
Translating Metabolic Reprogramming into New Targets for Kidney Cancer
Abstract
In the age of bioinformatics and with the advent of high-powered computation over the past decade or so the landscape of biomedical research has become radically altered. Whereas a generation ago, investigators would study their “favorite” protein or gene and exhaustively catalog the role of this compound in their disease of interest, the appearance of omics has changed the face of medicine such that much of the cutting edge (and fundable!) medical research now evaluates the biology of the disease nearly in its entirety. Couple this with the realization that kidney cancer is a “metabolic disease” due to its multiple derangements in biochemical pathways [1, 2], and clear cell renal cell carcinoma (ccRCC) becomes ripe for data mining using multiple omics approaches.
BODY
It is well known even from what has already been written in the pages of this new Journal that ccRCC is a disease which lacks sufficient therapeutic options. While in the past the search for new chemotherapies in cancer (and many other diseases) has been an exercise in the brute force trial-and-error approach of new toxic therapies, and sheer luck on the part of the patient and his/her physician, we now have the ability to predict what will likely work for each patient’s disease based on genomics or specific mutational analysis (as has been the case for several years) and, more recently, on metabolomics. While mutations have been targeted in other cancers with a high degree of success, this has not been the case for ccRCC and will not be further discussed in this commentary. On the other hand, the discovery largely using omics technologies of reprogrammed metabolism as part and parcel of this metabolic disease has resulted in several pathways that do not “follow the rules” and therefore are, or could be, exploited for new therapeutic paradigms with likely far less toxicity than non-specific brute force therapies. These pathways will be discussed in some detail later in this article.
Naturally, when discovering and validating novel or reprogrammed metabolism, the technique most likely to yield new insights is that of metabolomics as this field deals with the signatures of essentially all current and measurable metabolic processes [3, 4]. As with all omics methods, the practice of metabolomics (“non-targeted” in its most useful iteration) seeks to discover all of the small molecule metabolites present in a given biological system, yet of course just a relatively small fraction of these will actually be found and chemically identified. After biological validation (for example in cultured cells or, better, in whole organisms), those metabolites which are found to be altered can be studied in the context of their upstream and downstream neighbors [5]. Once this is done using in many cases commercially available software, established and novel reprogrammed pathways can be portrayed which will lead, on inspection, to pathway convergences or “nodes” conducive to intervention. In many of these cases, on examination of published lists of small molecules which have been shown to affect myriad pathways, it can be found that these pathways have previously been studied and targeted by pharmaceuticals designed for other diseases and, in many cases, have been long-discarded [6]. For these reasons, it is fair to say that metabolomics leads to reprogramming insights which result in new therapeutic targets.
While establishing reprogrammed pathways in RCC using metabolomics is a very powerful method with high clinical yield, it is a technique with more than its share of pitfalls in the main due to the fact that, with its commercial availability, it is readily available. Because any investigator in possession of tissue and/or biofluid samples (and sufficient funding!) can in theory perform a bare-bones metabolomics study and thereby generate hypotheses, it is important before dissemination of the work for the investigator to biologically validate any findings using in vitro or in vivo systems lest the findings be later found to be merely artefactual or of little to no biological significance. Thus, the proliferation of metabolomics publications purporting to show novel pathways and biomarkers need to be evaluated with a very critical eye and an open mind.
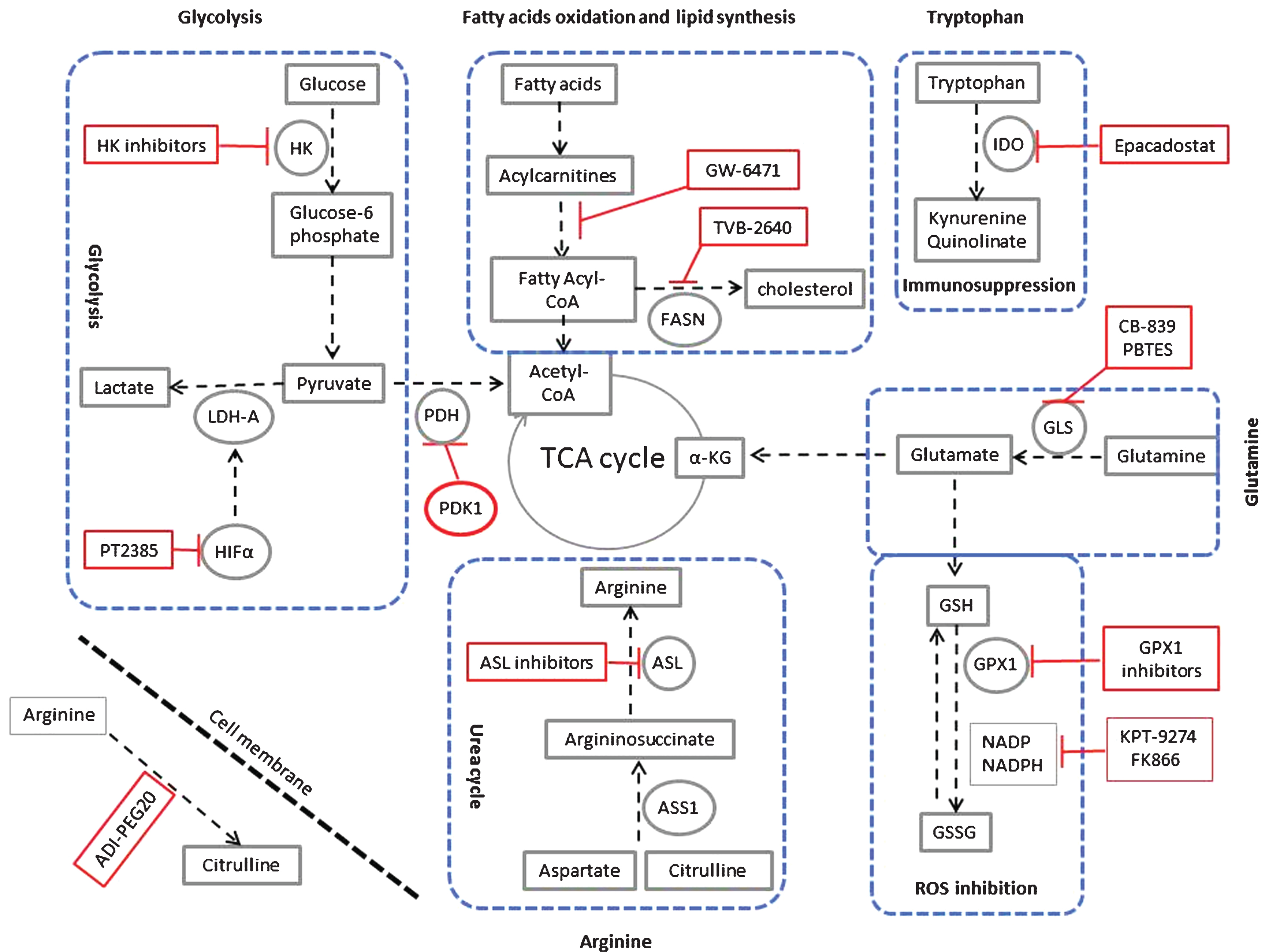
In light of the recent realization that RCC is in fact a metabolic disease, as discussed above, there have been a number of well-designed omics studies using RCC tissue and biofluids showing new and reprogrammed metabolic pathways (Fig. 1). We summarize the most relevant studies leading to possible new therapies in the following paragraphs.
Fig.1
Targeting metabolic reprogramming in ccRCC. Glycolysis can be inhibited using inhibitors for hexokinase (HK), the hypoxia-inducible factor α (HIF-α) such as PT2385, Lactate dehydrogenase (LDH-A). Glycolysis can also be inhibited by activators of pyruvate dehydrogenase kinase-1 (PDK-1), which inhibits pyruvate dehydrogenase (PDH). Fatty acid oxidation can be inhibited using peroxisome proliferator-activated receptor (PPARα) antagonist (GW-6471). Also fatty acid synthesis can be inhibited using the fatty acid synthase (FASN) inhibitor TVB-2640. In ccRCC, upregulation of tryptophan metabolism results in increased production of the immunosuppressive metabolites kynuernine and quinolinate. This pathway can be inhibited using the indoleamine 2,3-dioxygenase (IDO) inhibitor epacadostat. Glutamine metabolism can be inhibited using the glutaminase (GLS) inhibitor CB-839 and PBTES (Bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide). α – KG is α – ketoglutarate. Glutamine is also feeding the glutathione (GSH) pathway. Inhibitors of glutathione peroxidase (GPX1) and inhibitors of NADPH such as KPT-9274 and FK866 will inhibit changing GSH to GSSG. This reaction is responsible for scavenging reactive oxygen species (ROS) in the cell. Arginine is synthesized from citrulline in two steps of the urea cycle using the enzymes argininosuccinate synthase-1 (ASS1), and argininosuccinate lyase (ASL). As ASS1 levels are markedly decreased in all grades of ccRCC, the tumour cells are dependent on extracellular sources of arginine for their survival. Extracellular arginine can be depleted using the pegylated form of arginine deaminase enzyme (ADI-PEG20). ASL inhibitors can also be used for targeted therapy of ccRCC.
Glycolysis
Clear cell RCC (ccRCC) is now well established to utilize Warburg metabolism. Studies conducted on ccRCC cells and tissues from mice and patients using metabolomics [7, 8], proteomics [8–10] and transcriptomics [11] have shown a strong signature of increased glycolysis in ccRCC. Levels of enzymes involved in glycolysis such as hexokinase-1 (HK-1), pyruvate kinase (PK-2) and lactate dehydrogenase A (LDH–A) were significantly increased in ccRCC cells and tissues [9]. While the chemically modified glucose analog 2- deoxy-D-glucose (2-DG) acts as substrate to inhibit hexokinase [12], 3-bromopyruvate (3-BrPa) is believed to interfere directly with HK enzymatic activity and bind to hexokinase 2 [13]. In ccRCC, HIFα is playing a major role in activating LDH-A, which is a critical enzyme in glycolysis. Recent studies targeting HIFα using PT2385 resulted in inhibition of tumor growth in several different RCC cell lines [7] and now is being tested in clinical trials [14].
Fatty acid oxidation and lipid synthesis
Metabolomics studies in ccRCC have revealed increased utilization of fatty acids [15]. Indeed, inhibitors of fatty acids oxidation such as PPARα antagonist was tested in ccRCC models and found to inhibit tumor growth [16, 17]. In addition, fatty acid synthase (FASN) which is involved in building long chain fatty acids and cholesterol was found to be overexpressed in ccRCC and associated with tumor aggressiveness and poor prognosis in this disease [18]. Inhibition of FASN by cerulin or its derivative C75 induced a rapid increase in malonyl-coA with a marked reduction in lipogenesis in multiple cancer models, including RCC cells [19, 20].
Tryptophan
Tryptophan is an essential amino acid and is catabolized through the kynurenine pathway via the rate-limited enzyme activity of indoleamine 2,3–dioxygenase (IDO) [21]. One of the most reported effects of tryptophan metabolism is immunosuppression as a result of tryptophan depletion [22] accompanied by an increase in immunosuppressive metabolites in the kynurenine pathway [23]. In ccRCC, the level of tryptophan is reduced, suggesting upregulation of this pathway leading to immunosuppression [10]. Inhibition of IDO prevents the purported immunosuppressive effect and enables T-cell activation in a murine RCC model [24]. The selective IDO inhibitor epacadostat was tested in ccRCC and found to enhance the lytic ability of tumor-antigen-specific T cells in preclinical models [25].
Glutamine
Several independent studies have shown that glutamine utilization is increased in ccRCC compared to normal kidney tissues [10, 11, 26]. Targeting of enzymes involved in glutamine metabolism, such as glutaminase (GLS) is being tested. In a clinical trial, the small molecule inhibitor of glutaminase, CB-839, either alone or in combination with the mTOR inhibitor everolimus, showed favorable results in patients with ccRCC [27]. Furthermore glutamine in ccRCC is feeding the glutathione/oxidized glutathione pathway (GSH/GSSG), an important pathway for neutralizing ROS in cells [10, 11]. Inhibitors of NADPH synthesis such as KPT-9274 have showed decreased tumor burden in RCC mouse model [28].
Arginine
Argininosuccinate synthase-1 (ASS1) is the rate-limiting enzyme for the conversion of citrulline to arginine for ammonia detoxification through the urea cycle in the kidney [29]. In ccRCC, ASS1 is not expressed or is downregulated in tumors [30]. This finding is confirmed in all ccRCC grades in a proteomic study [9]. Consistent with this model, arginine deprivation inhibited tumor growth in the RENCA mouse model of RCC [30]. The pegylated form of arginine deaminase (ADI-PEG20) can be used to deplete arginine through deamination of arginine to citrulline [30, 31].
In summary, the intelligent use of the powerful technique of metabolomics for discovery and validation of reprogrammed metabolic pathways in RCC has thus far resulted in a plethora of new therapeutic targets for a disease in which they are seriously lacking. Given that such altered biochemical pathways are in most cases confined to the tumor and not essential for surrounding normal tissue increases the likelihood that interference with these pathways by the proposed therapeutic targets will not result in serious adverse effects. Thus, the stage is now set for clinical trials of these new compounds either alone or in combination with established therapies. While a somewhat high risk venture since in many cases the function of reprogramming in normal tissues is not known, the potential gain to the field of this approach has the potential to be revolutionary and to open up new therapeutic paradigms.
ACKNOWLEDGMENTS
This work was supported by NIH grants 1R01CA135401-01A1, 1R03CA181837-01, and 1R01DK082690-01A1, the Medical Service of the US Department of Veterans’ Affairs, and Dialysis Clinics, Inc. (DCI).
REFERENCES
[1] | Hu SL , Chang A , Perazella MA , Okusa MD , Jaimes EA , Weiss RH . The nephrologist’s tumor: Basic biology and management of renal cell carcinoma. J Am Soc Nephrol (2016) ;27: (8):2227–37. |
[2] | Wettersten HI , Aboud OA , Lara PN Jr , Weiss RH . Metabolic reprogramming in clear cell renal cell carcinoma. Nature reviews Nephrology (2017) ;13: (7):410–419. |
[3] | Weiss RH , Kim K . Metabolomics in the study of kidney diseases. Nat Rev Nephrology (2011) ;8: :22–33. |
[4] | Fan TW , Lane AN , Higashi RM . The promise of metabolomics in cancer molecular therapeutics. Curr Opin Mol Ther (2004) ;6: :584–92. |
[5] | Hwang VJ , Weiss RH . Metabolomic profiling for early cancer detection: current status and future prospects. Expert Opinion on Drug Metabolism & Toxicology (2016) :1–3. |
[6] | Held MA , Langdon CG , Platt JT , Graham-Steed T , Liu Z , Chakraborty A , et al. Genotype-selective combination therapies for melanoma identified by high-throughput drug screening. Cancer Discov (2013) ;3: :52–67. |
[7] | Chen W , Hill H , Christie A , Kim MS , Holloman E , Pavia-Jimenez A , et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature (2016) ;539: :112–7. |
[8] | Perroud B , Lee J , Valkova N , Dhirapong A , Lin PY , Fiehn O , et al. Pathway analysis of kidney cancer using proteomics and metabolic profiling. Mol Cancer (2006) ;5: :64. |
[9] | Perroud B , Ishimaru T , Borowsky AD , Weiss RH . Grade-dependent proteomics characterization of kidney cancer. Mol Cell Proteomics (2008) ;8: :971–85. |
[10] | Wettersten HI , Hakimi AA , Morin D , Bianchi C , Johnstone ME , Donohoe DR , et al. Grade-dependent metabolic reprogramming in kidney cancer revealed by combined proteomics and metabolomics analysis. Cancer Res (2015) ;75: :2541–52. |
[11] | Hakimi AA , Reznik E , Lee CH , Creighton CJ , Brannon AR , Luna A , et al. An integrated metabolic atlas of clear cellrenal cell carcinoma. Cancer Cell (2016) ;29: :104–16. |
[12] | Raez LE , Papadopoulos K , Ricart AD , Chiorean EG , Dipaola RS , Stein MN , et al. A phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemotherapy and Pharmacology (2013) ;71: :523–30. |
[13] | Chen Z , Zhang H , Lu W , Huang P . Role of mitochondria-associated hexokinase II in cancer cell death induced by 3-bromopyruvate. Biochimica et Biophysica Acta (2009) ;1787: :553–60. |
[14] | Medicine UNLo. https://clinicaltrials.gov/ct2/show/NCT02293980 (2017) . |
[15] | Ganti S , Taylor SL , Abu AO , Yang J , Evans C , Osier MV , et al. Kidney tumor biomarkers revealed by simultaneous multiple matrix metabolomics analysis. Cancer Res (2012) ;72: :3471–9. |
[16] | Abu Aboud O , Wettersten HI , Weiss RH . Inhibition of PPAR-alphainduces cell cycle arrest and apoptosis and synergizes withglycolysis inhibition in kidney cancer cells. PLoS One (2013) ;8: :e71115. |
[17] | Abu AO , Donohoe D , Bultman S , Fitch M , Riiff T , Hellerstein M , et al. PPARalpha inhibition modulates multiple reprogrammed metabolic pathways in kidney cancer and attenuates tumor growth. Am J Physiol Cell Physiol (2015) ;308: :C890–C8. |
[18] | Horiguchi A , Asano T , Asano T , Ito K , Sumitomo M , Hayakawa M . Fatty acid synthase over expression is an indicator of tumor aggressiveness and poor prognosis in renal cell carcinoma. J Urol (2008) ;180: :1137–40. |
[19] | Pizer ES , Thupari J , Han WF , Pinn ML , Chrest FJ , Frehywot GL , et al. Malonyl-coenzyme-A is a potential mediator of cytotoxicity induced by fatty-acid synthase inhibition in human breast cancer cells and xenografts. Cancer Research (2000) ;60: :213–8. |
[20] | Horiguchi A , Asano T , Asano T , Ito K , Sumitomo M , Hayakawa M . Pharmacological inhibitor of fatty acid synthase suppresses growth and invasiveness of renal cancer cells. The Journal of urology (2008) ;180: :729–36. |
[21] | Shimizu T , Nomiyama S , Hirata F , Hayaishi O . Indoleamine 2,3-dioxygenase. Purification and some properties. The Journal of Biological Chemistry (1978) ;253: :4700–6. |
[22] | Lee GK , Park HJ , Macleod M , Chandler P , Munn DH , Mellor AL . Tryptophan deprivation sensitizes activated T cells to apoptosis prior to cell division. Immunology (2002) ;107: :452–60. |
[23] | Fallarino F , Grohmann U , Vacca C , Bianchi R , Orabona C , Spreca A , et al. T cell apoptosis by tryptophan catabolism. Cell Death Differ (2002) ;9: :1069–77. |
[24] | Trott JF , Kim J , Aboud OA , Wettersten H , Stewart B , Berryhill G , et al. Inhibiting tryptophan metabolism enhances interferon therapy in kidney cancer. Oncotarget (2016) ;7: (41):66540–57. |
[25] | Jochems C , Fantini M , Fernando RI , Kwilas AR , Donahue RN , Lepone LM , et al. The IDO1 selective inhibitor epacadostat enhances dendritic cell immunogenicity and lytic ability of tumor antigen-specific T cells. Oncotarget (2016) ;7: :37762–72. |
[26] | Gameiro PA , Yang J , Metelo AM , Perez-Carro R , Baker R , Wang Z , et al. In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation. Cell Metabolism (2013) ;17: :372–85. |
[27] | Meric-Bernstam F , Tannir NMH JJ , Bennett MK . Phase 1 study of CB-839, a small molecule inhibitor of glutaminase (GLS), alone and in combination with everolimus (E) in patients (pts) with renal cell cancer (RCC). J Clin Oncol (2016) ;34. |
[28] | Abu Aboud O , Chen CH , Senapedis W , Baloglu E , Argueta C , Weiss RH . Dual and specific inhibition of NAMPT and PAK4 by KPT-9274 decreases kidney cancer growth. Mol Cancer Ther (2016) ;15: (9):2119–29. |
[29] | Haines RJ , Pendleton LC , Eichler DC . Argininosuccinate synthase: at the center of arginine metabolism. International journal of biochemistry and molecular biology (2011) ;2: :8–23. |
[30] | Yoon CY , Shim YJ , Kim EH , Lee JH , Won NH , Kim JH , et al. Renal cell carcinoma does not express argininosuccinate synthetase and is highly sensitive to arginine deprivation via arginine deiminase. International journal of cancer (2007) ;120: :897–905. |
[31] | Yoon JK , Frankel AE , Feun LG , Ekmekcioglu S , Kim KB . Arginine deprivation therapy for malignant melanoma. Clinical pharmacology: Advances and applications (2013) ;5: :11–9. |


