
The Body, the Brain, the Environment, and Parkinson’s Disease
Abstract
The brain- and body-first models of Lewy body disorders predict that aggregated alpha-synuclein pathology usually begins in either the olfactory system or the enteric nervous system. In both scenarios the pathology seems to arise in structures that are closely connected to the outside world. Environmental toxicants, including certain pesticides, industrial chemicals, and air pollution are therefore plausible trigger mechanisms for Parkinson’s disease and dementia with Lewy bodies.
Here, we propose that toxicants inhaled through the nose can lead to pathological changes in alpha-synuclein in the olfactory system that subsequently spread and give rise to a brain-first subtype of Lewy body disease. Similarly, ingested toxicants can pass through the gut and cause alpha-synuclein pathology that then extends via parasympathetic and sympathetic pathways to ultimately produce a body-first subtype.
The resulting spread can be tracked by the development of symptoms, clinical assessments, in vivo imaging, and ultimately pathological examination. The integration of environmental exposures into the brain-first and body-first models generates testable hypotheses, including on the prevalence of the clinical conditions, their future incidence, imaging patterns, and pathological signatures.
The proposed link, though, has limitations and leaves many questions unanswered, such as the role of the skin, the influence of the microbiome, and the effects of ongoing exposures. Despite these limitations, the interaction of exogenous factors with the nose and the gut may explain many of the mysteries of Parkinson’s disease and open the door toward the ultimate goal –prevention.
Twenty years ago, Braak and colleagues postulated that Parkinson’s disease (PD) begins outside the brain through “neuroinvasion” by an exogenous, unknown pathogen [1]. In the seminal paper, they indicated that the neuropathology of PD begins simultaneously in two sites: the dorsal motor nucleus of the vagus nerve and the olfactory system (olfactory bulb and anterior olfactory nucleus). They hypothesized that an unidentified pathogen passes the mucosal barrier of the gastrointestinal tract and enters the central nervous system via pre-ganglionic parasympathetic motor neurons. Spread from the olfactory system was deemed less likely.
Over the last two decades, refinements and alternatives to the Braak hypothesis have been developed [2–7]. Among them is a body-first versus brain-first model of Lewy body disorders, under which dementia with Lewy bodies and PD are two forms of the same disease [8, 9]. According to this model, which is supported by multi-modal imaging studies and post-mortem data [10–13], there are two principal ways by which the neuropathology of Lewy body disease spreads (Fig. 1). In body-first Lewy body disease, the initial alpha-synuclein pathology begins in the enteric nervous system and then spreads via the vagus nerve to the brainstem as envisioned by Braak. In addition, pathological propagation occurs via the sympathetic nervous system to the sympathetic trunk, heart, skin, and other organs. Consistent with this spread, body-first disease is characterized by early autonomic dysfunction (including in the heart) and REM sleep behavioral disorder. Motor manifestations (e.g., tremor, bradykinesia) occur later as the pathology spreads rostrally. Because of the bilateral autonomic innervation of the gut, the motor features may be more symmetric and the disease course progressive.
Fig. 1
Spread of Lewy pathology via body-first and brain-first models based on pathology severity profiles from postmortem data. Sacral SC, sacral spinal cord; IML, intermediolateral column; Symp Tr, sympathetic trunk; DMV, dorsal motor nucleus of vagus; LC, locus coeruleus; SN, substantia nigra; Amy, amygdala; OB, olfactory bulb; TrE, transentorhinal cortex; Temp Cortex, temporal cortex; Fron Cortex, frontal cortex; Par Cortex, parietal cortex
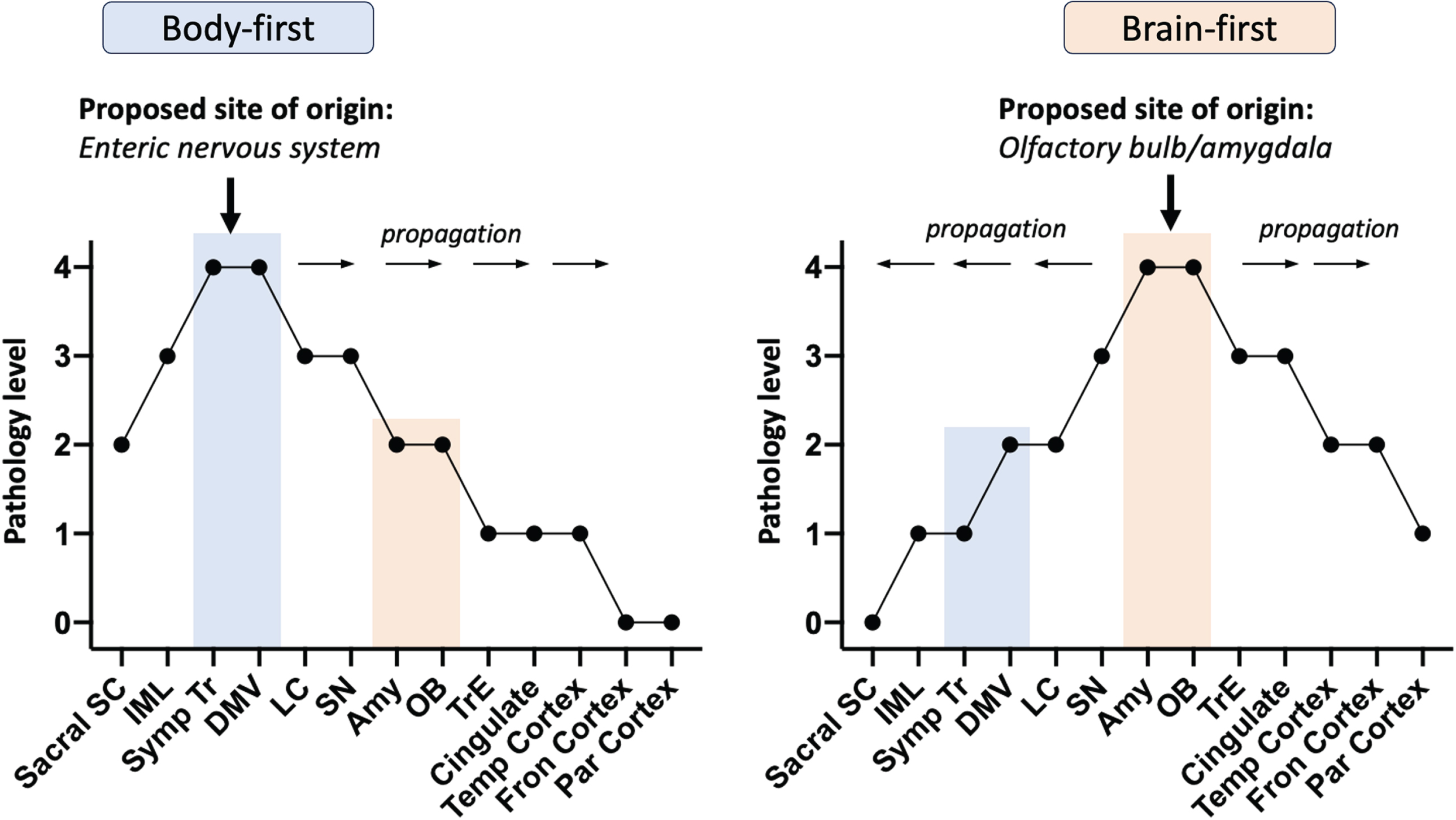
While progression, once diagnosed, can be fast, the prodromal phase of body-first disease is most likely long. Cardiac denervation, which often precedes REM sleep behavioral disorder, is estimated to begin 16–20 years before individuals with body-first Lewy body disease are clinically diagnosed. The initial gut pathology can thus begin decades before an individual comes to the attention of a neurologist [4]. This body-first model of Lewy body disorders is estimated to account for the majority of individuals with dementia with Lewy bodies and around one-third of individuals with PD.
By contrast, in most cases with brain-first Lewy body disease, the pathology is thought to commence in a single locus in the olfactory system, i.e., in one olfactory bulb unilaterally. From there, the pathology extends via neuron-to-neuron propagation to the olfactory and limbic systems and from there to the brainstem and cortex in a rostro-caudal manner leading to a more rapid involvement of the substantia nigra and a shorter prodromal phase. Consistent with this model, the disease is typically unilateral at diagnosis (as 99% of neurons connect to nerve cells in the same hemisphere), has early motor manifestations, and only later has autonomic and sleep dysfunction [4].
In this hypothesis paper, we seek to connect body- and brain-first models of the disease to one of the likely causes of PD, environmental toxicants [14]. In doing so, we focus on three common toxicants that have large effect sizes (e.g., certain pesticides, industrial chemicals) or are widely prevalent (e.g., air pollution) [15–17] and are implicated in alpha-synuclein misfolding and aggregation [18, 19]. Consistent with the brain- and body-first models of the disease, these toxicants are inhaled, ingested, or both (Fig. 2 and Table 1). As Chen and colleagues wrote, “Notably, the nasal cavity and gut are the two anatomic sites where the human mucosal surfaces directly interact with the environment, where inflammation commonly occurs, and where paths to the brain are well established” [20].
Fig. 2
A proposal on how environmental exposure to toxicants may cause either body-first or brain-first Lewy body disease. The size of the brown circles reflects the amount of Lewy pathology in each region. In body-first Lewy body disease, toxicants are most likely ingested through diet and drinking water, but swallowing inhaled toxicants trapped in mucous is also a possibility. In the gut, the toxicants trigger initial a-synuclein misfolding in the enteric nervous system, which then spread centripetally towards the central nervous system via parasympathetic and sympathetic neurons. Consequently, a-synuclein in the peripheral autonomic nervous system and brainstem will be dominating, and less pathology will be found in more rostral structures. The skin could be an alternative trigger site for body-first Lewy body disease. In brain-first Lewy body disease, inhaled toxicants may trigger initial pathology in the olfactory bulb, with subsequent spread to closely related structures. Consequently, a-synuclein in amygdala and olfactory-related structures such as the transentorhinal cortex display most pathology, and less will be found in the brainstem and peripheral autonomic nervous system. Sacral SC, sacral spinal cord; IML, intermediolateral column; Symp Tr, sympathetic trunk; DMV, dorsal motor nucleus of vagus; LC, locus coeruleus; SN, substantia nigra; Amy, amygdala; OB, olfactory bulb; TrE, transentorhinal cortex; PCBs, polychlorinated biphenyls; TCE, trichloroethylene; PCE, perchloroethylene.
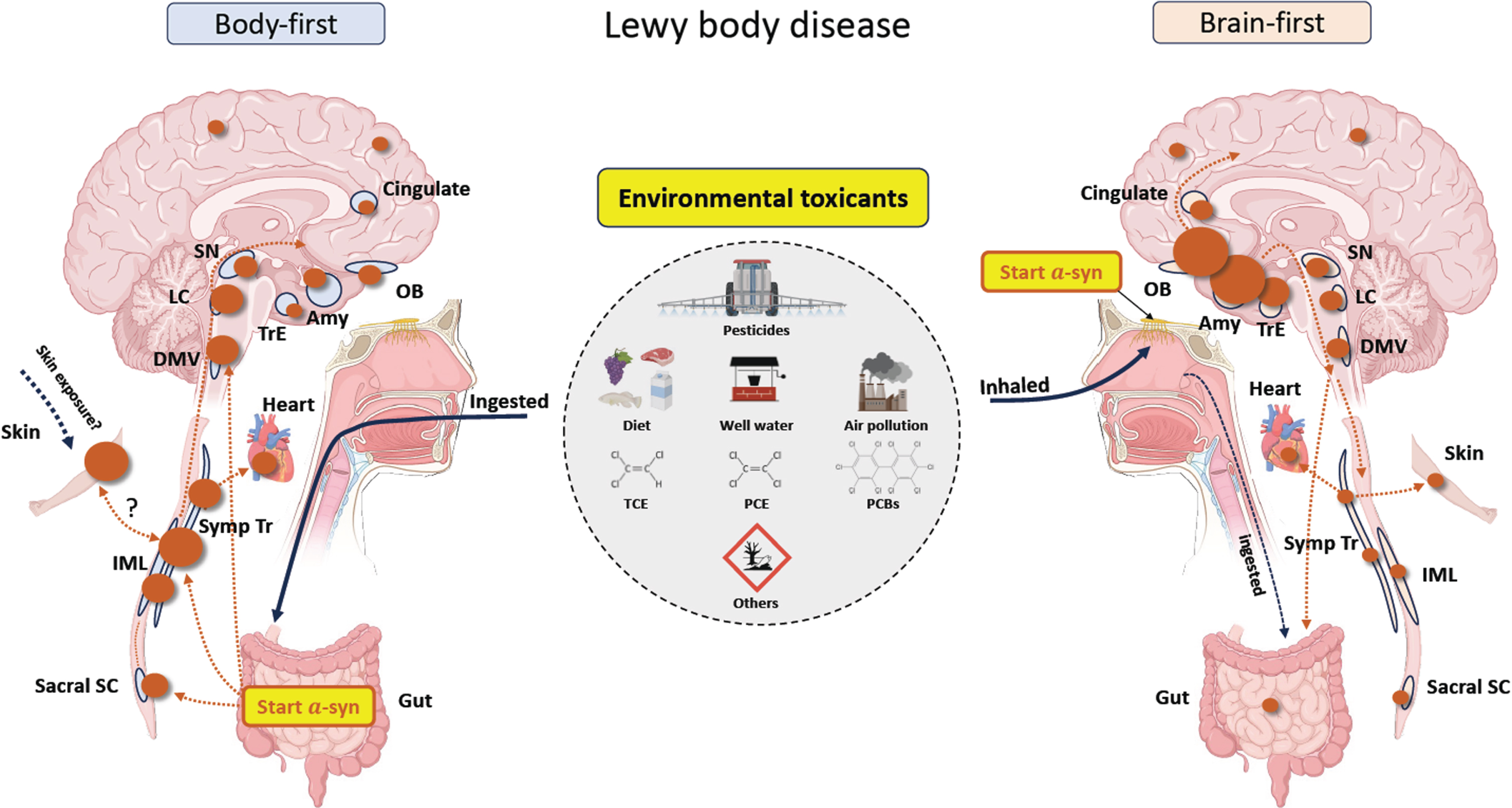
Table 1
Simplified model of the possible link between environmental toxicants and different models of Lewy body disease
| Lewy body disease | ||
| Body-first | Brain-first | |
| Principal disease | Dementia with Lewy bodies | Parkinson’s disease |
| Etiology | ||
| Pesticides | Ingested | Inhaled |
| Dry cleaning chemicals | Ingested | Inhaled |
| Air pollution | Ingested | Inhaled |
| Pathology | ||
| Cardiac denervation | Present early | Normal early |
| Dorsal motor nucleus pathology | Present early | Normal early |
| Degeneration of substantia nigra | Present late, symmetric degeneration | Present early, asymmetric degeneration |
| Clinical Features | ||
| Prodromal period | Very long | Long |
| Autonomic dysfunction | Early | Late |
| REM sleep behavioral disorder | Early | Late |
| Dementia | Early | Late |
| Motor features | More symmetric | More asymmetric |
| Progression post diagnosis | Faster | Slower |
Here we review the evidence, in some cases sparse, that tie toxicants to the body- and brain-first models of Lewy body disease. We begin with detailing how ingested toxicants (e.g., drinking water) may lead to a body-first model (section I) and then explain how inhaled toxicants (e.g., air pollution) can produce a brain-first model of Lewy body disorders (II). We then outline possible explanations for who develops the diseases (section III), propose testable hypotheses (IV), detail limitations (V), and discuss remaining questions and means to answer them (VI). The aim is not to provide an exhaustive review of any of the topics discussed. Rather, we suggest potential mechanisms by which widespread toxicants may be causing increasingly common neurodegenerative diseases, define a framework of ideas for investigating such potential mechanisms, and finally, lay the foundation for preventing these disabling diseases.
IBODY-FIRST MODEL AND INGESTED ENVIRONMENTAL TOXICANTS
Body-first Lewy body disease, including the majority of individuals with dementia with Lewy bodies, could be due (in part) to ingested environmental toxicants coming into contact with the enteric nervous system. Although it may have been unrecognized for years, dementia with Lewy bodies was only recently described [21]. In 1976, Kosaka, a Japanese psychiatrist, and colleagues reported an autopsy of a 65-year-old woman in his country with what he had clinically diagnosed as atypical presenile Alzheimer’s disease with parkinsonism [22, 23]. Her symptoms began at age 56 with involuntary movement of her neck and forgetfulness. Over nine years, she developed severe dementia and could not walk. On autopsy, the investigators reported numerous Lewy bodies in the brainstem and diencephalon and “Lewy-like bodies” in the cerebral cortices [22]. In 1984, Kosaka and colleagues subsequently reported affected individuals in Japan, Germany, and Austria, and twelve years later, the first consensus guidelines for its diagnosis were published [24, 25]. The causes of dementia with Lewy bodies remain to be elucidated but likely include some genetic risk factors, including many GBA mutations [26] and the APOE ɛ4 allele [27, 28], and possibly environmental factors, such as pesticides [29] and industrial chemicals.
Pesticides
Thirty years before Kosaka’s seminal report, following World War II, Japan began using fat-soluble organochlorine pesticides, such as DDT and chlordane, on its rice paddies [30]. This class of pesticides, which are associated with Lewy pathology, contaminated the food supply in Japan and elsewhere [28, 30]. In vitro, the chemicals can stimulate alpha-synuclein fibril formation, disrupt dopamine transport [31], and damage dopaminergic neurons through mitochondrial dysfunction. In humans, their levels in the adipose tissue of the Japanese population rose from the 1940s and peaked in the 1960s when regulatory action in Japan curbed their use [30, 32, 33]. A long latency between exposure to these toxicants and the diagnosis of dementia with Lewy bodies is consistent with the body-first model of Lewy body disease. While possible, the relationship between ingested pesticides and subsequent development of dementia with Lewy bodies is still speculative as little clinical history and no biological assays (to our knowledge) are available from these early reports.
While organochlorine pesticides have not been reported in dementia with Lewy bodies in Japan, Ross and colleagues found that Lewy pathology was nearly doubled in the presence of a metabolite of an organochlorine pesticide called heptachlor in Hawaii where the pesticide was used extensively [34]. Milk, consumption of which has been linked to PD [20, 35], was contaminated with heptachlor in Hawaii. The use of these pesticides is, of course, not limited to Japan and Hawaii but occurred throughout the industrialized world until their toxic effects led many countries to ban them [30]. However, they are still used to combat malaria and produce crops in Africa and beyond where they can be found in the breast milk of nursing mothers [36–39].
In addition to dementia with Lewy bodies, studies have found pesticide exposure to be associated with an increased risk of REM sleep behavioral disorder [40], which is a strong marker of early body-first Lewy body disease and often progresses to dementia with Lewy body disease [41]. A recent Chinese study found that non-occupational exposure to pesticides was associated with a doubling of the risk of REM sleep behavioral disorder [42]. However, conversion to dementia with Lewy bodies has yet to be tied to pesticide exposure [43, 44].
Beyond dementia with Lewy body disease, numerous studies have linked drinking well water to PD [45–47]. Many wells are shallow and thus prone to pollution from pesticides [45]. While drinking well water could also be a proxy for pesticide inhalation (e.g., from local spraying), at least one study has found that even after adjusting for such ambient exposure, the association with possible well water contamination remained [45]. When stratifying large cohorts of individuals with PD into benign and malignant subtypes, agricultural occupation and pesticide exposure are associated with a malignant subtype, characterized by autonomic symptoms, REM sleep behavior disorder, more symmetric parkinsonism, hallucinations, and faster progression [48, 49].
Ingestion of pesticides is thought to initiate peripheral Lewy pathology through several mechanisms. In one study, the oral administration of rotenone in mice promoted the release of alpha-synuclein from enteric and sympathetic neurons, and presynaptic sympathetic neurites took up the released protein and transported it retrogradely within neurons [50]. In those mice, hemivagotomy and partial sympathectomy delayed the appearance of motor but not gastrointestinal symptoms of PD, stopped progression of PD-like pathology into previously connected structures, and hemivagotomy alone prevented cell death in the ipsilateral substantia nigra. The authors concluded, “These results strongly suggest that pesticides can initiate the progression of PD pathology and that this progression is based on the transneuronal and retrograde axonal transport of alpha-synuclein” [50].
Intraperitoneal injection or oral ingestion of pesticides, such as rotenone, also reduces gastrointestinal motility and leads to alpha-synuclein accumulation in the gut [50, 51]. Ingested rotenone in mice directly damages enteric neurons which initiates the accumulation of alpha-synuclein and alters the sympathetic innervation of the gut [50, 52, 53]. Experimental evidence also suggests that rotenone damages enteric glial cell mitochondria, impairs autophagy, and induces inflammation, providing a potential mechanism that could drive protein accumulation, damage neurons, and alter the intestinal epithelium and gut permeability [54].
Industrial chemicals
In addition to pesticides, many other toxicants and chemical pollutants can be ingested, including those used commonly in degreasing and dry cleaning. Trichloroethylene (TCE) and perchloroethylene (PCE) are both simple six-atom molecules—one with three chlorine atoms, the other with four—that have been used for up to a century in consumer, commercial, industrial, and military applications [55, 56]. Case reports have linked occupational use of TCE to Parkinson’s since 1969 [57], but its major source of toxicity may be in the environment.
TCE has contaminated up to one-third of the groundwater in the U.S., including at the Marine base Camp Lejeune in North Carolina. There from 1953 to 1987, service members, civilians, and their families drank water with unsafe levels of TCE. In 2023, Goldman and colleagues found that those stationed at Camp Lejeune had a 70% increased risk of developing PD compared to those at a less polluted base in California [58]. Longitudinal follow-up of this cohort for PD and dementia with Lewy bodies as well as other features of these disorders, such as REM sleep behavioral disorder (not increased in the 2023 study) and autonomic dysfunction (mixed results) will be important [58, 59].
Other industrial chemicals may be ingested and also play a role in body-first models of Lewy body disorders. For example, polychlorinated biphenyls (PCBs) are non-flammable, insulating oils used for building materials and electrical equipment [60] that have chemical similarities to organochlorine pesticides [61] and are persistent halogenated organic pollutants. Although banned in the late 1970s, humans are still exposed to these lipophilic substances through diet (predominately meat and seafood) and ambient air (predominately from indoor evaporation in buildings containing PCBs) [62]. Since PCB accumulates in fat, eating marine animals at the top of the food chain may lead to high exposure [63, 64]. For example, the Inuit in Greenland, exposed through marine diet, exhibit elevated PCB plasma concentrations [65, 66], and the age-adjusted PD prevalence is higher among them (188 per 100,000) than on a Danish island (98 per 100,000) [67]. In addition, whale-meat consumption in the Faroe Islands in the North Atlantic has also been associated with increased risk of PD [68].
In post-mortem brain tissue, one of five tested PCB variants was significantly elevated in the caudate nucleus of individuals with PD [60]. In a separate part of that study, depigmentation of the substantia nigra in women (but not men) with PD was also associated with elevated concentrations of PCBs. A similar effect has been found in mice fed PCBs where striatal dopamine transporter loss was associated with high exposure [69]. In addition, mice that ingested PCBs accumulate the chemical primarily in their hearts, spleens, and large intestines suggesting that exposure through the gastrointestinal route could predispose to a body-first subtype [70]. Research on PD risk from PCBs has been inconsistent [71], but their high levels and continued persistence in the food supply raise concerns for their role in body-first Lewy body disorders [72–74].
Air pollution
While air pollution is generally inhaled, it can also be ingested by swallowing contaminated mucus [75, 76], eating contaminated food from animals (e.g., fish, cows) that feed on contaminated waters or plants, or drinking water with toxic air pollutants [77]. To our knowledge, assessments of such exposures have not been conducted for dementia with Lewy bodies or PD.
IIBRAIN-FIRST MODEL AND INHALED ENVIRONMENTAL TOXICANTS
Inhalation may be an even more compelling means of exposure for PD and the brain-first model of Lewy body disorders. Certain pesticides, industrial chemicals, and air pollution are all readily inhaled.
Pesticides
Chief among these inhaled toxicants is paraquat, which has been linked to PD in academic studies [17, 78, 79] and apparently the manufacturer’s own research [80]. In 2011 Tanner and colleagues found that farmers (primarily) who worked with paraquat had a 150% increased risk of having PD [17]. Occupational exposure likely leads to inhalation [81] and possibly a brain-first model of the disease. Consistent with this notion, all 23 of those with PD who were exposed to paraquat had an asymmetric onset, and all (who had tried) had a favorable response to dopaminergic therapy [17]. In the study, which also evaluated exposure to other pesticides, 115 individuals were diagnosed with PD, but no diagnoses of dementia with Lewy bodies were reported. A separate case report of PD in a farmer with antecedent exposure to paraquat also reported asymmetric onset of motor symptoms [82].
Replicating human inhalation exposure in in vivo models is challenging and requires controlled environments and proper safety measures, but emerging studies suggest that inhalation may be an important route of exposure. A recent investigation in mice found that inhaled paraquat “entered all examined regions of the brain (including the striatum and midbrain), with the highest concentration seen in the olfactory bulb” [81].
Industrial chemicals
TCE and PCE can be inhaled via occupational or environmental exposure. However, clinical descriptions of individuals with confirmed inhalation exposure through work are limited. Because both chemicals are highly volatile, they can evaporate from contaminated water and be inhaled during cooking and bathing. Like radon, they can also evaporate from contaminated soil and groundwater and pollute the indoor air of homes, barracks, schools, and hospitals [83].
In 2008, Gash and colleagues reported on 30 individuals who worked at a plant where TCE was used to degrease metal gauges [84]. Two men who placed the gauges in the open vat of TCE and one woman who dried them all developed PD. In addition, 14 of the 27 workers who were further from the source and thus presumably had exposure via inhalation “displayed many features of parkinsonism, including significant motor slowing.”[84]. The predominant features described in the cohort were motor. Case reports of PD following occupational exposure TCE have included asymmetric motor findings [56, 85, 86], and one reported asymmetric reduced uptake on a DAT scan [86]. Cognitive deficits have not been noted, and in at least one case, cognition was normal [86].
In experimental models, TCE inhalation appears to be more potent than oral ingestion in causing dopaminergic neurodegeneration. Animal studies that expose rodents to high levels of TCE via oral gavage, ranging from 200–1000 mg/kg, typically demonstrate the selective loss of nigrostriatal dopaminergic neurons over six weeks to three months [87]. In contrast, inhalation exposure in lower doses (up to 50 ppm TCE in rats and 100 ppm in mice) over similar time periods caused equivalent or greater dopaminergic neurodegeneration. Forgoing first-pass hepatic metabolism, olfactory and respiratory pathways may provide a more direct and potent route for TCE to reach the brain and damage dopaminergic neurons [88].
While likely less common than ingestion, inhalation of PCBs may also predispose to PD. In one study, researchers exposed ferrets to low concentrations of PCBs in the ambient air for five years [89]. They found that the olfactory bulb levels of PCBs were almost four times greater than in the remaining brain and double that in the peripheral tissues. The authors concluded that like heavy metals and solvents, “airborne PCBs enter olfactory sensory neurons directly and are transported to the olfactory bulb where they accumulate” [89].
Air pollution
Air pollution is a third toxicant that can readily be inhaled and thus implicated in the brain-first model of Lewy body disease. While synthetic pesticides and industrial solvents are largely products of the 20th century, air pollution predates Dr. Parkinson’s seminal description of the disease. Industrial air pollution, including the infamous London fog, was well established (Fig. 3) by the time Dr. Parkinson wrote his essay in 1817. London’s air quality in 1800 was twenty times worse than it is now, on par with the world’s most polluted cities (e.g., Delhi, India) today, and akin to what many North Americans experienced from Canadian wildfires in the summer of 2023 [90].
Fig. 3
Air pollution in London, 1700 –2016, and in Delhi, 1997–2010. Source: Our World in Data [146, 180].
![Air pollution in London, 1700 –2016, and in Delhi, 1997–2010. Source: Our World in Data [146, 180].](https://ip.ios.semcs.net:443/media/jpd/2024/14-3/jpd-14-3-jpd240019/jpd-14-jpd240019-g003.jpg)
Research increasingly links high levels of air pollution, especially particulate matter, to neurodegenerative diseases, including Parkinson’s disease [15, 91–93]. The smallest pieces of this dirt and soot can be 2.5 microns or smaller in diameter and penetrate nasal, gastrointestinal, and the blood-brain barriers [94]. Such ultra-fine particulate matter can carry along with it toxic heavy metals, such as iron, which are found in higher concentrations in the brains of individuals with PD [95, 96].
In his essay on six individuals, Parkinson describes the motor features of the disease. While determining the cause of these initial cases may be impossible (the first was a gardener who may have been exposed to naturally occurring pesticides [97]), all were almost certainly exposed to toxic levels of air pollution. In two of the three individuals he examined, he noted asymmetry in tremor onset. In only one does he comment on non-motor features of the disease, constipation, which was a late symptom in the last case he reported [98]. His famous description of the shaking palsy also found “the senses and intellects being uninjured” [98].
While air pollution has improved substantially in England since the times of Dr. Parkinson, other areas of the world have been similarly affected. In the early 1990s, Mexico City was the most polluted city on earth and had worse air quality than the most polluted cities in China today [99]. So bad was the air that birds reportedly fell dead from the sky. There, Calderón-Garcidueñas and colleagues examined the brains of children and young adults (ages 11 months to 40 years) who died prematurely from gun violence or car accidents. She found breakdown of the nasal epithelium with accumulation of particulate matter around nasal epithelial cells [100]. In addition, she saw Lewy neurites in the olfactory bulb of toddlers and in 68% of children [101]. She also found alpha-synuclein immunoreactivity in the brainstem nuclei of three young individuals (ages 17 to 35 years) [100].
IIITHE POSSIBLE FACTORS DETERMINING WHO DEVELOPS LEWY BODY DISORDERS
Given that these environmental toxicants are widespread and not everyone has PD or dementia with Lewy bodies, additional factors must determine who among the exposed develops a Lewy body disease and who does not. At least three sets of factors may be important: (1) exposure, (2) interactions, and (3) modifiers.
The timing, dose, and duration of exposure are key to determining who is most likely to develop the disease. In most cases, exposure occurs decades before diagnosis. For example, at Camp Lejeune, service members were diagnosed with PD on average 34 years (range 5 to 45) after likely exposure [58]. A three-year-old boy who lived at the base was diagnosed when he was 36 [56]. A twin study found that the lag between occupational or hobby exposure to TCE and PD diagnosis was 10 to 40 years [16].
The exposure can happen as a child or as an adult [102]. Pesticides, TCE, and tiny particulate matter can all cross the placenta suggesting that exposure could even begin in utero. TCE [103] and pesticides [39, 104] have been found in breast milk, and infants may be more susceptible to their toxic effects. Calderón-Garcidueñas found Lewy pathology in the brains of deceased teenagers exposed to high levels of air pollution.
The dose and duration of exposure are also likely important. Because most epidemiological studies have been retrospective, dose has been hard to quantify. Gatto and colleagues found that “high” levels of pesticide exposure in well water were generally more strongly associated with PD than lower levels [45]. In addition, exposure to more water-soluble pesticides also increased the risk of PD, and among those who drank well water, those with PD also had longer exposure (18 years) than those without (14 years) [45]. For TCE, the dose has been rarely quantified. At Camp Lejeune, the level of TCE contamination was more than 70 times the U.S. Environmental Protection Agency’s maximum contaminant level where the average duration of likely exposure was just over two years [58]. By contrast, other studies of hobby and occupational exposure to TCE have reported exposure durations of 15 to 25 years [16, 84].
The relevant dose and duration of exposure for air pollution remains uncertain [105]. A Danish study of traffic-related air pollution suggested that higher levels of air pollution (as measured by nitrogen dioxide concentrations) were more likely to be associated with PD [106]. A review of air pollution studies found that the exposure duration ranged from four to 32 years [91]. Most studies of air pollution and PD to date have suggested modest effect sizes [91, 105–107], but this may be because few studies [108, 109] in more densely polluted regions (e.g., early 20th century London, cities in India today [105]) have been conducted to date.
In addition to measures of exposure, interactions among genetic and other environmental factors are likely important [110, 111]. Numerous studies have linked GBA mutations across the spectrum of Lewy body disorders, especially those with cognitive impairment [26, 112]. One study in Greece found that individuals who carried GBA mutations and had PD were more likely to have bilateral onset of the disease (in 21% of cases) and exposure to pesticides [113]. GBA mutations also may predispose to a body-first pathological spread of Lewy bodies [4]. The APOE ɛ4 allele is also tied to an increased risk dementia with Lewy bodies [27] and may exacerbate the effects of air pollution, a known risk factor for Alzheimer’s disease [114–116]. While still early, such an association could help explain the predominance of Alzheimer’s pathology in individuals with dementia with Lewy bodies [117, 118].
Laboratory studies have also shown interactions between LRRK2 and environmental pollutants [119–121]. TCE, rotenone, and other toxicants linked to PD activate LRRK2 kinase activity in a manner similar to that seen with LRRK2 mutations [120, 122, 123]. The extent to which environmental exposures increase disease risk in this population remains to be determined [56]. Finally, mutations in genes encoding for the proteins that metabolize pesticides are likely important in determining who develops PD [124–126].
Toxicants also interact with other environmental factors. Some of these environmental-environmental interactions may be inherent to the exposure itself (e.g., multiple constituents of air pollution [15]) while others (e.g., pesticides and head trauma) may be entirely separate [127, 128].
Finally, chance may play a role in who develops the disease. As is likely with cancer [4], the initial misfolding of alpha-synuclein and subsequent development of Lewy body disease may be in part a stochastic event.
Other factors may modify the disease course. Aging, the time-related decline in physiological functioning, is likely critical [129, 130]. Aging reduces protein clearance [131], impairs lysosomal function [132], and contributes to oxidative stress [133], all of which may contribute to Lewy body disorders. Additional important modifiers likely include diet [134], exercise [135], other medical conditions (e.g., diabetes [136]), sex [137], concomitant co-pathology (e.g., Alzheimer’s), intercurrent illnesses, stress, and even ongoing toxicant exposure.
IVTHE HYPOTHESES
A paradigm by which environmental toxicants are ingested and inhaled and then induce a body- or brain-first Lewy body disease generates several testable hypotheses. These include the following:
Hypothesis 1: The prevalence of Lewy body disorders will vary based on the exposure to environmental toxicants in a given region –The age-adjusted prevalence of PD between countries varies 5-fold [138] and within countries 10-fold [139]. This variability may be a function of differences in environmental toxicants. In rural areas, for example, the prevalence of PD is almost perfectly correlated with pesticide use [140, 141]. The principal route of exposure, however, may vary. Some pesticides (e.g., paraquat) may exhibit their toxicity mainly through inhalation leading to a predominance of PD while others (e.g., organochlorines) may be ingested leading to more dementia with Lewy bodies.
Testing this hypothesis may be increasingly feasible as many toxicants are now measured. These measures on farms, in the air, water, food supply, and breast milk will vary between and within countries and could be evaluated alongside high quality incidence data. Geospatial mapping could even examine the prevalence of Lewy body disorders around potentially high risk areas, such as golf courses [142] (e.g., where chlorpyrifos and other pesticides are used) and dry cleaners (e.g., PCE).
Within a defined geography, the prevalence of disease may vary based on the exposure. Those with contaminated water (e.g., from private wells) will be more likely to have body-first Lewy body disorders. Those experiencing vapor intrusion [143–145] or high levels of air pollution could be at risk for brain-first disorders, especially PD [92].
Hypothesis 2: The future incidence of Lewy body disorders will decline in areas of the world with falling levels of environmental toxicants –Countries in western Europe that have banned neurotoxic pesticides, curbed their use of TCE, and improved their air quality are poised to see reductions in the rise of Lewy body diseases and even future declines. As early evidence for this hypothesis, the increase in the age-adjusted prevalence of PD appear to be much lower in western Europe (8.4%) than the world as a whole (21.7%) [138].
Hypothesis 3: The future incidence of Lewy body disorders will rise in areas of the world with increasing levels of toxicants –Historically, sub-Saharan Africa has had little pesticide use, limited air pollution [146, 147], and low prevalence of PD [138]. However, pesticide use in Africa has nearly tripled in the past 30 years [148]. Many that are used (e.g., organochlorines [36]) have been banned by other countries based on safety concerns [149]. This rise is especially concerning because ∼60% of the African population is engaged in agricultural production [147, 150]. Without better safeguards (e.g., on pesticides used, protection for those using them [151], and prevention of water and food contamination), many Africans could be a greater risk for Lewy body disorders in the future.
Hypothesis 4: Individuals with Lewy body disorders will display a certain imaging pattern depending on the route of toxicant exposure –Cases where ingested toxicants contribute to the disease will display a typical body-first imaging pattern including decreased cardiac sympathetic innervation (e.g., [123I]MIBG scintigraphy) and more symmetric dopaminergic degeneration. Conversely, cases where inhaled toxicants cause the disease will display a typical brain-first imaging pattern including relatively preserved cardiac sympathetic innervation and more asymmetric dopaminergic degeneration at diagnosis. These imaging patterns could then be correlated with pathological findings ideally in prospective cohort studies.
Hypothesis 5: Animal models will demonstrate a body-first pathological spread for ingested toxicants and a brain-first for inhaled ones
In laboratory animals exposed to toxicants, phenotypic similarities to humans can be studied. For example, ingested toxicants should lead to early accumulation of α-synuclein aggregates in the enteric and autonomic nervous system, early degeneration of the sympathetic and parasympathetic systems, and symmetric dopamine loss. In contrast, inhaled toxicants should lead to more rapid involvement of the nigrostriatal dopamine system followed by lower brainstem pathology and autonomic degeneration. Of note, if stochasticity plays a significant role in the onset of Lewy pathology in humans, this facet may be difficult to emulate in animal models. Thus brain-first animal models featuring inhaled toxicants might not show the asymmetric dopamine loss typically seen in many humans [152, 153].
Animal models also provide a convenient means to test gene-environment interactions that may drive PD phenotype. For example, genetic risk factors such as GBA could exacerbate Lewy pathology and result in cognitive dysfunction in a body- or brain-first mechanism depending on the exposure and route.
VLIMITATIONS
The hypothesis that toxicants, ingested and inhaled, enter the gut or the nose and trigger alpha-synuclein pathology that then spreads to structures as predicted by the body- and brain-first models is incomplete. Although much evidence supports that alpha-synuclein can misfold and then propagate from cell to cell in a prion-like manner, this pathogenic mechanism has not been conclusively proven to occur in the human disease. Alternative hypotheses have been proposed such as the threshold model [5], and the differences between that model, alternative hypotheses, and the body- and brain-first model were recently reviewed [4].
On the toxicant front, few individuals will have exposure limited solely to ingestion or inhalation. For example, a farmer working with a pesticide could inhale it in the field, swallow it in her saliva, absorb it through her skin, bring it into her home on her shoes where she could inhale it again, eat in her food, and drink it from a well.
Even presumed exposure to TCE or PCE via vapor intrusion can have multiple routes. For example, the indoor air in some apartments above dry cleaners are known to have unsafe levels of PCE [154]. However, because PCE is fat soluble, it can make its way into the butter and cheese in an apartment’s refrigerator [155] leading to both inhalation and ingestion.
Toxicants can also be absorbed via the skin [156], lung [157], and systemically. For example, organochlorine pesticides can enter the systemic circulation and because they are lipophilic, cross the blood-brain barrier [158]. However, based on the earliest distribution of Lewy pathology, a contribution from systemic circulation of toxicants seems less influential in the initiation sites of alpha-synuclein aggregation. Post-mortem cases with spatially very restricted Lewy pathology almost always display pathology in either the olfactory bulb or the autonomic nuclei innervating the gut [12, 159]. If PD were commonly caused by toxicants or other factors in systemic circulation that cross the blood-brain barrier, the first isolated pathology could begin in diverse locations such as the neocortex, substantia nigra, cingulum, or other regions. However, such cases are very rare or even non-existent [3, 13, 159]. This suggests that the pathology in most cases first arises in the olfactory bulb or enteric and autonomic nervous system at the “body-surface” interface [20].
In addition to multiple routes of exposure, almost all of us are exposed to multiple toxicants at multiple points in time. Many farmers are exposed to multiple pesticides at varying doses, perhaps from infancy until death. The same is likely true for most of us. In Italy in 1994, for example, three-quarters of a sample of the general population had detectable levels of TCE in their blood and urine [160].
The scarcity of data to substantiate some of our contentions is another limitation. This shortcoming is partly attributed to the failure to systematically assess a systemic disease. We have known now for at least two decades that PD may begin outside the brain [1, 10, 161], but whole body evaluations either by imaging [10, 161, 162] or autopsy are extremely limited [3, 159, 163, 164]. Similarly, we have known for a century that only 15% of individuals have a family history of the disease [165] and for a generation that environmental factors are a major cause of the disease [166], yet we have fundamentally underinvested [167, 168] in and under-investigated [169] the role environmental toxicants are playing.
VIUNANSWERED QUESTIONS
Table 2
Research needs to investigate the relationship between environmental toxicants and the body- and brain-first models of Lewy body diseases
| Research need | Rationale | Gaps/areas addressed | Proposed solution |
| Basic epidemiology studies | •Despite high prevalence, we only have crude measures of incidence of PD and dementia with Lewy bodies | •Geographic variation in prevalence of Lewy body disorders | •High quality incidence studies that track changes over time |
| •Few studies have examined incidence over time [181, 182], and those that have are limited to small (likely non-representative) areas | •Estimating the future incidence of PD | •Studies that cover large portions of the world, including areas exposed to different toxicants (e.g., air pollution, pesticides) | |
| Whole body investigations | •Given that the origins of PD may be outside the brain, systemic investigations are needed [183] | •Systemic assessment of a systemic disease | •Collection and analysis of brains and samples from other organs including nasal epithelium, gut, skin, sympathetic ganglia, the spinal cord, heart, and lungs from affected and unaffected populations [13, 159, 163] |
| •In vivo imaging of different organ systems and the peripheral and central nervous system can inform the likely routes of spread of the disease | •Initial location, spread, and extent of alpha-synuclein pathology | ||
| •Effect of multiple routes of exposure to toxicants | |||
| •Role of the skin, microbiome | |||
| Prospective cohort studies | •The time course of most PD and dementia with Lewy bodies is likely decades, but few studies have followed individuals over such time periods | •Initial location, spread, and extent of alpha-synuclein pathology | •Prospective, longitudinal studies of individuals likely exposed to toxicants (e.g., Marines at Camp Lejeune) |
| •Effect of multiple exposures to toxicants | •Detailed clinical assessments and their chronology, imaging, genetics, high resolution exposome (metabolomics), and post-mortem examination of cohorts | ||
| •Effects of current and future exposures | |||
| Biological measures of toxicant exposure | •We have measures of genetic risk; we need measures of toxicant exposure | •Effects of different toxicants on the prevalence of Lewy body disorders | •Assays in adipose tissue (e.g., for fat-soluble toxicants), breast milk, blood, urine, brain, including of likely metabolites |
| •Many individuals are unaware of their exposure (e.g., TCE) limiting the value of surveys | •Identifying individuals at risk for Lewy body disorders | ||
| •Predicting the future incidence of PD | |||
| Animal studies | •The mechanisms by which environmental toxicants contribute to Lewy body disease are still uncertain | •Biological response to different toxicants and different routes of exposure | •Evaluation of different routes of exposure for a broad range of toxicants |
| •The importance of dose, route, and timing of exposure and resulting pathological spread of disease requires further characterization | •Initial location, spread, and extent of alpha-synuclein pathology | •Sequential studies evaluating exposure at different time points and means to prevent or slow pathological spread | |
| •Optimal timing of therapeutic intervention in relationship to toxicant exposure and resulting pathology remains to be established | |||
| •Effect of exposure to toxicants at multiple points in time | |||
| •Role of the skin | |||
| •Means of slowing progression |
PD, Parkinson’s disease.
This paper leaves many important questions unanswered or unaddressed including the following:
1. Role of the skin –In addition to the nose and gut, the principal toxicants discussed here, pesticides, TCE, and air pollution, can enter via the lung and the skin [75]. A large portion of individuals with PD have misfolded alpha-synuclein in their skin is present early in the disease course [170]. The cause and implications for this pathology remain to be elucidated.
2. Influence of the microbiome – The role the gut and nasal microbiome play in developing Lewy body disorders is under active investigation [162, 171, 172]. Toxicants and many other exogenous and endogenous (e.g., genetic mutations) factors may influence its composition, resulting in microbiota dysbiosis including the reduction in short-chain fatty acid producing species [173] observed in PD and other chronic disorders. These changes could contribute to inflammation and dysfunction at the gut-brain axis [174]. Gene-environment interactions at the gut-brain axis also likely influence Lewy pathology although the direction of association is unclear. For example, elevated levels of opportunistic pathogens found in the PD gut microbiome correlated with SNCA genotype, suggesting that alpha-synuclein could have a bi-directional effect on microbiome dysbiosis [175]. The lung microbiome could also play a role, especially with inhaled toxicants [176].
3. Contributing factors –In most cases, individuals will be exposed to toxicants through the nose and the gut. The level and timing of these exposures, the interactions, and the steps required for an exposure to result in pathology are uncertain. Similarly, whether these factors differ based on the toxicant is unknown. Other remaining questions include the following: Does alpha-synuclein misfolding take place multiple times in multiple locations? If yes, how many times and in what locations? Which factors determine whether a locus of Lewy pathology propagates to other regions and at what rate? Are other sources of spread hiding in the background? Answers to the above will advance our understanding of the etiology of Lewy body diseases and provide powerful guidance on how to prevent and treat them.
4. Effect of ongoing exposure –The first recommendation to any smoker with lung cancer is to stop smoking. However, such advice is rarely given to individuals with Lewy body disorders. Reducing current or future exposure to toxicants could theoretically benefit affected individuals as continued exposure could accelerate the disease course. For example, high levels of air pollution may increase the risk of hospitalization among individuals with PD [177, 178].
4. A Korean study also found that individuals with PD who had undergone gastrectomy, which typically entails full truncal vagotomy, showed attenuated dopaminergic loss in the posterior putamen and a slower increase in the dose of PD medications compared to non-gastrectomized, matched individuals [179]. This finding suggests a benefit of gastrectomy plus vagotomy even after the diagnosis of PD is made. The result could be compatible with the elimination of an ongoing pathological process affecting the brain via the vagus nerve. Although surgical vagotomy is not recommended, such results indicate that it might be possible to modify the course of PD by reducing ongoing or future exposure to toxicants or ascending pathology. To our knowledge, there has been little or no research in experimental animal models examining this interesting prospect.
To answer the questions posed here, address the limitations presented, and test the proposed hypotheses will require additional research (Table 2) [169]. Among the needs are high quality studies on the incidence of PD, which should be tracked serially for different regions of the world. Whole body investigations of individuals with Lewy body disorders are also lacking. Such studies, perhaps as part of prospective cohort studies with body imaging, can help us define the timing and route of pathological spread of these diseases.
Biological measures of toxicants are also greatly needed. The ability to quantify smoking (e.g., pack-years) helped define the risk of lung cancer and related conditions. However, such measures are lacking for Lewy body disorders and are especially important since many are unaware of their exposure. Finally, animal studies can help evaluate the effects of toxicants, their relevant doses, different routes of exposure, and interactions with genetic factors.
CONCLUSION
In 2007, Hawkes, Del Tredici, and Braak wrote, “The enigma of PD is to explain why a condition that probably begins in anterior olfactory structures and portions of enteric nerve cell plexuses, has a presymptomatic period of several years, possibly decades, during which time the disease-related destruction inexorably advances retrogradely along fiber tracts in the brainstem and anterogradely along olfactory pathways in a predictable sequence to destroy the substantia nigra and then initiate a disease that runs a further course of approximately 15 additional years” [2]. Here, we outline how environmental toxicants –inhaled and ingested –could theoretically result in pathology that passes along these brain-first or body-first pathways to give rise to clinical Lewy body disorders decades later. While the enigma is far from solved, its mystery may be unfolding and a path toward prevention emerging.
ACKNOWLEDGMENTS
We thank Sarrah Hussain for her assistance in preparing the manuscript for submission.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
Dr. Dorsey has received honoraria from the American Neurological Association, Kairos Cinema, Texas Neurological Society, and University of Toronto; received compensation for consulting services from Abbvie, Adivo Associates, Bial-Biotech Investments, Inc., Biohaven Pharmaceuticals, Biosensics, Boehringer Ingelheim, California Pacific Medical Center, Caraway Therapeutics, Cerevance, Deallus, Genentech/Roche, Grand Rounds, HMP Education, Mediflix, MedRhythms, Medscape, Merck, Mitsubishi Tanabe Pharma, NACCME, NeuroDerm, NIH, Novartis, Otsuka, PRIME Education, Sanofi, Seelos Therapeutics, Sutter Home, and WebMD; research support from Averitas Pharma, Biogen, Burroughs Wellcome Fund, Department of Defense, Michael J. Fox Foundation, National Institutes of Health, PhotoPharmics, Roche, and Thomas Golisano Foundation; editorial services for Karger Publications; stock in Included Health and Mediflix and ownership interests in Synpaticure and SemCap.
Dr. De Miranda is funded by the National Institutes for Environmental Health Sciences (R00ES029986), the US Department of Defense (TX220241), the Parkinson’s Foundation, and the American Parkinson’s Disease Association. Dr. De Miranda is a scientific consultant for the Miller Firm.
Dr. Horsager is funded by the Lundbeck Foundation and has received compensation for lecture from Abbvie.
Dr. Borghammer is funded by the Lundbeck Foundation, Michael J Fox Foundation, Danish Parkinson’s disease association, Jascha Foundation, Riisfort Foundation, Independent Research Fund Denmark, Toyota Foundation, and F. Hoffmann-La Roche. He has received speakers’ fees from Eisai, Medtronic, and Roche.
REFERENCES
[1] | Braak H , Rüb U , Gai WP , Del Tredici K ((2003) ) Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm 110: , 517–536. |
[2] | Hawkes CH , Del Tredici K , Braak H ((2007) ) Parkinson’s disease: A dual-hit hypothesis. Neuropathol Appl Neurobiol 33: , 599–614. |
[3] | Beach TG , Adler CH , Lue L , Sue LI , Bachalakuri J , Henry-Watson J , Sasse J , Boyer S , Shirohi S , Brooks R , Eschbacher J , White CL 3rd , Akiyama H , Caviness J , Shill HA , Connor DJ , Sabbagh MN , Walker DG ((2009) ) Unified staging system for Lewy body disorders: Correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol 117: , 613–634. |
[4] | Borghammer P ((2023) ) The brain-first vs. body-first model of Parkinson’s disease with comparison to alternative models. J Neural Transm (Vienna) 130: , 737–753. |
[5] | Engelender S , Isacson O ((2017) ) The threshold theory for Parkinson’s disease. Trends Neurosci 40: , 4–14. |
[6] | Gonzalez-Rodriguez P , Zampese E , Surmeier DJ ((2020) ) Selective neuronal vulnerability in Parkinson’s disease. Prog Brain Res 252: , 61–89. |
[7] | Surmeier DJ , Obeso JA , Halliday GM ((2017) ) Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci 18: , 101–113. |
[8] | Borghammer P , Van Den Berge N ((2019) ) Brain-first versus gut-first Parkinson’s disease: A hypothesis. , S281-S. J Parkinsons Dis 9: , 295. |
[9] | Weintraub D ((2023) ) What’s in a name? The time has come to unify Parkinson’s disease and dementia with Lewy bodies. Mov Disord 38: , 1977–1981. |
[10] | Horsager J , Andersen KB , Knudsen K , Skjærbæk C , Fedorova TD , Okkels N , Schaeffer E , Bonkat SK , Geday J , Otto M , Sommerauer M , Danielsen EH , Bech E , Kraft J , Munk OL , Hansen SD , Pavese N , Göder R , Brooks DJ , Berg D , Borghammer P ((2020) ) Brain-first versus body-first Parkinson’s disease: A multimodal imaging case-control study. Brain 143: , 3077–3088. |
[11] | Borghammer P , Horsager J , Andersen K , Van Den Berge N , Raunio A , Murayama S , Parkkinen L , Myllykangas L ((2021) ) Neuropathological evidence of body-first vs. brain-first Lewy body disease. Neurobiol Dis 161: , 105557. |
[12] | Borghammer P , Just MK , Horsager J , Skjærbæk C , Raunio A , Kok EH , Savola S , Murayama S , Saito Y , Myllykangas L , Van Den Berge N ((2022) ) A postmortem study suggests a revision of the dual-hit hypothesis of Parkinson’s disease. NPJ Parkinsons Dis 8: , 166. |
[13] | Raunio A , Kaivola K , Tuimala J , Kero M , Oinas M , Polvikoski T , Paetau A , Tienari PJ , Myllykangas L ((2019) ) Lewy-related pathology exhibits two anatomically and genetically distinct progression patterns: A population-based study of Finns aged 85. Acta Neuropathol 138: , 771–782. |
[14] | Goldman SM ((2014) ) Environmental toxins and Parkinson’s disease. Annu Rev Pharmacol Toxicol 54: , 141–164. |
[15] | Murata H , Barnhill LM , Bronstein JM ((2022) ) Air pollution and the risk of Parkinson’s disease: A review. Mov Disord 37: , 894–904. |
[16] | Goldman SM , Quinlan PJ , Ross GW , Marras C , Meng C , Bhudhikanok GS , Comyns K , Korell M , Chade AR , Kasten M , Priestley B , Chou KL , Fernandez HH , Cambi F , Langston JW , Tanner CM ((2012) ) Solvent exposures and Parkinson disease risk in twins. Ann Neurol 71: , 776–784. |
[17] | Tanner CM , Kamel F , Ross GW , Hoppin JA , Goldman SM , Korell M , Marras C , Bhudhikanok GS , Kasten M , Chade AR , Comyns K , Richards MB , Meng C , Priestley B , Fernandez HH , Cambi F , Umbach DM , Blair A , Sandler DP , Langston JW ((2011) ) Rotenone, paraquat, and Parkinson’s disease. Environ Health Perspect 119: , 866–872. |
[18] | Rokad D , Ghaisas S , Harischandra DS , Jin H , Anantharam V , Kanthasamy A , Kanthasamy AG ((2017) ) Role of neurotoxicants and traumatic brain injury in α-synuclein protein misfolding and aggregation. Brain Res Bull 133: , 60–70. |
[19] | Wills J , Credle J , Oaks AW , Duka V , Lee JH , Jones J , Sidhu A ((2012) ) Paraquat, but not maneb, induces synucleinopathy and tauopathy in striata of mice through inhibition of proteasomal and autophagic pathways. PLoS One 7: , e30745. |
[20] | Chen H , O’Reilly E , McCullough ML , Rodriguez C , Schwarzschild MA , Calle EE , Thun MJ , Ascherio A ((2007) ) Consumption of dairy products and risk of Parkinson’s disease. Am J Epidemiol 165: , 998–1006. |
[21] | Okazaki H , Lipkin LE , Aronson SM ((1961) ) Diffuse intracytoplasmic ganglionic inclusions (Lewy type) associated with progressive dementia and quadriparesis in flexion. J Neuropathol Exp Neurol 20: , 237–244. |
[22] | Kosaka K , Oyanagi S , Matsushita M , Hori A ((1976) ) Presenile dementia with Alzheimer-, Pick- and Lewy-body changes. Acta Neuropathol 36: , 221–233. |
[23] | Kosaka K ((2014) ) Lewy body disease and dementia with Lewy bodies. Proc Jpn Acad Ser B Phys Biol Sci 90: , 301–306. |
[24] | McKeith IG , Galasko D , Kosaka K , Perry EK , Dickson DW , Hansen LA , Salmon DP , Lowe J , Mirra SS , Byrne EJ , Lennox G , Quinn NP , Edwardson JA , Ince PG , Bergeron C , Burns A , Miller BL , Lovestone S , Collerton D , Jansen EN , Ballard C , de Vos RA , Wilcock GK , Jellinger KA , Perry RH ((1996) ) Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): Report of the consortium on DLB international workshop. Neurology 47: , 1113–1124. |
[25] | Kosaka K , Yoshimura M , Ikeda K , Budka H ((1984) ) Diffuse type of Lewy body disease: Progressive dementia with abundant cortical Lewy bodies and senile changes of varying degree–a new disease? Clin Neuropathol 3: , 185–192. |
[26] | Tsuang D , Leverenz JB , Lopez OL , Hamilton RL , Bennett DA , Schneider JA , Buchman AS , Larson EB , Crane PK , Kaye JA , Kramer P , Woltjer R , Kukull W , Nelson PT , Jicha GA , Neltner JH , Galasko D , Masliah E , Trojanowski JQ , Schellenberg GD , Yearout D , Huston H , Fritts-Penniman A , Mata IF , Wan JY , Edwards KL , Montine TJ , Zabetian CP ((2012) ) GBA mutations increase risk for Lewy body disease with and without Alzheimer disease pathology. Neurology 79: , 1944. |
[27] | Dickson DW , Heckman MG , Murray ME , Soto AI , Walton RL , Diehl NN , van Gerpen JA , Uitti RJ , Wszolek ZK , Ertekin-Taner N , Knopman DS , Petersen RC , Graff-Radford NR , Boeve BF , Bu G , Ferman TJ , Ross OA ((2018) ) APOE ɛ4 is associated with severity of Lewy body pathology independent of Alzheimer pathology. Neurology 91: , e1182–e1195. |
[28] | Singleton AB , Wharton A , O’Brien KK , Walker MP , McKeith IG , Ballard CG , O’Brien J , Perry RH , Ince PG , Edwardson JA , Morris CM ((2002) ) Clinical and neuropathological correlates of apolipoprotein E genotype in dementia with Lewy bodies. Dement Geriatr Cogn Disord 14: , 167–175. |
[29] | Aloizou A-M , Siokas V , Vogiatzi C , Peristeri E , Docea AO , Petrakis D , Provatas A , Folia V , Chalkia C , Vinceti M , Wilks M , Izotov BN , Tsatsakis A , Bogdanos DP , Dardiotis E ((2020) ) Pesticides, cognitive functions and dementia: A review. Toxicol Lett 326: , 31–51. |
[30] | Loganathan BG , Tanabe S , Hidaka Y , Kawano M , Hidaka H , Tatsukawa R ((1993) ) Temporal trends of persistent organochlorine residues in human adipose tissue from Japan, 1928-1985. Environ Pollut 81: , 31–39. |
[31] | Hatcher JM , Delea KC , Richardson JR , Pennell KD , Miller GW ((2008) ) Disruption of dopamine transport by DDT and its metabolites. Neurotoxicology 29: , 682–690. |
[32] | Helley MP , Pinnell J , Sportelli C , Tieu K ((2017) ) Mitochondria: A common target for genetic mutations and environmental toxicants in Parkinson’s disease. Front Genet 8: , 177. |
[33] | Sharma H , Zhang P , Barber DS , Liu B ((2010) ) Organochlorine pesticides dieldrin and lindane induce cooperative toxicity in dopaminergic neurons: Role of oxidative stress. Neurotoxicol 31: , 215–222. |
[34] | Ross GW , Abbott RD , Petrovitch H , Duda JE , Tanner CM , Zarow C , Uyehara-Lock JH , Masaki KH , Launer LJ , Studabaker WB , White LR ((2019) ) Association of brain heptachlor epoxide and other organochlorine compounds with lewy pathology. Mov Disord 34: , 228–235. |
[35] | Park M , Ross GW , Petrovitch H , White LR , Masaki KH , Nelson JS , Tanner CM , Curb JD , Blanchette PL , Abbott RD ((2005) ) Consumption of milk and calcium in midlife and the future risk of Parkinson disease. Neurology 64: , 1047. |
[36] | Olisah C , Okoh OO , Okoh AI ((2020) ) Occurrence of organochlorine pesticide residues in biological and environmental matrices in Africa: A two-decade review. Heliyon 6: , e03518. |
[37] | Thompson LA , Darwish WS , Ikenaka Y , Nakayama SM , Mizukawa H , Ishizuka M ((2017) ) Organochlorine pesticide contamination of foods in Africa: Incidence and public health significance. J Vet Med Sci 79: , 751–764. |
[38] | Mekonen S , Ambelu A , Wondafrash M , Kolsteren P , Spanoghe P ((2021) ) Exposure of infants to organochlorine pesticides from breast milk consumption in southwestern Ethiopia. Sci Rep 11: , 22053. |
[39] | Pirsaheb M , Limoee M , Namdari F , Khamutian R ((2015) ) Organochlorine pesticides residue in breast milk: A systematic review. Med J Islam Repub Iran 29: , 228. |
[40] | Postuma RB , Montplaisir JY , Pelletier A , Dauvilliers Y , Oertel W , Iranzo A , Ferini-Strambi L , Arnulf I , Hogl B , Manni R , Miyamoto T , Mayer G , Stiasny-Kolster K , Puligheddu M , Ju Y , Jennum P , Sonka K , Santamaria J , Fantini ML , Zucconi M , Leu-Semenescu S , Frauscher B , Terzaghi M , Miyamoto M , Unger MM , Cock VCD , Wolfson C ((2012) ) Environmental risk factors for REM sleep behavior disorder. Neurology 79: , 428. |
[41] | Postuma RB , Iranzo A , Hu M , Högl B , Boeve BF , Manni R , Oertel WH , Arnulf I , Ferini-Strambi L , Puligheddu M , Antelmi E , Cochen De Cock V , Arnaldi D , Mollenhauer B , Videnovic A , Sonka K , Jung KY , Kunz D , Dauvilliers Y , Provini F , Lewis SJ , Buskova J , Pavlova M , Heidbreder A , Montplaisir JY , Santamaria J , Barber TR , Stefani A , St Louis EK , Terzaghi M , Janzen A , Leu-Semenescu S , Plazzi G , Nobili F , Sixel-Doering F , Dusek P , Bes F , Cortelli P , Ehgoetz Martens K , Gagnon JF , Gaig C , Zucconi M , Trenkwalder C , Gan-Or Z , Lo C , Rolinski M , Mahlknecht P , Holzknecht E , Boeve AR , Teigen LN , Toscano G , Mayer G , Morbelli S , Dawson B , Pelletier A ((2019) ) Risk and predictors of dementia and parkinsonism in idiopathic REM sleep behaviour disorder: A multicentre study. Brain 142: , 744–759. |
[42] | Hui Z , Zhuqin G , Chun Y , Yanning C , Yuan L , Wei M , Erhe X , Ronald BP , Piu C ((2020) ) Risk factors for possible REM sleep behavior disorders. Neurology 95: , e2214. |
[43] | Postuma RB , Iranzo A , Hogl B , Arnulf I , Ferini-Strambi L , Manni R , Miyamoto T , Oertel W , Dauvilliers Y , Ju Y-E , Puligheddu M , Sonka K , Pelletier A , Santamaria J , Frauscher B , Leu-Semenescu S , Zucconi M , Terzaghi M , Miyamoto M , Unger MM , Carlander B , Fantini M-L , Montplaisir JY ((2015) ) Risk factors for neurodegeneration in idiopathic rapid eye movement sleep behavior disorder: A multicenter study. Ann Neurol 77: , 830–839. |
[44] | Zhang H , Iranzo A , Högl B , Arnulf I , Ferini-Strambi L , Manni R , Miyamoto T , Oertel WH , Dauvilliers Y , Ju Y-EI , Puligheddu M , Sonka K , Pelletier A , Montplaisir JY , Stefani A , Ibrahim A , Frauscher B , Leu-Semenescu S , Zucconi M , Terzaghi M , Miyamoto M , Janzen A , Figorilli M , Fantini ML , Postuma RB ((2022) ) Risk factors for phenoconversion in rapid eye movement sleep behavior disorder. Ann Neurol 91: , 404–416. |
[45] | Gatto NM , Cockburn M , Bronstein J , Manthripragada AD , Ritz B ((2009) ) Well-water consumption and Parkinson’s disease in rural California. Environ Health Perspect 117: , 1912–1918. |
[46] | Jiménez-Jiménez FJ , Mateo D , Giménez-Roldán S ((1992) ) Exposure to well water and pesticides in Parkinson’s disease: A case-control study in the madrid area. Mov Disord 7: , 149–152. |
[47] | Tanner CM , Chen B , Wang W-Z , Peng M-L , Liu Z-L , Liang X-L , Kao LC , Gilley DW , Schoenberg BS ((1987) ) Environmental factors in the etiology of Parkinson’s disease. Can J Neurol Sci 14: , 419–423. |
[48] | Chase BA , Krueger R , Pavelka L , Chung SJ , Aasly J , Dardiotis E , Premkumar AP , Schoneburg B , Kartha N , Aunaetitrakul N , Frigerio R , Maraganore D , Markopoulou K ((2023) ) Multifactorial assessment of Parkinson’s disease course and outcomes using trajectory modeling in a multiethnic, multisite cohort - extension of the LONG-PD study. Front Aging Neurosci 15: , 1240971. |
[49] | Merola A , Romagnolo A , Dwivedi AK , Padovani A , Berg D , Garcia-Ruiz PJ , Fabbri M , Artusi CA , Zibetti M , Lopiano L , Pilotto A , Bonacina S , Morgante F , Zeuner K , Griewing C , Schaeffer E , Rodriguez-Porcel F , Kauffman M , Turcano P , de Oliveira LM , Palermo G , Shanks E , Del Sorbo F , Bonvegna S , Savica R , Munhoz RP , Ceravolo R , Cilia R , Espay AJ ((2020) ) Benign versus malignant Parkinson disease: The unexpected silver lining of motor complications. J Neurol 267: , 2949–2960. |
[50] | Pan-Montojo F , Schwarz M , Winkler C , Arnhold M , O’Sullivan GA , Pal A , Said J , Marsico G , Verbavatz J-M , Rodrigo-Angulo M , Gille G , Funk RHW , Reichmann H ((2012) ) Environmental toxins trigger PD-like progression via increased alpha-synuclein release from enteric neurons in mice. Sci Rep 2: , 898. |
[51] | Drolet RE , Cannon JR , Montero L , Greenamyre JT ((2009) ) Chronic rotenone exposure reproduces Parkinson’s disease gastrointestinal neuropathology. Neurobiol Dis 36: , 96–102. |
[52] | Arnhold M , Dening Y , Chopin M , Arévalo E , Schwarz M , Reichmann H , Gille G , Funk RH , Pan-Montojo F ((2016) ) Changes in the sympathetic innervation of the gut in rotenone treated mice as possible early biomarker for Parkinson’s disease. Clin Auton Res 26: , 211–222. |
[53] | Schaffernicht G , Shang Q , Stievenard A , Bötzel K , Dening Y , Kempe R , Toussaint M , Gündel D , Kranz M , Reichmann H , Vanbesien-Mailliot C , Brust P , Dieterich M , Funk RHW , Ravens U , Pan-Montojo F ((2021) ) Pathophysiological changes in the enteric nervous system of rotenone-exposed mice as early radiological markers for Parkinson’s disease. Front Neurol 12: , 642604. |
[54] | Palanisamy BN , Sarkar S , Malovic E , Samidurai M , Charli A , Zenitsky G , Jin H , Anantharam V , Kanthasamy A , Kanthasamy AG ((2022) ) Environmental neurotoxic pesticide exposure induces gut inflammation and enteric neuronal degeneration by impairing enteric glial mitochondrial function in pesticide models of Parkinson’s disease: Potential relevance to gut-brain axis inflammation in Parkinson’s disease pathogenesis. Int J Biochem Cell Biol 147: , 106225. |
[55] | De Miranda BR , Greenamyre JT ((2020) ) Trichloroethylene, a ubiquitous environmental contaminant in the risk for Parkinson’s disease. Environ Sci Process Impacts 22: , 543–554. |
[56] | Dorsey ER , Zafar M , Lettenberger SE , Pawlik ME , Kinel D , Frissen M , Schneider RB , Kieburtz K , Tanner CM , De Miranda BR , Goldman SM , Bloem BR ((2023) ) Trichloroethylene: An invisible cause of Parkinson’s disease? J Parkinsons Dis 13: , 203–218. |
[57] | Huber F ((1969) ) Clinical aspects and neuropathology of trichloroethylene poisoning. Z Unfallmed Berufskr 62: , 226–267. |
[58] | Goldman SM , Weaver FM , Stroupe KT , Cao L , Gonzalez B , Colletta K , Brown EG , Tanner CM ((2023) ) Risk of Parkinson Disease among service members at Marine Corps Base Camp Lejeune. JAMA Neurol 80: , 673–681. |
[59] | Ilieva NM , Wallen ZD , De Miranda BR ((2022) ) Oral ingestion of the environmental toxicant trichloroethylene in rats induces alterations in the gut microbiome: Relevance to idiopathic Parkinson’s disease. Toxicol Appl Pharmacol 451: , 116176. |
[60] | Hatcher-Martin JM , Gearing M , Steenland K , Levey AI , Miller GW , Pennell KD ((2012) ) Association between polychlorinated biphenyls and Parkinson’s disease neuropathology. Neurotoxicol 33: , 1298–1304. |
[61] | Korrick SA , Sagiv SK ((2008) ) Polychlorinated biphenyls, organochlorine pesticides and neurodevelopment. Curr Opin Pediatr 20: , 198–204. |
[62] | (2000) Agency for Toxic Substances and Disease Registry (ATSDR) Toxicological Profiles. In Toxicological Profile for Polychlorinated Biphenyls (PCBs), Agency for Toxic Substances and Disease Registry (US), Atlanta (GA). |
[63] | Megson D , Brown T , Jones GR , Robson M , Johnson GW , Tiktak GP , Sandau CD , Reiner EJ ((2022) ) Polychlorinated biphenyl (PCB) concentrations and profiles in marine mammals from the North Atlantic Ocean. Chemosphere 288: , 132639. |
[64] | Tanabe S , Mori T , Tatsukawa R , Miyazaki N ((1983) ) Global pollution of marine mammals by PCBs, DDTs and HCHs (BHCs). Chemosphere 12: , 1269–1275. |
[65] | Long M , Sonne C , Dietz R , Bossi R , Jørgensen N , Olsen TI , Bonefeld-Jørgensen EC ((2023) ) Diet, lifestyle and contaminants in three east Greenland Inuit municipalities. Chemosphere 344: , 140368. |
[66] | Bjerregaard P , Dewailly E , Ayotte P , Pars T , Ferron L , Mulvad G ((2001) ) Exposure of Inuit in Greenland to organochlorines through the Marine diet. J Toxicol Environ Health, Part A 62: , 69–81. |
[67] | Wermuth L , Pakkenberg H , Jeune B ((2004) ) High age-adjusted prevalence of Parkinson’s disease among Inuit in Greenland. Int J Circumpolar Health 63: , 369–370. |
[68] | Petersen MS , Halling J , Bech S , Wermuth L , Weihe P , Nielsen F , Jørgensen PJ , Budtz-Jørgensen E , Grandjean P ((2008) ) Impact of dietary exposure to food contaminants on the risk of Parkinson’s disease. Neurotoxicol 29: , 584–590. |
[69] | Caudle WM , Richardson JR , Delea KC , Guillot TS , Wang M , Pennell KD , Miller GW ((2006) ) Polychlorinated biphenyl-induced reduction of dopamine transporter expression as a precursor to Parkinson’s disease-associated dopamine toxicity. Toxicol Sci 92: , 490–499. |
[70] | Wang M-Y , Zhang L-F , Wu D , Cai Y-Q , Huang D-M , Tian L-L , Fang C-L , Shi Y-F ((2022) ) Simulation experiment on OH-PCB being ingested through daily diet: Accumulation, transformation and distribution of hydroxylated-2, 2′, 4, 5, 5′-pentachlorobiphenyl (OH-PCB101) in mice. Sci Tot Environ 802: , 149891. |
[71] | Weisskopf MG , Knekt P , O’Reilly EJ , Lyytinen J , Reunanen A , Laden F , Altshul L , Ascherio A ((2012) ) Polychlorinated biphenyls in prospectively collected serum and Parkinson’s disease risk. Mov Disord 27: , 1659–1665. |
[72] | Raffetti E , Donato F , De Palma G , Leonardi L , Sileo C , Magoni M ((2020) ) Polychlorinated biphenyls (PCBs) and risk of dementia and Parkinson disease: A population-based cohort study in a North Italian highly polluted area. Chemosphere 261: , 127522. |
[73] | Guo W , Pan B , Sakkiah S , Yavas G , Ge W , Zou W , Tong W , Hong H ((2019) ) Persistent organic pollutants in food: Contamination sources, health effects and detection methods. Int J Environ Res Public Health 16: , 4361. |
[74] | Steenland K , Hein MJ , Cassinelli RT 2nd , Prince MM , Nilsen NB , Whelan EA , Waters MA , Ruder AM , Schnorr TM ((2006) ) Polychlorinated biphenyls and neurodegenerative disease mortality in an occupational cohort. Epidemiol 17: , 8–13. |
[75] | Calderón-Garcidueñas L , Stommel EW , Rajkumar RP , Mukherjee PS , Ayala A ((2021) ) Particulate air pollution and risk of neuropsychiatric outcomes. What we breathe, swallow, and put on our skin matters. Int J Environ Res Public Health 18: , 11568. |
[76] | Particle Pollution Exposure, US Environmental Protection Agency,https://www.epa.gov/pmcourse/particle-pollution-exposureMay 8, 2023, Accessed December 2, 2023. |
[77] | Hazardous Air Pollutants: Sources and Exposure, US Environmental Protection Agency,https://www.epa.gov/haps/hazardous-air-pollutants-sources-and-exposureOctober 27, 2023, Accessed November 13, 2023.. |
[78] | Costello S , Cockburn M , Bronstein J , Zhang X , Ritz B ((2009) ) Parkinson’s disease and residential exposure to maneb and paraquat from agricultural applications in the central valley of California. Am J Epidemiol 169: , 919–926. |
[79] | Brooks AI , Chadwick CA , Gelbard HA , Cory-Slechta DA , Federoff HJ ((1999) ) Paraquat elicited neurobehavioral syndrome caused by dopaminergic neuron loss. Brain Res 823: , 1–10. |
[80] | Gillam C , Uteuova A ((2022) ) Secret files suggest chemical giant feared weedkiller’s link to Parkinson’s disease. The Guardian, –https://www.theguardian.com/us-news/2022/oct/20/syngenta-weedkiller-pesticide-parkinsons-disease-paraquat-documents. |
[81] | Anderson T , Merrill AK , Eckard ML , Marvin E , Conrad K , Welle K , Oberdörster G , Sobolewski M , Cory-Slechta DA ((2020) ) Paraquat inhalation, a translationally relevant route of exposure: Disposition to the brain and male-specific olfactory impairment in mice. Toxicol Sci 180: , 175–185. |
[82] | Juan RS-R , Franz H , William JW ((1987) ) Paraquat and Parkinson’s disease. Neurology 37: , 728. |
[83] | White AC , Bandlamudi S , Chang J , Kelly SD , Caudle WM , Sampson TR ((2021) ) Low-dose oral pyrethroid exposure induces gastrointestinal dysfunction and alters nigrostriatal dopamine signaling pathways in mice. ISEE Conference Abstracts 2021: , https://doi.org/10.1289/isee.2021.P-066. |
[84] | Gash DM , Rutland K , Hudson NL , Sullivan PG , Bing G , Cass WA , Pandya JD , Liu M , Choi D-Y , Hunter RL , Gerhardt GA , Smith CD , Slevin JT , Prince TS ((2008) ) Trichloroethylene: Parkinsonism and complex 1 mitochondrial neurotoxicity. Ann Neurol 63: , 184–192. |
[85] | Guehl D , Bezard E , Dovero S , Boraud T , Bioulac B , Gross C ((1999) ) Trichloroethylene and parkinsonism: A human and experimental observation. Eur J Neurol 6: , 609–611. |
[86] | Reis J , Benbrick E , Bonneterre V , Spencer PS ((2016) ) Parkinson’s disease and solvents: Is there a causal link? Rev Neurol (Paris) 172: , 761–765. |
[87] | Liu M , Choi DY , Hunter RL , Pandya JD , Cass WA , Sullivan PG , Kim HC , Gash DM , Bing G ((2010) ) Trichloroethylene induces dopaminergic neurodegeneration in Fisher 344 rats. J Neurochem 112: , 773–783. |
[88] | Adamson A , Ilieva N , Stone WJ , De Miranda BR ((2023) ) Low-dose inhalation exposure to trichloroethylene induces dopaminergic neurodegeneration in rodents. Toxicol Sci 196: , 218–228. |
[89] | Apfelbach R , Engelhart A , Behnisch P , Hagenmaier H ((1998) ) The olfactory system as a portal of entry for airborne polychlorinated biphenyls (PCBs) to the brain? Arch Toxicol 72: , 314–317. |
[90] | Bhatia A , Katz J , Sanger-Katz M (2023) Just How Bad Was the Pollution in New York? The New York Times. https://www.nytimes.com/interactive/2023/06/08/upshot/new-york-city-smoke.html. |
[91] | Palacios N ((2017) ) Air pollution and Parkinson’s disease –evidence and future directions. Rev Environ Health 32: , 303–313. |
[92] | Krzyzanowski B , Searles Nielsen S , Turner JR , Racette BA ((2023) ) Fine particulate matter and Parkinson disease risk among Medicare beneficiaries. Neurology 101: , e2058–e2067. |
[93] | Kwon D , Paul KC , Yu Y , Zhang K , Folle AD , Wu J , Bronstein JM , Ritz B ((2024) ) Traffic-related air pollution and Parkinson’s disease in central California. Environ Res 240: , 117434. |
[94] | Calderón-Garcidueñas L , Gónzalez-Maciel A , Reynoso-Robles R , Delgado-Chávez R , Mukherjee PS , Kulesza RJ , Torres-Jardón R , Ávila-Ramírez J , Villarreal-Ríos R ((2018) ) Hallmarks of Alzheimer disease are evolving relentlessly in Metropolitan Mexico City infants, children and young adults. APOE4 carriers have higher suicide risk and higher odds of reaching NFT stage V at ≤40 years of age. Environ Res 164: , 475–487. |
[95] | Cory-Slechta DA , Sobolewski M , Marvin E , Conrad K , Merrill A , Anderson T , Jackson BP , Oberdorster G ((2019) ) The impact of inhaled ambient ultrafine particulate matter on developing brain: Potential importance of elemental contaminants. Toxicol Pathol 47: , 976–992. |
[96] | Barnham KJ , Bush AI ((2008) ) Metals in Alzheimer’s and Parkinson’s diseases. Curr Opin Chem Biol 12: , 222–228. |
[97] | Olkowski W , Darr S , Olkowski H (2000) Pyrethrum-Based Insecticides from Chrysanthemums, Kitchen Gardener Magazine,https://www.finegardening.com/article/pyrethrum-based-insecticides-from-chrysanthemumsAccessed November 13, 2023. |
[98] | Parkinson J ((2002) ) An essay on the shaking palsy. 1817. J Neuropsychiatry Clin Neurosci 14: , 223–236. |
[99] | (1990) Mexico City: ProAire, Energy Policy Institute at the University of Chicago,https://aqli.epic.uchicago.edu/policy-impacts/mexico-city-proaire-1990/Accessed January 3, 2024. |
[100] | Calderón-Garcidueñas L , Solt AC , Henríquez-Roldán C , Torres-Jardón R , Nuse B , Herritt L , Villarreal-Calderón R , Osnaya N , Stone I , García R , Brooks DM , González-Maciel A , Reynoso-Robles R , Delgado-Chávez R , Reed W ((2008) ) Long-term air pollution exposure is associated with neuroinflammation, an altered innate immune response, disruption of the blood-brain barrier, ultrafine particulate deposition, and accumulation of amyloid β-42 and α-synuclein in children and young adults. Toxicol Pathol 36: , 289–310. |
[101] | Calderón-Garcidueñas L , González-Maciel A , Reynoso-Robles R , Kulesza RJ , Mukherjee PS , Torres-Jardón R , Rönkkö T , Doty RL ((2018) ) Alzheimer’s disease and alpha-synuclein pathology in the olfactory bulbs of infants, children, teens and adults ≤40 years in Metropolitan Mexico City. APOE4 carriers at higher risk of suicide accelerate their olfactory bulb pathology. Environ Res 166: , 348–362. |
[102] | Tanner CM , Chen B , Wang W , Peng M , Liu Z , Liang X , Kao LC , Gilley DW , Goetz CG , Schoenberg BS ((1989) ) Environmental factors and Parkinson’s disease: A case-control study in China. Neurology 39: , 660–664. |
[103] | Beamer PI , Luik CE , Abrell L , Campos S , Martínez ME , Sáez AE ((2012) ) Concentration of trichloroethylene in breast milk and household water from Nogales, Arizona. Environ Sci Technol 46: , 9055–9061. |
[104] | Weldon RH , Barr DB , Trujillo C , Bradman A , Holland N , Eskenazi B ((2011) ) A pilot study of pesticides and PCBs in the breast milk of women residing in urban and agricultural communities of California. J Environ Monit 13: , 3136–3144. |
[105] | Hu C-Y , Fang Y , Li F-L , Dong B , Hua X-G , Jiang W , Zhang H , Lyu Y , Zhang X-J ((2019) ) Association between ambient air pollution and Parkinson’s disease: Systematic review and meta-analysis. Environ Res 168: , 448–459. |
[106] | Ritz B , Lee P-C , Hansen J , Lassen CF , Ketzel M , Sørensen M , Raaschou-Nielsen O ((2016) ) Traffic-related air pollution and Parkinson’s disease in Denmark: A case–control study. Environ Health Perspect 124: , 351–356. |
[107] | Jo S , Kim Y-J , Park KW , Hwang YS , Lee SH , Kim BJ , Chung SJ ((2021) ) Association of NO2 and other air pollution exposures with the risk of Parkinson disease. JAMA Neurol 78: , 800–808. |
[108] | Yu Z , Wei F , Zhang X , Wu M , Lin H , Shui L , Jin M , Wang J , Tang M , Chen K ((2021) ) Air pollution, surrounding green road proximity and Parkinson’s disease: A prospective cohort study. Environ Res 197: , 111170. |
[109] | Zhu Z , Yang Z , Yu L , Xu L , Wu Y , Zhang X , Shen P , Lin H , Shui L , Tang M , Jin M , Wang J , Chen K ((2023) ) Residential greenness, air pollution and incident neurodegenerative disease: A cohort study in China. Sci Tot Environ 878: , 163173. |
[110] | Bogers JS , Bloem BR , Den Heijer JM ((2023) ) The etiology of Parkinson’s disease: New perspectives from gene-environment interactions. J Parkinsons Dis 13: , 1281–1288. |
[111] | Cannon JR , Greenamyre JT ((2013) ) Gene–environment interactions in Parkinson’s disease: Specific evidence in humans and mammalian models. Neurobiol Dis 57: , 38–46. |
[112] | Blauwendraat C , Reed X , Krohn L , Heilbron K , Bandres-Ciga S , Tan M , Gibbs JR , Hernandez DG , Kumaran R , Langston R , Bonet-Ponce L , Alcalay RN , Hassin-Baer S , Greenbaum L , Iwaki H , Leonard HL , Grenn FP , Ruskey JA , Sabir M , Ahmed S , Makarious MB , Pihlstrøm L , Toft M , van Hilten JJ , Marinus J , Schulte C , Brockmann K , Sharma M , Siitonen A , Majamaa K , Eerola-Rautio J , Tienari PJ , Team TaR , Pantelyat A , Hillis AE , Dawson TM , Rosenthal LS , Albert MS , Resnick SM , Ferrucci L , Morris CM , Pletnikova O , Troncoso J , Grosset D , Lesage S , Corvol J-C , Brice A , Noyce AJ , Masliah E , Wood N , Hardy J , Shulman LM , Jankovic J , Shulman JM , Heutink P , Gasser T , Cannon P , Scholz SW , Morris H , Cookson MR , Nalls MA , Gan-Or Z , Singleton AB ((2019) ) Genetic modifiers of risk and age at onset in GBA associated Parkinson’s disease and Lewy body dementia. Brain 143: , 234–248. |
[113] | Simitsi A , Koros C , Moraitou M , Papagiannakis N , Antonellou R , Bozi M , Angelopoulou E , Stamelou M , Michelakakis H , Stefanis L ((2018) ) Phenotypic characteristics in GBA-associated Parkinson’s disease: A study in a Greek population. J Parkinsons Dis 8: , 101–105. |
[114] | Calderón-Garcidueñas L , Reed W , Maronpot RR , Henriquez-Roldán C , Delgado-Chavez R , Calderón-Garcidueñas A , Dragustinovis I , Franco-Lira M , Aragón-Flores M , Solt AC , Altenburg M , Torres-Jardón R , Swenberg JA ((2004) ) Brain inflammation and Alzheimer’s-like pathology in individuals exposed to severe air pollution. Toxicol Pathol 32: , 650–658. |
[115] | Kilian J , Kitazawa M ((2018) ) The emerging risk of exposure to air pollution on cognitive decline and Alzheimer’s disease –Evidence from epidemiological and animal studies. Biomed J 41: , 141–162. |
[116] | Cacciottolo M , Wang X , Driscoll I , Woodward N , Saffari A , Reyes J , Serre ML , Vizuete W , Sioutas C , Morgan TE , Gatz M , Chui HC , Shumaker SA , Resnick SM , Espeland MA , Finch CE , Chen JC ((2017) ) Particulate air pollutants, APOE alleles and their contributions to cognitive impairment in older women and to amyloidogenesis in experimental models. Transl Psychiatry 7: , e1022–e1022. |
[117] | Chatterjee A , Hirsch-Reinshagen V , Moussavi SA , Ducharme B , Mackenzie IR , Hsiung G-YR ((2021) ) Clinico-pathological comparison of patients with autopsy-confirmed Alzheimer’s disease, dementia with Lewy bodies, and mixed pathology. Alzheimers Dement 13: , e12189. |
[118] | Kotzbauer PT , Trojanowski JQ , Lee VMY ((2001) ) Lewy body pathology in Alzheimer’s disease. J Mol Neuroscience 17: , 225–232. |
[119] | Rudyk C , Dwyer Z , Hayley S , Schurr E , Brown E , Gibbings D , Hayley S , Park D , Philpott DC , Rioux JD , Schlossmacher M , membership C ((2019) ) Leucine-rich repeat kinase-2 (LRRK2) modulates paraquat-induced inflammatory sickness and stress phenotype. J Neuroinflammation 16: , 120. |
[120] | Di Maio R , Hoffman EK , Rocha EM , Keeney MT , Sanders LH , De Miranda BR , Zharikov A , Van Laar A , Stepan AF , Lanz TA , Kofler JK , Burton EA , Alessi DR , Hastings TG , Greenamyre JT ((2018) ) LRRK2 activation in idiopathic Parkinson’s disease. Sci Transl Med 10: , eaar5429. |
[121] | Pajarillo E , Kim S , Digman A , Dutton M , Son DS , Aschner M , Lee E ((2023) ) The role of microglial LRRK2 kinase in manganese-induced inflammatory neurotoxicity via NLRP3 inflammasome and RAB10-mediated autophagy dysfunction. J Biol Chem 299: , 104879. |
[122] | De Miranda BR , Castro SL , Rocha EM , Bodle CR , Johnson KE , Greenamyre JT ((2021) ) The industrial solvent trichloroethylene induces LRRK2 kinase activity and dopaminergic neurodegeneration in a rat model of Parkinson’s disease. Neurobiol Dis 153: , 105312. |
[123] | Rocha EM , Keeney MT , Di Maio R , De Miranda BR , Greenamyre JT ((2022) ) LRRK2 and idiopathic Parkinson’s disease. Trends Neurosci 45: , 224–236. |
[124] | Dardiotis E , Xiromerisiou G , Hadjichristodoulou C , Tsatsakis AM , Wilks MF , Hadjigeorgiou GM ((2013) ) The interplay between environmental and genetic factors in Parkinson’s disease susceptibility: The evidence for pesticides. Toxicology 307: , 17–23. |
[125] | Goldman SM , Kamel F , Ross GW , Bhudhikanok GS , Hoppin JA , Korell M , Marras C , Meng C , Umbach DM , Kasten M , Chade AR , Comyns K , Richards MB , Sandler DP , Blair A , Langston JW , Tanner CM ((2012) ) Genetic modification of the association of paraquat and Parkinson’s disease. Mov Disord 27: , 1652–1658. |
[126] | Ritz BR , Paul KC , Bronstein JM ((2016) ) Of Pesticides and men: A California story of genes and environment in Parkinson’s disease. Curr Environ Health Rep 3: , 40–52. |
[127] | Pei-Chen L , Yvette B , Jeff B , Beate R ((2012) ) Traumatic brain injury, paraquat exposure, and their relationship to Parkinson disease. Neurology 79: , 2061. |
[128] | Sauerbeck A , Hunter R , Bing G , Sullivan PG ((2012) ) Traumatic brain injury and trichloroethylene exposure interact and produce functional, histological, and mitochondrial deficits. Exp Neurol 234: , 85–94. |
[129] | Gilbert SF (2000) Aging: The biology of senescence. In Developmental Biology, Sinauer Associates. |
[130] | Levy G ((2007) ) The relationship of Parkinson disease with aging. Arch Neurol 64: , 1242–1246. |
[131] | Vilchez D , Saez I , Dillin A ((2014) ) The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat Commun 5: , 5659. |
[132] | Collier TJ , Kanaan NM , Kordower JH ((2017) ) Aging and Parkinson’s disease: Different sides of the same coin? Mov Disord 32: , 983–990. |
[133] | Kumar H , Lim H-W , More SV , Kim B-W , Koppula S , Kim IS , Choi D-K ((2012) ) The role of free radicals in the aging brain and Parkinson’s disease: Convergence and parallelism. Int J Mol Sci 13: , 10478–10504. |
[134] | Agim ZS , Cannon JR ((2015) ) Dietary factors in the etiology of Parkinson’s disease. BioMed Res Int 2015: , 672838. |
[135] | Marras C , Canning CG , Goldman SM ((2019) ) Environment, lifestyle, and Parkinson’s disease: Implications for prevention in the next decade. Mov Disord 34: , 801–811. |
[136] | Chohan H , Senkevich K , Patel RK , Bestwick JP , Jacobs BM , Bandres Ciga S , Gan-Or Z , Noyce AJ ((2021) ) Type 2 diabetes as a determinant of Parkinson’s disease risk and progression. Mov Disord 36: , 1420–1429. |
[137] | Russillo MC , Andreozzi V , Erro R , Picillo M , Amboni M , Cuoco S , Barone P , Pellecchia MT ((2022) ) Sex differences in Parkinson’s disease: From bench to bedside. Brain Sci 12: , 917. |
[138] | Dorsey ER , Elbaz A , Nichols E , Abd-Allah F , Abdelalim A , Adsuar JC , Ansha MG , Brayne C , Choi J-YJ , Collado-Mateo D , Dahodwala N , Do HP , Edessa D , Endres M , Fereshtehnejad S-M , Foreman KJ , Gankpe FG , Gupta R , Hankey GJ , Hay SI , Hegazy MI , Hibstu DT , Kasaeian A , Khader YS , Khalil I , Khang Y-H , Kim YJ , Kokubo Y , Logroscino G , Massano J , Mohamed Ibrahim N , Mohammed AM , Mohammadi A , Moradi-Lakeh M , Naghavi M , Nguyen BT , Nirayo YL , Ogbo FA , Owolabi MO , Pereira DM , Postma MJ , Qorbani M , Rahman MA , Roba KT , Safari H , Safiri S , Satpathy M , Sawhney M , Shafieesabet A , Shiferaw MS , Smith M , Szoeke CEI , Tabarés-Seisdedos R , Truong NT , Ukwaja KN , Venketasubramanian N , Villafaina S , Weldegwergs KG , Westerman R , Wijeratne T , Winkler AS , Xuan BT , Yonemoto N , Feigin VL , Vos T , Murray CJL ((2018) ) Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 17: , 939 –953. |
[139] | Wright Willis A , Evanoff BA , Lian M , Criswell SR , Racette BA ((2010) ) Geographic and ethnic variation in Parkinson disease: A population-based study of US Medicare beneficiaries. Neuroepidemiology 34: , 143–151. |
[140] | Barbeau A , Roy M , Bernier G , Campanella G , Paris S ((1987) ) Ecogenetics of Parkinson’s disease: Prevalence and environmental aspects in rural areas. Can J Neurol Sci 14: , 36–41. |
[141] | Kab S , Spinosi J , Chaperon L , Dugravot A , Singh-Manoux A , Moisan F , Elbaz A ((2017) ) Agricultural activities and the incidence of Parkinson’s disease in the general French population. Eur J Epidemiol 32: , 203–216. |
[142] | Parrish ML , Gardner RE ((2012) ) Is living downwind of a golf course a risk factor for parkinsonism? Ann Neurol 72: , 984–984. |
[143] | Briscoe T (2023) Is a common industrial chemical fueling the spread of Parkinson’s disease? The Los Angeles Times,https://www.latimes.com/environment/story/2023-03-15/industrial-chemical-may-be-linked-to-parkinsons-disease. |
[144] | Geng C , Luo Q , Chen M , Li Z , Zhang C ((2010) ) Quantitative risk assessment of trichloroethylene for a former chemical works in Shanghai, China. Hum Ecol Risk Assess: Int J 16: , 429–443. |
[145] | Schwartz DM , LaRocco P (2020) The Grumman Plume: Decades of Deceit. Newsday,https://projects.newsday.com/long-island/plume-grumman-navy/Newsday. |
[146] | Ritchie H , Roser M Air Pollution, Our World in Datahttps://ourworldindata.org/air-pollution.January 2021,Accessed December 2, 2023. |
[147] | Sharma A , Kumar V , Shahzad B , Tanveer M , Sidhu GPS , Handa N , Kohli SK , Yadav P , Bali AS , Parihar RD , Dar OI , Singh K , Jasrotia S , Bakshi P , Ramakrishnan M , Kumar S , Bhardwaj R , Thukral AK ((2019) ) Worldwide pesticide usage and its impacts on ecosystem. SN Appl Sci 1: , 1446. |
[148] | Food and Agriculture Organization of the United Nations (1997) FAOSTAT Statistcal Database, Pesticides Usehttps://www.fao.org/faostat/en/#data/RP/visualize.Accessed January 2, 2024. |
[149] | Fuhrimann S , Wan C , Blouzard E , Veludo A , Holtman Z , Chetty-Mhlanga S , Dalvie MA , Atuhaire A , Kromhout H , Röösli M , Rother H-A ((2022) ) Pesticide research on environmental and human exposure and risks in Sub-Saharan Africa: A systematic literature review. Int J Environ Res Public Health 19: , 259–10.3390/ijerph19010259. |
[150] | Paice E (2022) Youthquake: Why African Demography Should Matter to the World, Apollo. |
[151] | Lekei EE , Ngowi AV , London L ((2014) ) Pesticide retailers’ knowledge and handling practices in selected towns of Tanzania. Environ Health 13: , 79. |
[152] | Fedorova TD , Knudsen K , Horsager J , Hansen AK , Okkels N , Gottrup H , Vang K , Borghammer P ((2023) ) Dopaminergic dysfunction is more symmetric in dementia with Lewy bodies compared to Parkinson’s disease. J Parkinsons Dis 13: , 515–523. |
[153] | Knudsen K , Fedorova TD , Horsager J , Andersen KB , Skjærbæk C , Berg D , Schaeffer E , Brooks DJ , Pavese N , Van Den Berge N , Borghammer P ((2021) ) Asymmetric dopaminergic dysfunction in brain-first versus body-first Parkinson’s disease subtypes. J Parkinsons Dis 11: , 1677–1687. |
[154] | McDermott Michael J , Mazor Kimberly A , Shost Stephen J , Narang Rajinder S , Aldous Kenneth M , Storm Jan E ((2005) ) Tetrachloroethylene (PCE, Perc) levels in residential dry cleaner buildings in diverse communities in New York City. Environ Health Perspect 113: , 1336–1343. |
[155] | Nijhuis N , Loh M , Harrison P (2010) Tetracholoroethylene. In WHO Guidelines for Indoor Air Quality, World Health Organization, Geneva. |
[156] | Hudson NL , Dotson GS (2017) NIOSH Skin Notation Profiles: Trichloroethylene (TCE). Centers for Disease Control and Prevention.https://www.cdc.gov/niosh/docs/2017-192/2017-192.pdf. |
[157] | Oberdörster G , Elder A , Rinderknecht A ((2009) ) Nanoparticles and the brain: Cause for concern? J Nanosci Nanotechnol 9: , 4996–5007. |
[158] | Dardiotis E , Aloizou AM , Sakalakis E , Siokas V , Koureas M , Xiromerisiou G , Petinaki E , Wilks M , Tsatsakis A , Hadjichristodoulou C , Stefanis L , Hadjigeorgiou GM ((2020) ) Organochlorine pesticide levels in Greek patients with Parkinson’s disease. Toxicol Rep 7: , 596–601. |
[159] | Tanei ZI , Saito Y , Ito S , Matsubara T , Motoda A , Yamazaki M , Sakashita Y , Kawakami I , Ikemura M , Tanaka S , Sengoku R , Arai T , Murayama S ((2021) ) Lewy pathology of the esophagus correlates with the progression of Lewy body disease: A Japanese cohort study of autopsy cases. Acta Neuropathol 141: , 25–37. |
[160] | Brugnone F , Perbellini L , Giuliari C , Cerpelloni M , Soave M ((1994) ) Blood and urine concentrations of chemical pollutants in the general population. Med Lav 85: , 370–389. |
[161] | Knudsen K , Fedorova TD , Hansen AK , Sommerauer M , Otto M , Svendsen KB , Nahimi A , Stokholm MG , Pavese N , Beier CP , Brooks DJ , Borghammer P ((2018) ) In-vivo staging of pathology in REM sleep behaviour disorder: A multimodality imaging case-control study. Lancet Neurol 17: , 618–628. |
[162] | Nishikawa N , Murata M , Hatano T , Mukai Y , Saitoh Y , Sakamoto T , Hanakawa T , Kamei Y , Tachimori H , Hatano K , Matsuda H , Taruno Y , Sawamoto N , Kajiyama Y , Ikenaka K , Kawabata K , Nakamura T , Iwaki H , Kadotani H , Sumi Y , Inoue Y , Hayashi T , Ikeuchi T , Shimo Y , Mochizuki H , Watanabe H , Hattori N , Takahashi Y , Takahashi R ((2022) ) Idiopathic rapid eye movement sleep behavior disorder in Japan: An observational study. Parkinsonism Relat Disord 103: , 129–135. |
[163] | Beach TG , Adler CH , Sue LI , Vedders L , Lue L , White Iii CL , Akiyama H , Caviness JN , Shill HA , Sabbagh MN , Walker DG ((2010) ) Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol 119: , 689–702. |
[164] | Gelpi E , Navarro-Otano J , Tolosa E , Gaig C , Compta Y , Rey MJ , Martí MJ , Hernández I , Valldeoriola F , Reñé R , Ribalta T ((2014) ) Multiple organ involvement by alpha-synuclein pathology in Lewy body disorders. Mov Disord 29: , 1010–1018. |
[165] | Gowers WR (1893) A manual of diseases of the nervous system, Blakiston, Philadelphia. |
[166] | Tanner CM , Ottman R , Goldman SM , Ellenberg J , Chan P , Mayeux R , Langston JW ((1999) ) Parkinson disease in TwinsAn Etiologic Study. JAMA 281: , 341–346. |
[167] | NIH RePORTER, https://reporter.nih.gov/search/TpAPMCuS7EmOd8irY02IdA/projects.Accessed January 9, 2024. |
[168] | NIH, NIH RePORTER, https://reporter.nih.gov/search/DQ4OWN_6KUCCFSYU6PQV7g/projects.Accessed January 8, 2024. |
[169] | De Miranda BR , Goldman SM , Miller GW , Greenamyre JT , Dorsey ER ((2022) ) Preventing Parkinson’s disease: An environmental agenda. J Parkinsons Dis 12: , 45–68. |
[170] | Wakabayashi K ((2020) ) Where and how alpha-synuclein pathology spreads in Parkinson’s disease. Neuropathology 40: , 415–425. |
[171] | Pereira PAB , Aho VTE , Paulin L , Pekkonen E , Auvinen P , Scheperjans F ((2017) ) Oral and nasal microbiota in Parkinson’s disease. Parkinsonism Relat Disord 38: , 61–67. |
[172] | Kulcsarova K , Bang C , Berg D , Schaeffer E ((2023) ) Pesticides and the microbiome-gut-brain axis: Convergent pathways in the pathogenesis of Parkinson’s disease. J Parkinsons Dis 13: , 1079–1106. |
[173] | Silva YP , Bernardi A , Frozza RL ((2020) ) The role of short-chain fatty acids from gut microbiota in gut-brain communication. Front Endocrinol 11: , 25. |
[174] | Scheperjans F , Aho V , Pereira PA , Koskinen K , Paulin L , Pekkonen E , Haapaniemi E , Kaakkola S , Eerola-Rautio J , Pohja M , Kinnunen E , Murros K , Auvinen P ((2015) ) Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov Disord 30: , 350–358. |
[175] | Wallen ZD , Stone WJ , Factor SA , Molho E , Zabetian CP , Standaert DG , Payami H ((2021) ) Exploring human-genome gut-microbiome interaction in Parkinson’s disease. NPJ Parkinsons Dis 7: , 74. |
[176] | Chen J , Li T , Ye C , Zhong J , Huang JD , Ke Y , Sun H ((2023) ) The lung microbiome: A new frontier for lung and brain disease. Int J Mol Sci 24: , 2170. |
[177] | Goria S , Pascal M , Corso M , Le Tertre A ((2021) ) Short-term exposure to air pollutants increases the risk of hospital admissions in patients with Parkinson’s disease –A multicentric study on 18 French areas. Atmospher Environ 264: , 118668. |
[178] | Lee H , Myung W , Kim DK , Kim SE , Kim CT , Kim H ((2017) ) Short-term air pollution exposure aggravates Parkinson’s disease in a population-based cohort. Sci Rep 7: , 44741. |
[179] | Chung SJ , Jeon S , Yoo HS , Yoon JH , Lee JE , Kim JS , Kim SJ , Oh JS , Kim JS , Ye BS , Sohn YH , Lee PH ((2019) ) Gastrectomy and nigrostriatal dopaminergic depletion in de novo Parkinson’s disease. Mov Disord 34: , 299–301. |
[180] | Ritchie H, What the history of London’s air pollution can tell us about the future of today’s growing megacities, OurWorld in Data, https://ourworldindata.org/london-air-pollution.June 20, 2017, Accessed January 5, 2024. |
[181] | Darweesh SKL , Koudstaal PJ , Stricker BH , Hofman A , Ikram MA ((2016) ) Trends in the incidence of Parkinson disease in the general population: The Rotterdam Study. Am J Epidemiol 183: , 1018–1026. |
[182] | Savica R , Grossardt BR , Bower JH , Ahlskog JE , Rocca WA ((2016) ) Time trends in the incidence of Parkinson disease. JAMA Neurol 73: , 981–989. |
[183] | Adler CH , Beach TG ((2016) ) Neuropathological basis of nonmotor manifestations of Parkinson’s disease. Mov Disord 31: , 1114–1119. |


