
Designing the First Trials for Parkinson’s Prevention
Abstract
For decades the greatest goal of Parkinson’s disease (PD) research has often been distilled to the discovery of treatments that prevent the disease or its progression. However, until recently only the latter has been realistically pursued through randomized clinical trials of candidate disease-modifying therapy (DMT) conducted on individuals after they received traditional clinical diagnosis of PD (i.e., tertiary prevention trials). Now, in light of major advances in our understanding of the prodromal stages of PD, as well as its genetics and biomarkers, the first secondary prevention trials for PD are beginning. In this review, we take stock of DMT trials to date, summarize the breakthroughs that allow the identification of cohorts at high risk of developing a traditional diagnosis of PD, and describe key design elements of secondary prevention trials and how they depend on the prodromal stage being targeted. These elements address whom to enroll, what interventions to test, and how to measure secondary prevention (i.e., slowed progression during the prodromal stages of PD). Although these design strategies, along with the biological definition, subtype classification, and staging of the disease are evolving, all are driven by continued progress in the underlying science and integrated by a broad motivated community of stakeholders. While considerable methodological challenges remain, opportunities to move clinical trials of DMT to earlier points in the disease process than ever before have begun to unfold, and the prospects for PD prevention are nowtangible.
Now is the time to start conducting clinical trials for the prevention of Parkinson’s disease (PD), a chronic multisystem progressive neurodegenerative condition. The rising prevalence of the disease with its substantial societal and economic burden1–5 adds to the urgency of pursuing its prevention. Although efforts to date have focused on modifying the disease course after its diagnosis, recent advances in our understanding of the very early stages of PD (i.e., preclinical and prodromal clinical) along with the establishment of at-risk cohorts, and the discovery of potential early diagnostic biomarkers for PD, have made it feasible to design and implement trials for PD prevention (which in this review refers to the prevention of traditionally diagnosed PD unless otherwise specified). In fact, several clinical protocols for trials of candidate PD preventatives are in the pipeline now, with a couple of them actively recruiting participants.6–8
In keeping with the circa 1735 adage of Benjamin Franklin that, “An ounce of prevention is worth a pound of cure”,9 preventing PD would have an enormous impact not only on the individual but also on society at large via reduced burden on the population, healthcare, government, etc. Prevention medicine, at its simplest, promotes health and prevents disease, and can be classified as primary, secondary, or tertiary in nature, as shown in Table 1 using the example of a dental cavity.
Given the advances in our understanding of PD pathophysiology, prevention medicine can also be applied to PD (Fig. 1) with:
1. primary prevention to reduce the risk of developing PD (prodromal or traditional motor PD) in individuals without PD;
2. secondary prevention to stop or delay the development of traditional motor PD (e.g., as defined by current clinical diagnostic criteria for PD) during the prodromal period of the disease, which defined as the time between the start of a discrete measurable pathophysiologic process (often characterized by α-synuclein-based biomarkers) and the traditionally diagnosable state of PD or related neurodegenerative synucleinopathy like dementia with Lewy bodies (DLB); and,
3. tertiary prevention to slow the progression of classical parkinsonian features and the development of complications like motor fluctuations and cognitive impairment in individuals with established PD (i.e., after traditional clinical diagnosis).
Table 1
Prevention medicine using the example of a dental cavity
| Goal | Interventions | |
| Primary prevention | To prevent the pathophysiological process (e.g., tooth demineralization, and enamel followed by dentin decay) that leads to the development of a cavity in the first place. | •Brushing and flossing teeth twice a day, •Eating a healthy diet with low sugary food consumption, •Drinking fluoridated water, •Applying topical fluoride to teeth in childhood. |
| Secondary prevention | To slow the rate of these pathophysiological processes or reduce the likelihood that they will lead to the disease (i.e., cavities). | •Regular dental checkups for early detection of cavity formation and to screen for the pathophysiological process that would warrant increased or improved brushing and flossing and dietary habits, or referral to a dental hygienist. |
| Tertiary prevention | To prevent complications of the disease (i.e., of the cavity) once it has developed. | •Dental fillings to prevent the development of an abscess, root canal, or tooth extraction, all of which are a consequence of an untreated, more severe dental cavity. |
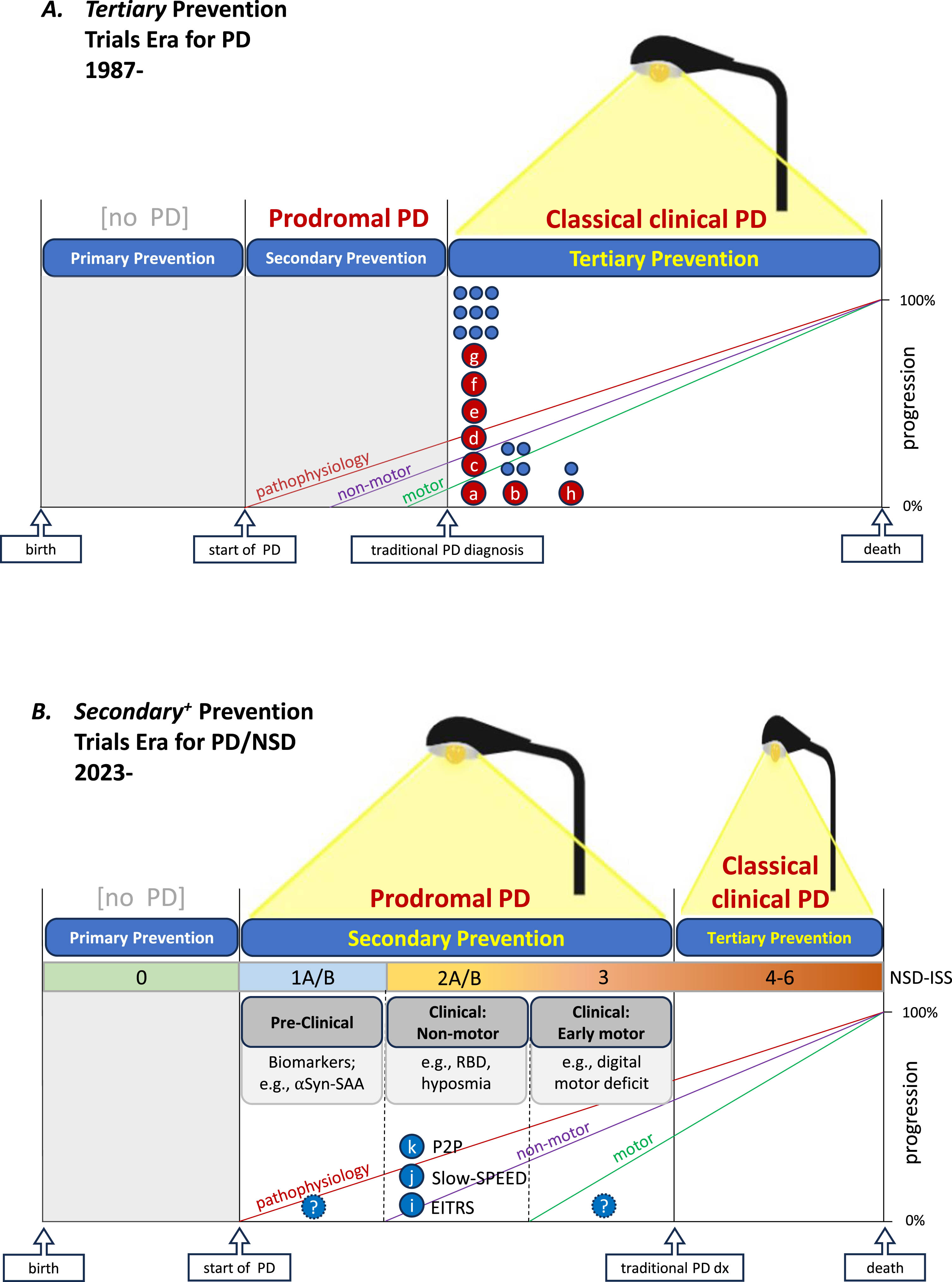
Fig. 1
Advancing prospects for prevention trials in Parkinson’s disease with new light shed onto its biology and prodromal period. A) Since 1987 and until recently ‘disease modification’ trials were plausible only during the classical clinical period of the disease, which begins with a traditional diagnosis based on established clinical criteria, and were designed to test for tertiary prevention. Preceding and generally sequential development of pathophysiology, progressive dopaminergic neuron loss (not shown but reflected by signal loss on dopamine transporter imaging), non-motor clinical features, and early motor (movement) difficulties had been known. However, they were not sufficiently illuminated to support trials assessing prevention prior to diagnosis but after the disease process has begun (secondary prevention), let alone before the onset of pathophysiological processes (primary prevention). Accordingly as depicted under a single streetlamp metaphor, since 1987 (when DATATOP the first phase 3 trial of proposed DMT began enrolling) only tertiary prevention trials had been conducted—in light of our detailed understanding of classical clinical PD and its dopaminergic deficits. B) Since 2023 growing knowledge of who is genetically and prodromally at substantially increased risk of developing (classical clinical) PD has provided adequate illumination to rationally design and initiate several relatively large-scale secondary prevention trials for the first time with others being actively developed. In both prevention trial eras, trial design strategies generally favor enrolling cohorts as early as feasible as reflected by the leftward distribution of major efficacy trials of candidate DMTs for tertiary prevention (A) and thus far for secondary prevention (where the non-motor preclinical stage of prodromal PD is the earliest that can be practically targeted at present.) Note that the simplified schematic timelines are based upon growing but evolving evidence for discernable periods of PD, including its prodromal and traditional clinical periods with the former comprising several conceptually discrete but practically overlapping stages. In panel A, maroon circles (a-h) represent completed (and ultimately negative) individual phase 3 efficacy trials of candidate DMT in PD,11–18 whereas unlettered blue circles approximate the number and timing of (unnamed) phase 2 efficacy trials of candidate DMT in PD. In panel B, blue circles (i-k) represent recently initiated or imminently starting individual phase 2 trials of candidate (secondary) preventatives in PD.6,42,43 This commonly employed prodromal vs traditional PD framework differs somewhat from a complementary and compelling integrated staging system (ISS) based on a broader disease scope of neuronal synuclein disease (NSD),37 comprising dementia with Lewy bodies as well as PD. This NSD-ISS has thus far restricted its biological anchors to synucleinopathic biomarkers (like CSF αSyn-SAA) and dopaminergic deficit biomarkers (like DAT deficit neuroimaging) but maps closely to the timeline depicted here (B). aDATATOP (selegiline, vitamin E, both) – PD, de novo.11 bELLDOPA (levodopa; https://clinicaltrials.gov/study/NCT00004733) – PD, late de novo.12 cPRECEPT (CEP-1347; https://clinicaltrials.gov/study/NCT00040404) – PD, de novo.13 dADAGIO (rasagiline; https://clinicaltrials.gov/study/NCT00256204) – PD, de novo.14 eQE3 (Co-Q10; https://clinicaltrials.gov/study/NCT00740714) – PD, de novo.15 fSTEADY-PD III (isradipine; https://clinicaltrials.gov/study/NCT02168842) – PD, de novo.16 gSURE-PD3 (inosine; https://clinicaltrials.gov/study/NCT02642393) – PD, de novo.17 hLS-1 (creatine; https://clinicaltrials.gov/study/NCT00449865) – PD, on treatment.18 iEITRS (idebenone; https://classic.clinicaltrials.gov/ct2/show/NCT04534023) – prodromal PD: RBD, pre-motor.6 jSlow-SPEED (exercise; https://clinicaltrials.gov/study/NCT06193252) – prodromal PD: RBD+/-motor.8 kP2P/PPMI (multiple drugs TBN) – prodromal PD: RBD or other non-motor,+/-motor.27 αSyn-SAA, α-synuclein-seed amplification assay; DMT, disease-modifying therapy; NSD-ISS, Neuronal Synuclein Disease-Integrated Staging System; PD, Parkinson’s disease; RBD, REM sleep behavior disorder; TBN, to be named.
PARKINSON’S DISEASE-MODIFICATION TRIALS TO DATE (TERTIARY PREVENTION)
To date, clinical trials in PD have pursued tertiary prevention through disease modification in individuals with a traditional diagnosis of PD. Since 1987 there have been numerous phase 3 tertiary prevention trials in PD (Fig. 1A). Most of these trials have enrolled participants with de novo PD (usually within 2 years of diagnosis, and not yet started on symptomatic dopaminergic therapy). Enrolling these participants as early as possible after diagnosis is in keeping with the broader principle that the earlier one intervenes with a disease-modifying therapy (DMT) in the pathophysiology and neurodegeneration of PD the more likely it will be effective. Intervening too late in the disease process, as proverbially presaged and discouraged by John Heywood through his 1546-published English aphorism, “Don’t shut the barn door after the horse has bolted” (as adapted from Old English), could be one reason why no tertiary prevention of PD has been established to date. Intervening with a DMT in participants with prodromal PD (Fig. 1B) may be more fruitful.
Although for decades there has been ample evidence that dopaminergic neuron deficits and various motor and non-motor symptoms precede the traditional clinical diagnosis of PD by years or decades,10 the inability to clearly define at-risk populations precluded the enrollment of earlier clinical cohorts until recently. Similarly, tertiary prevention trials of candidate DMT have typically based their primary trial outcome on imperfect but validated clinical measures of motor deficit progression (as determined by clinicians and/or patients).11–18 More sensitive motor and non-motor clinical measures of disease progression before the diagnosis, not to mention biomarkers of progression that would be needed to gauge the efficacy of DMT before the diagnosis, had not yet been developed and validated. The development of these tools and strategies for the design of the first (secondary) prevention trials for PD should benefit greatly from collaborative engagement across a community of invested stakeholders comprising newly mobilized advocates from at-risk populations,19 as well as from academic, government, industry, philanthropic, and regulatory partners committed to preventing the disease.
SECONDARY PREVENTION TRIALS
As noted above, advances in our understanding of the prodromal period of PD and the identification of prodromal features placing people at high risk for developing PD have provided an opportunity to modify the course of PD earlier, and enabled the field to design and implement secondary prevention trials now. The MDS Research Criteria for Prodromal PD were established in 2015 and updated in 2019, providing a framework to calculate an individual’s probability of being in the prodromal period of PD based on the presence of risk and prodromal PD markers.20 Polysomnogram-proven REM sleep Behavior Disorder (RBD), a “clearly abnormal dopaminergic PET/ SPECT scan”, “subthreshold parkinsonism”, hyposmia, and neurogenic orthostatic hypotension are some of the markers among these criteria with a high likelihood ratio for prodromal PD.20 Building on the elevated PD risk conferred by these prodromal features, PREDICT-PD, a UK-based predominantly online study used PD epidemiological risk factors (i.e., coffee consumer, NSAID use, head injury, etc.) to designate risk scores of low, medium, and high for later development of PD.21,22 They then validated the risk score result with the presence of other high-risk prodromal PD features: RBD, hyposmia, and reduced finger tapping speed.
In addition to those clinically at risk for PD development due to the presence of motor and non-motor symptoms, some individuals are instead (or in addition) genetically at risk for PD. These individuals have genetic variants associated with a risk of developing PD later in life. The most prevalent pathogenic genetic variants are those in the LRRK2 and the GBA genes, which may confer a PD risk of 43% and 19% by 80 years of age, respectively.23,24 Other more penetrant but less prevalent genetic variants, like several in the SNCA gene, are compelling to pursue but given their rarity, they are more difficult and less practical to target in early secondary prevention efforts.
In recent years, cohorts of clinically at-risk and genetically at-risk participants have been established. Given the high-risk nature of RBD and hyposmia, most of the cohorts to date have included participants with one or both of these features. The North American Prodromal Synucleinopathy (NAPS) Consortium for RBD has enrolled 361 participants with RBD, and these participants are undergoing longitudinal comprehensive clinical assessment.25 The Parkinson’s Progression Markers Initiative (PPMI) is currently enrolling a “prodromal cohort” of participants who meet the following sequential criteria: age 60 years or older; presence of risk factors or clinical features related to PD: have RBD, known genetic risk variants, or “other known PD risk criteria including those based on questionnaires in PPMI Online”; hyposmia based on UPSIT testing; and a positive dopamine transporter (DAT) SPECT on visual inspection.26 These participants undergo comprehensive clinical assessments along with the collection of biological fluids and serial neuroimaging. Participants in PPMI may be eligible to participate in the Path to Prevention (P2P) platform trial, a phase 2A, randomized, double-blind, placebo-controlled study to evaluate multiple investigational drugs in prodromal PD,27 which will be further discussed below.
In addition to the identification and further characterization of at-risk groups of participants, there has been ongoing research into biomarkers of risk. A recent and notable breakthrough in this area is the discovery and validation of early measures of PD pathophysiology centered on synucleinopathy, like the α-synuclein (αSyn) seed amplification assay (SAA) in cerebrospinal fluid (CSF) or phosphorylated α-synuclein detected by immunohistochemistry of the skin.28–31 The SAA has been studied in several different biological fluid samples (i.e., blood (plasma and serum), nasal secretions, and saliva) and has shown high diagnostic sensitivity and specificity in them for detecting PD.29,30,32
TRIAL DESIGN ELEMENTS: PRODROMAL STAGE-DEPENDENCE
With the identification of multiple at-risk populations, candidate therapeutics, and potential outcome measures suitable for prevention trials comes the challenge of their collective selection in designing a given prevention trial. In addition to their reliance on one another, key design elements for secondary prevention trials depend on the prodromal stage targeted, and vice versa33—as illustrated below and in Fig. 2. The prodromal period of PD (or more broadly, of neurodegenerative synucleinopathy) can be divided into three largely sequential stages: preclinical, clinical non-motor, and clinical early motor. (See Figs, 1B and 2).
Fig. 2
Key trial design elements for secondary prevention of Parkinson’s disease (PD) and their dependence on the stage within prodromal PD. For three key design questions several illustrative design elements are provided, including those selected for several emerging prevention trials: *EITRS (Efficacy of Idebenone in the Treatment of iRBD Into Synucleinopathies study).6 **Slow-SPEED-NL (Slowing Parkinson’s Early Through Exercise Dosage study).8 ***P2P (Path-to-Prevention)27. Note that the listed design elements are illustrative rather than exhaustive, as is the depicted sequence of clinical stages of prodromal PD (which may alternatively include early motor features preceding non-motor features). αSyn-SAA, α-synuclein-seed amplification assay; DAT, dopamine transporter; PD, Parkinson’s disease; RBD, REM sleep behavior disorder; Rx, drug or other treatment.

1) Selecting WHOM to enroll
a) Preclinical population defined by an early PD-specific biomarker: With the recent validation of CSF αSyn-SAA as a prototypic diagnostic biomarker,29 it is now plausible even if not yet practical to design an early-stage (preclinical) secondary prevention trial for PD. Cross-sectional data showing the vast majority of people with RBD to be αSyn-SAA+indicate that this biomarker may help identify an at-risk population prior to any clinical features. (See Fig. 2, row 1a.) However, longitudinal clinical and biomarker characterization of asymptomatic αSyn-SAA+ populations are first needed for the rational design of trials of candidate preventatives with the potential to slow or prevent the development of prodromal clinical features, and ultimately of classically manifest PD and related neurodegenerativesynucleinopathies.
Another practical impediment to establishing a trial-ready cohort for secondary prevention in preclinical PD based on an αSyn biomarker is the relative invasiveness of the lumbar puncture used for the best validated αSyn biomarker at present. However, the prospects for developing a more accessible, peripheral measure for αSyn-SAA are rapidly expanding (e.g., in neuronal extracellular vesicles from plasma32 or in αSyn immunoprecipitates from serum34), and other αSyn pathobiology measures such as phosphorylated forms of αSyn in skin biopsies,31 which have been associated with RBD35,36 as well as PD, may provide alternative αSyn markers for cohort enrichment in preclinical prodromal PD. Stage 1 of the recently proposed neuronal synuclein disease integrated staging system (NSD-ISS),37 which corresponds closely to the preclinical stage of prodromal PD (Fig. 1B), was inferred based on CSF αSyn-SAA data, yet this system similarly allows for alternative, adequately validated αSyn biomarkers to define active disease.37,58 Of note, while NSD-ISS considers early PD pathophysiology to be anchored in αSyn (neuronal αSyn, more specifically and by definition), other formulations rely on mechanistically less restrictive or more heterogeneous definitions of PD allowing for synuclein-independent pathophysiology. Thus, it remains plausible that novel, αSyn-unrelated biomarkers could help define a preclinical prodromal PD cohort at high risk of PD and its prodromal features (hypothetically, for example, a mitochondrial DNA damage marker).38
b) Genetically defined cohort: People with prodromal PD who also carry a relatively common and at least moderately penetrant pathogenic variant of the PD-associated genes LRRK2 and GBA comprise an attractive cohort for secondary prevention trials. (See Fig. 2, row 1b.) Their relative homogeneity may reduce outcome measure variability, and may be particularly well suited to therapeutics that target the putative pathogenic mechanism conferred by their particular gene variant. Recruitment may be enhanced by widening access to and use of direct-to-consumer genetic testing, identifying an expanding pool of potential participants.39 Their motivation to participate in prevention research may be enhanced by their knowledge of their genetic risk and potentially that of their children, and enhanced further when their own parents or siblings have been diagnosed and struggled with PD.
Genetically at-risk cohorts targeted for secondary prevention trials may be recruited from any stage of prodromal PD, but at present most practically from its clinical stages. PD gene carriers can be efficiently screened for non-motor features (e.g., RBD or hyposmia) or early parkinsonian motor deficits via simple questionnaires and/or at-home testing (e.g., scratch and sniff tests for hyposmia, smartphone assessments for slow gait and finger movements, rest tremor, and asymmetry.) The anticipated advent of non-invasive, blood-based αSyn markers (as above) would allow for enrollment at the preclinical stage, although such screening may exclude a substantial portion of LRRK2 variant carriers with imminent PD, given the evidence for an alternative pathophysiology in LRRK2 PD.29,40 Peripheral measures of LRRK2-linked lipids or -catalyzed phosphorylation products may eventually prove useful as alternative PD pathophysiology markers, but these are hypothetical at present.
c) RBD in pre-motor prodromal PD: Of all the prodromal PD features, polysomnography-confirmed RBD increases the likelihood of developing PD (or related neurodegenerative synucleinopathy) far more than any other.20 Its presence has been suggested to be sufficient on its own to drive the enrichment of an at-risk population for prevention trials.41 (See Fig. 2, row 1c.) Indeed, the first randomized clinical trial pursuing secondary prevention of PD or neurodegenerative synucleinopathy appears to be exclusively enrolling RBD patients prior to motor feature onset. The Efficacy of Idebenone in the Treatment of iRBD into Synucleinopathies (EITRS) trial7 is registered (NCT04534023) as a placebo-controlled, double-blind, multi-center, phase 2 trial of idebenone (a mitochondrial enhancer and coenzyme Q10 analog) in polysomnogram-confirmed RBD in active recruitment status since 2022. It seeks to randomize 142 participants 1 : 1 to idebenone:placebo treatment with a primary, efficacy outcome of conversion to synucleinopathy (rate of diagnosis of PD, DLB, or multiple system atrophy (MSA) over 5 years). Although the presence of any isolated parkinsonian motor feature is an exclusionary eligibility criterion for EITRS and thus reduces the likelihood of phenoconversion (to PD and MSA more than DLB), it helps ensure an earlier disease stage at enrollment and thus supports the foundational rationale for moving from tertiary to secondary prevention trials. The study’s long (5-year) follow-up plan, a potential advantage of non-commercial sponsorship, at least partially mitigates the challenge of a lower conversion rate. Complementary clinical (motor and cognitive scales) and biomarker (DAT neuroimaging) outcome measures will be collected serially to help gauge disease progression.
d) Non-motor prodromal PD, with or without motor features: Targeting the non-motor clinical stage (RBD, hyposmia) while allowing early parkinsonian motor features increases the sensitivity of quantitative motor measures to disease progression over a shorter follow-up period and aids recruitment by expanding eligibility. (See Fig. 2, row 1d.) This target population seems the current ‘sweet spot’ of prodromal PD stage selection for most secondary prevention trials, and has been adopted by two emerging prevention trials, the ‘Slowing Parkinson’s Early Through Exercise Dosage’ (Slow-SPEED-NL) trial8 and the P2P platform trial.42
Slow-SPEED (in its initial Dutch version Slow-SPEED-NL; NCT061932528), like EITRS is enrolling only RBD patients but in contrast allows those with isolated parkinsonian motor features (i.e., short of a PD/neurodegenerative disease diagnosis), with recruitment active as of January 2024. Although numerous serial motor and non-motor clinical outcome measures will be collected, the primary outcome assessment is the feasibility of increasing step count as an indicator of increased voluntary physical activity (exercise) over two years. Participants will be randomized 1 : 1 to receive guidance to gradually increase the volume and intensity of their step count-based exercise either by a modest or substantial amount relative to their personal baseline activity per instructions provided by a motivational Slow-SPEED-NL smartphone study app.
P2P proposes to enroll a more clinically heterogeneous prodromal cohort accepting RBD alone, hyposmia alone, or both as the requisite non-motor anchor (while allowing early motor features short of a PD/neurodegenerative disease diagnosis) beginning in early 2025. Conversely, it will more narrowly define the cohort biologically, with eligibility requiring αSyn-SAA positivity and a DAT deficit on neuroimaging (per most recently posted protocol information),27 consistent with Stage 2B or higher by the NSD-ISS.42
The requirement of a DAT deficit to enroll in P2P rather than allowing αSyn-SAA+/DAT- (formally a Scan Without Evidence of Dopaminergic Deficit (SWEDD) consistent with NSD-ISS Stage 2A) may reflect a difficult decision in defining a prodromal cohort. While excluding SWEDDs altogether increases the prospects for further DAT deficit during the follow-up period, there may be a favorable intermediate option to include DAT deficits that approach but fall below the current quantitative threshold that conservatively avoids false positives. A less stringent cutoff may facilitate recruitment while enhancing prospects for demonstrating efficacy with an earlier stage population, especially if a change in DAT signal was a primary outcome in this phase 2 study. That could better inform a decision of whether to advance to a clinically focused phase 3 prevention trial. Such nuanced design decisions may depend on a growing knowledge base on the dynamic range of imaging and other biomarkers across prodromal stages, as well as the specific intervention and outcomes, under study, and program or sponsor considerations.
e) Targeting the early-motor clinical stage of prodromal PD: This can be an important alternative design strategy despite its major limitation of approaching the point of traditional diagnosis, which minimizes the putative benefit of moving from a standard disease modification trial in de novo PD to prodromal PD. (See Fig. 2, row 1e.) Nevertheless, potential advantages include: 1) Maximizing the risk and rate of phenoconversion, which may be particularly important in justifying high-risk or invasive interventions (see below). 2) May be most efficient when relying on a sensitive digital motor readout. 3) May be more inclusive of broader PD population (though more exclusive of DLB) by enrolling the significant subpopulation of PD that does not pass through an RBD or other non-motor stage prior to diagnosis (including a higher proportion of LRRK2-PD, for example).
Challenges in selecting WHOM to enroll:
1. Risk of excluding populations commonly underrepresented in research: Identification of at-risk participants requires additional financial and technological resources, the lack of which may preclude prevention trial participation in many low and middle-income countries, and among disadvantaged groups within wealthier nations. Investigators, funding bodies, regulators, and sponsors should work to enhance local and global representation as lack of diversity in trials affects the generalizability and accuracy of their results along with increasing inequity and injustice. Thus, extra attention, effort, and cost will be required to ensure the representation among participants.43–45 The Global Parkinson’s Genetics Program (GP2), an initiative of the Aligning Science Across Parkinson’s (ASAP), is an excellent example of investing in a global research community, inclusive of underrepresented groups in PD genetics.46 In addition, global representation in prevention trials may be aided by innovative, decentralized, and accessible methods such as a smartphone-based exercise app used both for intervention implementation and outcome assessment. This approach employs relatively low-cost technology that is widely available even in many developing countries, and its ongoing validation may be scalable up for international, multi-lingual cohorts of individuals at high risk of or living with PD.47
Similarly, prevention trials should ensure proportional representation of women, a group who are also generally underrepresented in manifest PD.48,49 In at-risk individuals with iRBD, the ratio of male:female is comparable to that in manifest PD, in which a higher male prevalence is well established.50 Thus, strategies will be needed to recruit and enroll a greater percentage of women with iRBD than typically enrolled in PD studies. Unlike idiopathic disease, PD due to a pathogenic variant of the LRRK2 gene shows parity of prevalence between sexes, thus setting the goal at 1 : 1 for male:female enrollment in LRRK2 PD prevention trials.51
2. Risk of causing psychosocial harm: Understandably there is concern that these prevention studies in at-risk individuals may cause psychosocial harm to participants via learning about their elevated risk of PD (“risk disclosure”) or the intervention that they may receive during the trial. Risk disclosure of any type requires careful execution by an experienced professional. However, many at-risk individuals for PD feel empowered and are motivated to participate in clinical research.19 Furthermore, research on risk disclosure in a related neurodegenerative disorder, Alzheimer’s disease has not shown increased psychosocial harm.52–54 Current prevention studies, like Slow-SPEED-NL and P2P are recruiting adults who have undergone genetic testing for PD pathogenic variants or who were previously diagnosed with iRBD by their physician who may explain their increased PD risk before being recruited into the study. Interested individuals will also generally be engaged adults who can give informed consent to participate in research of their choosing.
2) Selecting WHAT interventions to test
It is plausibly reasoned that what initiates a neurodegenerative disease can also perpetuate it – especially when neuronal degeneration is known to be a common process that underlies progression prior to and after traditional diagnosis. On the other hand, there is a strong intuitive rationale and some empirical evidence to suggest an effective DMT for (secondary) prevention may differ markedly from an effective DMT for slowing classical PD (tertiary prevention). Genetically, while some pathogenic gene variants (e.g., GBA variant L444P) can predict a higher risk of PD and a faster rate of its clinical progression, others (e.g., LRRK2 variant G2019S) show a dissociation of increased risk from a slower rate of clinical decline.55 Thus these human data more directly support the rationale for secondary prevention trials of LRRK2 kinase inhibitors, while raising concerns over efficacy in PD trials for tertiary prevention (currently in overall encouraging phase 2 testing being conducted with clinical equipoise; NCT05348785).56
Similarly, epidemiological studies of large, prospectively followed, initially healthy cohorts have identified several dietary, pharmacological, or lifestyle factors (e.g., caffeine consumption, ibuprofen use, glucagon-like peptide-1 (GLP-1) agonist prescription, and smoking) that predict a reproducibly reduced rate of PD diagnosis but not of PD progression after diagnosis. For example, higher caffeine consumption in unaffected individuals consistently predicts a lower risk of PD, whereas its use in people with PD is not a reliable predictor of slower progression.57,58 Thus, even though caffeine has robust neuroprotective properties across animal models of PD, human data provide a stronger rationale to test it for PD prevention (primary or secondary) than for progression after a traditional diagnosis (tertiary).9
a) Lifestyle factors, and low-risk, repurposed treatment: The stage of the prodromal PD being targeted can also be a key determinant of intervention selection, and conversely the preventative of interest may help determine the optimal stage(s) to target. Safety or therapeutic index is of course of paramount importance in any trial design, but should be given greater weight at earlier, pre-symptomatic stages of disease. Thus, generally benign lifestyle, dietary, and repurposed drug candidates may offer a general advantage over new chemical entities with which there is limited human experience, and over more toxic, less tolerable, or more invasive treatments. Thus, interventions with well-established, relatively good safety records—like exercise, the non-specific adenosine antagonist caffeine, and the anti-inflammatory ibuprofen, which are all linked to reduced PD risk—would be more appropriate than new untested drugs for the earliest stages of prodromal PD; i.e., in people who have no appreciable disability or clinical disease and may never develop one.
While low-risk and repurposed interventions may be particularly appropriate for preclinical prodromal PD (when such studies become practical), they are at present practically best suited to non-motor prodromal PD states like RBD, hyposmia, and/or other non-motor prodromal features. (See Fig. 2, row 2a.) And indeed, the two therapies currently in the first actively enrolling randomized control trials for secondary prevention are widely available with the nutritional supplement idebenone in the EITRS trial NCT04534023,7 and titrated exercise guided via an interactive, smartphone-based app in the Slow-SPEED-NL trial NCT06193252.8 As noted above, Slow-SPEED-NL but not EITRS allows early motor as well as non-motor prodromal features, which will aid recruitment and enrich RBD for those at greater risk of PD versus DLB, and for better or worse shifts the cohort to a slightly more advanced diseasestate.
While these interventions target general oxidative and cellular stress pathways, other repurposable agents may credibly target the products of major PD genes and thus may be considered more compelling candidate preventatives. For example, ambroxol an over-the-counter mucolytic has been found to increase CSF levels of GCase (the GBA1 gene product) in PD,59 and is being pursued for tertiary prevention in people with GBA or idiopathic PD in a phase 3 RCT (NCT05778617). Given its long, favorable safety record60 ambroxol may also be well suited for a secondary prevention study in GBA+at-risk individuals with a positive αSyn-SAA even before any clinical features arise, as well as in those with RBD and/or hyposmia (i.e., a non-motor prodromal population).
Alternatively, PD gene-directed repurposed agents may act against neurotoxicity mediated by αSyn (encoded by SNCA), the core presumptive pathogenic pathway of PD. Albuterol, a beta-adrenergic receptor agonist commonly and chronically used for reactive airway disorders, has been found to reduce expression of αSyn, to confer neuroprotection in preclinical studies,61 and to be linked to reduced risk of PD in epidemiological studies, though not in all of them (and with residual controversy over role of prominent candidate confounding factors).61,62 Similarly, preclinical evidence63 has suggested that the potential neuroprotective effects of caffeine and other more specific adenosine A2A receptor antagonists in PD may reflect attenuated αSyn neurotoxicity, further supporting its consideration as a low-risk candidate preventative in early (e.g., αSyn-SAA+ preclinical, or non-motor) prodromal PD. With additional encouraging results of GLP-1 agonism as a repurposable anti-diabetic drug class in a recent phase 2 tertiary prevention trial of lixisenatide for PD,64 this approach may also be worthwhile pursuing for secondary prevention depending in part on anticipated upcoming results of ongoing phase 3 testing with exenatidein PD.65
b) Moderate-high risk agents: The increasingly realistic opportunity to intervene earlier in PD pathophysiology, combined with the disappointments to date of phase 3 tertiary prevention trials for PD, is garnering hope and associated investment from commercial, philanthropic, and public stakeholders. Recent scientific advances together with a receptive regulatory environment and a newly promulgated classification and staging frameworks for PD/synucleinopathy37,66,67 are encouraging industry programs to shift consideration for DMT for PD toward earlier prevention studies.
In an effort to facilitate this shift, the Michael J Fox Foundation’s PPMI, which has increasingly focused on characterizing biomarkers as well as clinical features of PD’s prodromal period, has developed the P2P platform trial initiative.27,42 P2P is inviting commercial therapeutic companies to conduct secondary prevention trials of their proprietary candidate therapeutics, and has announced its expectation that by early 2025 P2P participants with non-motor prodromal PD (with or without early motor features) will be randomized to receive the first of these as yet undisclosed treatments or placebo.
The effort to expedite industry pursuit of secondary prevention as spearheaded by PPMI/P2P will generally encourage the testing of new chemical entities (novel small molecules targeting pathogenic αSyn, GBA, LRRK2, inflammatory, etc. pathways) or biologics (e.g., αSyn-targeted antibodies, immunomodulating decoy receptors, mRNA vaccines, neurotrophic factors, etc.). The limited safety experience with these moderate-to-high risk agents warrants particular caution when administering them to a healthy population even if these participants are at substantial risk of developing a neurodegenerative synucleinopathy. (See Fig. 2, row 2b.) Thus such compounds at present are better suited to testing in the clinical phases of prodromal PD or NSD.
c) Invasive, high-risk interventions: More invasive novel experimental therapies (e.g., intracerebral infusion of cellular therapy or of viral vectors for gene therapy) for secondary prevention at present may be warranted only in those with the highest risk of developing NSD such as PD (in the early motor clinical stage of prodromal disease), and on the verge of substantial progressive disability. (See Fig. 2, row 2c.)
3) Selecting HOW to measure ‘prevention’
How to quantify prevention remains a major if not the greatest challenge in the design of PD prevention trials. While diagnosis of PD (or related neurodegenerative synucleinopathy) seems simplest, relying on phenoconversion can be limited practically (given the long follow-up periods required) and conceptually (given shifting definitions of PD). Preceding phenoconversion are a series of potentially informative, even if not yet definitive, biomarker and clinical metrics of PD progression (Fig. 2, rows 3a-d). Most of these are actively under investigation with their selection in early secondary prevention trials highly dependent on the prodromal stage being targeted.
a) Quantitative biomarker of prodromal disease state: A practical early prodromal stage outcome measure may have to await a quantitative, as well as a more accessible (e.g., blood-based), αSyn-SAA (or alternative biomarker of PD pathophysiology), as little is currently known about the natural history of αSyn-SAA+people in the preclinical stage of prodromal PD, including what proportion will develop clinical features and how long it will take. Nevertheless, the rapid pace of progress in αSyn-SAA related methods, suggests that it may not be long before quantitative68,69 and blood-based32,34 assays are available. (Fig. 2, row 3a.)
b) Composite prodromal load: Although all three highlighted RCTs pursuing secondary prevention of PD track non-motor and motor outcomes among secondary analyses, Slow-SPEED-NL has a major focus on developing an aggregate prodromal burden score from a wide range of serial standardized assessments of anxiety, depression, cognition, dysautonomia (including heart rate variability, orthostatic blood pressure changes), RBD, sleep, and olfaction.8 (See Fig. 2, row 3b.) An integration of prodromal features allows for the possibility of assessing progression more holistically as the accrual of clinical deficits beyond the cognitive and motor hallmarks of the two main neurodegenerative synucleinopathies, DLB and PD, respectively.
c) DAT Neuroimaging: In contrast to the currently validated utility of αSyn-SAA as a dichotomous diagnostic marker of PD, dopamine transporter (DAT) neuroimaging also has demonstrated value as a state biomarker of PD that reflects nigrostriatal neuron integrity and allows tracking of progression in PD—particularly in the immediate ‘de novo’ period following a traditional PD diagnosis, but likely also in the mid to late prodromal period.70,71 Despite the challenges of expense and participant burden of serial nuclear medicine testing, DAT neuroimaging remains a valuable marker. Its use in early-phase secondary prevention trials may allow smaller or shorter pilot studies guiding decisions on advancing to larger trials with more meaningful but ‘noisier’ clinical or patient-reported primary outcomes, as reflected in its inclusion as a repeated measure in the designs adopted by the EITRS and P2P trials (See Fig. 2, row 3c.)
d) Motor and cognitive measures: As motor deficits and cognitive decline are the clinical hallmarks of PD and DLB, quantitative measures of movement and memory are key outcomes in the later stages of prodromal PD or NSD. (See Fig. 2, row 3d.) These measures include long-established scales for parkinsonism (e.g., MDS-UPDRS, with patient- and clinician-reported components) and cognitive impairment (e.g., ADAS-Cog). In addition, a wide range of emerging quantitative digital options collect parallel data via apps on ubiquitous smartphones or home computers, various wearables (e.g., smartwatches), or invisibles (e.g., radio wave-based, passive home monitoring).72,73 These tech-fostered metrics offer potentially more sensitive and frequent assessments in a more natural environment, and thus may prove more effective than the traditional self- or clinician-administered surveys and tests for monitoring progression in the clinical stages of prodromal PD. EITRS, Slow-SPEED-NL, and P2P designs have all adopted multiple scales to cover these outcomes.
e) Phenoconversion: A traditional diagnosis of PD or another neurodegenerative synucleinopathy is currently the definitive endpoint for a secondary prevention trial enrolling people with prodromal PD, and may represent the gold standard for a registration or phase 3 trial until validation of a biomarker or clinical measure of prodromal PD progression occurs. However, prevention trials using phenoconversion as the primary outcome measure may be prohibitively long (or large), particularly for commercial development programs, even if targeting a late prodromal period (e.g., clinical early motor) with enrichment for a DAT deficit on neuroimaging. Moreover, determining phenoconversion (e.g., clinically diagnosing PD) is inherently subjective and may vary considerably between clinician investigators, even when employing specific diagnostic criteria.
Nevertheless, secondary prevention RCTs adopting phenoconversion as their primary outcome have recently been planned (e.g., with 732 RBD participants to be followed over 3 years in the SAHZJU_RBD study, registered but apparently delayed; NCT05611372)74 or initiated (e.g., with 142 participants with RBD to be followed over 5 years in the EITRS study, which is actively recruiting per NCT04534023)7 (See Figure 2, row 3e.)
CONCLUSION
The time has come to begin conducting rigorous secondary prevention trials in prodromal PD, while pursuing improved prevention trial designs. Advances in our understanding of the prodromal period of PD along with the establishment of at-risk cohorts and the recent validation of a synucleinopathy biomarker have led the way to the design and implementation of the first trials for the (secondary) prevention of PD.
ACKNOWLEDGMENTS
The authors have no acknowledgements to report.
FUNDING
The Michael J. Fox Foundation for Parkinson’s Research Award MJFF-022854 (MAS).
This publication was supported by the Food and Drug Administration (FDA) of the U.S. Department of Health and Human Services (HHS) as part of a financial assistance award, R13FD008015 totaling $60,000 with 100 percent funded by FDA. The contents are those of the author(s) and do not necessarily represent the official views of, nor an endorsement, by FDA/HHS, or the U.S. Government.
American Academy of Neurology Clinical Research Training Scholarship (GFC).
CONFLICT OF INTEREST
The authors have no conflicts of interest to report.
REFERENCES
1. | Rossi A , Berger K , Chen H , et al. Projection of the prevalence of Parkinson’s disease in the coming decades: Revisited. Mov Disord (2017) ; 33: : 156–159. |
2. | Ascherio A and Schwarzschild MA. The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol (2016) ; 15: : 1257–1272. |
3. | Dorsey ER , Okun MS and Tanner CM. Bad air and Parkinson disease—the fog may be lifting. JAMA Neurol (2021) ; 78: : 793. |
4. | Marras C , Beck JC , Bower JH , et al. Prevalence of Parkinson’s disease across North America. NPJ Parkinsons Dis (2018) ; 4: : 21. |
5. | Dorsey ER , Sherer T , Okun MS , et al. The emerging evidence of the Parkinson pandemic. J Parkinsons Dis (2018) ; 8: (s1), S3–S8. |
6. | Li Y , Wang C , Luo N , et al. Efficacy of idebenone in the treatment of irbd into synucleinopathies (EITRS): Rationale, design, and methodology of a randomized, double-blind, multi-center clinical study. Front Neurol (2022) ; 13: : 981249. |
7. | Liu J . A clinical study of the efficacy of idebenone in the treatment of iRBD into synucleinopathies. ClinicalTrials.gov, https://clinicaltrials.gov/study/NCT04534023 (2023, accessed 30 July 2024). |
8. | Darweesh S . Slow-SPEED-NL: Slowing Parkinson’s Early Through Exercise Dosage-Netherlands (Slow-SPEED-NL). ClinicalTrials.gov, https://clinicaltrials.gov/study/NCT06193252 (2024, accessed 30 July 2024). |
9. | Crotty GF and Schwarzschild MA. What to test in Parkinson disease prevention trials? Neurol (2022) ; 99: : 34–41. |
10. | Fearney J and Lees A. Ageing and Parkinson’s disease: Substantia nigra regional selectivity. Brain (1991) ; 114: : 2283–2301. |
11. | Parkinson Study Group. Effects of tocopherol and deprenyl on the progression of disability in early Parkinson’s disease. N Engl J Med (1993) ; 328: : 176–183. |
12. | Fahn S . Timing of Levodopa Treatment in Parkinson’s Disease. ClinicalTrials.gov, https://clinicaltrials.gov/study/NCT00004733 (2005, accessed 30 July 2024). |
13. | Parkinson Study Group. Safety and Efficacy Study of CEP-1347 in theTreatment of Parkinson’s Disease. ClinicalTrials.gov, https://clinicaltrials.gov/study/NCT00040404 (2012, accessed 30 July 2024). |
14. | Weiss Y . A Randomized Placebo Controlled Study to Show That Rasagiline May Slow Disease Progression for Parkinson’s Disease (ADAGIO). ClinicalTrials.gov, https://clinicaltrials.gov/study/NCT00256204 (2012, accessed 30 July 2024). |
15. | Beal F , Oakes D , Shoulson I . Effects of Coenzyme Q10 (CoQ) in Parkinson Disease (QE3). ClinicalTrials.gov, https://clinicaltrials.gov/study/NCT00740714 (2013, accessed 31 July 2024). |
16. | Simuni T , Holloway R . Efficacy of Isradipine in Early Parkinson Disease. ClinicalTrials.gov, https://clinicaltrials.gov/study/NCT02168842 (2020, accessed 31 July 2024). |
17. | Schwarzschild MA , Ascherio A , Oakes D , Macklin EA . Study of Urate Elevation in Parkinson’s Disease, Phase 3 (SURE-PD3). ClinicalTrials.gov, https://clinicaltrials.gov/study/NCT02642393 (2020, accessed 31 July 2024). |
18. | Writing group for the NINDS Exploratory Trials in Parkinson Disease (NET-PD) Investigators; Kieburtz K , Tilley BC , et al. Effect of creatine monohydrate on clinical progression in patients with Parkinson disease: A randomized clinical trial. JAMA (2015) ; 313: : 584–593. |
19. | Keavney JL , Mathur S , Schroeder K , et al. Perspectives of people at-risk on Parkinson’s prevention research. J Parkinsons Dis (2024) ; 14: : 399–414. |
20. | Heinzel S , Berg D , Gasser T , et al. Update of the MDS research criteria for prodromal Parkinson’s disease. Mov Disord (2019) ; 10: : 1464–1470. |
21. | Noyce AJ , R’Bibo L , Peress L , et al. PREDICT-PD: An online approach to prospectively identify risk indicators of Parkinson’s disease. Mov Disord (2017) ; 2: : 219–226. |
22. | Noyce AJ , Bestwick JP , Silveira-Moriyama L , et al. PREDICT-PD: Identifying risk of Parkinson’s disease in the community: Methods and baseline results. J Neurol Neurosurg Psychiatry (2014) ; 85: : 31–37. |
23. | Lee AJ , Wang Y , Alcalay RN , et al. Penetrance estimate of LRRK2 p.G2019S mutation in individuals of non-Ashkenazi Jewish ancestry. Mov Disord (2017) ; 10: : 1432–1438. |
24. | Smith LJ , Lee CY , Menozzi E , et al. Genetic variations in GBA1 and LRRK2 genes: Biochemical and clinical consequences in Parkinson disease. Front Neurol (2022) ; 13: : 971252. |
25. | Elliott JE , Lim MM , Keil AT , et al. Baseline characteristics of the North American Prodromal Synucleinopathy cohort. Ann Clin Transl Neurol (2023) ; 10: : 520–535. |
26. | Marek K . The Parkinson’s Progression Markers Initiative (PPMI) Clinical – Establishing a Deeply Phenotyped PD Cohort, https://www.ppmi-info.org/sites/default/files/docs/PPMI002_Clinical%20Protocol_AM3.2_30Jan2023_Final.pdf. (2023, accessed 30 July 2024). |
27. | Simuni T , Marek K , et al. P2P Master Protocol Synopsis & Schedule of Activities, https://www.ppmiinfo.org/sites/default/files/docs/P2P%20Master%20Protocol%20Synopsis-SOA_12July2022-DRAFT.pdf. (2022, accessed 30 July 2024). |
28. | Chahine LM , Beach TG , Adler CH , et al. Central and peripheral α-synuclein in Parkinson disease detected by seed amplification assay. Ann Clin Transl Neurol (2023) ; 10: : 696–705. |
29. | Siderowf A , Concha-Marambio L , Lafontant DE , et al. Parkinson’s Progression Markers Initiative. Assessment of heterogeneity among participants in the Parkinson’s Progression Markers Initiative cohort using α-synuclein seed amplification: A cross-sectional study. Lancet Neurol (2023) ; 22: : 407–417. |
30. | Zheng Y , Li S , Yang C , et al. Comparison of biospecimens for α-synuclein seed amplification assays in Parkinson’s disease: A systematic review and network meta-analysis. Eur J Neurol (2023) ; 30: : 3949–3967. |
31. | Gibbons CH , Levine T , Adler C , et al. Synuclein-one study: Skin biopsy detection of phosphorylated α-synuclein for diagnosis of Synucleinopathies. JAMA Neurol (2024) ; 331: : 1298–1306. |
32. | Kluge A , Bunk J , Schaeffer E , et al. Detection of neuron-derived pathological α-synuclein in blood. Brain (2022) ; 145: : 3058–3071. |
33. | Berg D , Crotty GF , Keavney JL , et al. Path to Parkinson disease prevention: Conclusion and outlook. Neurol (2022) ; 99: (7 Suppl 1): 76–83. |
34. | Okuzumi A , Hatano T , Matsumoto G , et al. Propagative α-synuclein seeds as serum biomarkers for synucleinopathies. Nat Med (2023) ; 29: : 1448–1455. |
35. | Doppler K , Jentschke HM , Schulmeyer L , et al. Dermal phospho-alpha-synuclein deposits confirm REM sleep behaviour disorder as prodromal Parkinson’s disease. Acta Neuropathol (2017) ; 133: : 535–545. |
36. | Liguori R , Donadio V , Wang Z , et al. A comparative blind study between skin biopsy and seed amplification assay to disclose pathological α-synuclein in RBD. NPJ Parkinsons Dis (2023) ; 9: : 34. |
37. | Simuni T , Chahine LM , Poston K , et al. T. A biological definition of neuronal α-synuclein disease: Towards an integrated staging system for research. Lancet Neurol (2024) ; 23: : 178–190. |
38. | Qi R , Sammler E , Gonzalez-Hunt CP , et al. A blood-based marker of mitochondrial DNA damage in Parkinson’s disease. Sci Transl Med (2023) ; 15: : 711. |
39. | Myers TL , Augustine EF , Baloga E , et al. Recruitment for remote decentralized studies in Parkinson’s disease. J Parkinsons Dis (2022) ; 12: : 371–380. |
40. | Garrido A , Fairfoul G , Tolosa ES , et al. α-synuclein RT-QuIC in cerebrospinal fluid of LRRK2-linked Parkinson’s disease. Ann Clin Transl Neurol (2019) ; 6: : 1024–1032. |
41. | Postuma RB . Neuroprotective trials in rem sleep behavior disorder: The way forward becomes clearer. Neurol (2022) ; 99: (7 Suppl 1): 19–25. |
42. | Simuni T , Coffey C , Siderowf A , et al. Path to prevention (P2P) therapeutics platform trial in biomarker defined Prodromal Parkinson’s Disease: Study Design. Zenodo, https://zenodo.org/records/8359811 (2023, accessed 30 July 2024). |
43. | Vaswani PA , Tropea TF and Dahodwala N. Overcoming barriers to Parkinson disease trial participation: Increasing diversity and novel designs for recruitment and retention. Neurotherapeutics (2020) ; 17: : 1724–1735. |
44. | Di Luca DG , Macklin EA , Hodgeman K , et al. Enrollment of participants from marginalized racial and ethnic groups: A comparative assessment of the STEADY-PD III and SURE-PD3 Trials. Neurol Clin Pract (2023) ; 13: : e200113. |
45. | Kelsey MD , Patrick-Lake B , Abdulai R , et al. Inclusion and diversity in clinical trials: Actionable steps to drive lasting change. Contemp Clin Trials (2022) ; 116: : 106740. |
46. | Rizig M , Bandres-Ciga S , Makarious MB , et al. Identification of genetic risk loci and causal insights associated with Parkinson’s disease in African and African admixed populations: A genome-wide association study. Lancet Neurol (2023) ; 22: : 1015–1025. |
47. | Schootemeijer S , de Vries NM , Macklin EA , et al. The STEPWISE study: Study protocol for a smartphone-based exercise solution for people with Parkinson’s Disease (randomized controlled trial). BMC Neurol (2023) ; 23: : 323. |
48. | Subramanian I , Mathur S , Oosterbaan A , et al. Unmet needs of women living with Parkinson’s disease: Gaps and controversies. Mov Disord (2022) ; 37: : 444–455. |
49. | Tosserams A , Araújo R , Pringsheim T , et al. Underrepresentation of women in Parkinson’s disease trials. Mov Disord (2018) ; 33: : 1825–1826. |
50. | Mano M , Nomura A and Sasanabe R. Gender difference in REM sleep behavior disorder in Japanese population: Polysomnography and sleep questionnaire study. J Clin Med (2024) ; 13: : 914. |
51. | Gan-Or Z , Leblond CS , Mallett V , et al. LRRK2 mutations in Parkinson disease; a sex effect or lack thereof? A meta-analysis. Parkinsonism Relat Disord (2015) ; 21: : 778–782. |
52. | de Wilde A , van Buchem MM , Otten RHJ , et al. Disclosure of amyloid positron emission tomography results to individuals without dementia: A systematic review. Alzheimers Res Ther (2018) ; 10: : 72. |
53. | Grill JD , Raman R , Ernstrom K , et al. Short-term psychological outcomes of disclosing amyloid imaging results to research participants who do not have cognitive impairment. JAMA Neurol (2020) ; 77: : 1504–1513. |
54. | Kim SY , Karlawish J and Berkman BE. Ethics of genetic and biomarker test disclosures in neurodegenerative disease prevention trials. Neurol (2015) ; 84: : 1488–1494. |
55. | Ahamadi M , Mehrotra N , Hanan N , et al. A disease progression model to quantify the nonmotor symptoms of Parkinson’s disease in participants with leucine-rich repeat kinase 2 mutation. Clin Pharmacol Ther (2021) ; 110: : 508–518. |
56. | West A and Schwarzschild MA. LRRK2-targeting therapies march through the valley of death. Mov Disord (2023) ; 38: : 361–365. |
57. | Simon DK , Swearingen CJ , Hauser RA , et al. Caffeine and progression of Parkinson disease. Clin Neuropharmacol (2008) ; 31: : 189–196. |
58. | Simon DK , Wu C , Tilley BC , et al. Caffeine, creatine, GRIN2A and Parkinson’s disease progression. J Neurol Sci (2017) ; 375: : 355–359. |
59. | Mullin S , Smith L , Lee K , et al. Ambroxol for the treatment of patients with Parkinson disease with and without glucocerebrosidase gene mutations: A nonrandomized, noncontrolled trial. JAMA Neurol (2020) ; 77: : 427–434. |
60. | Kantar A , Klimek L , Cazan D , et al. An overview of efficacy and safety of ambroxol for the treatment of acute and chronic respiratory diseases with a special regard to children. Multidiscip Respir Med (2020) ; 15: : 511. |
61. | Mittal S , Bjørnevik K , Im DS , et al. β2-Adrenoreceptor is a regulator of the α-synuclein gene driving risk of Parkinson’s disease. Science (2017) ; 357: : 891–898. |
62. | Singh A , Hussain S , Akkala S , et al. Beta-adrenergic drugs and risk of Parkinson’s disease: A systematic review and meta-analysis. Ageing Res Rev (2022) ; 80: : 101670. |
63. | Kachroo A , Schwarzschild MA . Adenosine A2A receptor gene disruption protects in an α-synuclein model of Parkinson’s disease. Ann Neurol (2011) ; 71: : 278–284. |
64. | Meissner WG , Remy P , Giordana C , et al. Trial of lixisenatide in early Parkinson’s disease. N Engl J Med (2024) ; 390: : 1176–1185. |
65. | Vijiaratnam N , Girges C , Auld G , et al. Exenatide once weekly over 2 years as a potential disease-modifying treatment for Parkinson’s disease: Protocol for a multicentre, randomised, double blind, parallel group, placebo controlled, phase 3 trial: The ‘Exenatide-PD3’ study. BMJ Open (2021) ; 11: : e047993. |
66. | Höglinger GU , Adler CH , Berg D , et al. A biological classification of Parkinson’s disease: The SynNeurGe research diagnostic criteria. Lancet Neurol (2024) ; 23: : 191–204. |
67. | Cardoso F , Goetz CG , Mestre TA , et al. A statement of the MDS on biological definition, staging, and classification of Parkinson’s disease. Mov Disord (2023) ; 39: : 259–266. |
68. | Vaneyck J , Yousif TA , Segers-Nolten I , et al. Quantitative seed amplification assay: A proof-of-principle study. J Phys Chem B (2023) ; 127: : 1735–1743. |
69. | Bentivenga GM , Mammana A , Baiardi S , et al. Performance of a seed amplification assay for misfolded alpha-synuclein in cerebrospinal fluid and brain tissue in relation to Lewy body disease stage and pathology burden. Acta Neuropathol (2024) ; 147: : 18. |
70. | Jennings D , Siderowf A , Stern M , et al. Conversion to Parkinson disease in the pars hyposmic and dopamine transporter-deficit prodromal cohort. JAMA Neurol (2017) ; 74: : 933–940. |
71. | Macklin EA , Coffey CS , Brumm MC , et al. Statistical considerations in the design of clinical trials targeting prodromal Parkinson disease. Neurol (2022) ; 99: (7 Suppl 1), 68–75. |
72. | Del Din S , Elshehabi M , Galna B , et al. Gait analysis with wearables predicts conversion to Parkinson disease. Ann Neurol (2019) ; 86: : 357–367. |
73. | O’Leary K . From ‘wearables’ to ‘invisibles’. Nat Med (2022) ; 28: : 2449. |
74. | Zhang B . Efficacy and Safety of Rasagiline in Prodromal Parkinson’s Disease. ClinicalTrials.gov, https://classic.clinicaltrials.gov/ct2/show/NCT05611372. Last updated November 10, 2022, accessed 14 April 2024). |


