
Approaches to Early Parkinson’s Disease Subtyping
Abstract
Parkinson’s disease (PD) unfolds with pathological processes and neurodegeneration well before the emergence of noticeable motor symptoms, providing a window for early identification. The extended prodromal phase allows the use of risk stratification measures and prodromal markers to pinpoint individuals likely to develop PD. Importantly, a growing body of evidence emphasizes the heterogeneity within prodromal and clinically diagnosed PD. The disease likely comprises distinct subtypes exhibiting diverse clinical manifestations, pathophysiological mechanisms, and patterns of α-synuclein progression in the central and peripheral nervous systems. There is a pressing need to refine the definition and early identification of these prodromal subtypes. This requires a comprehensive strategy that integrates genetic, pathological, imaging, and multi-omics markers, alongside careful observation of subtle motor and non-motor symptoms. Such multi-dimensional classification of early PD subtypes will improve our understanding of underlying disease pathophysiology, improve predictions of clinical endpoints, progression trajectory and medication response, contribute to drug discovery and personalized medicine by identifying subtype-specific disease mechanisms, and facilitate drug trials by reducing confounding effects of heterogeneity. Here we explore different subtyping methodologies in prodromal and clinical PD, focusing on clinical, imaging, genetic and molecular subtyping approaches. We also emphasize the need for refined, theoretical a priori disease models. These will be prerequisite to understanding the biological underpinnings of biological subtypes, which have been defined by large scale data-driven approaches and multi-omics fingerprints.
Plain Language Summary
Parkinson’s disease is an incurable neurodegenerative disorder characterized by a 20-year prodromal phase before an individual is formally diagnosed. During this prodromal or early disease phase, non-motor symptoms gradually accumulate including a reduced ability to smell, sleep disturbance, constipation, depression, anxiety, and memory problems. Subtle motor symptoms including slowness of movement, stiffness, and tremor may be present, but not to the extent required for a clinical diagnosis. Each individual with prodromal Parkinson’s disease is affected with a unique combination of these symptoms which progress at different rates. Subtyping attempts to understand this variability by defining groups of patients with sets of key features at a clinical, genetic, imaging, or molecular level. This article reviews subtyping approaches and how they might improve our understanding of how Parkinson’s disease evolves.
INTRODUCTION
Parkinson’s disease (PD) is a slowly progressive neurodegenerative disorder that is currently diagnosed and identified clinically, although both biological classification system (SynNeurGe)1 and biological definition of PD with integrated staging system (NSD-ISS)2 have recently been proposed, based on the underlying clinicopathological drivers of PD. Importantly, these biologically-driven PD classification systems make no distinction between prodromal or manifest PD, or between dementia with Lewy bodies (DLB) or PD dementia (PDD). Just as each person is unique, each person with Parkinson’s presents with a different range of motor and non-motor symptoms at varying severities, with a different individualized rate of subsequent progression. These differences, referred to as heterogeneity, have led many researchers to believe there are different subtypes of PD that can be defined using multiple statistical approaches (i.e., data-driven, machine learning) and different data sources (i.e., clinical, genetic, imaging).
Delineation of PD subtypes might (i) improve our understanding of underlying disease pathophysiology (ii) improve predictions of clinical endpoints, progression trajectory and medication response (iii) illuminate drug discovery by identifying perturbed cellular mechanisms underpinning each subtype, and (iv) deliver cheaper and improved drug trials by reducing potential confounding effects of heterogeneity.
PD does not start suddenly but progresses through early phases, during which neurodegeneration has commenced but has not yet reached the point where PD can be definitively diagnosed based on the emergence of several core clinical features.1,2 Early PD can be divided into preclinical PD (where neurodegeneration has commenced but no evident symptoms or signs), prodromal PD (where motor/nonmotor symptoms and signs are present but are yet insufficient to define disease), and clinical PD (diagnosis based on classical motor signs)1. For this subtyping review we focus on MDS-criteria diagnosed prodromal and clinical PD within 5 years of diagnosis as representing the key stages of early PD. Here we explore different subtyping methodologies with a focus on clinical, imaging, genetic, and molecular subtyping approaches. Lastly, we explore how current and future personalized medicine approaches, including molecular subtyping could be employed on an individual basis for PD patients to determine treatment choices and deliver neuroprotective therapies that slow down disease progression.
CLINICAL SUBTYPING IN PD AND PRODROMAL PD
Clinical subtyping studies tend to fall into two main categories: 1) hypothesis-driven (top-down) analysis based on pre-determined groups and 2) data-driven (bottom-up) analyses. Most hypothesis-driven analysis have included analysis of only a single clinical or demographic characteristic, such as age of onset, motor subtyping (e.g., akinetic-rigid versus tremor dominant motor complications or prominent non-motor symptoms in untreated and early PD).3
By contrast, data-driven approaches tend to consider multiple domains of data in a hypothesis-free, unbiased subtyping approach. Examples include cluster analysis4,5 and machine learning modelling.6 In 2021 the Movement Disorders Society (MDS) Task Force for PD subtypes critically appraised 38 available PD subtyping studies, finding that 68% were cross sectional (32% longitudinal), with 66% using exclusively a data-driven approach and 21% using a hypothesis-driven approach.7 Participants across all studies had approximately five years of disease duration. Most data-driven studies used at least 3 phenotypic domains, while hypothesis-driven domains used a single domain (e.g., age or sex). Only 5 studies included non-clinical biomarkers, for example imaging. Only 4/38 studies received best quality ratings, being multi-center, longitudinal, data-driven using more than 1 clinical domain (motor and non-motor) and only 1 study used additional CSF and neuroimaging biomarker data.8 There is therefore a future shift-change needed from PD subtyping to precision medicine approaches, which utilize omics and clinical data via machine learning and other statistical approaches to interrogate disease mechanisms on an individual basis.
There is limited knowledge on subtyping of prodromal forms of PD. However, a recent review suggested that the heterogeneity seen in PD is likely to start in the prodromal phase, manifesting through the variable onset, progression of motor and non-motor symptoms and imaging markers seen in prodromal phase.9 Possible strategies for prodromal subtyping can be based on symptoms such as REM sleep behavior disorder, genotypes, or on a priori biologically based disease models, such as the brain-first vs. body-first model of α-synuclein propagation.9 A recent, large multicenter study of 1160 polysomnogram-confirmed REM sleep behavior disorder subjects assessed for prodromal PD using MDS criteria demonstrated faster progression in motor compared to non-motor (cognitive, olfactory and autonomic) variables. Overall, phenoconvertors showed faster progression than non-convertors in motor, olfactory, cognitive and autonomic markers.10
IMAGING HETEROGENEITY IN PRODROMAL PD
Radiotracer imaging of the dopamine transporter (DaT) plays a decisive role in PD diagnostics.11 In early PD, the reduction is typically most pronounced in the posterior putamen leading to a low putamen-to-caudate ratio.12 Importantly, patients with early RBD-positive PD or dementia with Lewy bodies (DLB) often show more symmetric striatal reductions and DLB patients also display early caudate involvement.13–15 Thus, the appearance of these features during the prodromal phase may associate to a more severe clinical phenotype with early dementia.
Importantly, dopamine imaging has value relatively close to the appearance of parkinsonism. In prodromal PD, a pathological DaT scan is a strong marker of imminent phenoconversion.16 However, in a cross-sectional sample of prodromal iRBD patients, who will be at different “prodromal disease stages”, the majority of dopamine scans (63%–69%) are still normal.17,18 Similarly, in LRRK2 G2019S mutation carriers, DAT deficits were present in all who phenoconverted within 2 years, but the vast majority of non-symptomatic LRRK2 carriers had normal DAT scans.19 Thus, variable patterns of dopamine reduction will mainly add to prodromal subtyping close to phenoconversion.
In community-based groups of subjects with relevant non-motor symptoms, DaT imaging is an important tool for enrichment, due to its ability to predict short-term phenoconversion. Two studies assembled relatively large groups of patients with idiopathic hyposmia.20–22 Approximately 10–15% of these cases had pathological DaT scans at baseline, and they nearly all converted to PD within 5 years. In a subgroup of cases with hyposmia, male sex, and constipation, more than 40% had a pathological DAT scan at baseline.23
Such studies demonstrate the power of combining several symptoms and biomarkers, but they also run the risk of introducing subtype-specific biases. For instance, RBD, constipation, hyposmia, orthostatism, and pathological MIBG scans tend to cluster together, and probably reflect a relatively homogeneous “body-first” subtype of Lewy body disease in RBD positive cases.5,24 In contrast, RBD-negative cases generally have fewer and milder non-motor symptoms including hyposmia, so these cases will be more difficult to identify during the prodromal phase, and they will generally have normal imaging markers.5,25,26 However, if hyposmia is more common in one subtype (i.e., in RBD-positive body-first cases), using hyposmia to define a prodromal group will lead to an enriched body-first cohort. In other words, a subtype-specific bias is introduced.
PET imaging with cholinergic tracers has shown marked reduction in colon uptake in iRBD patients,27 and in newly diagnosed DLB patients, most of whom had RBD.28 These alterations point to early parasympathetic denervation. In contrast, de novo PD patients without RBD showed much more normal colon signal compatible with a “brain-first” subtype 25. Additionally, de novo DLB patients exhibit profound cholinergic cortical denervation in an almost universal pattern compared to non-demented PD patients, in whom reductions are seen primarily in posterior cortical regions.29,30 Interestingly, GBA-mutations are associated with more rapid cognitive decline, and a similar universal pattern of cholinergic cortical reduction was seen de novo PD cases with GBA-mutations, when compared to de novo sporadic PD.31 Since these different patterns most likely developed gradually during the prodromal stage, it seems probable that cholinergic PET could be useful to stratify prodromal PD patients into subtypes with slower and faster progression to dementia, respectively.
Imaging of the cardiac sympathetic system with [123I]MIBG has shown potential for separating meaningful subtypes at the prodromal stage. Nearly all cases with iRBD develop severely pathological MIBG scans during the prodromal phase, in many cases more than 10 years before the nigrostriatal dopamine system starts degenerating.18,27,32–34 Conversely, most early PD cases without RBD exhibit normal or only mildly affected MIBG scans.25 Both RBD and pathological MIBG scans are associated with a more severe, progressive clinical phenotype. On MRI, prodromal cases with iRBD display reductions in the neuromelanin-sensitive signal of the locus coeruleus, whereas de novo PD without RBD have more normal scans.27,35–37
Imaging of co-pathologies may also be highly relevant for prodromal subtyping. Rapid cognitive decline in Lewy body disease is strongly associated with Alzheimer-type co-pathology.38–40 Approximately 40–50% of patients with PD dementia and 75% of DLB patients exhibit marked amyloid-β pathology at postmortem.40 On amyloid PET, a meta-analysis demonstrated that around 38% of PDD and 68% of DLB patients had positive amyloid scans.41 Given that build-up of amyloid-β takes many years, these findings suggest that amyloid PET during the prodromal phase can identify patients at high risk of rapid cognitive decline due to amyloid pathology. This also presents an interesting opportunity for disease-modifying trials using anti-amyloid treatments. In patients with isolated RBD, who are positive on amyloid PET, anti-amyloid immunotherapy could theoretically delay or reduce cognitive decline.
GENETIC SUBTYPING IN PRODROMAL PD
The most common PD-associated gene mutations are found in GBA and LRRK2, and different mutations in these genes have variable effects on risk for PD.42,43 GBA mutations can be divided into severe or mild mutations, and individuals with severe mutations have higher risk for PD and earlier age at onset than mild mutation carriers. Recent genetic studies specifically focusing on RBD have shown the strongest genetic association is with GBA mutations.44,45 The association between RBD and GBA mutations is even stronger than the association with PD in a similar population46, suggesting that GBA mutations are specifically associated with probable RBD. Clinically, both GBA-associated PD and RBD-associated PD are associated with rapid motor progression, cognitive decline and faster dementia progression, and autonomic dysfunction. From a pathological point of view, both RBD-associated PD and GBA-associated PD probably have a more diffused spread of α-synuclein accumulation.47 LRRK2-associated PD has a more benign course, with less cognitive decline, hyposmia and autonomic dysfunction than PD.42 Unsurprisingly, LRRK2 mutations have not been associated with RBD.48 Other examples of the genetic overlap between RBD and PD include an association with SCARB2 and MAPT loci49, with SCARB2 encoding the transporter responsible for glucocerebrosidase transport from endoplasmic reticulum and lysosome. Lastly, a study by the same group performed in-depth analysis of the SNCA locus in 1076 RBD, 1013 PD, 415 DLB and 6,155 control subjects. A distinct pattern of association at the SNCA locus in RBD as compared to PD was found, with an opposite direction of effect at the 3’ of SNCA. Several 5’ SNCA variants were associated with iRBD and with pRBD in overt synucleinopathies.50
In summary, there is convincing evidence that the genetic basis of PD and RBD overlap, at least partially.42 These observations are important to note for future studies and clinical trials, as screening for RBD should be considered for early detection of conversion to a synucleinopathy in asymptomatic carriers of GBA mutations followed longitudinally. Cohorts of GBA mutation carriers with RBD would be an ideal at-risk group to consider for future clinical trial interventions, given their high risk of future conversion to PD or a related synucleinopathy. Drugs that aim to modify the enzymatic activity of glucocerebrosidase, which is encoded by the GBA1 gene could be tested in polysomnogram-diagnosed RBD patients carrying GBA mutations.42
MOLECULAR SUBTYPING AND FUTURE PRECISION MEDICINE APPROACHES
Key findings of the MDS Task Force for PD subtypes literature review include that the majority of clinical subtyping studies had questionable clinical applicability and unknown biological relevance. Only 1 study out of 38 using additional CSF and neuroimaging biomarker data to inform subtyping, for example to understand the underlying cellular mechanisms driving PD and prodromal PD progression.7 This sets the stage for future precision medicine (PM) approaches, and how these might be applied on an individual basis for those with PD and other complex diseases caused by a combination of genetic, lifestyle, and environmental factors. PM can be broadly defined as the use of diagnostic tools and treatments targeted to individual patient needs based on genetic, biomarker, or psychosocial characteristics.
Within PD, a focus on key pathophysiological mechanisms including impaired lysosomal and mitochondrial function, neuroinflammation and α-synuclein deposition using as tools proteomics and metabolomics of blood and spinal fluid, patient-derived fibroblasts from skin cells, peripheral blood-derived mononuclear cells and exosomes, induced pluripotent-stem cell derived cell models and α-synuclein seeding aggregation measured in spinal fluid, skin and blood (Fig. 1). The aim of such multi-omics approaches is to understand the perturbed molecular mechanisms driving clinically-defined PD subtypes and subsequent motor/cognitive progression4,51,52 before then applying this on an individual precision medicine basis.
Fig. 1
Molecular subtyping in PD. Figure 1A: The ‘palette’ model of the multifactorial etiology of a complex disease such as PD. The different colors under ‘Disease pathways’ represent four key hypothetical disease pathways or pathophysiological mechanisms relevant to PD. Each of the four individuals (A–D) has different contributions by the colors representing the various pathways contributing to their disease at a specific time point. The contribution of each pathway at the specific timepoint is represented by an ‘X’, so that individuals with the same clinically defined disease can have very different underlying pathophysiological mechanisms, as illustrated by the difference in colors in Fig. 1B. Moving from prodromal to manifest PD, different pathways and contributions from each pathway will occur over time. Source: the figure is adapted from Johansson.58
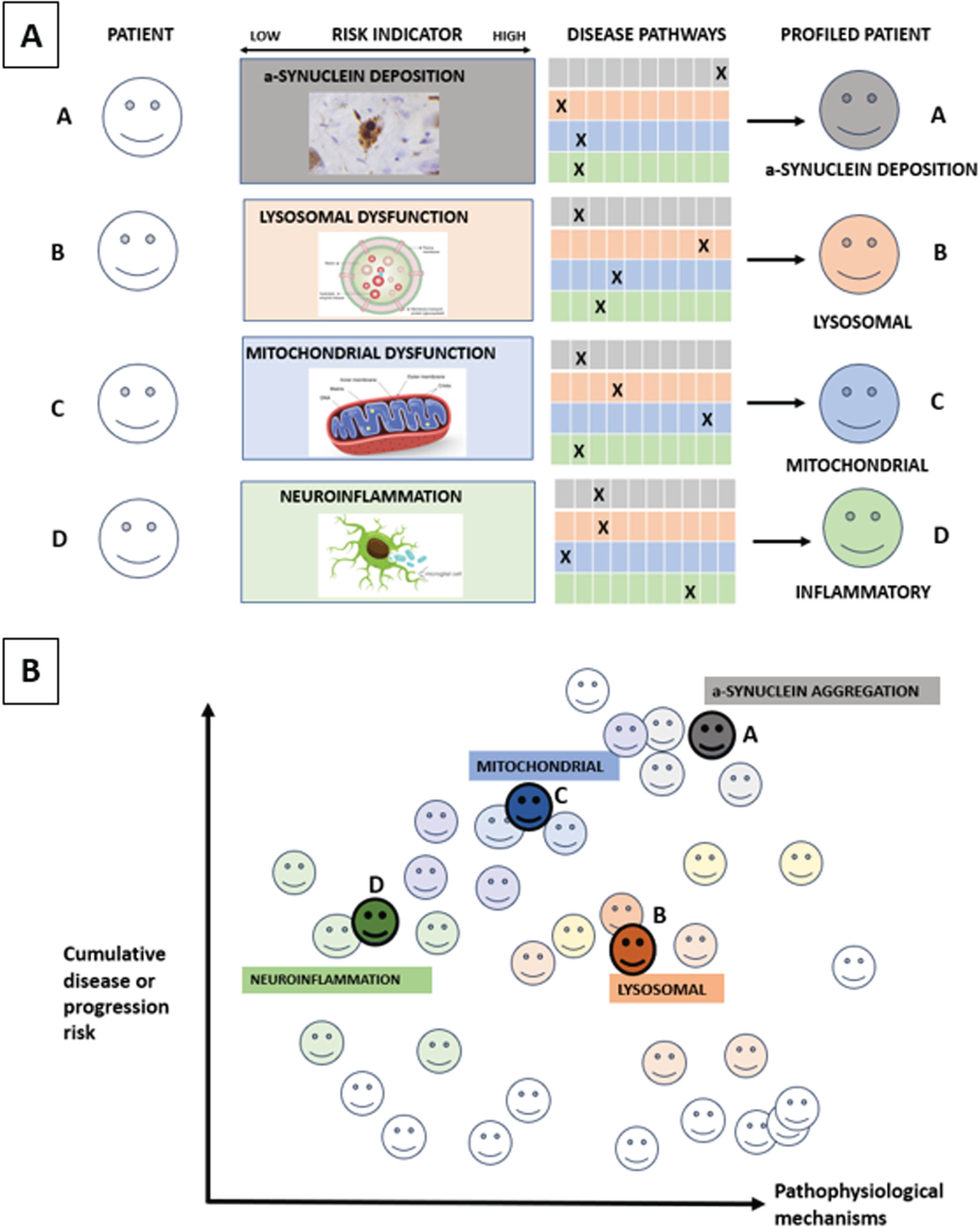
The MDS task force on PD subtyping recently proposed that the key purposes of PD subtyping are to: 1) Predict disease progression; 2) To predict treatment response; and 3) Identify therapeutic targets for disease modification.53 Recommendations for sensitive and specific biomarkers of pathophysiology-defined subtypes54,55 for future proof-of-concept disease modifying therapies (DMT) consistent with the proposed mechanism of action of the investigation product included: a) Genetic subtyping (e.g., LRRK2, GBA1) as covered above; b) α-synuclein seeding status;54,55 c) lysosomal dysfunction assays and neuro-inflammatory markers. Here, a particular lysosomal function assay relevant for prodromal or clinical PD biology or progression could be deployed to identify a particular biological subtype that would benefit from treatment designed to improve lysosomal function. As is the case for cancer treatments however, it is important to recognize that most patients with prodromal or clinical PD will experience multiple perturbations across multiple cell mechanisms relevant to PD, that will require a combination of different treatments at different disease time points designed to optimize their clinical function. Furthermore, we currently lack trial-ready prodromal and clinical PD cohorts with comprehensive biological characterization, using reproducible assays that are expensive and time-consuming to deliver.
So far, in clinical and prodromal PD, genomic-based precision medicine has been mainly used in patients with highly penetrant monogenic disease forms or those carrying common PD risk genes including GBA, LRRK2, and PRKN (Parkin). Trials in clinical PD patients carrying a heterozygous GBA mutation include the oral Venglustat trial (MOVES-PD)56 and BIA-28-6156 (a GCase allosteric activator) both designed to improve GCase activity,57 alongside trials in LRRK2-PD using small molecule LRRK2 kinase inhibitors,58 and Parkin gene activators aimed at improving mitophagy, a key impaired pathway.59
A NEED FOR THEORETICAL DISEASE MODELS IN EARLY PD
Large GWAS and multi-omics datasets will in combination with detailed clinical information allow more robust and granular subtyping of prodromal and manifest patients and will simultaneously pave the way for developing diagnostic algorithms to diagnose such subtypes. However, these strategies are typically post hoc and cannot generally offer explanations as to how the different patient subtypes arise. Thus, if we manage to define robust subtypes of Lewy body disease based on reproducible multi-omics fingerprints, it does not automatically guarantee that we will know, which pharmaceutical intervention is likely to benefit each of those subtypes. It therefore remains important to develop a priori models of disease mechanisms, which have clear, testable predictions and then refine or refute such models using the increasingly rich multi-omics datasets.
Several a priori models of disease mechanisms have been proposed, including the Braak hypothesis, the threshold theory, and the cortical pathogenic theory of PD.60–62 Figure 2 summarizes the brain-first vs. body-first hypothesis of Lewy body disease, another example of an a priori disease model.26 Some of these models are more well developed than others, but each of them is in principle testable and falsifiable. For example, the brain- vs. body-first model proposes that α-synuclein aggregation and propagation is the core pathogenic feature in Lewy body disease. Other pathogenic factors such as genetic mutations, toxins, lysosomal and mitochondrial dysfunction, and neuroinflammation serve as important triggers or aggravators of the α-synuclein pathology.
Fig. 2
A priori disease modelling exemplified by brain- vs. body-first Lewy body disease. Figure 2 (A) A priori disease modelling exemplified by the brain-first vs. body-first model of Lewy body disease.64 This model posits that (i) α-synuclein aggregation starts in a single place in most patients and propagates from there. (ii) The two most probable origin sites are the olfactory epithelium (brain-first) and gastro-intestinal nervous system (body-first). (iii) Propagating α-synuclein pathology thereby leads to two overarching clinical types of Lewy body disease. Body-first cases typically develop dysautonomia and RBD before a relatively symmetric dopamine degeneration occurs. In contrast, brain-first cases often develop more asymmetric dopamine loss, have a shorter prodromal phase, and display few non-motor symptoms at diagnosis. (iv) Histologically, early body-first cases display Lewy pathology mainly in the autonomic system and brainstem, whereas early brain-first cases exhibit pathology mainly restricted to the olfactory bulb and limbic system. (v) With time, the two types converge on a common end-stage phenotype characterized by widespread peripheral and central pathology, parkinsonism, non-motor symptoms including RBD, and dementia. (B) The brain- vs. body-first model proposes that α-synuclein aggregation and propagation is the core pathogenic feature but acknowledges that many other pathogenic factors are important to the pathogenesis of Lewy body disease. The panel shows a non-exhaustive list of potential factors, which may (i) trigger the aggregation of α-synuclein in the first place or allow it to be triggered, (ii) facilitate the propagation of α-synuclein aggregates through the nervous system, and (iii) increase the vulnerability of neurons to the presence of α-synuclein pathology leading to neuronal dysfunction and eventually degeneration. Amy, amygdala; DMV, dorsal motor nucleus of vagus; IML, intermediolateral column; LC, locus coeruleus; OB, olfactory bulb; SN, substantia nigra; ST, sympathetic trunk; Tox, transentorhinal cortex.
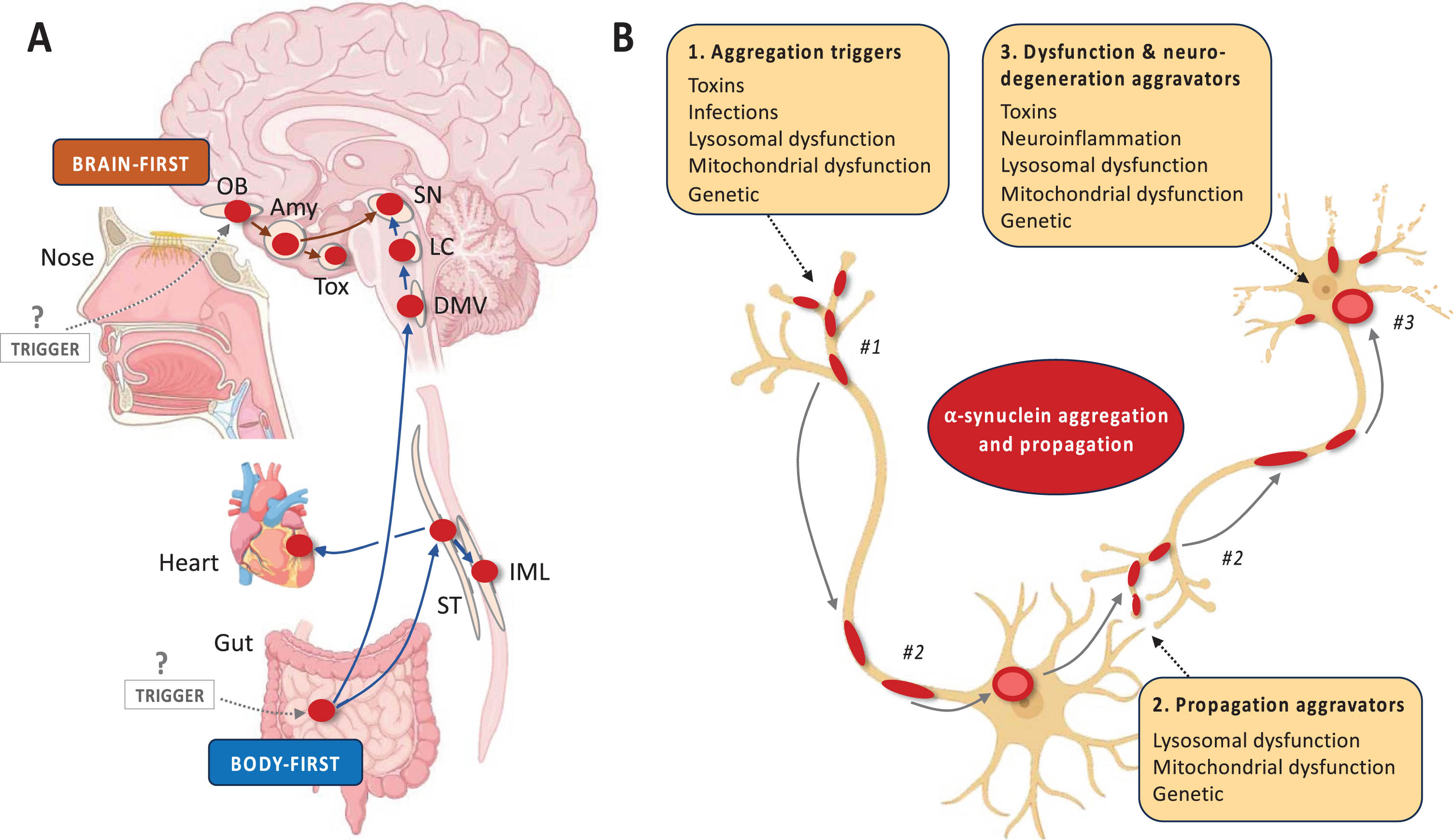
As explained in Fig. 2, the model predicts that certain clinical phenotypes with a stereotypical evolution of symptoms will appear (i.e., clinical brain-first and body-first subtypes) and that these types map onto specific patterns of Lewy pathology during the earlier disease stages (i.e., amygdala-olfactory predominant vs. brainstem-autonomic predominant patterns, respectively). These predictions are testable, especially once an α-synuclein PET tracer becomes available, but can be tested by other means. For example, one study showed patterns of top-down vs. bottom-up neurodegeneration using multi-modal imaging, which was compatible with the model.25 Another recent study showed that some early PD patients display pathological α-synuclein aggregates in skin but not in the nose, compatible with body-first etiology, whereas other cases showed the opposite pattern, compatible with brain-first etiology.63
If such studies demonstrate that the model has predictive power, it will support that localized α-synuclein aggregation and propagation is a key factor in shaping the evolution of symptoms and neurodegeneration in Lewy body disease—and therefore a likely treatment target. If on the other hand, studies were to show significant decoupling between temporal spreading of α-synuclein and neurodegeneration, it would suggest that the model is false or at least incomplete.
CONCLUSION
The first steps towards a biological definition and staging system of PD have recently been made,54,55 and it has been proposed to define PD based on the presence of aggregated α-synuclein and associated neurodegeneration. Likewise, future subtyping of PD will also need to move towards a biological foundation. Multi-omics and other studies defining perturbed cellular mechanisms on deeply phenotyped cohorts should allow us to define subtypes, which are more homogenous from a pathophysiological and biochemical point of view.
Initially, clinical characteristics, progression rates, and prognosis can be mapped onto particular omics-fingerprints in well-defined patient cohorts without a full understanding of the underlying disease mechanisms responsible for producing such homogenous clusters. However, continued research will be needed to understand and model these disease mechanisms, as this will provide clues to new treatment targets, which can be implemented in the prodromal phase.
ACKNOWLEDGMENTS
The Oxford Discovery cohort of prodromal RBD participants is funded by Parkinson’s UK and the National Institute of Health Research (NIHR) Oxford Biomedical Research Centre
FUNDING
MH receives support from the NIHR Oxford Biomedical Research Centre, Parkinson’s UK, Horizon 2020, Oxford University, Cure Parkinson’s Trust. PB received support from the Lundbeck Foundation, Michael J. Fox Foundation, Danish Parkinson’s association, Jascha foundation, Riisfort foundation.
CONFLICT OF INTEREST
MH is an academic founder member of NeuHealth Digital Ltd (company number: 14492037), a digital biomarker platform to remotely manage condition progression for Parkinson’s. She has received payment for Advisory Board attendance/consultancy for Lundbeck, ESCAPE Bio, Evidera, Manus Neurodynamica, Biogen MA, CuraSen Therapeutics, Roche Products Ltd, JAZZ Pharma, Aventis Pharma. CS declares no conflicts of interest. PB is a paid consultant of Novo Nordisk A/S, Abbvie, Curium, TreeFrog therapeutics.
REFERENCES
1. | Berg D , Postuma RB , Adler CH , et al. MDS research criteria for prodromal Parkinson’s disease. Mov Disord (2015) ; 30: : 1600–1611. |
2. | Postuma RB , Berg D , Stern M , et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord (2015) ; 30: : 1591–1601. |
3. | Sauerbier A , Jenner P , Todorova A , et al. Non motor subtypes and Parkinson’s disease. Parkinsonism Relat Disord (2016) ; 22: (Suppl 1): S41–S46. |
4. | Lawton M , Ben-Shlomo Y , May MT , et al. Developing and validating Parkinson’s disease subtypes and their motor and cognitive progression. J Neurol Neurosurg Psychiatry (2018) ; 89: : 1279–1287. |
5. | Fereshtehnejad SM , Romenets SR , Anang JB , et al. New clinical subtypes of Parkinson disease and their longitudinal progression: A prospective cohort comparison with other phenotypes. JAMA Neurol (2015) ; 72: : 863–873. |
6. | Dadu A , Satone V , Kaur R , et al. Identification and prediction of Parkinson’s disease subtypes and progression using machine learning in two cohorts. NPJ Parkinsons Dis (2022) ; 8: : 172. |
7. | Mestre TA , Fereshtehnejad SM , Berg D , et al. Parkinson’s disease subtypes: Critical appraisal and recommendations. J Parkinsons Dis (2021) ; 11: : 395–404. |
8. | Zhang X , Chou J , Liang J , et al. Data-driven subtyping of Parkinson’s disease using longitudinal clinical records: A cohort study. Sci Rep (2019) ; 9: : 797. |
9. | Berg D , Borghammer P , Fereshtehnejad SM , et al. Prodromal Parkinson disease subtypes - key to understanding heterogeneity. Nat Rev Neurol (2021) ; 17: : 349–361. |
10. | Joza S , Hu MT , Jung KY , et al. Progression of clinical markers in prodromal Parkinson’s disease and dementia with Lewy bodies: a multicentre study. Brain (2023) ; 146: : 3258–3272. |
11. | Kaasinen V , Vahlberg T , Stoessl AJ , et al. Dopamine receptors in Parkinson’s disease: A meta-analysis of imaging studies. Mov Disord (2021) ; 36: : 1781–1791. |
12. | Kong Y , Zhang C , Liu K , et al. Imaging of dopamine transporters in Parkinson disease: A meta-analysis of (18) F/(123) I-FP-CIT studies. Ann Clin Transl Neurol (2020) ; 7: : 1524–1534. |
13. | Walker Z , Costa DC , Walker RW , et al. Striatal dopamine transporter in dementia with Lewy bodies and Parkinson disease: a comparison. Neurology (2004) ; 62: : 1568–1572. |
14. | Knudsen K , Fedorova TD , Horsager J , et al. Asymmetric dopaminergic dysfunction in brain-first versus body-first Parkinson’s disease subtypes. J Parkinsons Dis (2021) ; 11: : 1677–1687. |
15. | Fedorova TD , Kundsen K , Horsager J , et al. Dopaminergic dysfunction is more symmetric in dementia with Lewy bodies compared to Parkinson’s disease. J Parkinsons Dis (2023) ; 13: : 515–523. |
16. | Arnaldi D , Chincarini A , Hu MT , et al. Dopaminergic imaging and clinical predictors for phenoconversion of REM sleep behaviour disorder. Brain (2021) ; 144: : 278–287. |
17. | Postuma RB , Iranzo A , Hu M , et al. Risk and predictors of dementia and parkinsonism in idiopathic REM sleep behaviour disorder: a multicentre study. Brain (2019) ; 142: : 744–759. |
18. | Nishikawa N , Murata M , Hatano T , et al. Idiopathic rapid eye movement sleep behavior disorder in Japan: An observational study. Parkinsonism Relat Disord (2022) ; 103: : 129–135. |
19. | Simuni T , Merchant K , Brumm MC , et al. Longitudinal clinical and biomarker characteristics of non-manifesting LRRK2 G2019S carriers in the PPMI cohort. NPJ Parkinsons Dis (2022) ; 8: : 140. |
20. | Jennings D , Siderowf A , Stern M , et al. Conversion to Parkinson disease in the PARS hyposmic and dopamine transporter–deficit prodromal cohort. JAMA Neurol (2017) ; 74: : 933–940. |
21. | Ponsen MM , Stoffers D , Booij J , et al. Idiopathic hyposmia as a preclinical sign of Parkinson’s disease. Ann Neurol (2004) ; 56: : 173–181. |
22. | Ponsen MM , Stoffers D , Wolters E , et al. Olfactory testing combined with dopamine transporter imaging as a method to detect prodromal Parkinson’s disease. J Neurol Neurosurg Psychiatry (2010) ; 81: : 396–399. |
23. | Jennings D , Siderowf A , Stern M , et al. Imaging prodromal Parkinson disease: the Parkinson Associated Risk Syndrome Study. Neurology (2014) ; 83: : 1739–1746. |
24. | Kim JS , Park HE , Park IS , et al. Normal ‘heart’ in Parkinson’s disease: Is this a distinct clinical phenotype? Eur J Neurol (2017) ; 24: : 349–356. |
25. | Horsager J , Andersen KB , Knudsen K , et al. Brain-first versus body-first Parkinson’s disease: A multimodal imaging case-control study. Brain (2020) ; 143: : 3077–3088. |
26. | Borghammer P . The alpha-Synuclein Origin and Connectome Model (SOC Model) of Parkinson’s disease: Explaining motor asymmetry, non-motor phenotypes, and cognitive decline. J Parkinsons Dis (2021) ; 11: : 455–474. |
27. | Knudsen K , Fedorova TD , Hansen AK , et al. In-vivo staging of pathology in REM sleep behaviour disorder: a multimodality imaging case-control study. Lancet Neurol (2018) ; 17: : 618–628. |
28. | Okkels N , Horsager J , Fedorova TD , et al. Impaired cholinergic integrity of the colon and pancreas in dementia with Lewy bodies. Brain (2023) ; 147: : 255–266. |
29. | Okkels N , Horsager J , Labrador-Espinosa M , et al. Severe cholinergic terminal loss in newly diagnosed dementia with Lewy bodies. Brain (2023) ; 146: : 3690–3704. |
30. | Shimada H , Hirano S , Shinotoh H , et al. Mapping of brain acetylcholinesterase alterations in Lewy body disease by PET. Neurology (2009) ; 73: : 273–278. |
31. | Slingerland S , van der Zee S , Carli G , et al. Cholinergic innervation topography in GBA-associated de novo Parkinson’s disease patients. Brain (2023) ; 147: : 900–910. |
32. | Miyamoto T , Miyamoto M , Iwanami M , et al. Follow-up study of cardiac (1)(2)(3)I-MIBG scintigraphy in idiopathic REM sleep behavior disorder. Eur J Neurol (2011) ; 18: : 1275–1278. |
33. | Nagayama H , Hamamoto M , Ueda M , et al. Reliability of MIBG myocardial scintigraphy in the diagnosis of Parkinson’s disease. J Neurol Neurosurg Psychiatry (2005) ; 76: : 249–251. |
34. | Borghammer P . The brain-first vs. body-first model of Parkinson’s disease with comparison to alternative models. J Neural Transm (Vienna) (2023) ; 130: : 737–753. |
35. | Ehrminger M , Latimier A , Pyatigorskaya N , et al. The coeruleus/subcoeruleus complex in idiopathic rapid eye movement sleep behaviour disorder. Brain (2016) ; 139: : 1180–1188. |
36. | Garcia-Lorenzo D , Longo-Dos Santos C , Ewenczyk C , et al. The coeruleus/subcoeruleus complex in rapid eye movement sleep behaviour disorders in Parkinson’s disease. Brain (2013) ; 136: : 2120–2129. |
37. | Sommerauer M , Fedorova TD , Hansen AK , et al. Evaluation of the noradrenergic system in Parkinson’s disease: an 11C-MeNER PET and neuromelanin MRI study. Brain (2018) ; 141: : 496–504. |
38. | Donaghy P , Firbank M , Petrides G , et al. The relationship between plasma biomarkers and amyloid PET in dementia with Lewy bodies. Parkinsonism Relat Disord (2022) ; 101: : 111–116. |
39. | Donaghy PC , Firbank MJ , Thomas AJ , et al. Amyloid imaging and longitudinal clinical progression in dementia with Lewy bodies. Am J Geriatr Psychiatry (2020) ; 28: : 573–577. |
40. | Irwin DJ , Grossman M , Weintraub D , et al. Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: a retrospective analysis. Lancet Neurol (2017) ; 16: : 55–65. |
41. | Petrou M , Dwamena BA , Foerster BR , et al. Amyloid deposition in Parkinson’s disease and cognitive impairment: a systematic review. Mov Disord (2015) ; 30: : 928–935. |
42. | Gan-Or Z , Alcalay RN , Rouleau GA , et al. Sleep disorders and Parkinson disease; lessons from genetics. Sleep Med Rev (2018) ; 41: : 101–112. |
43. | Gan-Or Z , Liong C and Alcalay RN. GBA-associated Parkinson’s disease and other synucleinopathies. Curr Neurol Neurosci Rep (2018) ; 18: : 1–10. |
44. | Barber TR , Lawton M , Rolinski M , et al. Prodromal parkinsonism and neurodegenerative risk stratification in REM sleep behavior disorder. Sleep (2017) ; 40: : zsx071. |
45. | Gan-Or Z , Mirelman A , Postuma RB , et al. GBA mutations are associated with rapid eye movement sleep behavior disorder. Ann Clin Transl Neurol (2015) ; 2: : 941–945. |
46. | Noreau A , Riviere JB , Diab S , et al. Glucocerebrosidase mutations in a French-Canadian Parkinson’s disease cohort. Can J Neurol Sci (2011) ; 38: : 772–773. |
47. | Postuma RB , Adler CH , Dugger BN , et al. REM sleep behavior disorder and neuropathology in Parkinson’s disease. Mov Disord (2015) ; 30: : 1413–1417. |
48. | Fernandez-Santiago R , Iranzo A , Gaig C , et al. Absence of LRRK2 mutations in a cohort of patients with idiopathic REM sleep behavior disorder. Neurology (2016) ; 86: : 1072–1073. |
49. | Gan-Or Z , Girard SL , Noreau A , et al. Parkinson’s disease genetic loci in rapid eye movement sleep behavior disorder. J Mol Neurosci (2015) ; 56: : 617–622. |
50. | Krohn L , Wu RYJ , Heilbron K , et al. Fine-mapping of SNCA in rapid eye movement sleep behavior disorder and overt synucleinopathies. Ann Neurol (2020) ; 87: : 584–598. |
51. | Lawton M , Tan MM , Ben-Shlomo Y , et al. Genetics of validated Parkinson’s disease subtypes in the Oxford Discovery and Tracking Parkinson’s cohorts. J Neurol Neurosurg Psychiatry (2022) ; 93: : 952–959. |
52. | Lawton M , Baig F , Toulson G , et al. Blood biomarkers with Parkinson’s disease clusters and prognosis: The oxford discovery cohort. Mov Disord (2020) ; 35: : 279–287. |
53. | Marras C , Fereshtehnejad SM , Berg D , et al. Transitioning from subtyping to precision medicine in Parkinson’s disease: a purpose-driven approach. Mov Disord (2024) ; 39: : 462–471. |
54. | Hoglinger GU , Adler CH , Berg D , et al. A biological classification of Parkinson’s disease: the SynNeurGe research diagnostic criteria. Lancet Neurol (2024) ; 23: : 191–204. |
55. | Simuni T , Chahine LM , Poston K , et al. A biological definition of neuronal alpha-synuclein disease: towards an integrated staging system for research. Lancet Neurol (2024) ; 23: : 178–190. |
56. | Peterschmitt MJ , Saiki H , Hatano T , et al. Safety, pharmacokinetics, and pharmacodynamics of oral venglustat in patients with Parkinson’s disease and a GBA mutation: results from part 1 of the randomized, double-blinded, placebo-controlled MOVES-PD Trial. J Parkinsons Dis (2022) ; 12: : 557–570. |
57. | den Heijer JM , Kruithof AC , Moerland M , et al. A phase 1B trial in GBA1-associated Parkinson’s disease of BIA-28-6156, a glucocerebrosidase activator. Mov Disord (2023) ; 38: : 1197–1208. |
58. | Jennings D , Huntwork-Rodriguez S , Henry AG , et al. Preclinical and clinical evaluation of the LRRK2 inhibitor DNL201 for Parkinson’s disease. Sci Transl Med (2022) ; 14: : eabj2658. |
59. | Miller S and Muqit MMK. Therapeutic approaches to enhance PINK1/Parkin mediated mitophagy for the treatment of Parkinson’s disease. Neurosci Lett (2019) ; 705: : 7–13. |
60. | Braak H , Del Tredici K , Rub U , et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging (2003) ; 24: : 197–211. |
61. | Engelender S and Isacson O. The threshold theory for Parkinson’s disease. Trends Neurosci (2017) ; 40: : 4–14. |
62. | Foffani G and Obeso JA. A cortical pathogenic theory of Parkinson’s disease. Neuron (2018) ; 99: : 1116–1128. |
63. | Kuzkina A , Rossle J , Seger A , et al. Combining skin and olfactory alpha-synuclein seed amplification assays (SAA)-towards biomarker-driven phenotyping in synucleinopathies. NPJ Parkinsons Dis (2023) ; 9: : 79. |
64. | Borghammer P and Van Den Berge N. Brain-first versus gut-first Parkinson’s disease: A hypothesis. J Parkinsons Dis (2019) ; 9: : S281–S295. |


