
B Lymphocytes in Parkinson’s Disease
Abstract
It is well known that B lymphocytes differentiate into plasma cells that produce antibodies. B cells also perform a number of less well-known roles including antigen presentation, regulation of T cells and innate immune cells, cytokine production, and maintenance of subcapsular sinus macrophages. Given that there is clear evidence of inflammation in Parkinson’s disease (PD) both in the central nervous system and in the periphery, it is almost certain that B lymphocytes are involved. This involvement is likely to be complicated given the variety of roles B cells play via a number of distinct subsets. They have received less attention to date than their counterparts, T cells, and monocytes. B lymphocytes are decreased in PD overall with some limited evidence that this may be driven by a decrease in regulatory subsets. There is also evidence that regulatory B cells are protective in PD. There is evidence for a role played by antibodies to alpha-synuclein in PD with a possible increase in early disease. There are many exciting potential future avenues for further exploration of the role of B lymphocytes including improving our understanding of the role of meningeal and calvarial (skull bone marrow) based B cells in health and disease, the use of larger, well phenotyped clinical cohorts to understand changes in peripheral and cerebrospinal fluid B cells over time and the potential application of B cell targeted therapies in PD.
AN INTRODUCTION TO B LYMPHOCYTES
B lymphocytes perform a variety of roles as part of the adaptive immune system with complex interactions with other branches of the innate and adaptive immune systems. Recent evidence suggests that immune cells including B lymphocytes are likely to interact with the central nervous system in complex ways via the meningeal lymphatic [1, 2] system and via more recently described channels in the skull bone marrow that allow egress of calvarial immune cells including B lymphocytes [3, 4]. These discoveries suggest that B lymphocytes are likely to be involved with the inflammation in Parkinson’s disease (PD) in complex ways, both centrally and peripherally, and are potentially a relevant target for disease modifying therapies.
It is now relatively well established that B lymphocytes are reduced in PD [5–8], as this has been replicated across a number of studies (although not in all) but the mechanism behind this observation remains unclear. Scott et al. showed that B cells were also reduced in transgenic alpha-synuclein mouse models of PD suggesting that this may be related to alpha-synuclein pathology [9]. It is also possible that this represents migration of B cells to other compartments (e.g., the cerebrospinal fluid (CSF) or meninges) or alternatively the end result of chronic stimulation.
To get a clearer understanding of these and other observations, it is necessary to first review B cell development and function to appreciate that their role in PD is likely to be complex as their functions are diverse.
B LYMPHOCYTE DEVELOPMENT
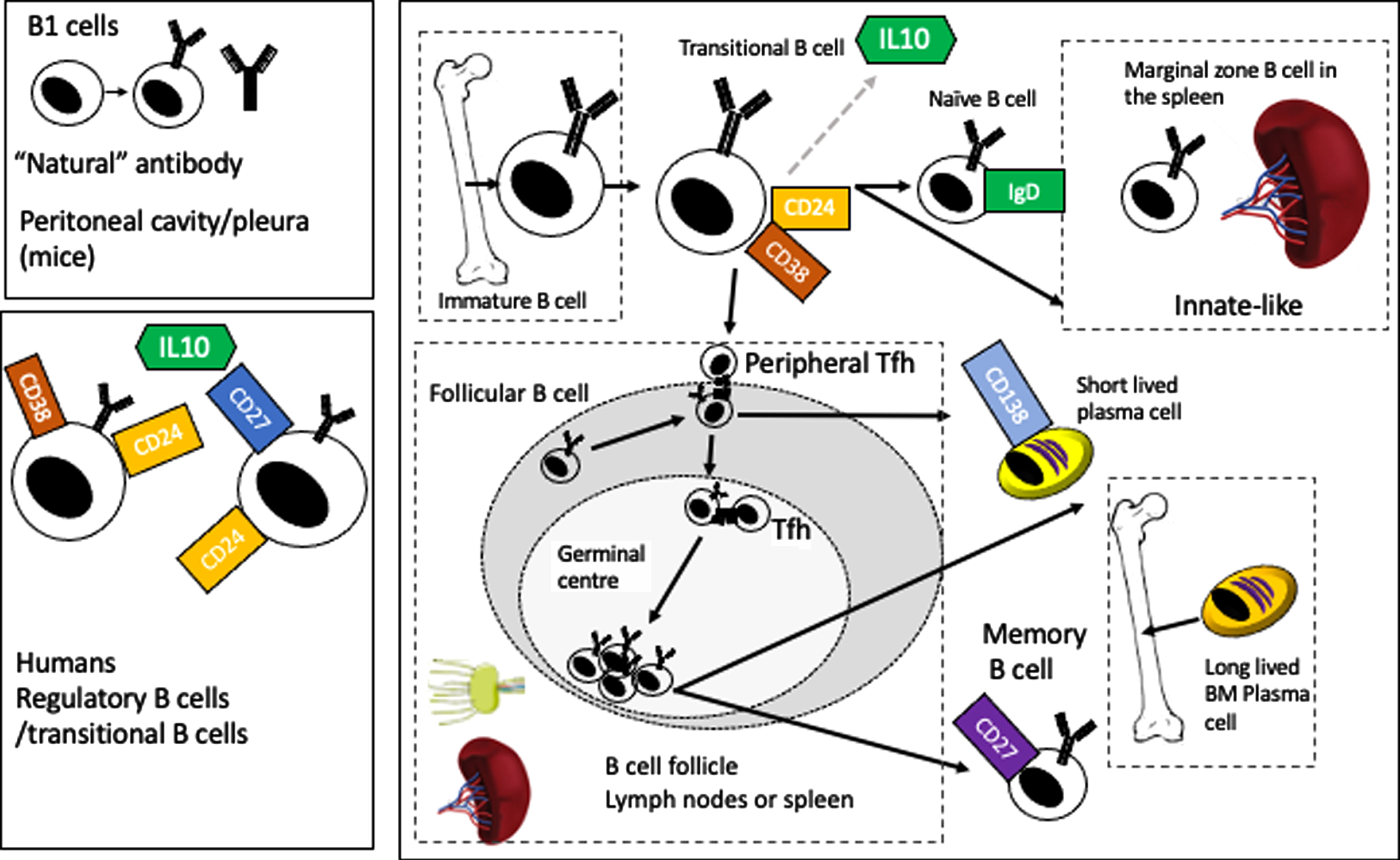
B lymphocytes are derived from a common lymphoid progenitor in the bone marrow. At the pre-B cell stage, the nascent IgM molecule is only found in the cytoplasm, moving to the cell surface in the immature B lymphocyte to form the B cell receptor (BCR). These cells then leave the bone marrow as transitional B lymphocytes to circulate in the blood to secondary lymphoid tissues (the spleen and lymph nodes mainly but also mucosal tissues, Peyer’s patches and possibly the meninges) where they differentiate into naive, follicular (circulating or in lymph nodes) or marginal zone B cells (in the spleen) [10]. Chemokine and cytokine gradients drive subsequent movement of B cells back into circulation (e.g., sphingosine-1-phosphate) or back into lymph nodes or germinal centers (e.g., CXCL13). Figure 1 gives a broad overview of this process, showing the major developmental points, from an immature B cell to a transitional B cell (which is also one of the B cell subsets that produces the regulatory cytokine IL10) (figure and summary modified from Clatworthy et al. [11]). Subsequently, the B cell may become a follicular B cell, where upon encountering antigen and costimulatory signals from follicular T helper cells (Tfh) (including CD40L/CD40, ICOS/ICOSL, and CD28/CD86), it can enter the germinal center reaction where, via a process of clonal selection and somatic hypermutation, more specific high affinity antibodies are generated to the antigen (with subsequent class switching from IgM to IgG, IgA, or IgG). The B cells then either differentiate into long lived plasma cells (homing back to bone marrow) or to memory B cells which retain the capacity to recognize antigen and proliferate in response to re-challenge for decades (at least in humans) [11]. The roles of B lymphocytes in central nervous system (CNS) disease more generally are reviewed elsewhere [12, 13].
Fig. 1
Overview of B cell development and maturation.
B LYMPHOCYTE FUNCTIONS
B lymphocytes are usually defined by their ability to produce antibodies following terminal differentiation to plasma cells. B lymphocytes also secrete cytokines such as IL6 and tumor necrosis factor (TNF) [14] which result in activation of IL-17 producing T helper 17 cells or interferon-γ secreting type 1 T helper cells. B lymphocytes can also regulate T cells and the innate response (via production of largely anti-inflammatory cytokines IL10 and IL35 from so called ‘B10’ or regulatory B cells) [15–18], they can act as antigen presenting cells [19] and regulate subcapsular sinus macrophages in lymph nodes [20]. More recently, meningeal B lymphocytes have been described in the dural lymphatics draining the brain [1] where they are the predominant immune cell [21]. B lymphocytes are also implicated in linking the gut with the brain: in mouse models of disseminated fungal infection B lymphocytes are ‘trained’ in the gut and home to the meninges to protect the brain [22]. Blocking meningeal drainage in mouse models of PD also leads to worsening pathology [2].
The inhibitors, or “regulatory” B cells provide a brake on immune activation. Unlike their well described counterparts, T regulatory cells, they are not defined by a single transcription factor (FoxP3 in the case of Tregs) and work via several different pathways to inhibit either T cell or innate immune activation. There are an increasing number of B cell subsets and markers that have been described as regulatory B cells (which act via several pathways in additional to IL10). In mice, the most commonly used surface markers are CD1d and CD5, but studies have also described IL10 producing, marginal zone B cells, TIM-1+ B cells, plasmablasts, plasma cells (also acting via IL-35) PDL1hi cells (acting via PDL1). In humans, the IL10 producing subset is usually identified by an immature transitional subset (CD27–) CD24hi CD38hi (acting via PDL1 or IL10), or as B10 cells CD24hi, CD27+ (IL10) but also as being CD25hi, CD71hiCD73lo (acting via both IL10 and IgG4) (markers in both humans and mice are reviewed in Mauri and Menon [23]). As one might expect, deficiencies of regulatory B cells in mouse models and human disease are associated with increased severity of autoimmune or inflammatory diseases (e.g., systemic lupus erythematosus and rheumatoid arthritis) while offering some protection from infection [18, 23].
ANTIBODIES IN PARKINSON’S DISEASE
Many studies to date have now looked for disease specific antibodies in PD, mainly focusing on alpha-synuclein. Approximately 30% of neurons in the substantia nigra were bound by IgG in a postmortem study [24]. The Fc (or constant) portion of IgG is capable of binding to Fc receptors (both activating and inhibitory) which are present on microglia, astrocytes, oligodendrocytes, and even neurons in the CNS [25]. Immunoglobulins also initiate the classical complement cascade via C1q [26]. It is therefore mechanistically plausible that antibodies to alpha-synuclein (or other disease relevant proteins) could contribute to CNS inflammation associated with disease progression. A meta-analysis and systematic review by us in 2018 suggested that there was weak evidence for increased serum antibodies in early disease (within the first 6 years) but noted significant heterogeneity across studies including wide variation in disease stage, age, clinical parameters, and the types of antibodies measured [27]. These initial studies were largely aimed at finding disease biomarkers rather than exploring pathophysiology. More recent studies have taken a more sophisticated approach [28] looking at IgG subtypes (1–4) as well as IgM antibodies against alpha-synuclein finding more complex signatures that differed between multiple systems atrophy (MSA) and PD (both alpha-synucleinopathies) and also noting that most of the previous studies did not discriminate between high affinity and low affinity antibodies. The former are likely to be the result of a T cell dependent germinal center reaction following engagement of the BCR with its cognate antigen (in this case alpha-synuclein in a pathological state). Low affinity antibodies are more likely to result from T independent processes (e.g., activation of B cells by molecular patterns via toll-like receptors or via the BCR) and be produced by ‘innate like’ B cells (known as B1 cells which have mainly been described in mice) (possible mechanisms are reviewed in more detail in Scott et al. [27]). The same authors also recently looked at prodromal PD and MSA, finding that there is an increase in anti-alpha-synuclein total IgG (including high and low affinity antibodies) in prodromal PD compared to MSA and healthy controls, driven by a relative increase in IgG1 alpha-synuclein antibodies [29]. At the same time, they describe an overall decrease in the high affinity antibodies in prodromal disease, noting that this occurs at an early prodromal stage (> 4 years) in PD. They make a similar observation in established disease, noting that there is an overall decrease in high affinity antibodies in PD and MSA [28]. They also find similar decreases in all IgG subtypes in the CSF of PD patients [30].
Consistent with their work, we have also found increased alpha-synuclein antibodies in prodromal PD patients at high risk of conversion to PD [9] (but did not find these antibodies in early PD patients). There is therefore some evidence that alpha-synuclein antibodies may play a pathological role. It is, however, unclear where in the disease process this is most relevant and what role is played by different antibody subtypes with varying affinities for different alpha-synuclein epitopes.
It has also been suggested that the antibodies could be protective, playing a role in clearance of pathological proteins (rather than just contributing to inflammation). Currently trials of therapeutic antibodies are underway with the hypothesized mechanism being improved clearance of pathological extracellular alpha-synuclein (reviewed in Castonguay et al. [31]). One possible mechanism for this is via neuronal uptake of unbound antibody via FcγRI which is present on the neuronal cell surface. This could then stimulate degradation within the cell. Alternatively, alpha-synuclein fragments in complex with immunoglobulin could be internalized by microglia via FcγRIII [31]. In line with this idea, Li et al. showed recently that some alpha-synuclein antibodies derived from patients were able to inhibit seeding of alpha-synuclein in vitro (and that this was epitope dependent). These same mechanisms are likely to be at play with naturally produced antibodies although the effects may be more complex depending on the concentration and subtype of the antibody (which affects which downstream Fc receptors are engaged, for example, as some Fc receptors are high affinity whilst others are low affinity and they can be either inhibitory or excitatory [32]). It is interesting to note that at least one candidate for therapeutic vaccination (ABBV-0805) uses the IgG4 molecule to avoid the more extensive complement binding seen with IgG1 as well as weaker engagement with Fcγ receptors [33].
The results of phase II antibody trials are therefore eagerly awaited. If early trials fail to show efficacy this may be due to a number of factors including the choice of IgG subtype, the affinity of the antibodies for alpha-synuclein, the epitopes that they bind and the resulting activation of other, possibly off target effects, such as complement activation. Patient factors such as disease stage are also likely to be relevant.
The role of antibodies to alpha-synuclein and the downstream pathways involved is therefore in need of further more sophisticated investigation, looking to understand disease pathogenesis rather than an overly simplistic search for biomarkers.
ANTIBODY INDEPENDENT FUNCTIONS OF B LYMPHOCYTES
It is only very recently that studies have looked at B cell subsets in PD. Li et al. found that B lymphocyte proliferation was reduced in patients compared to controls and that proportions of regulatory B lymphocytes (transitional B lymphocytes) were also reduced while proportions of B lymphocytes producing pro-inflammatory cytokines were increased [8]. They also found that T follicular helper cells were reduced in patients. This study was done across two cohorts with most of their findings replicated in both cohorts. They conclude that there are aberrant T follicular:B lymphocyte interactions with a shift in the B cell compartment to a more pro-inflammatory phenotype. Similarly, Yan et al. showed a decrease in circulating naïve B cells in PD patients compared to controls with an associated increase in ‘non-switched memory’ (CD27+IgD+) and ‘double negative’ (CD27–IgD–) B cells [34]. These changes were associated with an increase in pro-inflammatory T-cells (including IFNγ producing CD8 T cells, IL17 producing Th 7 CD4+ cells and IL4 producing Th2 T cells). The CD27+IgD+ ‘non-switched’ memory cells are thought to be part of an early inflammatory response (being likely to home to lymph nodes to take part in a germinal center reaction) [35]. Double negative B cells have been associated with aging and senescence and are thought to produce increased levels of pro-inflammatory cytokines [36]. They also found that the B cells produced more TNF-α in vitro [34]. TNF-α has recently been established as a relevant CSF biomarker predicting PD in a cohort of LRRK2 carriers (comparing symptomatic versus non-symptomatic) along with decreased levels of total alpha-synuclein and increased levels of oligomeric alpha-synuclein [37]. Further work to establish the source of TNF-α in the CSF is required but it could be hypothesized that this is derived at least partially from B lymphocytes. Lastly they showed that there was also a decrease in circulating CXCR3+ memory B cells: these cells respond to chemotaxic signals from CXCR3 to migrate to secondary lymphoid organs and this may include the meninges.
There is one published study looking at peripheral B cell phenotypes in PD by using single cell sequencing rather than flow cytometry (although only with 8 patients and 6 controls with minimal clinical information) [38]. They found decreased naive and increased memory B cells and increased IgG and IgA B cells as well as clonal expansion of memory B cells with upregulation of MHC class II and transcription activator protein-1, both of which are involved in antigen presentation. This again suggests that B cells are playing a role in activating T cells (by presenting antigen, likely alpha-synuclein fragments).
We have also described decreased B lymphocytes, particularly in patients at high risk of an early dementia [9], and we showed a protective effect of transitional B cells within a patient cohort: higher proportions of these regulatory cells were associated with better motor outcomes. Like previous studies above, we also found that B lymphocytes from PD patients produced more cytokine in vitro than controls. Finally, in a 6-OHDA mouse model of dopaminergic cell death we showed that knocking out B cells (either genetically or with a monoclonal antibody) lead to worse outcomes. Our study added to the existing literature by showing a protective effect of B lymphocytes, in particular the transitional or regulatory B lymphocytes, within a patient cohort (rather than just showing a difference between patients and control). This finding is consistent with other studies showing a protective effect of T regulatory cells in PD [39, 40].
These recent manuscripts are relatively consistent in their description of a more pro-inflammatory state in the B cell compartment in PD but remain limited by a lack of longitudinal data confirming their role in disease progression. There is also a focus on peripheral blood lymphocytes which give little information about the phenotypes or behavior of B lymphocytes in other relevant compartments such as the CSF, meninges, or lymphoid tissue, and no studies to date have attempted to correlate imaging based measures of neuroinflammation with peripheral blood phenotyping.
FUTURE PERSPECTIVES
There is a clear need now for larger longitudinal studies in clinically well-phenotyped cohorts using data driven approaches (e.g., single cell RNA sequencing) to identify whether changes in the B lymphocyte compartment are associated with neuroinflammation (using imaging biomarkers) and shifts in other immune cell populations. Adequately powered studies addressing this question should be able to identify relevant pathways for intervention.
We also need a more sophisticated understanding of the role of alpha-synuclein antibodies in PD, again using longitudinal cohorts to identify changes over time. This also requires robust assays looking not only at IgG subtypes but also at IgA and IgM antibodies in both serum and CSF and potentially looking at downstream targets such as Fc receptors on peripheral and CSF immune cells to understand the consequences of changes in antibodies titers across CSF blood compartments. Differentiating between high and low affinity antibodies may also help to explain some of the differences in the literature as these are likely to arise due to separate processes (germinal center reactions in the case of high affinity antibodies and via T independent mechanisms such as engagement of toll like receptors by alpha-synuclein in the case of low affinity receptors). It would be helpful to establish whether the presence of these antibodies is associated with markers of neuroinflammation or disease progression or whether they are playing a role in clearance of pathological proteins and at what stage in disease they are most relevant. The field needs to more away from a very simplistic biomarker-based approach (looking for a simple diagnostic biomarker in cross-sectional studies) to attempting to understand the mechanisms of disease.
We are only at the beginning of understanding the role of the relatively newly described B cell niches in the meninges and the skull bone marrow [13]. Most of the work to date in this area has been carried out using mouse models. Additional animal work is required to demonstrate the role of B cells in these niches in models of PD. The biggest future challenge is to work out how to demonstrate the relevance of these findings in clinical populations by combining neuroimaging and clinico-pathological correlations using postmortem tissue from patients who have been phenotyped in life. Animal models would then be required to find out whether we can change disease outcomes by altering the behavior of B cells (and other immune cells) in these niches in order to identify approaches to take forward into clinical trials. There are a large number of existing drugs that target B lymphocytes with subsequent downstream effects on other immune cell types and there is therefore the potential to take these into trials once appropriate targets have been clearly demonstrated.
Lastly, despite the title of this review, it does not makes sense to study B lymphocytes on their own. They need to be studied in the complex context of the immune system where both effectors and inhibitors interact in a fine balance across different anatomical niches and have differing effects at different disease stages.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Louveau A , Herz J , Alme MN , Salvador AF , Dong MQ , Viar KE , Herod SG , Knopp J , Setliff JC , Lupi AL , Da Mesquita S , Frost EL , Gaultier A , Harris TH , Cao R , Hu S , Lukens JR , Smirnov I , Overall CC , Oliver G , Kipnis J ((2018) ) CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat Neurosci 21: , 1380–1391. |
[2] | Zou W , Pu T , Feng W , Lu M , Zheng Y , Du R , Xiao M , Hu G ((2019) ) Blocking meningeal lymphatic drainage aggravates Parkinson’s disease-like pathology in mice overexpressing mutated α-synuclein. Transl Neurodegener 8: , 7. |
[3] | Herisson F , Frodermann V , Courties G , Rohde D , Sun Y , Vandoorne K , Wojtkiewicz GR , Masson GS , Vinegoni C , Kim J , Kim DE , Weissleder R , Swirski FK , Moskowitz MA , Nahrendorf M ((2018) ) Direct vascular channels connect skull bone marrow and the brain surface enabling myeloid cell migration. Nat Neurosci 21: , 1209–1217. |
[4] | Kolabas ZI , Kuemmerle LB , Perneczky R , Förstera B , Büttner M , Caliskan OS , Ali M , Rong Z , Mai H , Hummel S , Bartos LM , Biechele G , Zatcepin A , Albert NL , Unterrainer M , Gnörich J , Zhao S , Khalin I , Rauchmann B-S , Molbay M , Sterr M , Kunze I , STanic K , Besson-Gerard S , Kopczak A , Katzdobler S , Palleis C , Gokce O , Lickert H , Steinke H , Bechmann I , Buerger K , Levin J , Haass C , Dichgans M , Havla J , Kümpfel T , Kerschensteiner M , Simons M , Plesnila N , Krahmer N , Singh Bhatia H , Erener S , Hellal F , Brendel M , Theis FJ , Erturk A (2021) Multi-omics and 3D-imaging reveal bone heterogeneity and unique calvaria cells in neuroinflammation. bioRxiv. doi: 10.1101/2021.12.24.473988. |
[5] | Gruden MA , Sewell RD , Yanamandra K , Davidova TV , Kucheryanu VG , Bocharov EV , Bocharova OA , Polyschuk VV , Sherstnev VV , Morozova-Roche LA ((2011) ) Immunoprotection against toxic biomarkers is retained during Parkinson’s disease progression. J Neuroimmunol 233: , 221–227. |
[6] | Kedmi M , Bar-Shira A , Gurevich T , Giladi N , Orr-Urtreger A ((2011) ) Decreased expression of B cell related genes in leukocytes of women with Parkinson’s disease. Mol Neurodegener 6: , 66. |
[7] | Stevens CH , Rowe D , Morel-Kopp MC , Orr C , Russell T , Ranola M , Ward C , Halliday GM ((2012) ) Reduced T helper and B lymphocytes in Parkinson’s disease. J Neuroimmunol 252: , 95–99. |
[8] | Li R , Tropea TF , Baratta LR , Zuroff L , Diaz-Ortiz ME , Zhang B , Shinoda K , Rezk A , Alcalay RN , Chen-Plotkin A , Bar-Or A ((2021) ) Abnormal B-cell and Tfh-cell profiles in patients with Parkinson disease: A cross-sectional study. Neurol Neuroimmunol Neuroinflamm 9: , e1125. |
[9] | Scott KM , Chong YT , Park S , Wijeyekoon R , Hayat S (2022) B lymphocyte responses in Parkinson–s disease and their possible significance in disease progression. Brain Commun, in press. |
[10] | Pieper K , Grimbacher B , Eibel H ((2013) ) B-cell biology and development. J Allergy Clin Immunol 131: , 959–971. |
[11] | Clatworthy MR ((2014) ) B-cell regulation and its application to transplantation. Transpl Int 27: , 117–128. |
[12] | Sabatino JJ , Pröbstel AK , Zamvil SS ((2019) ) B cells in autoimmune and neurodegenerative central nervous system diseases. Nat Rev Neurosci 20: , 728–745. |
[13] | Posner DA , Lee CY , Portet A , Clatworthy MR ((2022) ) Humoral immunity at the brain borders in homeostasis. Curr Opin Immunol 76: , 102188. |
[14] | Barr TA , Shen P , Brown S , Lampropoulou V , Roch T , Lawrie S , Fan B , O’Connor RA , Anderton SM , Bar-Or A , Fillatreau S , Gray D ((2012) ) B cell depletion therapy ameliorates autoimmune disease through ablation of IL-6-producing B cells. J Exp Med 209: , 1001–1010. |
[15] | Blair PA , Noreña LY , Flores-Borja F , Rawlings DJ , Isenberg DA , Ehrenstein MR , Mauri C ((2010) ) CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic Lupus Erythematosus patients. Immunity 32: , 129–140. |
[16] | DiLillo DJ , Matsushita T , Tedder TF ((2010) ) B10 cells and regulatory B cells balance immune responses during inflammation, autoimmunity, and cancer. Ann N Y Acad Sci 1183: , 38–57. |
[17] | Matsushita T , Horikawa M , Iwata Y , Tedder TF ((2010) ) Regulatory B cells (B10 cells) and regulatory T cells have independent roles in controlling experimental autoimmune encephalomyelitis initiation and late-phase immunopathogenesis. J Immunol 185: , 2240–2252. |
[18] | Mauri C , Bosma A ((2012) ) Immune regulatory function of B cells. Ann Rev Immunol 30: , 221–241. |
[19] | Yuseff M-I , Pierobon P , Reversat A , Lennon-Duménil A-M ((2013) ) How B cells capture, process and present antigens: A crucial role for cell polarity. Nat Rev Immunol 13: , 475–486. |
[20] | Moseman EA , Iannacone M , Bosurgi L , Tonti E , Chevrier N , Tumanov A , Fu YX , Hacohen N , von Andrian UH ((2012) ) B cell maintenance of subcapsular sinus macrophages protects against a fatal viral infection independent of adaptive immunity. Immunity 36: , 415–426. |
[21] | Korin B , Ben-Shaanan TL , Schiller M , Dubovik T , Azulay-Debby H , Boshnak NT , Koren T , Rolls A ((2017) ) High-dimensional, single-cell characterization of the brain’s immune compartment. Nat Neurosci 20: , 1300–1309. |
[22] | Fitzpatrick Z , Frazer G , Ferro A , Clare S , Bouladoux N , Ferdinand J , Tuong ZK , Negro-Demontel ML , Kumar N , Suchanek O , Tajsic T , Harcourt K , Scott K , Bashford-Rogers R , Helmy A , Reich DS , Belkaid Y , Lawley TD , McGavern DB , Clatworthy MR ((2020) ) Gut-educated IgA plasma cells defend the meningeal venous sinuses. Nature 587: , 472–476. |
[23] | Mauri C , Menon M ((2015) ) The expanding family of regulatory B cells. Int Immunol 27: , 479–486. |
[24] | Orr CF , Rowe DB , Mizuno Y , Mori H , Halliday GM ((2005) ) A possible role for humoral immunity in the pathogenesis of Parkinson’s disease. Brain 128: (Pt 11), 2665–2674. |
[25] | Okun E , Mattson MP , Arumugam TV ((2010) ) Involvement of Fc receptors in disorders of the central nervous system. Neuromolecular Med 12: , 164–178. |
[26] | Dunkelberger JR , Song W-C ((2010) ) Complement and its role in innate and adaptive immune responses. Cell Res 20: , 34–50. |
[27] | Scott KM , Kouli A , Yeoh SL , Clatworthy MR , Williams-Gray CH ((2018) ) A systematic review and meta-analysis of alpha synuclein auto-antibodies in Parkinson’s disease. Front Neurol 9: , 815. |
[28] | Folke J , Rydbirk R , Løkkegaard A , Salvesen L , Hejl AM , Starhof C , Bech S , Winge K , Christensen S , Pedersen LØ , Aznar S , Pakkenberg B , Brudek T ((2019) ) Distinct autoimmune anti-α-synuclein antibody patterns in multiple system atrophy and Parkinson’s disease. Front Immunol 10: , 2253. |
[29] | Folke J , Bergholt E , Pakkenberg B , Aznar S , Brudek T ((2022) ) Alpha-synuclein autoimmune decline in prodromal multiple system atrophy and Parkinson’s disease. Int J Mol Sci 23: , 6554. |
[30] | Folke J , Rydbirk R , Løkkegaard A , Hejl AM , Winge K , Starhof C , Salvesen L , Pedersen LØ , Aznar S , Pakkenberg B , Brudek T ((2021) ) Cerebrospinal fluid and plasma distribution of anti-α-synuclein IgMs and IgGs in multiple system atrophy and Parkinson’s disease. Parkinsonism Relat Disord 87: , 98–104. |
[31] | Castonguay A-M , Gravel C , Lévesque M ((2021) ) Treating Parkinson’s disease with antibodies: Previous studies and future directions. J Parkinsons Dis 11: , 71–92. |
[32] | Bruhns P , Iannascoli B , England P , Mancardi DA , Fernandez N , Jorieux S , Daëron M ((2009) ) Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood 113: , 3716–3725. |
[33] | Nordström E , Eriksson F , Sigvardson J , Johannesson M , Kasrayan A , Jones-Kostalla M , Appelkvist P , Söderberg L , Nygren P , Blom M , Rachalski A , Nordenankar K , Zachrisson O , Amandius E , Osswald G , Moge M , Ingelsson M , Bergström J , Lannfelt L , Möller C , Giorgetti M , Fälting J ((2021) ) ABBV-0805, a novel antibody selective for soluble aggregated α-synuclein, prolongs lifespan and prevents buildup of α-synuclein pathology in mouse models of Parkinson’s disease. Neurobiol Dis 161: , 105543. |
[34] | Yan Z , Yang W , Wei H , Dean MN , Standaert DG , Cutter GR , Benveniste EN , Qin H ((2021) ) Dysregulation of the adaptive immune system in patients with early-stage Parkinson disease. Neurol Neuroimmunol Neuroinflamm 8: , e1036. |
[35] | Seifert M , Przekopowitz M , Taudien S , Lollies A , Ronge V , Drees B , Lindemann M , Hillen U , Engler H , Singer BB , Küppers R ((2015) ) Functional capacities of human IgM memory B cells in early inflammatory responses and secondary germinal center reactions. , E546-E. Proc Natl Acad Sci U S A 112: , 555 |
[36] | Frasca D ((2018) ) Senescent B cells in aging and age-related diseases: Their role in the regulation of antibody responses. Exp Gerontol 107: , 55–58. |
[37] | Majbour NK , Aasly JO , Hustad E , Thomas MA , Vaikath NN , Elkum N , van de Berg WDJ , Tokuda T , Mollenhauer B , Berendse HW , El-Agnaf OMA ((2020) ) CSF total and oligomeric α-Synuclein along with TNF-α as risk biomarkers for Parkinson’s disease: A study in LRRK2 mutation carriers. Transl Neurodegener 9: , 15. |
[38] | Wang P , Luo M , Zhou W , Jin X , Xu Z , Yan S , Li Y , Xu C , Cheng R , Huang Y , Lin X , Yao L , Nie H , Jiang Q ((2022) ) Global characterization of peripheral B cells in Parkinson’s disease by single-cell RNA and BCR sequencing. Front Immunol 13: , 814239. |
[39] | He F , Balling R ((2013) ) The role of regulatory T cells in neurodegenerative diseases. Wiley Interdiscip Rev Syst Biol Med 5: , 153–180. |
[40] | Saunders JA , Estes KA , Kosloski LM , Allen HE , Dempsey KM , Torres-Russotto DR , Meza JL , Santamaria PM , Bertoni JM , Murman DL , Ali HH , Standaert DG , Mosley RL , Gendelman HE ((2012) ) CD4+ regulatory and effector/memory T cell subsets profile motor dysfunction in Parkinson’s disease. J Neuroimmune Pharmacol 7: , 927–938. |


