analysis of this class of conformational changes based on molecular dynamics simulations starting from the open-apo, closed-holo and closed-apo conformations, respectively. The closed-apo RBP and closed-holo RBP were in the same original closed state. However, closed-apo RBP was in an open state after the first 4 ns, and closed-holo RBP was in a closed state during the whole 50 ns MD simulation. There are more H-bonds and more hydrophobic contacts between RBP and ribose in the closed state. This makes the closed-ligand conformation more stable than the open-apo and closed-apo conformations. The first three major movements that emerge from the closed-apo trajectories are rotation, bending and torsion, which explain the large-scale redesign of domains from ligand-bound (closed) to ligand-free (open) conformations. Significantly, it is found that the motions associated with the first three principal motions show a strongly one-to-one correspondence with the slowest modes in our previous study of RBP by the Anisotropic Network Model (ANM). The results we obtained from the RBP contribute to the generalization of robust for the study of protein domain motions, using either ANM or PCA of trajectories from the MD.
Acknowledgments
We acknowledge the support from the Youth Innovation Team Lead Education Project of Shandong Educational Committee. This work was financially supported by the Natural Science Foundation of China (nos 31670727, 62071085 and 61671107) and the Shandong Provincial Natural Science Foundation (ZR2019MA040), which is gratefully acknowledged.
Conflict of interest
None to report.
References
[1] | Bjorkman AJ, Binnie RA, Zhang H, Cole LB, Hermodson MA, and Mowbray SL. Probing protein-protein interactions. The ribose-binding protein in bacterial transport and chemotaxis. The Journal of Biological Chemistry. (1994) ; 269: : 30206-30211. |
[2] | Bjorkman AJ, and Mowbray SL. Multiple open forms of ribose-binding protein trace the path of its conformational change. Journal of Molecular Biology. (1998) ; 279: : 651-664. |
[3] | Gerstein M, Lesk AM, and Chothia C. Structural mechanisms for domain movements in proteins. Biochemistry. (1994) ; 33: : 6739-6749. |
[4] | Mowbray SL, and Cole LB. 1.7 A X-ray structure of the periplasmic ribose receptor from Escherichia coli. Journal of Molecular Biology. (1992) ; 225: : 155-175. |
[5] | Anderson C, Zucker F, and Steitz T. Space-filling models of kinase clefts and conformation changes. Science. (1979) ; 204: : 375-380. |
[6] | Evenas J, Tugarinov V, Skrynnikov NR, Goto NK, Muhandiram R, and Kay LE. Ligand-induced structural changes to maltodextrin-binding protein as studied by solution NMR spectroscopy. Journal of Molecular Biology. (2001) ; 309: : 961-974. |
[7] | Luck LA, and Falke JJ. Open conformation of a substrate-binding cleft: 19F NMR studies of cleft angle in the D-galactose chemosensory receptor. Biochemistry. (1991) ; 30: : 6484-6490. |
[8] | Tang C, Schwieters CD, and Clore GM. Open-to-closed transition in apo maltose-binding protein observed by paramagnetic NMR. Nature. (2007) ; 449: : 1078-1082. |
[9] | Loeffler HH, and Kitao A. Collective dynamics of periplasmic glutamine binding protein upon domain closure. Biophysical Journal. (2009) ; 97: : 2541-2549. |
[10] | Ravindranathan KP, Gallicchio E, and Levy RM. Conformational equilibria and free energy profiles for the allosteric transition of the ribose-binding protein. Journal of Molecular Biology. (2005) ; 353: : 196-210. |
[11] | Stockner T, Vogel HJ, and Tieleman DP. A salt-bridge motif involved in ligand binding and large-scale domain motions of the maltose-binding protein. Biophysical Journal. (2005) ; 89: : 3362-3371. |
[12] | Chang G, Still W, and Guida W. An internal coordinate Monte Carlo method for searching conformational space. Journal of the American Chemical Society; (USA). (1989) ; 111. |
[13] | Li HY, Cao ZX, Zhao LL, and Wang JH. Analysis of conformational motions and residue fluctuations for Escherichia coli ribose-binding protein revealed with elastic network models. International Journal of Molecular Sciences. (2013) ; 14: : 10552-10569. |
[14] | Su JG, Jiao X, Sun TG, Li CH, Chen WZ, and Wang CX. Analysis of domain movements in glutamine-binding protein with simple models. Biophysical Journal. (2007) ; 92: : 1326-1335. |
[15] | Keskin O. Comparison of full-atomic and coarse-grained models to examine the molecular fluctuations of c-AMP dependent protein kinase. Journal of Biomolecular Structure & Dynamics. (2002) ; 20: : 333-345. |
[16] | Vercillo NC, Herald KJ, Fox JM, Der BS, and Dattelbaum JD. Analysis of ligand binding to a ribose biosensor using site-directed mutagenesis and fluorescence spectroscopy. Protein science: A Publication of the Protein Society. (2007) ; 16: : 362-368. |
[17] | Shilton BH, Flocco MM, Nilsson M, and Mowbray SL. Conformational changes of three periplasmic receptors for bacterial chemotaxis and transport: the maltose-, glucose/galactose- and ribose-binding proteins. Journal of Molecular Biology. (1996) ; 264: : 350-363. |
[18] | Zhang Y, Gardina PJ, Kuebler AS, Kang HS, Christopher JA, and Manson MD. Model of maltose-binding protein/chemoreceptor complex supports intrasubunit signaling mechanism. Proceedings of the National Academy of Sciences of the United States of America. (1999) ; 96: : 939-944. |
[19] | Cuneo MJ, Beese LS, and Hellinga HW. Ligand-induced conformational changes in a thermophilic ribose-binding protein. BMC Structural Biology. (2008) ; 8: : 50. |
[20] | Pronk S, Páll S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, and van der Spoel D. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. (2013) ; 29: : 845. |
[21] | Mudi A, and Chakravarty C. Effect of the Berendsen thermostat on the dynamical properties of water. Molecular Physics. (2004) ; 102: : 681-685. |
[22] | Petersen HG. Accuracy and efficiency of the particle mesh Ewald method. The Journal of Chemical Physics. (1995) ; 103: : 3668-3679. |
[23] | Hess B, Bekker H, Berendsen H, and Fraaije J. 3 LINCS: a linear constraint solver for molecular simulations. |
[24] | David CC, and Jacobs DJ. Principal component analysis: a method for determining the essential dynamics of proteins. In: Protein dynamics Springer, (2014) , pp. 193-226. |
[25] | Atilgan AR, Durell SR, Jernigan RL, Demirel MC, Keskin O, and Bahar I. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophysical Journal. (2001) ; 80: : 505-515. |


