
Pharmacotherapy for Disease Modification in Early Parkinson’s Disease: How Early Should We Be?
Abstract
Slowing or halting progression continues to be a major unmet medical need in Parkinson’s disease (PD). Numerous trials over the past decades have tested a broad range of interventions without ultimate success. There are many potential reasons for this failure and much debate has focused on the need to test ‘disease-modifying’ candidate drugs in the earliest stages of disease. While generally accepted as a rational approach, it is also associated with significant challenges around the selection of trial populations as well as trial outcomes and durations. From a health care perspective, intervening even earlier and before at-risk subjects have gone on to develop overt clinical disease is at the heart of preventive medicine. Recent attempts to develop a framework for a biological definition of PD are aiming to enable ‘preclinical’ and subtype-specific diagnostic approaches. The present review addresses past efforts towards disease-modification, including drug targets and reasons for failure, as well as novel targets that are currently being explored in disease-modification trials in early established PD. The new biological definitions of PD may offer new opportunities to intervene even earlier. We critically discuss the potential and challenges around planning ‘disease-prevention’ trials in subjects with biologically defined ‘preclinical’ or prodromal PD.
INTRODUCTION
| Panel: Glossary of terms surrounding early disease-modification in Parkinson’s disease (PD) |
| –Preclinical PD |
| A phase of disease where biochemical and pathological changes are present, but not have yet led to any clinical manifestations. This phase can only be identified through biomarkers. |
| –Premotor PD |
| A phase of the disease prior to the emergence of disease-related motor symptoms, may overlap with ‘preclinical PD’ and ‘Prodromal PD’. |
| –Prodromal PD |
| A phase of disease characterized by the presence of disease-related nonmotor and subtle motor symptoms that are subthreshold to the current definition of ‘clinical PD’ (see [18] for the MDS research diagnostic criteria for prodromal PD). |
| –Clinical PD |
| A phase of disease characterized by presence of the cardinal motor features as defined by current diagnostic criteria [17]. The term ‘early PD’ refers to newly diagnosed patients are not yet on medication or on stable dopaminergic medication and without functional impact of the disease. |
| –Disease-modification |
| Any therapy that alters the clinical course (‘natural history’) of a disease can be regarded as ‘disease-modifying’. Such a broad definition would also include symptomatic therapies for PD as they reduce the severity and functional impact of motor and non-motor symptoms and, thus, positively influence the progression of disability. However, in regulatory science, the term disease-modification is used in a narrower sense, i.e., for a therapy that is capable of positively influencing the course of the disease by biological mechanisms that revert disease-specific pathophysiological changes [13,19]. |
| –Neuroprotection |
| The term ‘neuroprotection’ was introduced to capture beneficial (protective) effects of an intervention on neuronal survival and function. While such neuroprotective effects can be expected to translate into clinically detectable disease-modification, the presumed underlying biological effect on neuronal survival cannot be proven during lifetime without validated biomarkers that are closely linked to the disease-specific neuronal pathology. In the context of PD, such biomarkers are currently largely lacking. While alpha-synuclein imaging is still under development, presynaptic dopaminergic imaging has been used as a surrogate for such effects in some of the randomized controlled trials outlined in Table 1. |
| –Disease-prevention |
| The term ‘disease-prevention’ is tempting to use in the context of trials in presymptomatic or prodromal cohorts, as prevention of clinically established PD can be seen as the ultimate goal of disease-modifying interventions in such subjects. However, there are conceptual problems even here, given that presymptomatic or prodromal disease stages are, by definition, already a disease state. Nonetheless, in a broader sense, the term can be used to describe effects of an intervention that forestall the development of clinically overt PD. |
| –Regulatory definitions of disease-modification in PD |
| The European Medical Agency requires a two-step procedure to demonstrate disease-modification in PD— first a delay in clinical measures of disease progression should be shown and second an effect on the underlying pathophysiological process which correlates to a meaningful, and persistent changes in clinical function [20]. The Food and Drug Administration (FDA) has not published guidance related to PD. However, in their latest guidance related to drug development in early Alzheimer’s disease, the term ‘disease-modifying’ has been replaced by ‘persistent effect on disease course’ that should be accompanied by a ‘direct effect on the underlying disease pathophysiology’ [21]. |
Modified from Mahlknecht et al. (2022) [10], as permitted under the applying Creative Commons Attribution 4.0 International License.
Parkinson’s disease (PD) is unique among the neurodegenerative diseases for the availability of highly effective symptomatic therapies. The clinical efficacy of levodopa and other dopaminergic drugs is striking and, in many cases, able to almost completely control the cardinal motor features of the disease [1–4]. However, none of the available drugs to treat the symptoms of PD are able to slow the underlying progression of the disease and the increase in overall disability. The latter is driven by a combination of motor and non-motor features that characterize advanced PD and include levodopa-related response fluctuations and drug-induced dyskinesias, the emergence of drug resistant motor symptoms like freezing of gait and falls, as well as a plethora of increasingly severe non-motor problems including cognitive decline, dysautonomia and sleep-wake dysregulation [5]. Despite symptomatic therapy 50% of PD patients have been shown to meet pre-defined disability milestones and 22% are functionally dependent after only 5 years of disease [6, 7]. After 10–15 years more than >50% have developed hallucinations and/or dementia and >40% require institutional care [8, 9].
Disease-modification defined by preventing or delaying the progression of disability beyond symptomatic treatment effects is, thus, generally accepted as a key unmet need in the treatment of PD. From a regulatory perspective evidence for ‘disease-modification’ requires demonstration of effects of an intervention not only on clinical progression but also on underlying pathophysiological disease mechanisms, although it has been argued that demonstration of delay or prevention of clinical decline should be the prime anchor of definitions of disease-modification [10, 11] (see Panel).
The history of disease-modification trials in PD now spans about three decades during which a large number of well-designed and often large studies have used a broad range of drugs targeting different pathways potentially or definitely involved in the molecular pathology of the disease [12, 13] (Table 1). With the exception of two phase 2 trials of GLP1 agonists [14, 15], all of these efforts have yielded negative or, as in the case of the ADAGIO trial of the MAO-B inhibitor rasagilin [16], inconclusive results. Here we review potential reasons why so far almost all drug trials aiming to show disease-modification have failed, with a focus on novel targets and emerging perspectives of intervening at the earliest stages of disease.
Table 1
Completed disease-modification trials in Parkinson’s disease (non-exhaustive list)
| Trial [Reference] | Year | Trial design | Intervention | Patient population | Trial duration | Primary Endpoint | Result (with regard to disease-modification) |
| DATATOP [66] | 1993 | 2 X 2 factorial, double-blind | Selegiline and tocopherol | Early, untreated PD | 24 months | Time to need for levodopa | Negative |
| ELLDOPA [67] | 2004 | Two-arm double-blind | Levodopa | Early, untreated PD | 40 (+2) weeks | UPDRS after 2-week washout | Negative |
| ADAGIO [16] | 2009 | Phase III; two-arm double-blind delayed-start | Rasagiline | Early, untreated PD | 72 weeks | three hierarchical end points* | Conflicting results (1 mg dose positive, 2 mg dose negative) |
| PROUD [68] | 2013 | Phase IV; two-arm double-blind delayed-start | Pramipexole | Early, untreated PD | 15 months | UPDRS change | Negative (DAT-SPECT substudy negative) |
| NET-PD [69] | 2015 | Phase III; double-blind, parallel-group, placebo-controlled | Creatine | Early, treated PD | 5 years | Clinical decline on global statistical test | Negative |
| FS-ZONE [70] | 2015 | Phase II; double-blind, placebo-controlled, parallel group | Pioglitazone | Early, untreated PD | 44 weeks | Change in total UPDRS | Negative |
| EXENATIDE-PD [14] | 2017 | Phase II; two-arm double-blind | Exenatide | Moderately advanced PD (H&Y<3) | 48 (+12) weeks | UPDRS after 12-week washout | 3.5 points benefit on MDS-UPDRS III, but secondary outcomes not supportive |
| LEAP [71] | 2019 | two-arm double-blind delayed-start | Levodopa | Early, untreated PD | 80 weeks | Change in UPDRS | Negative |
| STEADY-PD-III [72] | 2020 | Phase III; parallel-group, double-blind, placebo-controlled | Isradipine | Early, untreated PD | 36 months | Change in UPDRS parts I to III score on medication | Negative |
| SURE-PD3 [73] | 2021 | Phase III; two-arm double-blind | Inosine | Early, untreated PD | 24 (+3) months | Annualized change in MDS-UPDRS III; DAT-Scan substudy | Negative (DAT-Scan substudy negative) |
| NILO-PD [74] | 2021 | Phase IIa; parallel-group, double-blind, placebo-controlled | Nilotinib | Moderately advanced PD (H&Y 2.5–3, disease duration >5y) | 6 (+2) months | Safety and Tolerability; change in MDS-UPDRS as secondary endpoint (2 months washout) | Negative (low CSF penetration, lack of biomarkers effect, and change in MDS-UPDRS trending in the negative direction) |
| PD STAT [75] | 2022 | Simvastatin | Moderately advanced PD (H&Y<4) with wearing OFFs | 24 months (+2 months washout) | 24-month OFF medication MDS-UPDRS part III scores | Negative (primary outcome indicated faster deterioration with simvastatin) | |
| FAIRPARK-II [76] | 2022 | Phase II; parallel-group, double-blind, placebo-controlled | Deferiprone | Early, untreated PD | 36 weeks | change in MDS-UPDRS at 36 weeks (and after 4 weeks washout) | Negative (worse clinical outcome with deferiprone); In MRI substudy nigrostriatal iron content decreased more with deferiprone; in DAT-Scan Substudy no difference between groups |
| PASADENA [22] | 2022 | Phase II; double-blind, placebo-controlled | Prasinezumab | Early, untreated PD (or MAO-B inhibitor only) | 52 weeks | Change from baseline to week 52 in the MDS-UPDRS total score | Negative (DAT-SPECT substudy negative) |
| SPARK [23] | 2022 | Phase II; double-blind, placebo-controlled | Cinpanemab | Early, untreated PD | 52 and 72 weeks | Change from baseline to week 52 (and 72 for the active-treatment dose-blinded extension phase) in the MDS-UPDRS total score | Negative (DAT-SPECT substudy negative |
| NCT02953665 [77] | 2022 | Phase II; double-blind, placebo-controlled | Liraglutide | Early, treated PD | 54 weeks | Change from baseline to week 54 in the MDS-UPDRS III, Non-Motor Symptoms Scale (NMSS), and Mattis Dementia Rating Scale (MADRS-2) | Positive regarding change in the NMSS and the MDS-UPDRS II (secondary outcome), but negative regarding change in the MDS-UPDRS III and MADRS-2 |
| MOVES-PD [26] | 2023 | Phase II; parallel-group, double-blind, placebo-controlled | Venglustat | Early PD with pathogenic GBA1 variants | 52 weeks | Change from baseline to week 52 in the MDS-UPDRS parts II and III combined score in the practically defined OFF condition | Negative |
| LIXIPARK NCT03439943 [15] | 2023 | Phase II; double-blind, placebo-controlled | Lixisenatide | Early PD (<3 years) on stable symptomatic medications | 12 months, followed by a 2-month wash-out period | Primary: change over 12 month in the MDS-UPDRS III ON scores; Secondary: mean MDS-UPDRS III OFF scores at month-14 (end of wash-out) | Both primary and secondary endpoints positive |
*Superiority to placebo in the rate of change in the UPDRS score between weeks 12 and 36, superiority to delayed-start treatment in the change in the score between baseline and week 72, and noninferiority to delayed-start treatment in the rate of change in the score between weeks 48 and 72. DAT, dopamine transporter; GBA1, glucocerebrosidase gene; H&Y, Hoehn and Yahr stage; MDS, Movement Disorder Society; PD, Parkinson’s disease; UPDRS, Unified Parkinson’s disease rating scale; MAO-B, monoamine oxidase Type B.
DISEASE MODIFICATION TRIALS IN PD: WHICH PATIENTS TO TARGET WITH WHAT INTERVENTION?
Numerous articles over the past decade have reviewed obstacles to demonstrate ‘neuroprotection’ or ‘disease-modification’ and potential reasons for the fact that no trial has yet led to the approval of a disease-modifying drug for PD [10–13, 17–21]. These include limitations in the translatability of preclinical findings of neuroprotection into the human disease, lack of reliable biomarkers for target engagement in early phase clinical development, challenges around selecting the right dose in clinical trials, as well as multiple trial design issues (see Table 2).
Table 2
Challenges for disease-modification trials in Parkinson’s disease
| Obstacles | Main problem |
| Drug target selection | |
| –Multiple pathways involved in PD pathogenesis –Differentiating upstream vs. downstream event in pathogenetic cascades –Single vs. multiple targets | Imperfect animal models to test targets, difficult to prioritize among agents, largely unknown safety profiles for new drugs |
| Candidate drug | |
| –Demonstration target engagement –Dose selection –Single drug vs. combinations | Lack of reliable biomarkers |
| Target Population | |
| –Early vs. later disease stage –Selecting disease subtypes (e.g., genetic PD) | Difficulty demonstrating clinically meaningful effects in early disease, neuropathology may be too far advanced in later stages Large cohorts of genetic PD hard to recruit, may not be representative for sporadic PD |
| Trial Design | |
| –Sample size –Trial duration –Outcome measures | Variability in outcomes demands large samples Slow progression in PD requires long duration to show clinically meaningful effects Outcomes should be clinically meaningful |
Foremost among the latter are the selection of the right target population in a stage of disease that seems most accessible to disease-modification as well as the challenge of achieving sufficient trial durations required to detect clinically meaningful effects on disease progression.
Target populations for disease-modification trials in PD: early vs. later stages
With very few exceptions all trials of potentially disease-modifying agents have been conducted in newly diagnosed patients with disease durations of less than 2 to 3 years since the time of diagnosis with or without symptomatic drug therapy who were free of functional impairments (Table 1). This type of population is commonly referred to as ‘early’ PD as opposed to patients on treatment with levodopa plus other agents who have developed motor complications and are classified as ‘advanced’ PD.
Selecting subjects with early untreated PD has been accepted as a plausible strategy to ensure that the underlying pathology has not progressed too far for the respective pharmacological agent to still exert meaningful effects and at the same time allow for clinical comparisons with a placebo arm that are not confounded by effects of symptomatic therapies. One major reason why this approach has failed most of the time could be related to the slow progression of the severity of motor as well as non-motor symptoms in early PD. This leads to sensitivity issues of the ‘gold-standard’ scales that have been developed to assess symptom severity as well as functional impact. Most trials have used a primary endpoint of worsening of motor symptoms as measured by the UPDRS or MDS-UPDRS, which, because of its slow decline in early untreated PD, may be not sufficiently sensitive to detect statistically significant differences between active and placebo arms in this type of populations. Recent examples are the passive anti-synuclein immunotherapy trials of the monoclonal antibodies prasinezumab and cinpanemab, which failed to meet their primary endpoints of significant differences in progression of combined MDS-UPDRS parts I, II, and III scores over one year to 18 months [22, 23]. Over follow-up periods of 1 to 2 years the motor examination section of the MDS-UPDRS (part III) seems to be more sensitive compared to the patient report-based ‘motor experiences of daily living’ section (part II). This has been observed in several of the trials listed in Table 1 including the Prasinezumab trial, where progression of part III but not of part II scores (or combined part I, II, and III scores) was reduced in the active arms as compared to placebo [22].
Recent trials have moved into target populations with more advanced disease and recruited treated subjects with disease durations of up to 3 years or even beyond (see Table 1). Two of these assessing the efficacy and safety of the GLP-1 agonists exenatide [14] and lixisenatide [15] have indeed been positive showing significant differences in favor of active drug on a primary endpoint of motor worsening over 12 months as assessed by the MDS-UPDRS part III (as opposed to part II).
Selecting target populations in more advanced PD stages also improves chances to demonstrate clinical meaningfulness of effects from a putative disease-modifying intervention by using primary outcomes like time to development of functional disability or the occurrence of disability milestones in the motor or non-motor domains [13], although time-to event endpoints might require longer trial durations beyond the 12 to 24 months horizon of most previous trials (Table 1). An ongoing immunotherapy trial of prasinezumab in patients on stable symptomatic medication with disease duration of up to 3 years is using a primary outcome of time to a 5-point worsening of the MDS-UPDRS part III (NCT04777331).
One concern around testing disease-modifying agents in more advanced PD is related to the risk of the underlying pathology being too far advanced for the intervention to still exert clinical effects in spite of positive target engagement.
Selecting the right pharmacological targets
Identifying critical drug target within the pathogenetic cascade driving PD progression is another major challenge and the path to disease modification in PD is flanked by numerous failures of translation from preclinical proof-of-concept to clinical efficacy. Not least through the advances made in understanding the genetic architecture of PD multiple novel targets for disease-modifying pharmacological interventions have emerged and many of these are currently addressed in ongoing drug development programs [12, 13, 24] (Fig. 1 and Table 3).
Fig. 1
Mechanisms in the pathophysiological cascade of PD and potential treatment targets. See text and Table 3 for more details regarding drug candidates. ASO, antisense oligonucleotide; Cav1.3, Calcium channel, voltage-dependent; GBA, gene encoding for Glucocerebrosidase; GLP-1, Glucagon-like peptide-1; iNOS, Nitric oxide synthases; LRRK2, Leucine rich repeat kinase 2; ROS, reactive oxygen species; siRNA, small interfering RNAs.
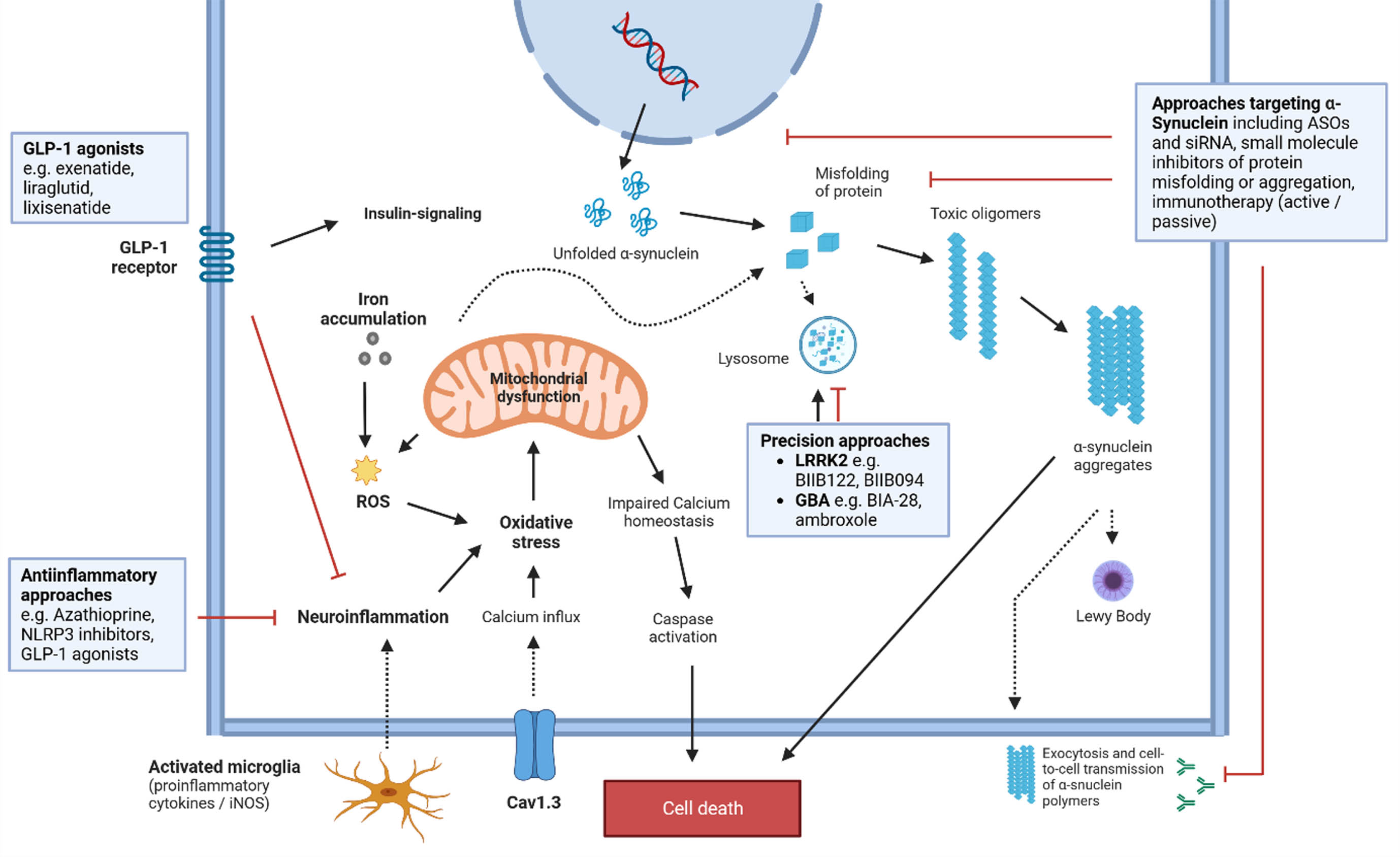
Table 3
Candidate drugs currently tested for disease-modification in PD (non-exhaustive list)
| Target | Drug | Mechanism | Stage of development | Outcome [REF]/registration number |
| Insulin signaling | Exenatide | GLP-1 agonist | Phase 3 | Ongoing; NCT04232969 |
| NLY01 | GLP-1 agonist | Phase 2 | Ongoing; NCT04154072 | |
| Liraglutid | GLP-1 agonist | Phase 2 | Completed, see Table 1; NCT02953665 | |
| Lixisenatide | GLP-1 agonist | Phase 2 | Completed, see Table 1; NCT03439943 | |
| Semaglutide | GLP-1 agonist | Phase 2 | Ongoing; NCT03659682 | |
| α-Synuclein proteostasis | Antisense oligonucleotides (ASO)Small interfering RNAs (siRNAs) | Reducing α-synuclein production | Preclinical | [78] |
| anle138b | Inhibition of α-synuclein aggregation | Phase 1 | Ongoing; NCT04685265 | |
| Radotinib | c-Abl tyrosin kinase inhibitor; Inhibition of α-synuclein aggregation | Phase 2 | Ongoing; NCT04691661 | |
| IkT-148009 | c-Abl tyrosin kinase inhibitor | Phase 2 | Ongoing; NCT05424276 | |
| Buntanetap | Small molecule inhibitor of neurotoxic proteins | Phase 3 | NCT05357989 | |
| UCB0599 | Small-molecule inhibitor of α-synuclein misfolding | Phase 2 | Ongoing; NCT04658186, NCT05543252 | |
| KM-819 | Small molecule inhibitor of FAF1 | Phase 2 | NCT05670782 | |
| Prasinezumab | Anti-α-synuclein antibody | Phase 2 | Ongoing; NCT04777331 (PADOVA) | |
| Lu AF82422 | Anti-α-synuclein antibody | Phase 1 | Completed, results pending; NCT03611569 | |
| MEDI1341 | Anti-α-synuclein antibody | Phase 1 | Completed, results pending; NCT03272165 | |
| Lysosomal function/LRRK2 | DNL 201 | LRRK2 inhibitor | Phase 1b | Favorable safety, biomarker evidence for target engagement in PD subjects; NCT03710707 |
| BIIB122 (DNL151) | LRRK2 inhibitor | Phase 2 | Ongoing; NCT05348785 (LUMA) | |
| BIIB094 | LRRK2 inhibitor (ASO; intrathecal administration) | Phase 1 | Ongoing; NCT03976349 | |
| Lysosomal function/β-Glucocerebrosidase | Ambroxole | Modulator of GCase activity | Phase 1 (published) | CSF penetration and biochemical effects demonstrated [79] |
| Phase 2 | Ongoing; NCT02914366 | |||
| Phase 3 | Ongoing; NCT05778617, NCT05830396 | |||
| BIA-28 | Modulator of GCase activity | Phase 2 | Ongoing; NCT05819359 | |
| Mitochondrial function | Terazosin | alpha-1 antagonist; enhances glycolysis and ATP levels | Phase 2 | Ongaoing; NCT05109364, NCT05855577 |
| Ursodeoxycholic acid | Naturally occurring bile acid; improves mitochondrial function | Phase 2 | Completed; NCT03840005Safe and well-tolerated, signals of target engagement on magnetic resonance spectroscopy; Phase III trials warranted [80] | |
| Neuroinflammation(partially including GLP-1 agonists listed above) | Inzomelid (IZD174) | NLRP3 inhibitor | Phase 1 | Completed, results pending; NCT04015076 |
| RO7486967 | NLRP3 inhibitor | Phase 1 | Ongoing; NCT05924243 | |
| Sargramostim | Granulocyte macrophage colony-stimulating factor (GM-CSF) | Phase 1 | Ongoing; NCT05677633 and NCT03790670 | |
| Azathioprine | Immunosuppressant | Phase 2 | EudraCT Number: Ongoing; 2018-003089-14 |
GCase, β-Glucocerebrosidase; FAF1, Fas-associated factor 1; HV, healthy volunteers; LRRK2, Leucine-rich repeat kinase 2; PD, Parkinson’s disease; PPAR γ, peroxisome proliferator-activated receptor; DAT, Dopamine transporter; GBA1, glucocerebrosidase gene; H&Y, Hoehn and Yahr stage; MDS, Movement Disorder Society; PD, Parkinson’s disease; UPDRS, Unified Parkinson’s disease rating scale; MAO-B, monoamine oxidase Type B.
Targeting specific pathogenetic pathways introduces the option of ‘personalized’ or ‘precision-medicine’ approaches to disease modification in PD, exemplified by recent drug trials targeting GCase or LRRK2 activity in PD populations harboring mutations in these genes [25]. The first large disease-modification trial phase 2 study in a genetic PD subtype enrolled 221 participants with one or more GBA1 gene variants to test if venglustat, a brain penetrant glucosylceramide synthase inhibitor, could slow the progression of combined MDS-UPDRS part II and III scores over 52 weeks [26]— following the rationale of ‘substrate-reduction’ that is a standard therapy for people with Gaucher’s disease and also been able to reduce α-synuclein pathology and behavioral deficits in rodent models. Patients were on dopaminergic therapy and had average disease durations of 4 to 5 years from time of diagnosis. The trial failed its primary endpoint and change from baseline to week 52 in combined MDS-UPDRS part II and II scorers was even numerically greater in the venglustat group, although there was target engagement with a 75% reduction of CSF glucosylceramide levels. While the negative results of this trial show that the principle of substrate-reduction for Gcase is unlikely to provide benefit in GBA-PD, it does not invalidate targeting Gcase activity directly by molecular chaperones. Several such agents are currently tested in phase 2 and 3 trials including ambroxol (NCT05778617, NCT05830396) and BIA-28 (NCT05819359).
LRRK2 inhibitors are another group of candidate drugs for a ‘personalized’ approach to disease-modification in PD. BIIB122 is an oral selective, brain penetrant inhibitor of LRRK2 for which safety and target engagement have been shown in both healthy volunteers and subjects with PD [27]. A phase 3 trial of this drug in PD patients carrying a pathogenic LRRK2 mutations was set up to measure effects on time to predefined worsening in the MDS-UPDRS II and III (NCT05418673) and a corresponding phase 2 trial is enrolling PD patients without mutation (NCT05348785).
DISEASE PREVENTION TRIALS IN PD: CHALLENGES AND FUTURE OPPORTUNITIES
Preventing or delaying ‘phenoconversion’ in subjects meeting criteria for ‘prodromal’ PD has been discussed as a new approach of ‘disease-modification’ trials that would conceptually become a type of ‘disease-prevention’ effort [10, 28].
Such target groups can now be defined by established multifactorial screening algorithms for prodromal PD such as the MDS criteria [29], although— despite their high specificity— their sensitivity and positive predictivity for early conversion in population-based cohorts seem suboptimal [30–32]. Other approaches to identify such individuals target single specific prodromal markers with subsequent enrichment steps like hyposmia followed by DAT-Scan as exemplified in the PARS study [33]. In idiopathic RBD, the strongest and most specific marker for PD and other α-synucleinopathies, conversion rates are only around 6% per year [34] and long-term series have reported median latencies from presumed RBD onset to clinically overt disease of 12–14 years and from diagnosis of idiopathic RBD to clinically overt disease of 6 years [35, 36]. These drawbacks of long delays to phenoconversion can potentially be overcome by enrichment strategies like adding further risk-markers such as hyposmia, subtle motor dysfunction, or subtle cognitive decline, which have all been shown to indicate higher conversion rates over shorter periods of time in subjects with idiopathic RBD [34, 37]. Despite these obstacles, prodromal cohorts are highly appealing as targets for disease-modification trials. On one hand the neurodegenerative process has already caused clinical symptoms that can be monitored for study purposes and that may also enhance motivation for individuals to participate in clinical trials. On the other hand, neurodegeneration may not have progressed too far to be modified by putatively neuroprotective interventions. All the approaches mentioned above in prodromal cohorts have yielded a maximum conversion rate of approximately 60% over 5 years or less [10, 33, 34]. Before inclusion of participants into disease-modification trials it would be desirable to have an additional highly specific and ‘confirming’ biomarker to further enhance likelihood of a true prodromal state and to homogenize groups of prodromal individuals. Evidence of pathogenic α-synuclein on SAA would lend itself to such a purpose as it shows presence of the pathological hallmarks of synucleinopathies and it is highly specific for these disorders. Indeed, in line with the overall conversion rates in idopathic RBD of >80%, positive SAAs from CSF have been detected in around 90% in different series of idiopathic RBD patients [38–41].
Targeting asymptomatic or ‘preclinical’ disease stages could come even closer to an ultimate goal of ‘disease-prevention’ in the sense of completely preventing development of clinical disease in a subject’s lifetime [42]. Genetic PD subtypes are of particular interest in relation to disease-prevention trials for a number of reasons. First, these patients have defined molecular defects that are linked to the pathophysiology of their disease and thus allow for a precision-medicine approach to treatment [25]. In addition to reduced pathophysiological heterogeneity there is also less clinical heterogeneity in terms of disease progression in such cohorts [43]. Finally, asymptomatic individuals harboring mutations in PD genes are conceptualized as being in a ‘preclinical’ state of disease and intervening in this period could be more effective compared to later stages where pathology has progressed to override compensation and causes clinical symptoms [44]. Nonetheless, there are significant obstacles in the way of implementing disease-prevention trials in healthy carriers of PD-associated genes. These include the problem of recruitment of a trial population given that even GBA1 mutations, which represent the most common risk gene for PD, only affect around 5–10% of PD subjects globally and the overall carrier frequency of the G2019S mutation of the LRRK2 gene has been estimated at 0.5% [43, 45]. Reduced penetrance is another significant problem. As many as 70% of LRRK2 or 90% of GBA1 mutation carriers will never develop PD [43, 44] and for those who will the time to developing clinical symptoms is unknown. This would pose great difficulty in selecting meaningful outcome measures for trials in these types of target population and lead to long trial durations, which would also be true for the subsequent group of preclinical PD subjects.
Recent attempts to develop frameworks for a biological definition of PD may offer new opportunities to better define and enrich target populations for both disease-modification as well as ‘disease-prevention’ trials. Two recently published proposals both anchor the diagnosis of PD (subsumed under the term of ‘Neuronal Synuclein Disease’ (NSD) in one of them) on the presence of biomarkers independent of clinical symptoms. The proposed biological anchors rely on the demonstration of disease-specific alpha-synuclein pathology by seed amplification assays (SAAs) in the CSF, the presence of highly penetrant PD mutations, and evidence for dopaminergic neurodegeneration by molecular imaging [46, 47]. These approaches follow the example of biological diagnostic concepts initially put forward as the ‘ATN’ system for Alzheimer’s disease [48] and allow for a PD or ‘NSD’ diagnosis in the earliest stages of disease, when affected individuals have not yet developed any clinical symptoms or signs. Asymptomatic individuals with positive a-synuclein SAAs are classified as ‘Parkinson’s type synucleinopathy’ [46] or stage 1 NSD [47], while the appearance of ‘prodromal’ motor or non-motor signs with sufficient likelihood of being disease-related are classified as ‘stage 2’ in the NSD integrated staging system (NSD-ISS) proposed by Simuni and colleagues. Such biological diagnostic definitions would both allow for a more accurate diagnosis of ‘prodromal’ PD then is currently possible by using clinical features as the main anchors and would also offer an objectively measurable tool to detect preclinical disease. The latter, although conceptualized for a long time [49], has only recently become practically tangible –first by genetic markers and now through the availability of highly sensitive in-vivo assays to detect disease-specific α-synuclein pathology [50, 51]. A ‘biological’ definition of disease not only allows to detect preclinical disease but also has the potential to delineate pathogenetic subtypes and further reduce heterogeneity in future trial populations. The most attractive vision for the future use of these approaches relates to the prospect of implementing disease-prevention trials in populations of biomarker defined preclinical disease, i.e., people with ‘Parkinson’s type synucleinopathy’ [46] or ‘stage 1 NSD’ [47]. The recruitment base for such trials would presumably be much larger than is the case for trials selecting mutation carriers (see above). However, there are considerable challenges to be addressed when trying to screen for and select individuals in the population for such efforts.
OUTLOOK: WILL DISEASE-PREVENTION TRIALS IN PRECLINICAL PD BECOME FEASIBLE?
Using the newly proposed criteria for a biological disease definition there are two potential groups of preclinical subjects that could be recruited for disease-modification trials, those defined by harboring fully penetrant PD gene mutations or those with positive α-synuclein SAAs. Healthy subjects with such genetic variants are, however, very rare and recruitment for clinical trials would pose a major challenge. More common pathogenic mutations such as in the LRRK2 and GBA genes are associated with incomplete penetrance and much lower disease risk. Designing disease-prevention trials in healthy carriers of the latter group of mutations would require enrichment by additional molecular or imaging biomarkers for early conversion in order to arrive at sufficient endpoints in time frames that are feasible for trials and relevant for patients. Similar problems around recruitment and outcomes have to be addressed when targeting preclinical subjects that are defined by a positive result on α-synuclein SAAs (corresponding to stage 1 NSD [47] or ‘Parkinson’s type synucleinopathy’[46]). Even if reliable large scale SAA testing became feasible through non-invasive sampling via plasma- or serum-based assays [52, 53], the significance of a positive test in the population is currently uncertain. There are no studies dedicated to the assessment of SAA in the elderly community and prevalence of positive SAAs as well as the risk for subsequent development of α-synucleinopthies are unknown. In addition, identifying such individuals is not trivial even if the rate of SAA positivity in healthy control groups was around 5% to a maximum of 10% across the different hospital-based cohorts [54]. This seems to be in line with earlier studies finding incidental Lewy bodies and nigral neuronal loss in more than 10% of individuals free of PD above age 60 in histopathological population-based cohorts [55, 56]. Based on these numbers trial screen failure rates of up to 20%, α-synuclein SAA screening would have to be performed in more than 7000 subjects to arrive at a total sample size of 300 subjects for a disease prevention trial. In addition, lag times to clinical symptoms and/or to markers of neurodegeneration such as striatal dopaminergic loss in preclinical SAA positive individuals are expected to be very long. For these reasons alone SAAs testing would make more sense in preselected individuals with prodromal features like RBD or hyposmia (see above), who may have α-synuclein SAA positivity rates of around 80%. Once such individuals are identified, there is a need for additional biomarkers that indicate early progression or conversion to clinical disease. Imaging evidence for neurodegeneration such as DAT-SPECT has been proposed for such purposes [46]. Enrichment strategies will be essential to reduce numbers needed for disease-modification trials and to avoid employing unnecessary interventions that may be associated to psychological stress in individuals that may never go on to develop clinically relevant disease.
Selecting outcome measures for disease-modification trials in preclinical target populations would also mean entering largely unchartered territory. Time to ‘phenoconversion’ has been previously discussed in relation to disease-modification trials in prodromal PD but its use in biologically defined asymptomatic individuals poses even greater challenge in the latter compared to the former (see above). Alternative outcomes like on composite scores of motor, cognitive and other non-motor progression combined with evidence for progression of neurodegeneration from imaging or molecular markers could be viable and powerful alternatives [10, 57–61]. Achieving quantification of α-synuclein SAAs or the introduction of α-synuclein PET-imaging and establishing correlations with such quantitative measures with clinical progression might have an impact for outcome measures in disease-prevention and disease-modification trials alike, although it is yet unclear whether disease-modifying compounds would or should indeed have an influence on such measures or not.
Selecting agents to be tested also raises additional issues in preclinical cohorts. Testing interventions in people free of any clinical signs of disease and disability requires a level of safety that would have to practically exclude any risk of serious adversity. While this appears feasible for non-pharmacological approaches like lifestyle interventions, e.g., structured physical activity or nutritional programs [62], it is a very high bar for agents other than repurposed drugs with very well-established safety profiles and almost precludes trials of new and experimental interventions in preclinical individuals. The uncertainty if and when subjects meeting criteria for biomarker-based ‘preclinical’ disease will develop early clinical disease with functional impairment means that the burden of any ‘disease-preventing’ intervention should be minimal in order to be acceptable to patients and authorities alike [42].
Apart from safety risks of experimental therapies there are important additional ethical considerations to be addressed [63]. One of these concerns the phenomenon of ‘overdiagnosis’, which is a well-known problem for controversial screening tests and refers to the detection of subclinical disease (sometimes called pseudodisease), which would not have become manifest clinically in someone’s remaining life time (e.g., prostate cancer through prostate-specific antigen screening) [64]. ‘False-positive’ results of a screening procedure give rise to unnecessary further diagnostic tests and create worries of having a disease that may never manifest. Furthermore, depending on a given health care system, a positive screening result, e.g., a diagnosis of NSD, can have significant impact on the access to health care or life insurances [65]. Risk disclosure strategies should therefore take many factors into account, related to the screening test itself (e.g., its evidence, accuracy, reliability etc.) as well as to the subjects including education, social background, comorbidities, age, cognitive status, etc. Studies or trials in presymptomatic PD should therefore offer careful individualized counseling of participants and an option for psychosocial support.
Despite of all these challenges defining disease by the detection of underlying molecular pathology (‘biological definition’) marks a major step forward for efforts to achieve a reliable prodromal or even preclinical diagnosis which will eventually open the door to prevention also in the field of PD.
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
P.M. reports lecture fees from AbbVie outside the submitted work. W.P. reports consultancy and lecture fees in relation to clinical drug development programmes for PD from AC Immune, Alterity, AbbVie, Affiris, BIAL, Biogen, Britannia, Lilly, Lundbeck, Merz, Neuroderm, Neurocrine, Roche, Sunovion, Stada, Takeda, UCB and Zambon, all outside the submitted work.
REFERENCES
[1] | Fox SH , Katzenschlager R , Lim S-Y , Barton B , de Bie RMA , Seppi K , Coelho M , Sampaio C , Movement Disorder Society Evidence-Based Medicine Committee ((2018) ) International Parkinson and movement disorder society evidence-based medicine review: Update on treatments for the motor symptoms of Parkinson’s disease. Mov Disord 33: , 1248–1266. |
[2] | LeWitt PA , Fahn S ((2016) ) Levodopa therapy for Parkinson disease: A look backward and forward. , . Neurology 86: , S3–S12. |
[3] | Espay AJ , Lang AE ((2017) ) Common myths in the use of levodopa in Parkinson disease: When clinical trials misinform clinical practice. JAMA Neurol 74: , 633–634. |
[4] | Poewe W , Mahlknecht P ((2020) ) Pharmacologic treatment of motor symptoms associated with Parkinson disease. Neurol Clin 38: , 255–267. |
[5] | Poewe W , Seppi K , Tanner CM , Halliday GM , Brundin P , Volkmann J , Schrag AE , Lang AE ((2017) ) Parkinson disease. Nat Rev Dis Prim 3: , 1–21. |
[6] | Brumm MC , Siderowf A , Simuni T , Burghardt E , Choi SH , Caspell-Garcia C , Chahine LM , Mollenhauer B , Foroud T , Galasko D , Merchant K , Arnedo V , Hutten SJ , O’Grady AN , Poston KL , Tanner CM , Weintraub D , Kieburtz K , Marek K , Coffey CS , Parkinson’s Progression Markers Initiative ((2023) ) Parkinson’s Progression Markers Initiative: A milestone-based strategy to monitor Parkinson’s disease progression. J Parkinsons Dis 13: , 899–916. |
[7] | Valent D , Krismer F , Grossauer A , Peball M , Heim B , Mahlknecht P , Djamshidian A , Poewe W , Seppi K ((2023) ) Nomogram to predict the probability of functional dependence in early Parkinson’s disease. J Parkinsons Dis 13: , 49–55. |
[8] | Hely MA , Morris JGL , Reid WGJ , Trafficante R ((2005) ) Sydney Multicenter Study of Parkinson’s disease: Non-L-dopa-responsive problems dominate at 15 years. Mov Disord 20: , 190–199. |
[9] | Hely MA , Reid WGJ , Adena MA , Halliday GM , Morris JGL ((2008) ) The Sydney Multicenter Study of Parkinson’s disease: The inevitability of dementia at 20 years. Mov Disord 23: , 837–844. |
[10] | Mahlknecht P , Marini K , Werkmann M , Poewe W , Seppi K ((2022) ) Prodromal Parkinson’s disease: Hype or hope for disease-modification trials? . Transl Neurodegener 11: , 11. |
[11] | Kieburtz K , Katz R , McGarry A , Olanow CW ((2021) ) A new approach to the development of disease-modifying therapies for PD; fighting another pandemic. Mov Disord 36: , 59–63. |
[12] | Poewe W , Seppi K , Marini K , Mahlknecht P ((2020) ) New hopes for disease modification in Parkinson’s disease. Neuropharmacology 171: , 108085. |
[13] | Vijiaratnam N , Simuni T , Bandmann O , Morris HR , Foltynie T ((2021) ) Progress towards therapies for disease modification in Parkinson’s disease. Lancet Neurol 20: , 559–572. |
[14] | Athauda D , Maclagan K , Skene SS , Bajwa-Joseph M , Letchford D , Chowdhury K , Hibbert S , Budnik N , Zampedri L , Dickson J , Li Y , Aviles-Olmos I , Warner TT , Limousin P , Lees AJ , Greig NH , Tebbs S , Foltynie T ((2017) ) Exenatide once weekly versus placebo in Parkinson’s disease: A randomised, double-blind, placebo-controlled trial. Lancet 6736: , 1–12. |
[15] | Meissner W , Remy P , Giordana C , Maltete D , Damier P , Houeto J , Geny C , Hopes L , Durif F , Defer G , Tranchant C , Corvol J , Carriere N , Azulay J , Drapier S , Krystkowiak P , Thalamas C , Benard A , Rascol O ((2023) ) Multicenter, randomized, placebo-controlled, double-blind, parallel group proof-of-concept study of lixisenatide in patients with early Parkinson’s disease (PD): The LIXIPARK Trial (NCT03439943) –Abstract 94. Mov Disord 38: , S41. |
[16] | Olanow CW , Rascol O , Hauser R , Feigin PD , Jankovic J , Lang A , Langston W , Melamed E , Poewe W , Stocchi F , Tolosa E , ADAGIO Study Investigators ((2009) ) A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med 361: , 1268–1278. |
[17] | Lang AE , Espay AJ ((2018) ) Disease modification in Parkinson’s disease: Current approaches, challenges, and future considerations. Mov Disord 33: , 660–677. |
[18] | Salat D , Noyce AJ , Schrag A , Tolosa E ((2016) ) Challenges of modifying disease progression in prediagnostic Parkinson’s disease. Lancet Neurol 15: , 637–648. |
[19] | Thibault L , Rascol O , Corvol J-C , Ferreira J , Defebvre L , Deplanque D , Bordet R , Moreau C , Devos D ((2017) ) New perspectives on study designs for evaluating neuroprotection in Parkinson’s disease. Mov Disord 32: , 1365–1370. |
[20] | Cummings J ((2017) ) Disease modification and Neuroprotection in neurodegenerative disorders. Transl Neurodegener 6: , 25. |
[21] | Devos D , Hirsch E , Wyse R ((2021) ) Seven solutions for neuroprotection in Parkinson’s disease. Mov Disord 36: , 306–316. |
[22] | Pagano G , Taylor KI , Anzures-Cabrera J , Marchesi M , Simuni T , Marek K , Postuma RB , Pavese N , Stocchi F , Azulay J-P , Mollenhauer B , López-Manzanares L , Russell DS , Boyd JT , Nicholas AP , Luquin MR , Hauser RA , Gasser T , Poewe W , Ricci B , Boulay A , Vogt A , Boess FG , Dukart J , D’Urso G , Finch R , Zanigni S , Monnet A , Pross N , Hahn A , Svoboda H , Britschgi M , Lipsmeier F , Volkova-Volkmar E , Lindemann M , Dziadek S , Holiga Š , Rukina D , Kustermann T , Kerchner GA , Fontoura P , Umbricht D , Doody R , Nikolcheva T , Bonni A , PASADENA Investigators Prasinezumab Study Group ((2022) ) Trial of prasinezumab in early-stage Parkinson’s disease. N Engl J Med 387: , 421–432. |
[23] | Lang AE , Siderowf AD , Macklin EA , Poewe W , Brooks DJ , Fernandez HH , Rascol O , Giladi N , Stocchi F , Tanner CM , Postuma RB , Simon DK , Tolosa E , Mollenhauer B , Cedarbaum JM , Fraser K , Xiao J , Evans KC , Graham DL , Sapir I , Inra J , Hutchison RM , Yang M , Fox T , Budd Haeberlein S , Dam T , SPARK Investigators ((2022) ) Trial of cinpanemab in early Parkinson’s disease. N Engl J Med 387: , 408–420. |
[24] | McFarthing K , Buff S , Rafaloff G , Fiske B , Mursaleen L , Fuest R , Wyse RK , Stott SRW ((2023) ) Parkinson’s disease drug therapies in the clinical trial pipeline: 2023 update. J Parkinsons Dis 13: , 427–439. |
[25] | von Linstow CU , Gan-Or Z , Brundin P ((2020) ) Precision medicine in Parkinson’s disease patients with LRRK2 and GBA risk variants –Let’s get even more personal. Transl Neurodegener 9: , 39. |
[26] | Giladi N , Alcalay RN , Cutter G , Gasser T , Gurevich T , Höglinger GU , Marek K , Pacchetti C , Schapira AHV , Scherzer CR , Simuni T , Minini P , Sardi SP , Peterschmitt MJ ((2023) ) Safety and efficacy of venglustat in GBA1-associated Parkinson’s disease: An international, multicentre, double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Neurol 22: , 661–671. |
[27] | Jennings D , Huntwork-Rodriguez S , Vissers MFJM , Daryani VM , Diaz D , Goo MS , Chen JJ , Maciuca R , Fraser K , Mabrouk OS , van de Wetering de Rooij J , Heuberger JAAC , Groeneveld GJ , Borin MT , Cruz-Herranz A , Graham D , Scearce-Levie K , De Vicente J , Henry AG , Chin P , Ho C , Troyer MD ((2023) ) LRRK2 inhibition by BIIB122 in healthy participants and patients with Parkinson’s disease. Mov Disord 38: , 386–398. |
[28] | Molsberry SA , Hughes KC , Schwarzschild MA , Ascherio A ((2022) ) Who to enroll in Parkinson disease prevention trials? The case for composite prodromal cohorts. Neurology 99: , 26–33. |
[29] | Heinzel S , Berg D , Gasser T , Chen H , Yao C , Postuma RB , MDS Task Force on the Definition of Parkinson’s Disease ((2019) ) Update of the MDS research criteria for prodromal Parkinson’s disease. Mov Disord 34: , 1464–1470. |
[30] | Giagkou N , Maraki MI , Yannakoulia M , Kosmidis MH , Dardiotis E , Hadjigeorgiou GM , Sakka P , Ntanasi E , Anastasiou CA , Xiromerisiou G , Stefanis L , Scarmeas N , Stamelou M ((2020) ) A prospective validation of the updated Movement Disorders Society Research Criteria for prodromal Parkinson’s disease. Mov Disord 35: , 1802–1809. |
[31] | Mahlknecht P , Gasperi A , Djamshidian A , Kiechl S , Stockner H , Willeit P , Willeit J , Rungger G , Poewe W , Seppi K ((2018) ) Performance of the Movement Disorders Society criteria for prodromal Parkinson’s disease: A population-based 10-year study. Mov Disord 33: , 405–413. |
[32] | Pilotto A , Heinzel S , Suenkel U , Lerche S , Brockmann K , Roeben B , Schaeffer E , Wurster I , Yilmaz R , Liepelt-Scarfone I , von Thaler A-K , Metzger FG , Eschweiler GW , Postuma RB , Maetzler W , Berg D ((2017) ) Application of the movement disorder society prodromal Parkinson’s disease research criteria in 2 independent prospective cohorts. Mov Disord 32: , 1025–1034. |
[33] | Jennings D , Siderowf A , Stern M , Seibyl J , Eberly S , Oakes D , Marek K , PARS Investigators ((2017) ) Conversion to Parkinson disease in the PARS hyposmic and dopamine transporter-deficit prodromal cohort. JAMA Neurol 74: , 933–940. |
[34] | Postuma RB , Iranzo A , Hu M , Högl B , Boeve BF , Manni R , Oertel WH , Arnulf I , Ferini-Strambi L , Puligheddu M , Antelmi E , Cochen De Cock V , Arnaldi D , Mollenhauer B , Videnovic A , Sonka K , Jung K-Y , Kunz D , Dauvilliers Y , Provini F , Lewis SJ , Buskova J , Pavlova M , Heidbreder A , Montplaisir JY , Santamaria J , Barber TR , Stefani A , St Louis EK , Terzaghi M , Janzen A , Leu-Semenescu S , Plazzi G , Nobili F , Sixel-Doering F , Dusek P , Bes F , Cortelli P , Ehgoetz Martens K , Gagnon J-F , Gaig C , Zucconi M , Trenkwalder C , Gan-Or Z , Lo C , Rolinski M , Mahlknecht P , Holzknecht E , Boeve AR , Teigen LN , Toscano G , Mayer G , Morbelli S , Dawson B , Pelletier A ((2019) ) Risk and predictors of dementia and parkinsonism in idiopathic REM sleep behaviour disorder: A multicentre study. Brain 142: , 744–759. |
[35] | Iranzo A , Tolosa E , Gelpi E , Molinuevo JL , Valldeoriola F , Serradell M , Sanchez-Valle R , Vilaseca I , Lomeña F , Vilas D , Lladó A , Gaig C , Santamaria J ((2013) ) Neurodegenerative disease status and post-mortem pathology in idiopathic rapid-eye-movement sleep behaviour disorder: An observational cohort study. Lancet Neurol 12: , 443–453. |
[36] | Schenck CH , Boeve BF , Mahowald MW ((2013) ) Delayed emergence of a parkinsonian disorder or dementia in 81% of older men initially diagnosed with idiopathic rapid eye movement sleep behavior disorder: A 16-year update on a previously reported series. Sleep Med 14: , 744–748. |
[37] | Mahlknecht P , Iranzo A , Högl B , Frauscher B , Müller C , Santamaría J , Tolosa E , Serradell M , Mitterling T , Gschliesser V , Goebel G , Brugger F , Scherfler C , Poewe W , Seppi K , Sleep Innsbruck Barcelona Group ((2015) ) Olfactory dysfunction predicts early transition to a Lewy body disease in idiopathic RBD. Neurology 84: , 654–658. |
[38] | Iranzo A , Fairfoul G , Ayudhaya ACN , Serradell M , Gelpi E , Vilaseca I , Sanchez-Valle R , Gaig C , Santamaria J , Tolosa E , Riha RL , Green AJE ((2021) ) Detection of α-synuclein in CSF by RT-QuIC in patients with isolated rapid-eye-movement sleep behaviour disorder: A longitudinal observational study. Lancet Neurol 20: , 203–212. |
[39] | Siderowf A , Concha-Marambio L , Lafontant D-E , Farris CM , Ma Y , Urenia PA , Nguyen H , Alcalay RN , Chahine LM , Foroud T , Galasko D , Kieburtz K , Merchant K , Mollenhauer B , Poston KL , Seibyl J , Simuni T , Tanner CM , Weintraub D , Videnovic A , Choi SH , Kurth R , Caspell-Garcia C , Coffey CS , Frasier M , Oliveira LMA , Hutten SJ , Sherer T , Marek K , Soto C , Parkinson’s Progression Markers Initiative ((2023) ) Assessment of heterogeneity among participants in the Parkinson’s Progression Markers Initiative cohort using α-synuclein seed amplification: A cross-sectional study. Lancet Neurol 22: , 407–417. |
[40] | Rossi M , Candelise N , Baiardi S , Capellari S , Giannini G , Orrù CD , Antelmi E , Mammana A , Hughson AG , Calandra-Buonaura G , Ladogana A , Plazzi G , Cortelli P , Caughey B , Parchi P ((2020) ) Ultrasensitive RT-QuIC assay with high sensitivity and specificity for Lewy body-associated synucleinopathies. Acta Neuropathol 140: , 49–62. |
[41] | Singer W , Schmeichel AM , Shahnawaz M , Schmelzer JD , Sletten DM , Gehrking TL , Gehrking JA , Olson AD , Suarez MD , Misra PP , Soto C , Low PA ((2021) ) Alpha-synuclein oligomers and neurofilament light chain predict phenoconversion of pure autonomic failure. Ann Neurol 89: , 1212–1220. |
[42] | Berg D , Crotty GF , Keavney JL , Schwarzschild MA , Simuni T , Tanner C ((2022) ) Path to Parkinson disease prevention: Conclusion and outlook. Neurology 99: , 76–83. |
[43] | Niotis K , West AB , Saunders-Pullman R ((2022) ) Who to enroll in Parkinson disease prevention trials? The case for genetically at-risk cohorts. Neurology 99: , 10–18. |
[44] | Crotty GF , Keavney JL , Alcalay RN , Marek K , Marshall GA , Rosas HD , Schwarzschild MA ((2022) ) Planning for prevention of Parkinson disease: Now is the time. Neurology 99: , 1–9. |
[45] | Bryant N , Malpeli N , Ziaee J , Blauwendraat C , Liu Z , AMP PD Consortium and West AB ((2021) ) Identification of LRRK2 missense variants in the accelerating medicines partnership Parkinson’s disease cohort. Hum Mol Genet 30: , 454–466. |
[46] | Höglinger G , Adler C , Berg D , Klein C , Outeiro O , Poewe W , Postuma R , Stoessl J , Lang A ((2024) ) Towards a biological definition of Parkinson’s disease.. Lancet Neurol 23: , 191–204. |
[47] | Simuni T , Chahine L , Poston K , Brumm M , Buracchio T , Campbell M , Chowdhury S , Coffey C , Concha-Marambio L , Dam T , DiBiaso P , Foroud T , Frasier M , Gochanour C , Jennings D , Kieburtz K , Kopil C , Merchant K , Mollenhauer B , Montine T , Nudelman K , Pagano G , Seibyl J , Sherer T , Singleton A , Stephenson D , Stern M , Soto C , Tanner C , Tolosa E , Weintraub D , Xiao Y , Siderowf A , Dunn B , Marek K ((2024) ) Biological definition of neuronal alpha-synuclein disease: Towards an integrated staging system for research.. Lancet Neurol 23: , 178–190. |
[48] | Jack CR , Bennett DA , Blennow K , Carrillo MC , Dunn B , Haeberlein SB , Holtzman DM , Jagust W , Jessen F , Karlawish J , Liu E , Molinuevo JL , Montine T , Phelps C , Rankin KP , Rowe CC , Scheltens P , Siemers E , Snyder HM , Sperling R , Contributors ((2018) ) NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement 14: , 535–562. |
[49] | Stern MB , Lang A , Poewe W ((2012) ) Toward a redefinition of Parkinson’s disease. Mov Disord 27: , 54–60. |
[50] | Kluge A , Iranzo A ((2024) ) Biofluid detection of pathological α-synuclein in the prodromal phase of the synucleinopathies.. J Parkinsons Dis–10.3233/JPD-230429. |
[51] | Lim S-Y , Klein C ((2024) ) Parkinson’s disease is predominantly a genetic disease.. J Parkinsons Dis–10.3233/JPD-230376. |
[52] | Kluge A , Bunk J , Schaeffer E , Drobny A , Xiang W , Knacke H , Bub S , Lückstädt W , Arnold P , Lucius R , Berg D , Zunke F ((2022) ) Detection of neuron-derived pathological α-synuclein in blood. Brain 145: , 3058–3071. |
[53] | Okuzumi A , Hatano T , Matsumoto G , Nojiri S , Ueno S , Imamichi-Tatano Y , Kimura H , Kakuta S , Kondo A , Fukuhara T , Li Y , Funayama M , Saiki S , Taniguchi D , Tsunemi T , McIntyre D , Gérardy J , Mittelbronn M , Kruger R , Uchiyama Y , Nukina N , Hattori N ((2023) ) Propagative α-synuclein seeds as serum biomarkers for synucleinopathies. Nat Med 29: , 1448–1455. |
[54] | Bellomo G , De Luca CMG , Paoletti FP , Gaetani L , Moda F , Parnetti L ((2022) ) α-synuclein seed amplification assays for diagnosing synucleinopathies: The way forward. Neurology 99: , 195–205. |
[55] | Buchman AS , Shulman JM , Nag S , Leurgans SE , Arnold SE , Morris MC , Schneider JA , Bennett DA ((2012) ) Nigral pathology and parkinsonian signs in elders without Parkinson disease. Ann Neurol 71: , 258–266. |
[56] | Ross GW , Petrovitch H , Abbott RD , Nelson J , Markesbery W , Davis D , Hardman J , Launer L , Masaki K , Tanner CM , White LR ((2004) ) Parkinsonian signs and substantia nigra neuron density in decendents elders without PD. Ann Neurol 56: , 532–539. |
[57] | Alotaibi F , Pelletier A , Gagnon J-F , Montplaisir JY , Postuma RB ((2019) ) Prodromal marker progression in idiopathic rapid eye movement sleep behavior disorder: Sample size for clinical trials. Mov Disord 34: , 1914–1919. |
[58] | Joza S , Hu MT , Jung K-Y , Kunz D , Stefani A , Dušek P , Terzaghi M , Arnaldi D , Videnovic A , Schiess MC , Hermann W , Lee J-Y , Ferini-Strambi L , Lewis SJG , Leclair-Visonneau L , Oertel WH , Antelmi E , Sixel-Döring F , Cochen De Cock V , Liguori C , Liu J , Provini F , Puligheddu M , Nicoletti A , Bassetti CLA , Bušková J , Dauvilliers Y , Ferri R , Montplaisir JY , Lawton M , Kim H-J , Bes F , Högl B , Šonka K , Fiamingo G , Mattioli P , Lavadia ML , Suescun J , Woo KA , Marelli S , Martens KE , Janzen A , Plazzi G , Mollenhauer B , Fernandes M , Li Y , Cortelli P , Figorilli M , Cicero CE , Schaefer C , Guiraud L , Lanza G , Gagnon J-F , Sunwoo J-S , Ibrahim A , Girtler N , Trenkwalder C , Baldelli L , Pelletier A , Postuma RB , International REM Sleep Behavior Disorder Study Group ((2023) ) Progression of clinical markers in prodromal Parkinson’s disease and dementia with Lewy bodies: A multicentre study. Brain 146: , 3258–3272. |
[59] | Seibyl JP , Kuo P ((2022) ) What is the role of dopamine transporter imaging in parkinson prevention clinical trials? . Neurology 99: , 61–67. |
[60] | Mirelman A , Siderowf A , Chahine L ((2022) ) Outcome assessment in Parkinson disease prevention trials: Utility of clinical and digital measures. Neurology 99: , 52–60. |
[61] | Schwarzschild M ((2024) ) Trial design for early Parkinson’s disease. J Parkinsons Dis, in press. |
[62] | Janssen Daalen JM , Schootemeijer S , Richard E , Darweesh SKL , Bloem BR ((2022) ) Lifestyle Interventions for the Prevention of Parkinson Disease: A Recipe for Action. Neurology 99: , 42–51. |
[63] | Schaeffer E , Toedt I , Köhler S , Rogge A , Berg D ((2021) ) Risk Disclosure in Prodromal Parkinson’s Disease. Mov Disord 36: , 2833–2839. |
[64] | Kramer BS , Croswell JM ((2009) ) Cancer screening: The clash of science and intuition. Annu Rev Med 60: , 125–137. |
[65] | Eaden J , Mayberry MK , Sherr A , Mayberry JF ((2001) ) Screening: The legal view. Public Health 115: , 218–221. |
[66] | Parkinson Study Group ((1993) ) Effects of tocopherol and deprenyl on the progression of disability in early Parkinson’s disease. N Engl J Med 328: , 176–183. |
[67] | Fahn S , Oakes D , Shoulson I , Kieburtz K , Rudolph A , Lang A , Olanow CW , Tanner C , Marek K , Parkinson Study Group ((2004) ) Levodopa and the progression of Parkinson’s disease. N Engl J Med 351: , 2498–508. |
[68] | Schapira AHV , McDermott MP , Barone P , Comella CL , Albrecht S , Hsu HH , Massey DH , Mizuno Y , Poewe W , Rascol O , Marek K ((2013) ) Pramipexole in patients with early Parkinson’s disease (PROUD): A randomised delayed-start trial. Lancet Neurol 12: , 747–755. |
[69] | Writing Group for the NINDS Exploratory Trials in Parkinson Disease (NET-PD) Investigators, Kieburtz K , Tilley BC , Elm JJ , Babcock D , Hauser R , Ross GW , Augustine AH , Augustine EU , Aminoff MJ , Bodis-Wollner IG , Boyd J , Cambi F , Chou K , Christine CW , Cines M , Dahodwala N , Derwent L , Dewey RB , Hawthorne K , Houghton DJ , Kamp C , Leehey M , Lew MF , Liang GSL , Luo ST , Mari Z , Morgan JC , Parashos S , Pérez A , Petrovitch H , Rajan S , Reichwein S , Roth JT , Schneider JS , Shannon KM , Simon DK , Simuni T , Singer C , Sudarsky L , Tanner CM , Umeh CC , Williams K , Wills A-M ((2015) ) Effect of creatine monohydrate on clinical progression in patients with Parkinson disease: A randomized clinical trial. JAMA 313: , 584–93. |
[70] | NINDS Exploratory Trials in Parkinson Disease (NET-PD) FS-ZONE Investigators ((2015) ) Pioglitazone in early Parkinson’s disease: A phase 2, multicentre, double-blind, randomised trial. Lancet Neurol 14: , 795–803. |
[71] | Verschuur CVM , Suwijn SR , Boel JA , Post B , Bloem BR , van Hilten JJ , van Laar T , Tissingh G , Munts AG , Deuschl G , Lang AE , Dijkgraaf MGW , de Haan RJ , de Bie RMA , LEAP Study Group ((2019) ) Randomized delayed-start trial of levodopa in Parkinson’s disease. N Engl J Med 380: , 315–324. |
[72] | Parkinson Study Group STEADY-PD III Investigators ((2020) ) Isradipine versus placebo in early Parkinson disease: A randomized trial. Ann Intern Med 172: , 591–598. |
[73] | Parkinson Study Group SURE-PD3 Investigators, Schwarzschild MA , Ascherio A , Casaceli C , Curhan GC , Fitzgerald R , Kamp C , Lungu C , Macklin EA , Marek K , Mozaffarian D , Oakes D , Rudolph A , Shoulson I , Videnovic A , Scott B , Gauger L , Aldred J , Bixby M , Ciccarello J , Gunzler SA , Henchcliffe C , Brodsky M , Keith K , Hauser RA , Goetz C , LeDoux MS , Hinson V , Kumar R , Espay AJ , Jimenez-Shahed J , Hunter C , Christine C , Daley A , Leehey M , de Marcaida JA , Friedman JH , Hung A , Bwala G , Litvan I , Simon DK , Simuni T , Poon C , Schiess MC , Chou K , Park A , Bhatti D , Peterson C , Criswell SR , Rosenthal L , Durphy J , Shill HA , Mehta SH , Ahmed A , Deik AF , Fang JY , Stover N , Zhang L , Dewey RB , Gerald A , Boyd JT , Houston E , Suski V , Mosovsky S , Cloud L , Shah BB , Saint-Hilaire M , James R , Zauber SE , Reich S , Shprecher D , Pahwa R , Langhammer A , LaFaver K , LeWitt PA , Kaminski P , Goudreau J , Russell D , Houghton DJ , Laroche A , Thomas K , McGraw M , Mari Z , Serrano C , Blindauer K , Rabin M , Kurlan R , Morgan JC , Soileau M , Ainslie M , Bodis-Wollner I , Schneider RB , Waters C , Ratel AS , Beck CA , Bolger P , Callahan KF , Crotty GF , Klements D , Kostrzebski M , McMahon GM , Pothier L , Waikar SS , Lang A , Mestre T ((2021) ) Effect of urate-elevating inosine on early parkinson disease progression: The SURE-PD3 randomized clinical trial. JAMA 326: , 926–939. |
[74] | Simuni T , Fiske B , Merchant K , Coffey CS , Klingner E , Caspell-Garcia C , Lafontant D-E , Matthews H , Wyse RK , Brundin P , Simon DK , Schwarzschild M , Weiner D , Adams J , Venuto C , Dawson TM , Baker L , Kostrzebski M , Ward T , Rafaloff G , Parkinson Study Group NILO-PD Investigators and Collaborators ((2021) ) Efficacy of nilotinib in patients with moderately advanced Parkinson disease: A randomized clinical trial. JAMA Neurol 78: , 312–320. |
[75] | Stevens KN , Creanor S , Jeffery A , Whone A , Zajicek J , Foggo A , Jones B , Chapman R , Cocking L , Wilks J , Webb D , Carroll C , PD STAT Study Group ((2022) ) Evaluation of simvastatin as a disease-modifying treatment for patients with Parkinson disease: A randomized clinical trial. JAMA Neurol 79: , 1232–1241. |
[76] | Devos D , Labreuche J , Rascol O , Corvol J-C , Duhamel A , Guyon Delannoy P , Poewe W , Compta Y , Pavese N , Růžička E , Dušek P , Post B , Bloem BR , Berg D , Maetzler W , Otto M , Habert M-O , Lehericy S , Ferreira J , Dodel R , Tranchant C , Eusebio A , Thobois S , Marques A-R , Meissner WG , Ory-Magne F , Walter U , de Bie RMA , Gago M , Vilas D , Kulisevsky J , Januario C , Coelho MVS , Behnke S , Worth P , Seppi K , Ouk T , Potey C , Leclercq C , Viard R , Kuchcinski G , Lopes R , Pruvo J-P , Pigny P , Garçon G , Simonin O , Carpentier J , Rolland A-S , Nyholm D , Scherfler C , Mangin J-F , Chupin M , Bordet R , Dexter DT , Fradette C , Spino M , Tricta F , Ayton S , Bush AI , Devedjian J-C , Duce JA , Cabantchik I , Defebvre L , Deplanque D , Moreau C , FAIRPARK-II Study Group ((2022) ) Trial of deferiprone in Parkinson’s disease. N Engl J Med 387: , 2045–2055. |
[77] | Malatt C , Wu T , Bresee C , Hogg E , Wertheimer J , Tan E , Pomeroy H , Obialisi G , Tagliati M ((2022) ) Liraglutide improves non-motor function and activities of daily living in patients with Parkinson’s disease: A randomized, double-blind, placebo-controlled trial (P9-11.005). Neurology 98: , 3068. |
[78] | Nakamori M , Junn E , Mochizuki H , Mouradian MM ((2019) ) Nucleic acid-based therapeutics for Parkinson’s disease. Neurotherapeutics 16: , 287–298. |
[79] | Mullin S , Smith L , Lee K , D’Souza G , Woodgate P , Elflein J , Hällqvist J , Toffoli M , Streeter A , Hosking J , Heywood WE , Khengar R , Campbell P , Hehir J , Cable S , Mills K , Zetterberg H , Limousin P , Libri V , Foltynie T , Schapira AHV ((2020) ) Ambroxol for the treatment of patients with Parkinson disease with and without glucocerebrosidase gene mutations: A nonrandomized, noncontrolled trial. JAMA Neurol 77: , 427–434. |
[80] | Payne T , Appleby M , Buckley E , van Gelder LMA , Mullish BH , Sassani M , Dunning MJ , Hernandez D , Scholz SW , McNeill A , Libri V , Moll S , Marchesi JR , Taylor R , Su L , Mazzà C , Jenkins TM , Foltynie T , Bandmann O ((2023) ) A double-blind, randomized, placebo-controlled trial of ursodeoxycholic acid (UDCA) in Parkinson’s disease. Mov Disord 38: , 1493–1502. |


