
The Role of Endophilin A1 in Lipopolysaccharide-Induced Parkinson’s Disease Model Mice
Abstract
Background:
Endophilin A1 (EPA1) is encoded by the SH3GL2 gene, and SH3GL2 was designated as a Parkinson’s disease (PD) risk locus by genome-wide association analysis, suggesting that EPA1 may be involved in the occurrence and development of PD.
Objective:
To investigate the role of EPA1 in lipopolysaccharide (LPS)-induced PD model mice.
Methods:
The mice PD model was prepared by injecting LPS into the substantia nigra (SN), and the changes in the behavioral data of mice in each group were observed. The damage of dopaminergic neurons, activation of microglia, and reactive oxygen species (ROS) generation were detected by immunofluorescence method; calcium ion concentration was detected by calcium content detection kit; EPA1 and inflammation and its related indicators were detected by western blot method. EPA1 knockdown was performed by an adeno-associated virus vector containing EPA1-shRNA-eGFP infusion.
Results:
LPS-induced PD model mice developed behavioral dysfunction, SN dopaminergic nerve damage, significantly increased calcium ion, calpain 1, and ROS production, activated NLRP1 inflammasome and promoted pro-inflammatory cell release, and SN EPA1 knockdown improves behavioral disorders, alleviates dopaminergic neuron damage, reduces calcium, calpain 1, ROS generation, and blocks NLRP1 inflammasome-driven inflammatory responses.
Conclusion:
The expression of EPA1 in the SN of LPS-induced PD model mice was increased, and it played a role in promoting the occurrence and development of PD. EPA1 knockdown inhibited the NLRP1 inflammasome activation, decreased the release of inflammatory factors and ROS generation, and alleviated dopaminergic neuron damage. This indicated that EPA1 may participating in the occurrence and development of PD.
INTRODUCTION
Parkinson’s disease (PD) is a common neurodegenerative disease. Clinical manifestations include bradykinesia, tremor, rigidity, and postural instability. A number of studies have shown that the pathogenesis of PD is related to various factors such as genetics, body aging, environment, and oxidative stress [1]. Although the pathogenesis of PD has been studied from many aspects, its specific pathogenesis is still not fully understood.
Endophilin A1 (EPA1) plays a crucial role in the regulation of nervous system diseases and tumor diseases [2, 3]. Its structure includes an amino-terminal N-BIN/amphiphysin/Rvs (BAR) domain and a carboxy-terminal Src homology region 3 (SH3) domain, encoded by the SH3GL2 gene and expressed almost exclusively in the brain, and is associated with epilepsy and schizophrenia [4, 5]. In addition to being associated with Alzheimer’s disease (AD) [6, 7], SH3GL2 was designated as a risk locus for PD by genome-wide association analysis [8].
Recent literature reports that EPA1 can act as an effector of Ca2 +/calmodulin, thereby affecting calcium homeostasis [9]. At the same time, EPA1 affects oxidative stress and inflammatory response [3, 10]. The above findings suggest that EPA1 may be involved and play an important role in the occurrence and development of PD. However, the role and mechanism of EPA1 in PD remain unclear.
Oxidative stress plays a contributing role in the development of neurodegenerative diseases. Excessive reactive oxygen species (ROS) will lead to an increase in the level of oxidative stress in the body, and is closely related to the occurrence and development of various diseases, such as AD, PD, myocardial infarction, tumors, etc. [11–13]. The NOD-like receptor protein 1 (NLRP1) inflammasome plays an important role in the pathogenesis of neurodegenerative diseases and contains the Caspase activation recruitment domain recruiting proteins ASC and Caspase-1 [14, 15]. Activated NLRP1 inflammasome will lead to the production and release of pro-inflammatory factors IL-1β, IL-18, and TNF-α [16], and its role in depression, PD, and other diseases has attracted more and more attention. It has been reported in the literature that ROS is one of the canonical pathways to activate the inflammasome, and increased ROS production leads to the activation of the NLRP1 inflammasome and induces neuroinflammation [17, 18]. Therefore, the ROS/NLRP1 inflammasome signaling pathway is involved in the occurrence of various neurological diseases.
In the present study, we established a mice PD model by local injection of LPS into substantia nigra (SN) to investigate changes in EPA1/ROS/NLRP1 inflammasome signaling in LPS-induced PD model mice. Injected adeno-associated virus (AAV) into SN to knock down EPA1 to further explore the role and mechanism of EPA1 in LPS-induced PD model mice.
MATERIALS AND METHODS
Reagents
LPS was purchased from Sigma-Aldrich (St. Louis, MO, USA, L4391). The antibodies of Calpain 1, IL-18, and α-Syn were obtained from Proteintech Group (Hubei, China). The primary antibody for NLRP1 was purchased from Abcam (Cambridge, UK). Tyrosine hydroxylase (TH), Iba-1, and Caspase 1 were obtained from Servicebio (Hubei, China). Other general reagents were commercially available.
Animals
Male C57BL/6 mice (aged 12-13 weeks and weighing 25–30 g) were obtained from the Experimental Animal Center of Anhui Medical University. They were kept in a controlled environment with a temperature of 22±2°C and humidity of 60% under a 12 h light/dark cycle. Food and water were available ad libitum. All animal procedures were approved by the Committee for Experimental Animal Use and Care of Anhui Medical University.
Animal models
All mice were randomly separated into two groups: sham group and LPS-treated 7d group. The animals were allowed 7 days to adapt to the new environment before the experiment. The mice were anesthetized with 1% Pentobarbital Sodium and fixed in the stereotaxic apparatus (RWD Life Science Co. Ltd, Shenzhen, China). A single dose of LPS (5.0μg in 2.5μL of sterile saline; 5.0μg/mice, Sigma, L4391) was injected into SN through Hamilton syringe (Hamilton Co., 1701RN/SYR 10μl/ga26 s/51 mm/pst2, Reno, NV) at a rate of 0.5μl/min. After each injection, the needle was left in situ for 10 min, then retracted at the same rate. The sham-operated mice were injected with saline solution. (stereotaxic coordinates: 3.0 mm posterior to bregma, 1.3 mm lateral to the midline, 4.7 mm ventral to the surface of the cortex). On the 7th day after the injection of LPS, behavioral tests were performed.
To explore the effect and possible mechanism of EPA1, in the second part of the experiment, male C57 mice were randomly divided into the following four groups: control group, LPS model group, LPS+NC shRNA group, LPS+EPA1 shRNA group. The PD model was established four weeks after virus injection, and related indicators were detected.
Open-field test
Open-field test was performed in a black box (60 cm×60 cm×60 cm), which was located in an experimental room with controlled conditions of light, temperature, and noise. Firstly, the mice were placed in the center area, allowing them to adapt to the environment for 2 min. Then the moving distance, Mean speed, Line crossing and Number of stand up were recorded over a 3-min period. At each stress session, the floor surfaces and walls of the OFT apparatus were thoroughly cleaned with 75% ethanol to abolish any sign of olfactory cues.
Grip forced test
Place the grip tester on a stable operating table, and then gently place the mice on the grip plate to avoid disturbing the mice. Adjust the position of the mice so that its two front paws firmly grasp the grip lever on the grip plate. Under the premise of confirming that the mice’s two paws are firmly grasped, pull the right hand horizontally to the back of the mice until the mice’s paws are stretched out. After the maximum grip is obtained, the mice automatically release the grip lever and the machine automatically determines Maximum grip. The maximum grip value of each mice needs to be measured three times to get the average value.
Immunofluorescence staining
The anesthetized mice were given cardiac perfusion with precooled PBS and 4% paraformaldehyde, then brain tissue was quickly removed and fixed overnight with 4% paraformaldehyde. The brain tissue was embedded in paraffin and the thickness of continuous sections was 4μm. The brain sections were dewaxed, hydrated, and incubated with 3% H2O2 for 10 min to quench endogenous peroxidase. The sections were then sealed with 5% BSA for 30 min and incubated with primary antibody at 4°C overnight. The next day, the sections were placed in corresponding fluorescent secondary antibodies and incubated for 1 h at room temperature and under dark conditions. Then, the cell nuclei were stained with DAPI at room temperature and kept away from light for 5 min and observed and photographed under the microscope. The average fluorescence intensity of positive neurons in each section was analyzed by Image-Pro Plus 6.0 analysis system.
Calcium content detection
The Ca2 + content in hippocampus of mice is detected by the calcium content chromogenic detection kit (Beyotime, S1063S). The principle is to use Ca2 + to react with an o-phthalein complex ketone in an alkaline environment and produce a purple complex. First, 20 mg of substantia nigra tissue was weighed, according to the ratio of tissue: sample lysate = 1:5∼10 (the specific ratio is adjusted according to the results of the pre-experiment), and then the sample is placed into a tissue homogenizer to fully grind and aspirate the supernatant. Then, according to the instructions, the standard is prepared and added to the 96-well plate, placed in a microplate reader, and incubated at room temperature while avoiding light for 5–10 min. Finally, the corresponding absorbance value was measured, drawing the standard curve. The absorbance value of the sample was brought into the standard curve to obtain the Ca2 + concentration of the sample.
ROS detection
The mice were sacrificed via cervical dislocation and the brains were fixed with 4% paraformaldehyde. Then, the brains were embedded in OCT (Sakura, Torrance, CA) at –20°C to be sectioned into 5μm slices with a freezing microtome (Leica CM3050, Germany). Sections were incubated with the ROS staining solution for 30 min and washed three times with PBS, then incubated with DAPI staining solution for 10 min and washed three times with PBS. The sections were examined by a fluorescence microscope (Olympus IX71, Japan). The mean fluorescence density of ROS was quantified from 3 random brain SNby using an Image-Pro Plus 6.0 analysis software to indicate the ROS production.
Western blotting
The animals were sacrificed by decapitation and brain tissue was dissected. About 50 mg SN of brain tissue was homogenized in a homogenizer containing 400μl of lysate and 10μl of PMSF. After being lysed for 30 min on ice, the samples were centrifuged at 12,000 r/min at 4°C for 15 min, the supernatant was separated. The protein concentration was determined by BCA Protein Assay Kit (Beyotime Biotechnology, China). The same protein was separated by 10% SDS-polyacrylamide gel electrophoresis and then transferred onto PVDF membranes (Millipore Corp., Billerica, MA, USA). The membranes were soaked in blocking buffer (5% skimmed milk in TBST) for 1 h and then probed with primary antibodies of EPA1 (CST, 65169S, 1:1000), NLRP1 (Abcam, ab36852, 1:1000), Caspase 1, TNF-α (Bioss bs-0078R,1:1000), IL-1β (Bioss, bs-20449R, 1:1000), Calpain 1 (Proteintech, 10538-1-AP, 1:1000), IL-18 (Proteintech, 10663-1-AP, 1:1000), α-Syn (Proteintech, 10842-1-AP, 1:1000), or β-actin (1:1000) overnight at 4°C, followed by horseradish peroxidase (HRP)-conjugated secondary antibody (1:10,000) for 1 h at room temperature. After washing three times, bands corresponding to the proteins were detected by using the Super Signal West Femto chemiluminescence substrate kit (Bridgen, China), and the optical density analysis was performed using Image J software. The β-actin of the same sample was used as a control to calculate the relative expression of the target protein.
Injection of adenovirus-associated vector
To study the role of EPA1 in LPS-induced PD model mice, injecting AAV vectors containing AAV-EPA1 shRNA or AAV-NC shRNA (Hanbio, Shanghai, China) was employed. The sequence was CATCAACTATGTAGAAATT. AAV-EPA1 shRNA or AAV-NC shRNA was injected into the SN. After a 4-week recovery period, fluorescence microscopy and western blotting were used to observe transfection in vivo. After successful virus transfection, a PD model was established, and male C57 mice were randomly divided into the following four groups: control group, LPS model group, LPS+NC shRNA group, and LPS+EPA1 shRNA group. Behavioral testing was performed on day 7 after LPS injection.
Statistical analysis
All data were analyzed with the statistical program SPSS 17.0 (Chicago, IL, USA). Data are expressed as means±SEM. Unpaired two-tailed Student’s t-test or one-way analysis of variance (ANOVA) was used to evaluate differences. p < 0.05 was considered statistically significant.
RESULTS
Expression of Endophilin A1 in LPS-induced PD mice model
To study the role of EPA1 in PD, we first established a PD animal model using LPS. The results of the open field experiment showed that compared with the control group, the LPS model group had the number of lines crossing, the Maximum grip strength, the mean moving speed, the total Moving distance and the number of stand up were significantly decreased, the results showed that the mice had obvious behavioral dysfunction, suggesting that the PD model was successfully established (Fig. 1B–F). Then, we detected the changes of DA neurons and microglia in the SN of mice, as well as the localization and expression level of EPA1 in the SN of mice by immunofluorescence co-localization technology and western blot. As shown in Fig. 1G, the co-localization of the EPA1 probe can be observed in TH-positive cells. Meanwhile, LPS treatment induced a decrease in the number of TH-positive cells. microglia were activated in the LPS model group, but the co-expression of EPA1 and Iba1 was not observed. The above results indicated that EPA1 expressed and localized in TH-positive cells between PD model mice and normal mice. These results showed that LPS treatment attenuated the spontaneous activity and motor ability of mice and induced DA neuron damage in the SN of model mice.
Fig. 1
Expression of Endophilin A1 in LPS-induced PD mice model. A) Drug treatment schedule. B) The number of lines crossing. C) The Maximum grip strength (g). D) The mean moving speed (m/s). E) The total Moving distance (m). F) The number of stand up. Data are expressed as mean±SEM (n = 12). G) double labeling for TH, Iba1, and EPA1 was performed with immunofluorescence. TH, Iba1 colocalized with EPA1. Scale bar = 200μm and Scale bar = 40μm. H) Quantification of TH-positive expression in the SN. I) Quantification of EPA1-positive expression in the SN. J) Western blot for EPA1 protein level. Data are expressed as mean±SEM (n = 6). **p < 0.01 vs. the control group.
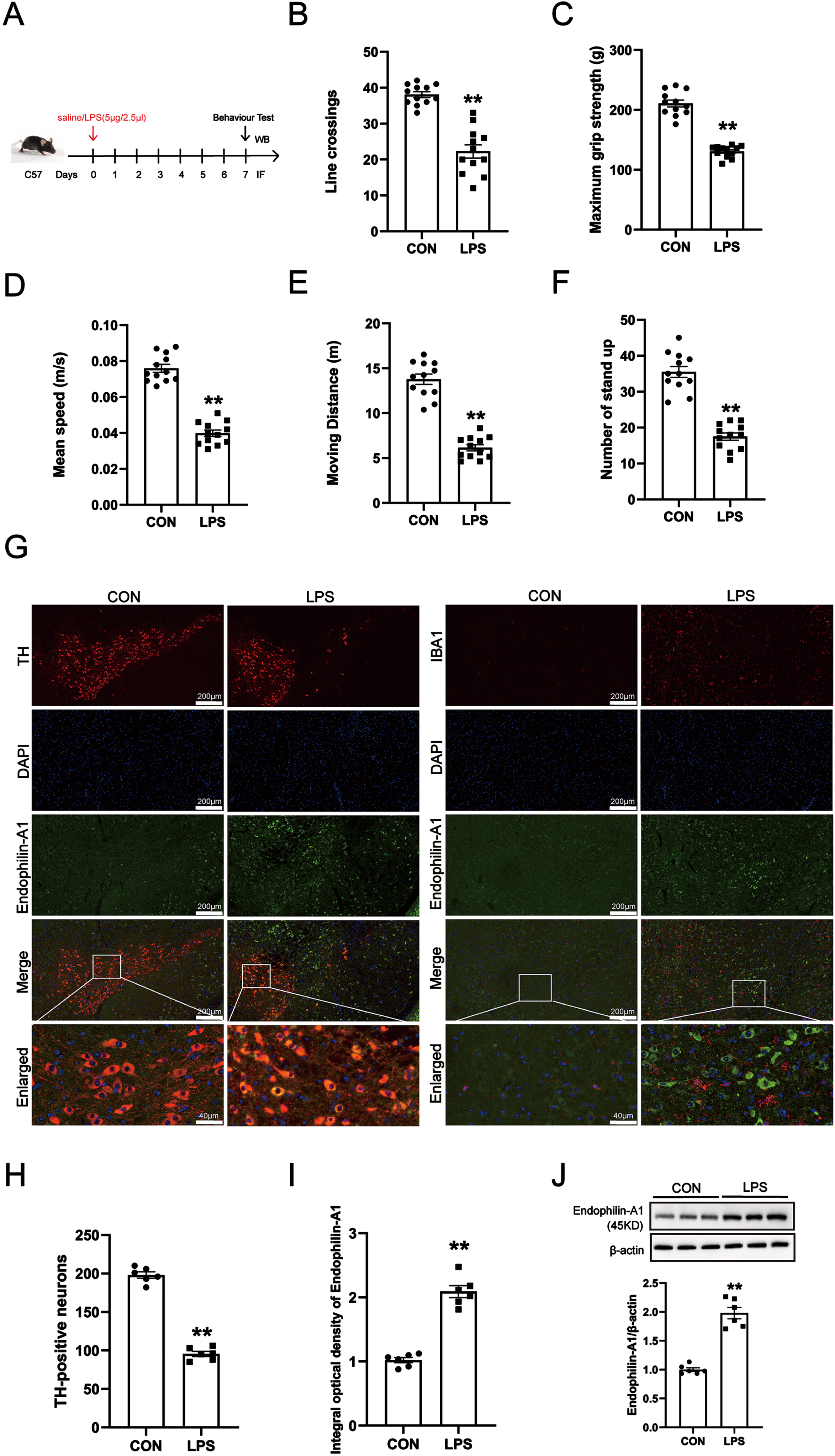
Next, the results of immunofluorescence quantitative detection and western blot showed that compared with the control group, the expression level of EPA1 protein in the SN of mice in the LPS model group was significantly increased (Fig. 1I, J).
Changes of calcium ions, calpain 1, and ROS in LPS-induced PD mice model
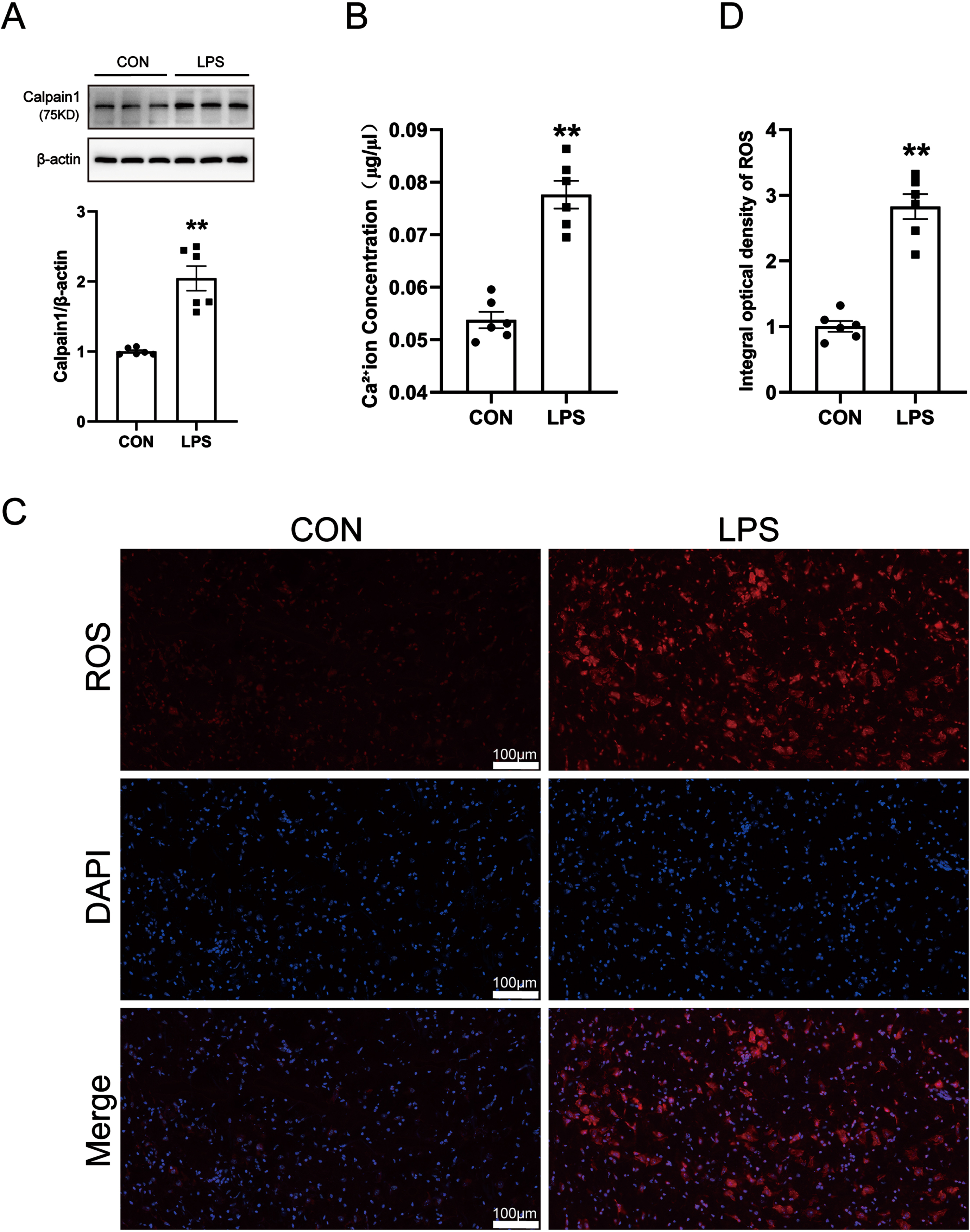
Calcium ions are involved in the occurrence of various neurological diseases. When the stable state of calcium ions is destroyed, abnormal changes in the level of calpain 1 will be caused. Activated calpain 1 is closely related to oxidative stress and neuroinflammation. A large amount of highly active ROS is generated in the body, which will cause damage to DA neurons in a variety of ways. Therefore, we detected the levels of calpain 1, calcium ions, and ROS in the SN of PD model mice by western blot, kits, and immunofluorescence techniques. The results are shown in Fig. 2A–C. Compared with the control group, the levels of calcium ions, calpain 1, and ROS in the SN of mice in the LPS model group were significantly increased. The results showed that LPS-induced increases in calcium levels and further activation of calpain 1, along with increased ROS generation.
Fig. 2
Changes of calcium ions, calpain 1 and ROS in LPS-induced PD mice model. A) Calcium Colorimetric Assay Kit for calcium level. B) Western blot for calpain 1 protein level. C) ROS-Immunofluorescence images. The bar is 100μm. D) The mean density of ROS production in the SN. Data are expressed as mean±SEM (n = 6). **p < 0.01 vs. the control group.
NLRP1 inflammasome activation and related inflammatory response in LPS-induced PD mice model
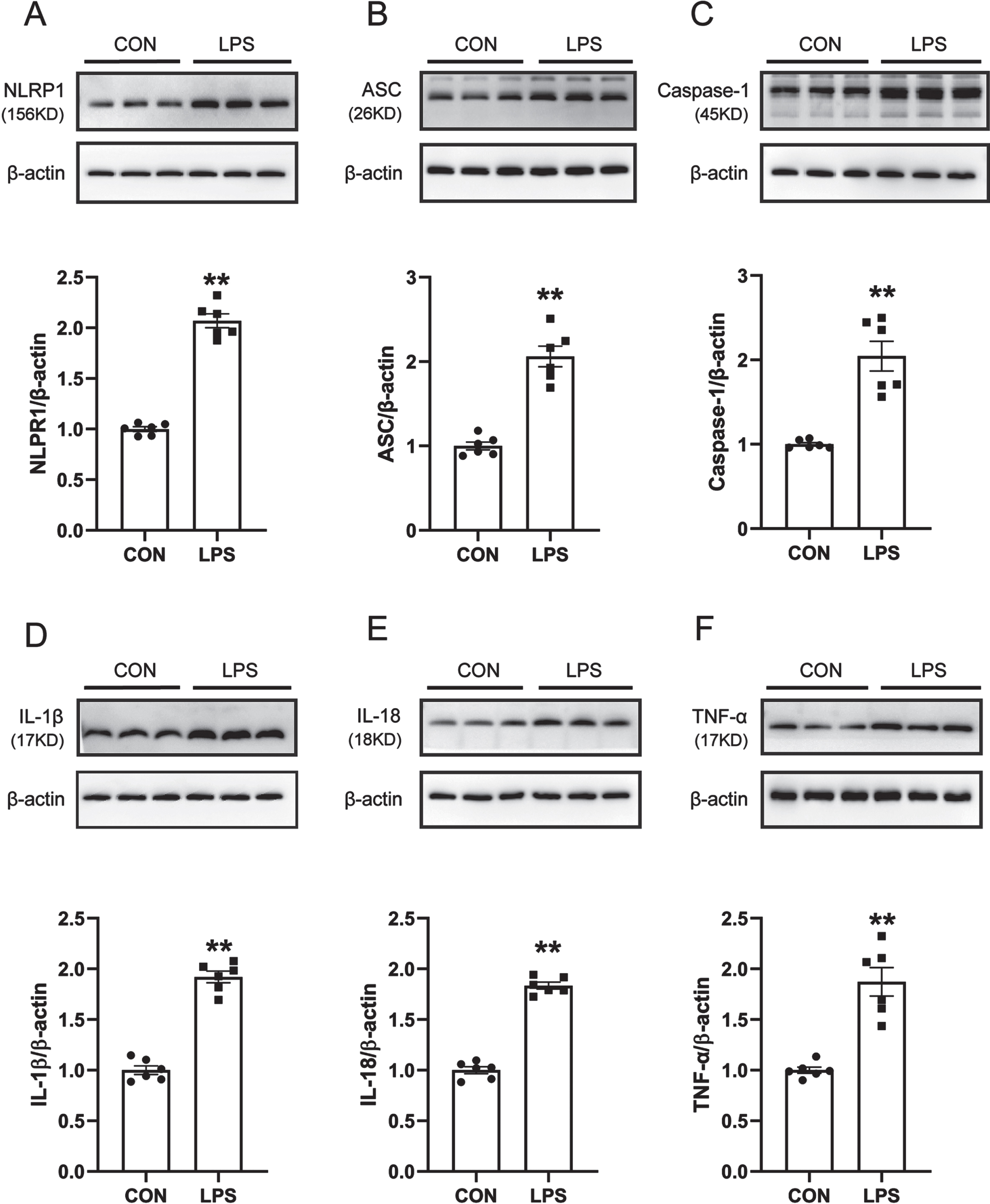
ROS is one of the canonical pathways to activate the inflammasome, and increased production of ROS leads to the activation of the NLRP1 inflammasome and induces neuroinflammation. Therefore, western blot was used to detect the protein expression levels of NLRP1, Caspase-1, ASC, IL-1β, IL-18, and TNF-α in the SN of mice. The results are shown in Fig. 3A–F. Compared with the control group, the expression levels of NLRP1, Caspase-1, ASC, IL-1β, IL-18, and TNF-α in the SN of mice in the LPS model group were significantly increased. It is suggested that the NLRP1 inflammasome in the SN of LPS-induced PD model mice is activated and causes the release of a large number of inflammatory factors.
Fig. 3
NLRP1 inflammasome activation and related inflammatory response in LPS-induced PD mice model. A) Western blot for NLRP1 protein level. B) Western blot for ASC protein level. C) Western blot for Caspase1 protein level. D) Western blot for IL-1β protein level. E) Western blot for IL-18 protein level. F) Western blot for TNF-α protein level. Data are expressed as mean±SEM (n = 6). **p < 0.01 vs. the control group.
EPA1 knockdown analysis
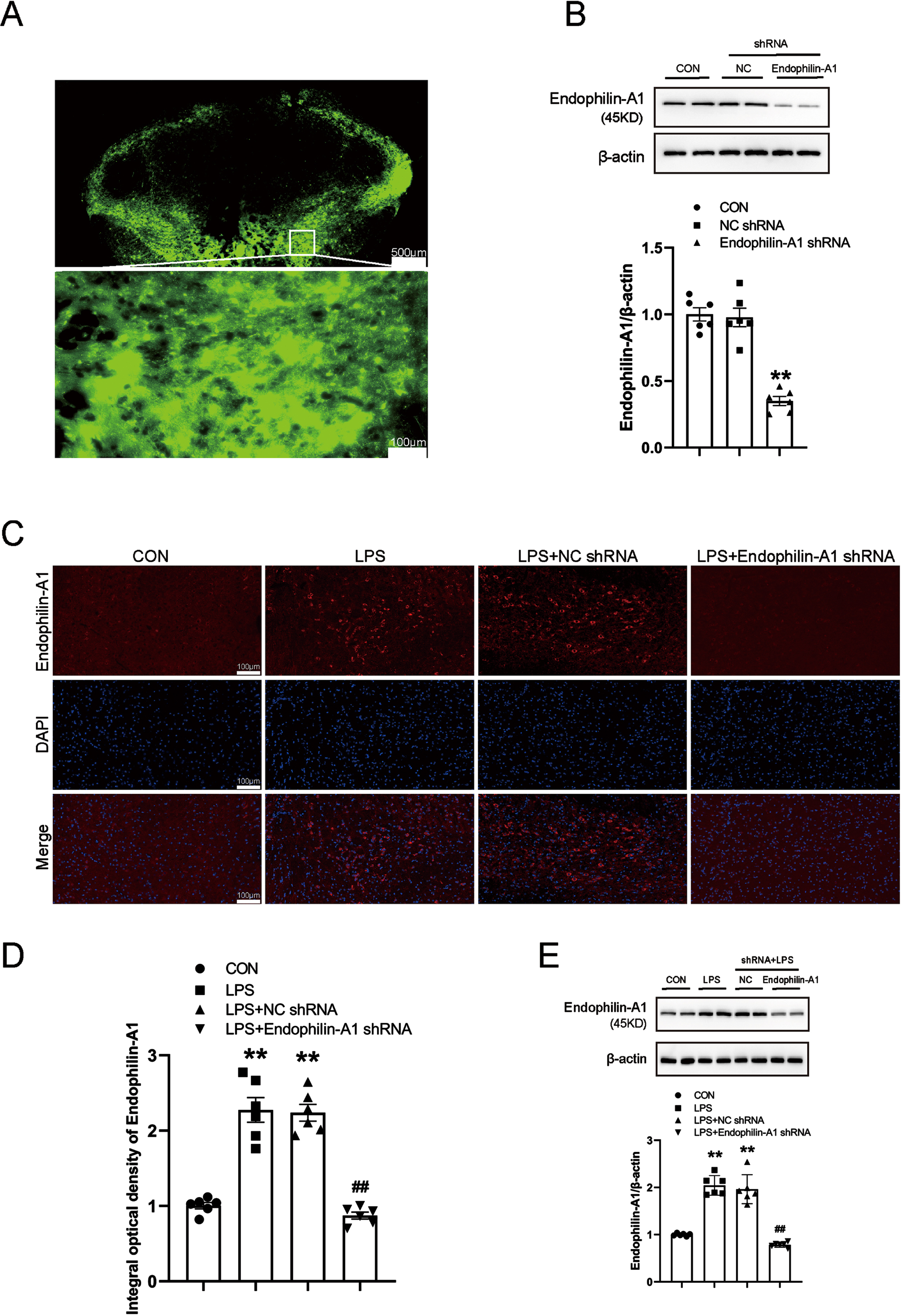
To clarify the specific role and possible mechanism of EPA1 in the occurrence and development of PD. We knockdown EPA1 by injecting AAV-EPA1 shRNA into the SN, and injecting AAV-EPA1 shRNA virus through stereotaxic brain. The results showed that EPA1 was successfully knockdowned (Fig. 4A, B). At the same time, we further detected the expression level of EPA1 in the SN of mice in the control group, LPS model group, LPS+NC shRNA group, and LPS+EPA1 shRNA group by immunofluorescence technique and western blot. Compared with the control group, the expression level of EPA1 protein was significantly increased in the LPS+NC shRNA group and the LPS model group. Compared with the LPS model group, the expression level of EPA1 was no significant difference in the LPS+NC shRNA group. Compared with the LPS+NC shRNA group, the expression level of EPA1 protein in the LPS+EPA1 shRNA group was significantly decreased (Fig. 4C–E). The above results indicated that EPA1 was successfully silenced and significantly reduced the expression level of EPA1 in the SN region.
Fig. 4
EPA1 knockdown analysis. A) Fluorescence images that expressed AAV- EPA1-EGFP in the SN four weeks after infusion. The bar is 500μm. B) Western blot for EPA1 protein level. C) EPA1-Immunofluorescence images. The bar is 100μm. D) The mean density of EPA1 production in the SN. E) Western blot for EPA1 protein level. Data are expressed as mean±SEM (n = 6). **p < 0.01 vs. the control group; # #p < 0.01 vs. the LPS+NC shRNA group.
EPA1 knockdown ameliorated abnormal behaviors and DA neurons damage in LPS-induced PD model mice
Then, to investigate the effect of EPA1 on motor activity in PD mice, LPS model establishment was performed 4 weeks after NC shRNA or EPA1 shRNA infusion. OFT was used to evaluate the locomotor activity. Figure 5B–F shows the number of times crossing the grid, the maximum gripping force, the average moving speed of the mice in the LPS+NC shRNA group and the LPS model group, as compared with the control group. The total moving distance and standing times showed a downward trend, compared with the LPS+NC shRNA group, the times of crossing the grid, the maximum grasping force, and the average moving speed of the mice in the LPS+EPA1 shRNA group, moving distance, and standing times were increased. The above results suggest that EPA1 knockdown can improve the behavioral disorders of LPS-induced PD model mice. Besides, compared with the LPS+NC shRNA group, the LPS+EPA1 shRNA group attenuated LPS-induced neuronal damage and decreased the number of neurons (Fig. 5G, H). These results suggest that EPA1 gene silencing in SN can alleviate LPS-induced loss of neuronal damage in PD model mice. There was no significant difference between LPS model group and LPS+NC shRNA group.
Fig. 5
EPA1 knockdown ameliorated abnormal behaviors and DA neurons damage in LPS-induced PD model mice. A) Drug treatment schedule. B) The number of lines crossing. C) The maximum grip strength (g). D) The mean moving speed (m/s). E) The total moving distance (m). F) The number of stand up. Data are expressed as mean±SEM (n = 12). G) TH-Immunofluorescence images. The bar is 200μm. H) Quantification of TH-positive expression in the SN. Data are expressed as mean±SEM (n = 6). **p < 0.01 vs. the control group, #p < 0.05, # #p < 0.01 vs. the LPS+NC shRNA group.
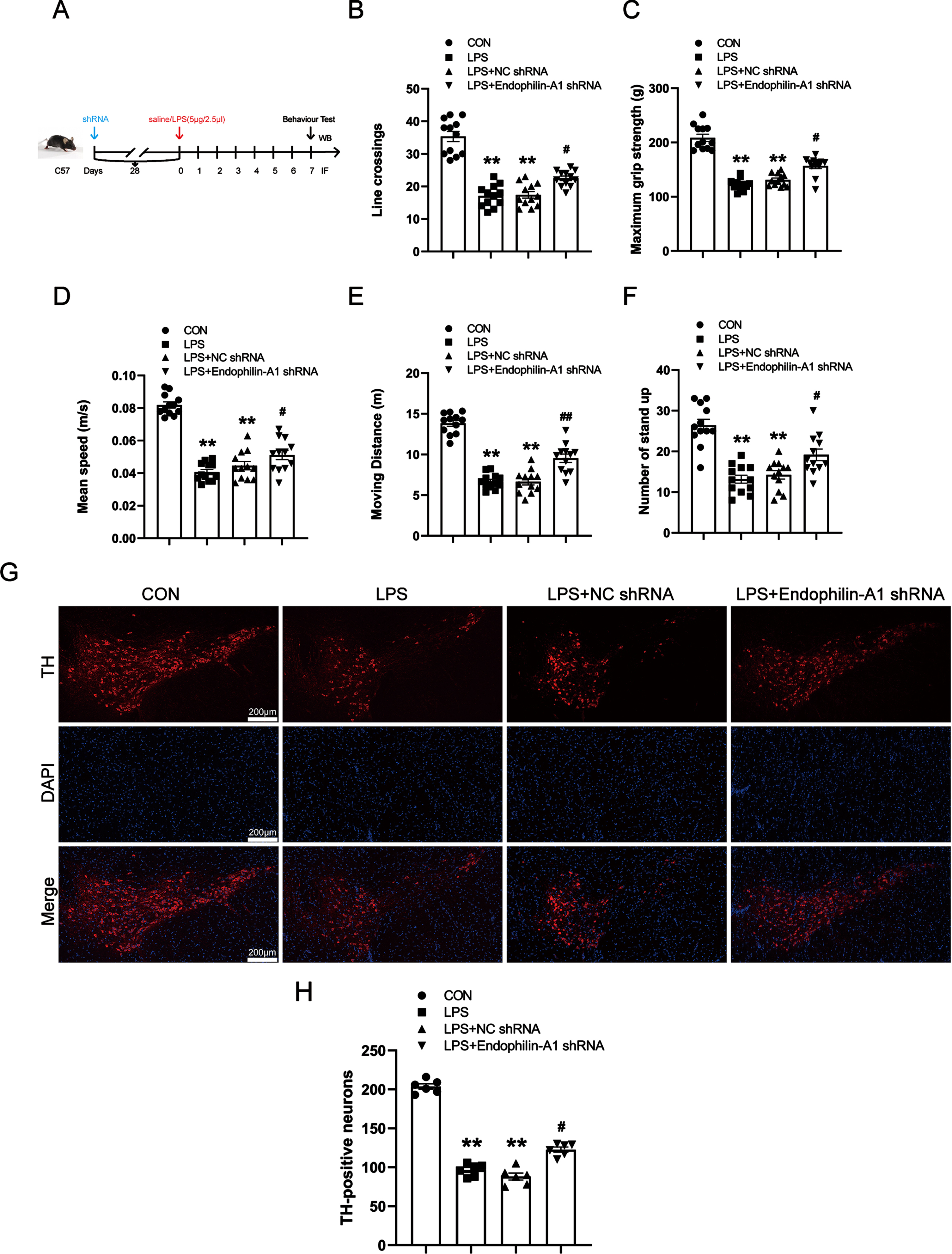
EPA1 knockdown reduces the production of calcium ions, calpain 1, and ROS in the SN of LPS-induced PD model mice
To confirm whether EPA1 gene silencing can improve calcium homeostasis and inhibit the activation of calpain 1, as well as the effect on ROS. We detected the levels of calpain 1, calcium ions, and ROS in the SN of PD model mice by western blot, kits, and immunofluorescence techniques. The results are shown in Fig. 6A–D. Compared with the control group, the levels of calcium ion and calpain 1 in the SN of mice in the LPS model group and LPS+NC shRNA group were significantly increased. Compared with the LPS+NC shRNA group, the calcium ion, calpain 1, and ROS levels in the LPS+EPA1 shRNA group were significantly decreased. The results showed that EPA1 knockdown could ameliorate the disruption of calcium ion homeostasis and inhibit the activation of calpain 1 in the SN of LPS-induced PD model mice while alleviating oxidative stress. There was no significant difference between LPS model group and LPS+NC shRNA group.
Fig. 6
EPA1 knockdown reduces the production of calcium ions, calpain 1, and ROS in the SN of LPS-induced PD model mice. A) Calcium Colorimetric Assay Kit for calcium level. B) Western blot for calpain 1 protein level. C) ROS-Immunofluorescence images. The bar is 100μm. D) The mean density of ROS production in the SN. Data are expressed as mean±SEM (n = 6). **p < 0.01 vs. the control group, # #p < 0.01 vs. the LPS+NC shRNA group.
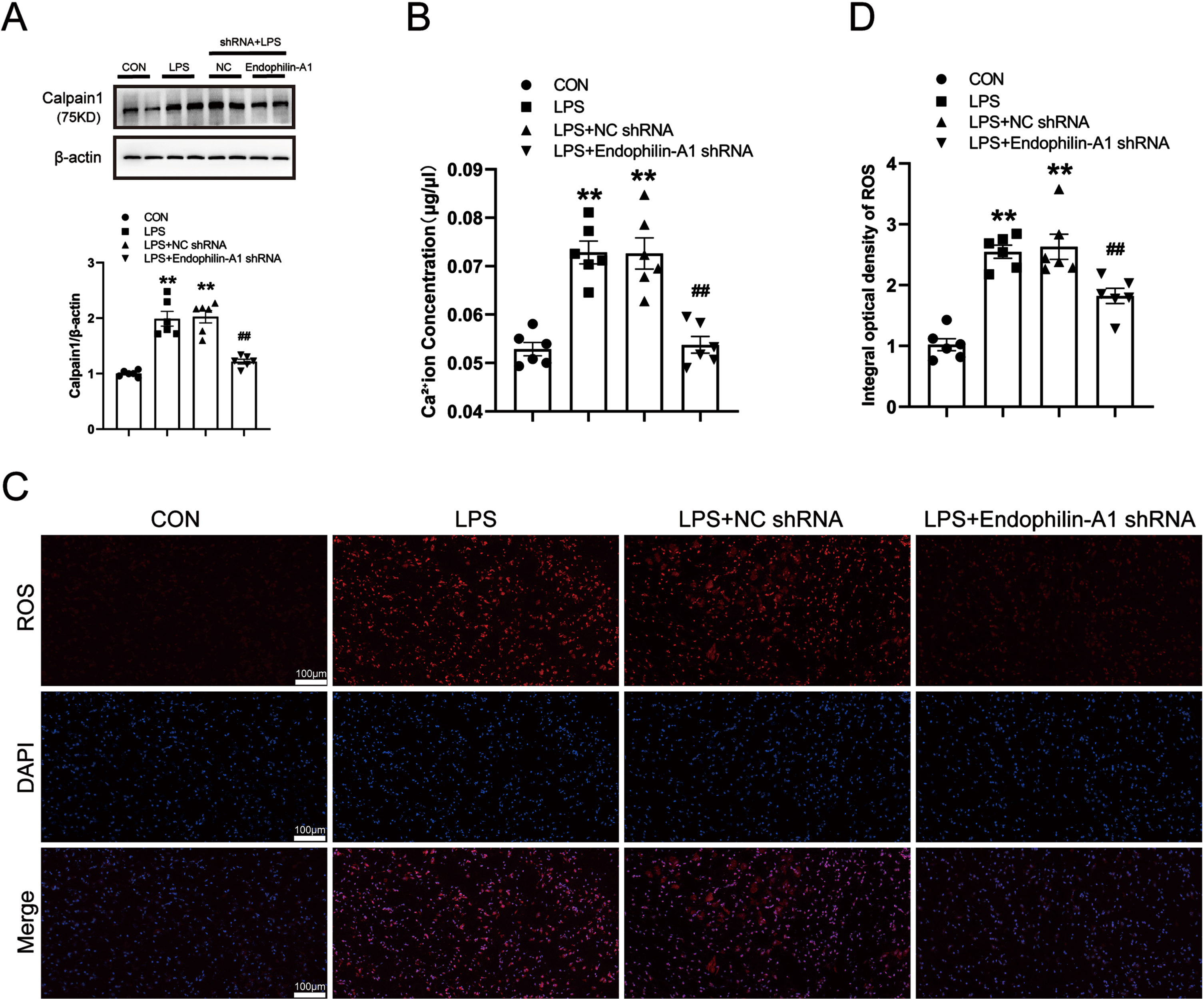
EPA1 knockdown alleviates NLRP1 inflammasome activation and related inflammatory responses in the SN of LPS-induced PD model mice
As a key regulatory platform of the inflammatory response, the activation of the inflammasome can promote the release of pro-inflammatory cytokines. To investigate the mechanism of EPA1, we tested the SN proinflammatory cytokine levels in mice. Western blot was used to detect the protein expression levels of NLRP1, Caspase-1, ASC, IL-1β, IL-18, and TNF-α in the SN of mice. The results are shown in Fig. 7A–F. Compared with the control group, the expression levels of NLRP1, Caspase-1, ASC, IL-1β, IL-18, and TNF-α in the SN of mice in the LPS model group and the LPS+NC shRNA group were significantly increased. Compared with the LPS+NC shRNA group, the protein expression levels of NLRP1, Caspase-1, ASC, IL-1β, IL-18, and TNF-α in the LPS+EPA1 shRNA group were significantly decreased. There was no significant difference between LPS model group and LPS+NC shRNA group. The results showed that EPA1 gene silencing effectively attenuated NLRP1 inflammasome activation and its mediated inflammatory response.
Fig. 7
EPA1 knockdown alleviates NLRP1 inflammasome activation and related inflammatory responses in the SN of LPS-induced PD model mice. A–F) Western blot for NLRP1, Caspase-1, ASC, IL-1β, IL-18, and TNF-α protein level. Data are expressed as mean±SEM (n = 6). **p < 0.01 vs. the control group, #p < 0.05, # #p < 0.01 vs. the LPS+NC shRNA group.
DISCUSSION
As a member of the synaptic vesicle endocytic recycling protein family, the mechanism of action of EPA1 is very complex. The abnormal expression of EPA1 in the brain is involved in the occurrence and development of various neurodegenerative diseases and psychiatric diseases, such as AD, epilepsy, etc. However, the role of EPA1 in PD is unclear. Previous studies have shown that EPA1 is involved in calcium homeostasis, inflammatory response, and oxidative stress; therefore, we speculate that EPA1 may play an important role in PD.
To test this hypothesis, we injected LPS into the SN of mice through a brain stereotaxic instrument, and after successfully establishing a PD model, we studied the expression of EPA1 in PD model mice. The experimental results showed that compared with the control group, the expression level of EPA1 in the SN of mice in the LPS model group was increased, suggesting that the abnormal changes in the expression level of EPA1 may be related to PD. Localized expression, but the co-localization of EPA1 and microglia was not observed in this experiment.
Activation of Calpain 1 is considered to be a key pathogenic factor in neurodegenerative diseases such as AD [19, 20]. It has been reported in the literature that when the steady state of calcium ions is disrupted, it leads to the activation of Calpain 1, which will lead to the occurrence of many diseases including neuroinflammation. Therefore, the neuronal death caused by the activation of Calpain 1 may also be related to the mechanism of PD. The experimental results in this study showed that LPS-induced disruption of calcium homeostasis and activation of Calpain 1.
ROS are produced in large quantities under the influence of oxidative stress and destroy cellular components such as intracellular proteins, nucleic acids, and lipids. Studies have found that ROS is associated with neurodegenerative diseases and aging-related neurosenescence, and the increased production of ROS leads to the activation of the NLRP1 inflammasome and induces neuroinflammation [21–23]. However, in PD, how will ROS affect the occurrence and development of the disease. In this experiment, after modeling with LPS, it was found that the production of ROS in the SN of mice was significantly increased. The results showed that the induction of LPS increased the production of ROS.
The NLRP1 inflammasome plays an important role in the pathogenesis of neurodegenerative diseases. As a member of the NLRP family, the NLRP1 inflammasome consists of NLRP1, caspase-1, and ASC. After the activation of the NLRP1 inflammasome, pro-caspase-1 is cleaved into active caspsase-1, which subsequently leads to the massive production of inflammatory factors such as IL-1β and IL-18, resulting in the occurrence of inflammatory responses [18, 24, 25]. The results of western blot in this experiment showed that compared with the control group, the expression levels of NLRP1, caspase-1, and ASC proteins in the SN of the LPS model group were significantly increased, suggesting that the NLRP1 inflammasome was activated. The levels of inflammatory cytokines IL-1β, IL-18, and TNF-α also increased in the model group. The above results indicate that the NLRP1 inflammasome is activated and triggers an inflammatory response.
However, we silenced EPA1 by injecting AAV-EPA1 shRNA into the SN, and the results showed that AAV adenovirus was successfully transfected and significantly reduced the expression level of EPA1 in the SN region. On this basis, we found that compared with the LPS model group, the LPS+EPA1 shRNA group had significantly higher grid crossing times, maximum grasping force, average moving speed, total moving distance, and standing times. The above results suggest that EPA1 gene silencing can improve LPS-induced behavioral dysfunction in PD model mice. At the same time, we also observed DA neurons in the SN, and the results showed that the damage of TH-positive neurons in the SN of mice in the EPA1 gene silencing group decreased and the number increased.
These results indicate that EPA1 gene silencing has a effect on the occurrence and development of PD. At the same time, when the EPA1 gene was silenced, LPS-induced PD model mice improved calcium homeostasis in the SN and inhibited the activation of calpain 1; significantly reduced ROS generation; effectively attenuated the NLRP1 inflammasome activation and its mediated effects on inflammation.
Conclusion
EPA1 play a role in the pathogenesis of PD, and its mechanism may be related to the inflammatory response mediated by promoting ROS generation and activating the NLRP1 inflammasome. Therefore, EPA1 is expected to become a new therapeutic target.
ACKNOWLEDGMENTS
This work was supported by: Natural Science Fund of Anhui Provincial Department of Education (Grant No. KJ2018A171); The Natural Science Fund of Anhui Province (Grant No.1908085MH270); Research Fund of Anhui Institute of translational medicine (Grant No.2021zhyx-C18); Anhui Medical University Undergraduate Innovation and Entrepreneurship Training Program (Grant No. AYDDCxj2021057); Anhui Medical University Early Exposure to Scientific Research Program (Grant No.2021-ZQKY-200)
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
DATA AVAILABILITY
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
REFERENCES
[1] | Schneider RB , Iourinets J , Richard IH ((2017) ) Parkinson’s disease psychosis: Presentation, diagnosis and management. Neurodegener Dis Manag 7: , 365–376. |
[2] | Huang EW , Liu CZ , Liang SJ , Zhang Z , Lv XF , Liu J , Zhou JG , Tang YB , Guan YY ((2016) ) Endophilin-A2-mediated increase in scavenger receptor expression contributes to macrophage-derived foam cell formation. Atherosclerosis 254: , 133–141. |
[3] | Murdoch JD , Rostosky CM , Gowrisankaran S , Arora AS , Soukup SF , Vidal R , Capece V , Freytag S , Fischer A , Verstreken P , Bonn S , Raimundo N , Milosevic I ((2016) ) Endophilin-A deficiency induces the Foxo3a-Fbxo32 network in the brain and causes dysregulation of autophagy and the ubiquitin-proteasome system. Cell Rep 17: , 1071–1086. |
[4] | Yu X , Xu T , Ou S , Yuan J , Deng J , Li R , Yang J , Liu X , Li Q , Chen Y ((2018) ) Endophilin A1 mediates seizure activity via regulation of AMPARs in a PTZ-kindled epileptic mouse model. Exp Neurol 304: , 41–57. |
[5] | Corponi F , Bonassi S , Vieta E , Albani D , Frustaci A , Ducci G , Landi S , Boccia S , Serretti A , Fabbri C ((2019) ) Genetic basis of psychopathological dimensions shared between schizophrenia and bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry 89: , 23–29. |
[6] | Yin Y , Cha C , Wu F , Li J , Li S , Zhu X , Zhang J , Guo G ((2019) ) Endophilin 1 knockdown prevents synaptic dysfunction induced by oligomeric amyloid beta. Mol Med Rep 19: , 4897–4905. |
[7] | Yu Q , Wang Y , Du F , Yan S , Hu G , Origlia N , Rutigliano G , Sun Q , Yu H , Ainge J , Yan SF , Gunn-Moore F , Yan SS ((2018) ) Overexpression of endophilin A1 exacerbates synaptic alterations in a mouse model of Alzheimer’s disease. Nat Commun 9: , 2968. |
[8] | Keo A , Mahfouz A , Ingrassia A , Meneboo JP , Villenet C , Mutez E , Comptdaer T , Lelieveldt B , Figeac M , Chartier-Harlin MC , van de Berg W , van Hilten JJ , Reinders M ((2020) ) Transcriptomic signatures of brain regional vulnerability to Parkinson’s disease. Commun Biol 3: , 101. |
[9] | Yang Y , Chen J , Chen X , Li D , He J , Wang S , Zhao S , Yang X , Deng S , Tong C , Wang D , Guo Z , Li D , Ma C , Liang X , Shi YS , Liu JJ ((2021) ) Endophilin A1 drives acute structural plasticity of dendritic spines in response to Ca2+/calmodulin. J Cell Biol 220: , e202007172. |
[10] | Yao Y , Xue Y , Ma J , Shang C , Wang P , Liu L , Liu W , Li Z , Qu S , Li Z , Liu Y ((2014) ) MiR-330-mediated regulation of SH3GL2 expression enhances malignant behaviors of glioblastoma stem cells by activating ERK and PI3K/AKT signaling pathways. PLoS One 9: , e95060. |
[11] | Chen H , Xu J , Lv Y , He P , Liu C , Jiao J , Li S , Mao X , Xue X ((2018) ) Proanthocyanidins exert a neuroprotective effect via ROS/JNK signaling in MPTPinduced Parkinson’s disease models in vitro and in vivo. Mol Med Rep 18: , 4913–4921. |
[12] | Shi Z , Zhang K , Zhou H , Jiang L , Xie B , Wang R , Xia W , Yin Y , Gao Z , Cui D , Zhang R , Xu S ((2020) ) Increased miR-34c mediates synaptic deficits by targeting synaptotagmin 1 through ROS-JNK-p53 pathway in Alzheimer’s disease. Aging Cell 19: , e13125. |
[13] | Kennel KB , Greten FR ((2021) ) Immune cell - produced ROS and their impact on tumor growth and metastasis. Redox Biol 42: , 101891. |
[14] | Martinon F , Tschopp J ((2005) ) NLRs join TLRs as innate sensors of pathogens. Trends Immunol 26: , 447–454. |
[15] | Schroder K , Tschopp J ((2010) ) The inflammasomes. Cell 140: , 821–832. |
[16] | Yu CH , Moecking J , Geyer M , Masters SL ((2018) ) Mechanisms of NLRP1-mediated autoinflammatory disease in humans and mice. J Mol Biol 430: , 142–152. |
[17] | Qin YY , Li M , Feng X , Wang J , Cao L , Shen XK , Chen J , Sun M , Sheng R , Han F , Qin ZH ((2017) ) Combined NADPH and the NOX inhibitor apocynin provides greater anti-inflammatory and neuroprotective effects in a mouse model of stroke. Free Radic Biol Med 104: , 333–345. |
[18] | Freeman LC , Ting JP ((2016) ) The pathogenic role of the inflammasome in neurodegenerative diseases. J Neurochem 136 Suppl 1: , 29–38. |
[19] | Fan YG , Guo T , Han XR , Liu JL , Cai YT , Xue H , Huang XS , Li YC , Wang ZY , Guo C ((2019) ) Paricalcitol accelerates BACE1 lysosomal degradation and inhibits calpain-1 dependent neuronal loss in APP/PS1 transgenic mice. EBioMedicine 45: , 393–407. |
[20] | Muruzheva ZM , Traktirov DS , Zubov AS , Pestereva NS , Tikhomirova MS , Karpenko MN ((2021) ) Calpain activity in plasma of patients with essential tremor and Parkinson’s disease: A pilot study. Neurol Res 43: , 314–320. |
[21] | Islam MT ((2017) ) Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol Res 39: , 73–82. |
[22] | Sun D , Gao G , Zhong B , Zhang H , Ding S , Sun Z , Zhang Y , Li W ((2021) ) NLRP1 inflammasome involves in learning and memory impairments and neuronal damages during aging process in mice. Behav Brain Funct 17: , 11. |
[23] | Singh A , Kukreti R , Saso L , Kukreti S ((2019) ) Oxidative stress: A key modulator in neurodegenerative diseases. Molecules 24: , 1583. |
[24] | Yap J , Pickard BS , Chan E and Gan SY ((2019) ) The role of neuronal NLRP1 inflammasome in Alzheimer’s disease: Bringing neurons into the neuroinflammation game. Mol Neurobiol 56: , 7741–7753. |
[25] | Sun L , Chen Y , Shen X , Xu T , Yin Y , Zhang H , Ding S , Zhao Y , Zhang Y , Guan Y and Li W ((2019) ) Inhibition of NOX2-NLRP1 signaling pathway protects against chronic glucocorticoids exposure-induced hippocampal neuronal damage. Int Immunopharmacol 74: , 105721. |


