
Unraveling Parkinson’s Disease Neurodegeneration: Does Aging Hold the Clues?
Abstract
Aging is the greatest risk factor for Parkinson’s disease (PD), suggesting that mechanisms driving the aging process promote PD neurodegeneration. Several lines of evidence support a role for aging in PD. First, hallmarks of brain aging such as mitochondrial dysfunction and oxidative stress, loss of protein homeostasis, and neuroinflammation are centrally implicated in PD development. Second, mutations that cause monogenic PD are present from conception, yet typically only cause disease following a period of aging. Third, lifespan-extending genetic, dietary, or pharmacological interventions frequently attenuate PD-related neurodegeneration. These observations support a central role for aging in disease development and suggest that new discoveries in the biology of aging could be leveraged to elucidate novel mechanisms of PD pathophysiology. A recent rapid growth in our understanding of conserved molecular pathways that govern model organism lifespan and healthspan has highlighted a key role for metabolism and nutrient sensing pathways. Uncovering how metabolic pathways involving NAD+ consumption, insulin, and mTOR signaling link to the development of PD is underway and implicates metabolism in disease etiology. Here, we assess areas of convergence between nervous system aging and PD, evaluate the link between metabolism, aging, and PD and address the potential of metabolic interventions to slow or halt the onset of PD-related neurodegeneration drawing on evidence from cellular and animal models.
AGING IS THE GREATEST RISK FACTOR FOR PARKINSON’S DISEASE
Development and progression of Parkinsonism, the movement disorder of Parkinson’s disease (PD), is driven primarily by the degeneration of dopaminergic neurons within the substantia nigra (SN) [1, 2]. Established contributors to this progressive neurodegeneration include mitochondrial dysfunction [3], oxidative stress [4], inflammation [5], and the misfolding and aggregation of α-synuclein into protein deposits such as Lewy bodies [6].
It is now clear that aging is the greatest risk factor for developing PD [7–9]. While this connection had long been presumed, detailed studies characterizing the aging-PD link emerged in the 1980s and 1990s. One of the most influential studies from that time was the Rotterdam Study, a door-to-door population-based survey of nearly 7,000 persons of 55 years of age or older living in a Netherlands suburb [10]. The authors reported quantitative evidence of the connection between aging and PD, with prevalence in males and females combined increasing from 0.3% in those aged 55–64 to 1.0% in those aged 65–74, 3.1% in those aged 75–84, and 4.3% in those aged 85–94. Census studies like the Rotterdam study were conducted in several other countries as well, including Italy [11], Greece [12], Spain [13], France [14], China [15], Taiwan [16], and USA [17], all of which reported similar age-associated increases in PD prevalence. These studies consistently confirmed the close association of aging with PD25;) development.
Besides aging, PD has been associated with genetic risk factors including highly penetrant familial PD gene mutations and common risk variants, as well as environmental risk factors such as pesticide exposure [18]. Epidemiological evidence indicates that exposure to the pesticides paraquat or rotenone increases PD risk 2-3-fold due to the neurotoxic action of these hazardous materials [19]. However, the reported increase in PD risk associated with known environmental factors pales in comparison to that of aging. Recent analyses of older populations have observed that PD incidence increases approximately 100-fold when comparing people aged 45–49 to those aged 85–89 [20].
The strong relationship between aging and PD has several major implications for the future outlook of disease burden and on approaching the study of PD development and treatment. First, population analyses conducted by the World Health Organization (WHO) have estimated that the number of Americans aged 65 and older will approximately double by 2050, with the world population of those aged 85 and older projected to triple in the same timeframe [21]. This trend will inevitably lead to a rapidly increasing incidence of PD and other aging-related pathologies over the upcoming decades, resulting in greater public health burdens. Second, new advances made in understanding the drivers of CNS aging may offer an important roadmap to delineating mechanisms of PD development and vice versa. These fields may inform each other and synergy may be derived from an approach encompassing both aspects. This concept forms the foundation of geroscience, which strives to understand the mechanisms that make aging a key driver for diseases such as PD. Third, there is rationale for incorporating aging into preclinical animal testing of PD therapeutics. Preclinical studies of candidate disease-modifying PD therapies continue to rely on non-aged acute animal disease models for practical reasons related to the cost and duration involved in such studies. While there are numerous challenges in translating therapies to the clinic, the profound physiologic changes associated with aging may negate the efficacy of disease-modifying therapies that work well in young animals but fail in aged PD patients.
Here, to examine the aging-PD link further, we will first assess points of convergence in the hallmarks of CNS aging and PD that constitute abundant correlative evidence linking the two. We will then discuss how metabolism has emerged as a potential nexus connecting aging to PD neurodegeneration, and evaluate evidence that altered expression of lifespan-regulating metabolic genes influences PD-related phenotype development, potentially demonstrating a causal link between aging and PD.
AGING AND PD: POINTS OF CONVERGENCE
The process of aging is highly complex and spans many integral biological pathways and systems within organisms. While the details differ across species, the underlying phenomenon is always the same: a progressive accumulation of molecular and cellular dysfunction that ultimately results in the breakdown of tissues necessary to sustain life. Several major molecular hallmarks of brain aging overlap with mechanisms implicated in PD neurodegeneration, including oxidative damage and mitochondrial dysfunction, loss of protein homeostasis, neuroinflammation, genomic instability, and impaired stress responses. Accumulating age-related dysfunction in these areas likely renders neurons vulnerable to PD-associated environmental and genetic factors that affect the same processes, thus compounding dysfunction and promoting α-synuclein pathology. Additionally, dopaminergic neurons may be more vulnerable to age-related loss of mitochondrial function and resulting bioenergetic stress due to their highly ramified processes that harbor dense mitochondria to sustain energy-requiring processes at distal sites [22]. Hence, it is conceivable that some effects of aging may be more deleterious in dopamine neurons and may contribute to regional vulnerability in PD. The convergence of aging hallmarks with PD is consistent with aging being the predominant risk factor for disease. To an extent, PD-related neurodegeneration resembles an exacerbated form of aging in that disease-associated mitochondrial dysfunction, loss of protein homeostasis, and neuroinflammation occur in a more pronounced manner within vulnerable regions such as the SNpc than generally manifests across the brain as a result of aging. It is important to note, however, that the degree to which these functional deficits are actually causal in the physiological declines of aging and neurodegeneration of PD has not yet been resolved. An alternative hypothesis is conceivable, in which PD neurodegeneration occurs independently of aging but on a matching time-scale. For example, if the seeding and spread of toxic α-synuclein aggregates across the brain are slow and take decades to reach the point where they trigger the manifestation of disease, they could be misattributed to aging. However, it seems unlikely that these phenomena would not be promoted by the breakdown in protein folding, trafficking and turnover that occurs with age, and in this context it seems more plausible that they would be influenced by aging if not governed by it.
Compelling evidence from cell and animal disease models supports convergent roles for 1) mitochondrial dysfunction and oxidative stress; 2) loss of protein homeostasis and protein aggregate formation; and 3) chronic inflammation in aging and PD (Fig. 1). Similarities between aging and PD for each of these areas are discussed below.
Fig. 1
Convergent effects of aging and Parkinson’s disease development on key biological functions. Major overlapping effects of aging and PD on mitochondrial homeostasis, protein homeostasis and inflammation are described. See text for additional details. ROS, reactive oxygen species.
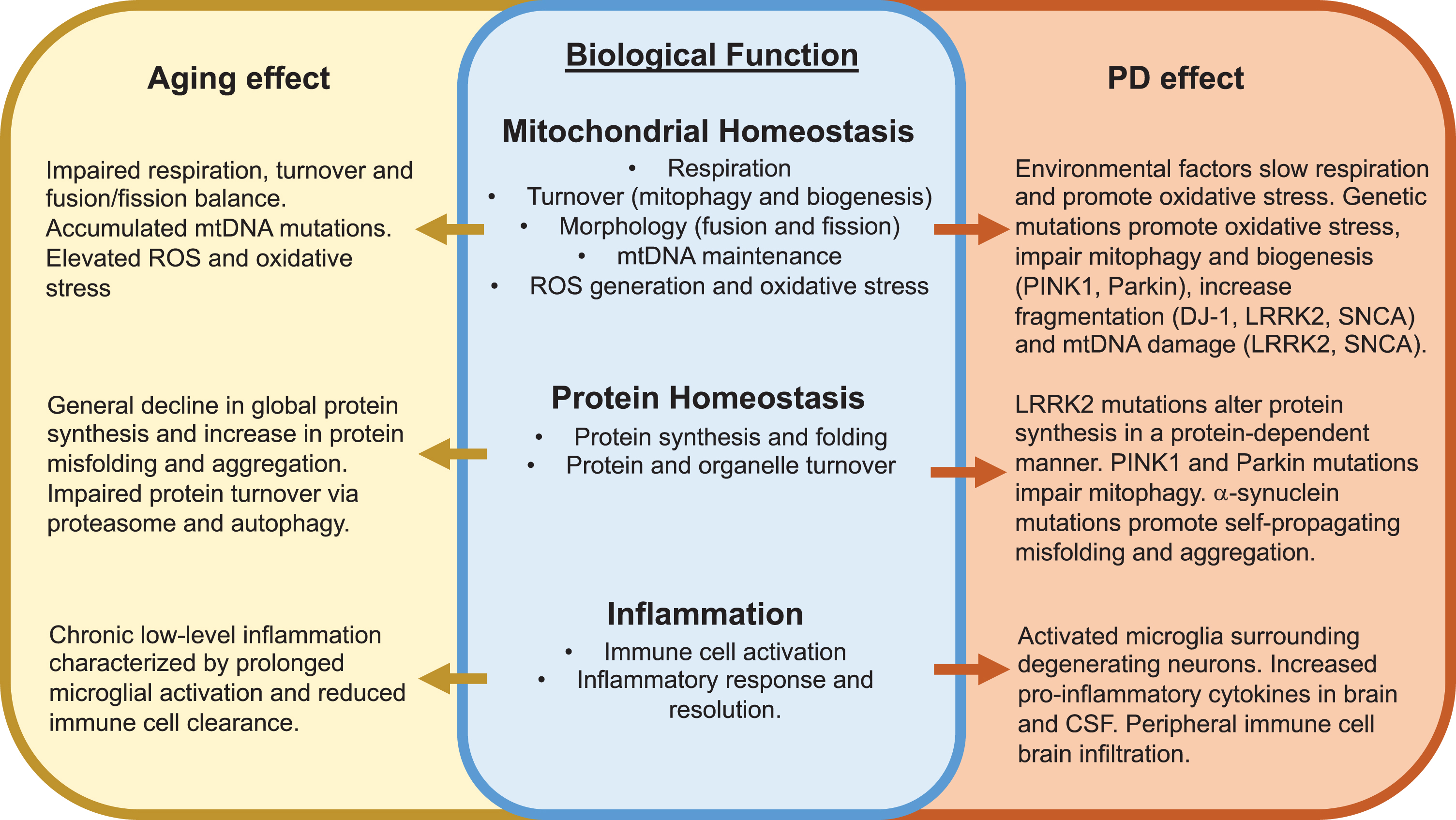
Table 1
Effect of familial PD gene mutations on mitochondrial function. The mitochondrial role and effect of PD-associated loss or gain of function mutations on mitochondrial morphology and function are described
| Gene | Inheritance Pattern and Disease Onset | Protein Function and Mitochondrial Role | Mitochondrial effect of PD Mutation | References |
| Parkin | Autosomal recessive, early onset | E3 ubiquitin ligase, interacts with PINK1, involved in mitophagy and mitochondrial biogenesis | Deficient mitophagy and mitochondrial biogenesis | [34–36, 38, 39, 160, 162, 181, 183] |
| PINK1 | Autosomal recessive, early onset | Mitochondria-targeted kinase, interacts with Parkin, involved in mitophagy and mitochondrial biogenesis | Deficient mitophagy and mitochondrial biogenesis | [37, 39, 160, 161, 182] |
| DJ-1 | Autosomal recessive, early onset | Redox-regulated chaperone, DJ-1 oxidation promotes mitochondrial interaction and protection | Mitochondrial fragmentation, increased vulnerability to oxidative stress | [51–60, 71] |
| SNCA | Autosomal dominant, early onset | Role in neurotransmission, regulates mitochondrial morphology | Mitochondrial fragmentation, mitochondrial DNA damage | [45–50] |
| LRRK2 | Autosomal dominant, late onset | Kinase, protective mitochondrial role via unclear mechanism | Mitochondrial fragmentation, mitochondrial DNA damage | [37, 40–44] |
Mitochondrial dysfunction and oxidative stress
Mitochondrial oxidative phosphorylation relies on the electron transport chain (ETC) utilizing oxygen molecules for their electron accepting potential. This use of oxygen creates the opportunity for the formation of superoxide (O2-) radicals that can give rise to other reactive oxygen species (ROS), such as hydrogen peroxide (H2O2) and reactive nitrogen species (RNS) such as peroxynitrite (ONOO-). While recent research has supported that these ROS or RNS molecules may have essential signaling functions within the cell [23–25], their high biochemical reactivities can also cause disruption through oxidative or nitrosative stress, respectively [26]. Mitochondria harboring dysfunctional ETC complex component(s) have increased potential for generating O2- (produced predominantly by complexes I and III) which can elevate overall oxidative stress in the cell.
The accumulation of oxidative stress has been implicated in the normal processes of aging. In fact, the oxidative stress theory of aging was one of the most popular until recent evidence has begun to cast doubt on its validity [26–28]. Originally conceived as the mitochondrial free radical theory of aging, the oxidative stress theory of aging posits that the growing presence of reactive species within the cell derived from both endogenous (e.g., mitochondrial) and exogenous sources causes structural damage to macromolecules including lipids, proteins, and nucleic acids. Since these ROS/RNS could target the mitochondria themselves, a putative feed-forward cycle of mitochondrial dysfunction and ROS/RNS generation has been proposed to account for the progressive nature of cellular aging. The widely-held hypothesis that oxidative damage alone is sufficient to account for functional losses associated with aging has recently been challenged [29]. An alternative hypothesis posits that an age-related shift toward a pro-oxidizing cellular redox state via mitochondrial ROS production leads to a disruption of redox-regulated signaling pathways that in turn promotes cell senescence and death [29].
Mitochondrial dysfunction has been implicated in PD for over thirty years, initially arising from studies examining the effects of exposure to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) [30, 31] and certain pesticides such as rotenone and paraquat that selectively inhibit mitochondrial ETC complex I [32, 33]. This evidence was supported by the identification of reduced complex I levels in PD patient dopamine neurons [34] and more recently reinforced by studies on familial PD-linked genes and from the PD-like phenotypes arising from genetic deletion of a catalytic ETC complex I subunit [35], thus establishing mitochondrial dysfunction and bioenergetic failure as a primary candidate mechanism for PD development.
Familial PD genes and mitochondrial dysfunction
Investigations into highly penetrant mutations in genes including leucine-rich repeat kinase 2 (LRRK2), Parkin, PTEN-induced kinase 1 (PINK1), synuclein alpha (SNCA), and DJ-1 have revealed a robust association of neurodegeneration with markers of mitochondrial damage, dysfunction, and oxidative stress in genetic disease models, marking a clear parallel with mitochondrial hallmarks of aging (Table 1). Parkin encodes an E3-ubiquitin ligase that can monoubiquitinate and polyubiquitinate a number of cellular substrates and PD-associated Parkin mutations are generally thought to result in a loss of function via one of several mechanisms [36–38]. PINK1 is a mitochondria-targeted kinase whose functional silencing has also been linked to PD. Recent work has shed light on a coordinated role for PINK1 and Parkin in regulating mitochondrial quality control via mitophagy and in mitochondrial biogenesis via transcriptional control of Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) [39]. Quality control deficits occurring through loss of Parkin or PINK1 function enables dysfunctional mitochondrial to remain within the overall mitochondrial pool thus promoting ROS generation and bioenergetic deficits [40, 41] reminiscent of aging cells.
Mutations in the leucine-rich repeat kinase 2 (LRRK2) gene cause late-onset PD (>60 years old) consistent with neurodegeneration occurring following a major intersection with aging. The common LRRK2 G2019S mutation increases LRRK2 kinase activity which appears to be a key route to LRRK2-induced neurotoxicity. Cells expressing pathogenic LRRK2 mutations exhibit mitophagy deficits through unclear mechanisms [39, 42, 43], hence impaired clearance of damaged mitochondria may be a common feature of multiple familial PD genetic mutations. Pharmacologically inhibiting LRRK2 kinase activity restores mitophagy, thus linking aberrant LRRK2 kinase activity to altered mitochondrial homeostasis [39, 43]. iPSC-derived neural cells from PD patients carrying LRRK2 G2019S mutations or at-risk individuals harboring the R1441C mutation display elevated levels of mtDNA damage that can be blocked by correcting the gene mutation [44]. LRRK2 G2019S was also reported to hyperphosphorylate the essential H2O2 scavenger peroxiredoxin 3 (PRDX3) in Drosophila brains, reducing overall peroxide activity and resulting in skeletal muscle mitochondrial degeneration and dopaminergic neurodegeneration that could be reversed by PRDX3 overexpression or treatment with the peroxidase mimic ebselen [45]. Lastly, LRRK2 G2019S overexpression can induce mitochondrial fragmentation in cultured neurons, severely impairing their energetic homeostasis and increasing ROS levels [46].
Further intersection between aging and PD within the context of mitochondrial health can be found in studies of α-synuclein and DJ-1. Point mutations and common variants in SNCA, the gene encoding for α-synuclein are linked to familial and sporadic forms of PD, respectively [47, 48]. α-synuclein is a small, natively unfolded, cytosolic protein that has been observed to cause mitochondrial fragmentation in dopaminergic neurons in vivo that may be dependent on its binding to and disruption of mitochondrial membranes [49, 50]. These effects were reported to occur independently of the mitochondrial fission protein Drp1 [50], although in Drosophila, α-synuclein overexpression led to disruption of the spectrin cytoskeleton resulting in actin-mediated Drp1 mislocalization and aberrant mitochondrial dynamics [51]. In transgenic mice expressing the PD-linked α-synuclein A53T mutation, degenerating mitochondria were observed harboring significant mitochondrial DNA (mtDNA) damage [52]. DJ-1 is a ubiquitous and highly-conserved protein critical for cellular responses to oxidative stress, where upon it becomes relocalized to mitochondria [53]. While the specific mechanism of action is still obscure, loss of DJ-1 results in mitochondrial fragmentation in a human dopaminergic cell line [54]. Specific pathogenic mutations in DJ-1 have been associated with autosomal recessive early-onset Parkinsonism, likely due to a lack of protection from oxidative stress-induced cytotoxicity [55–57]. In mice, overexpression of wild-type DJ-1 has conferred resistance to MPTP, a mitochondrial complex 1 inhibitor that can cause a PD-like clinical syndrome with dopaminergic neurodegeneration [58]. Conversely, mice lacking DJ-1 show increased susceptibility to MPTP toxicity and display nigrostriatal dopaminergic deficits [59, 60]. Despite its effectiveness at neutralizing oxidative stress threats in the cell, over time DJ-1 undergoes oxidative modifications that impair its functionality in the cell [61]. One study found that the percentage of modified DJ-1 in fly brains increased from 0.8% in 1-day-old flies to 17.9% in 40-day-old flies; similar significant age-related changes were also observed in mouse and human brain samples [62]. Therefore, both aging-related modification and PD-associated mutations have the potential to impair DJ-1 functionality and lead to the development of PD.
POLG and mtDNA mutations
Aging cells accumulate mtDNA mutations [63], potentially via oxidative damage and clonal expansion of mtDNA replication errors that occur early in life [64]. Deficiencies in the polymerase gamma (POLG) gene, encoding mtDNA polymerase gamma results in accelerated parameters of aging and reduced lifespan in mice accompanied by mutations in mtDNA [65, 66]. Likewise, POLG1 mutations are associated with PD in humans [67, 68] and POLG mutator mice that express a proofreading-deficient variant of POLG exhibit comparable mtDNA mutations in substantia nigra pars compacta (SNpc) neurons by 1 year of age to that of aged human SNpc [69–71]. Despite the manifestation of brain metabolic and neurotransmitter abnormalities [72], these mice do not develop overt dopaminergic neurodegeneration [71, 73], even when challenged with loss-of-function in the familial PD gene DJ-1 [73] or the PD-associated toxin MPTP [74]. Closer examination indicates that mtDNA deletions in these mice trigger a neuroprotective response within dopaminergic neurons that may stave off neuronal death [71]. This does not appear to be the case in flies, however, where expression of proofreading-deficient mtDNA polymerase leads to somatic mtDNA mutations, mitochondrial dysfunction, and PD-related phenotypes such as dopamine neuron loss and locomotor dysfunction [75].
Loss of protein homeostasis and protein aggregate formation
Protein homeostasis is achieved upon the balance and fidelity of protein synthesis, folding, and degradation. Impaired proteolysis and the accumulation of misfolded protein aggregates, implicated in both aging and PD etiology, can cause significant stress on cell resources and ability to function. The aggregation of specific proteins are directly linked to aging-related diseases such amyloid-β-containing plaques in Alzheimer’s disease [76] and α-synuclein-containing Lewy bodies in PD [6]. While it still remains to be conclusively determined if these protein aggregates are the cause of their respective disease’s clinical progression, it is clear that such a phenomenon can be promoted by the aging process.
Healthy cells produce vast amounts of proteins on a daily basis to maintain homeostasis amid constant protein turnover. Errors in translation or folding can render proteins dysfunctional or actively disruptive via toxic gain of function. These misfolded proteins are dealt with by molecular chaperones and degradation machinery within the cell to prevent damage and stress. As cells age, however, these proteome-stabilizing elements lose effectiveness which results in a growing loss of protein homeostasis [77, 78]. Additionally, levels of protein oxidation and nitrosylation, modifications that can impair protein folding and increase aggregation potential, increase over aging and add additional pressure to the proteostasis machinery [79, 80]. Given enough time and accumulation, aging cells may reach their threshold of proteome stress tolerance leading to cell death [81, 82].
While protein aggregation is commonly associated with age-related disease, recent studies have shown that the phenomenon occurs during normal aging as well. In C. elegans, it was shown that hundreds of proteins become insoluble upon aging, spanning a wide range of biological systems including proteostasis itself [83]. Other studies in C. elegans as well as cultured mammalian cells suggests that osmotic stress results in the formation of age-related unstable polyglutamine (polyQ)-repeat-containing proteins that have the potential to self-aggregate, without any mutation needed [84, 85]. These data led to even further work that found that for a large portion of these unstable polyQ proteins, the increased aggregation potential is conferred by aging-related post-translational carbonyl modification [86, 87].
α-synuclein point mutations or gene multiplications linked to PD promote the formation of oligomeric and higher-order aggregates within neurons [88–90]. Interestingly, brainstem Lewy bodies and Lewy neurites have been repeatedly documented in a small subset (∼10%) of aged individuals without PD [91], consistent with a baseline degree of synuclein aggregation during normal aging. Haploinsufficiency of GBA1, the gene that encodes for glucocerebrosidase (GCase), is one of the most common genetic risk factors for PD [92–94]. GCase is a lysosomal hydrolase critical for the metabolism of glycosphingolipids, and recent studies have suggested that its impaired activity can contribute to increased aggregation of α-synuclein and the formation of Lewy bodies within the cell [95–97]. This impairment can be caused by PD-associated GBA1 mutations but may also appear as a natural result of aging; in wild-type mice, levels of GCase activity in the brain were found to decrease with aging and were correlated with an increase in glycosphingolipids [96]. Patients with a PD-linked GBA1 mutation exhibit earlier age of onset and a more rapid disease progression compared to non-carrier PD patients [98, 99].
Chaperones assist in intracellular protein folding by providing the space and protection for proteins to undergo the conformational changes needed to achieve the proper configuration and functionality. Chaperones are also able to recognize misfolded proteins that are beyond repair and target them for degradation, a process known as chaperone-mediated autophagy (CMA) [100]. Several PD-related mutations interfere with CMA, likely contributing to the disease pathology. The disease-associated mutants of α-synuclein and LRRK2, while recognized by chaperones and marked for CMA, fail to be fully degraded by the lysosomes due to their mutation-derived association with lysosomal-associated membrane protein 2a (Lamp2a) [101]. Even more interestingly, this degradation failure instead results in mutant α-synuclein accumulating at lysosomal surfaces, promoting its subsequent aggregation and further impairing the overall CMA proteostasis of the cell [102]. Overexpression of Lamp2a was found to be neuroprotective against α-synuclein aggregation, as CMA function was maintained [103]. Taken together, multiple lines of evidence indicate that the ability of a cell to maintain proper protein folding and correct for misfolding is impaired similarly with old age and with PD-associated mutations, illustrating the convergence between the two and highlighting the relevance for investigation.
Chronic inflammation
A chronic low-grade inflammation associated with aging, dubbed “inflammaging” may result from elevated innate immune system triggers such as pro-inflammatory damaged or dysfunctional cells [104]. Aging also increases the likelihood of developing conditions that promote chronic inflammation, such as obesity, cardiovascular disease, and diabetes [105]. A major biochemical root of aging-induced chronic inflammation has to do with immune response resolution. In the prime of adult life, the human immune system can clear out threats efficiently while also ramping down the inflammatory response to reduce unnecessary stress on the body. With aging, however, inflammatory triggers from damaged and dying cells are chronically elevated resulting in prolonged inflammation with increased collateral damage to the body, damage that can leave the immune system hypervigilant and overreactive to future insults [104].
The role of inflammation in PD has been extensively studied since the connection was first posited several decades ago and has recently garnered even greater attention as a causative factor with the proposed influence of intestinal inflammation on disease development via a putative gut-brain transmission of α-synuclein [106]. PD genetic studies support a role for inflammation, at the level of penetrant familial PD genes and low-risk common variants. Genome-wide association studies (GWAS) have linked late-onset PD with variants at the HLA (human leukocyte antigen) locus [107, 108] and several of the familial PD genes discussed above are expressed in immune cells and involved in their function (reviewed in [109]), particularly LRRK2. LRRK2 is highly expressed in cells of the innate immune system and is elevated in the peripheral immune cells of idiopathic PD patients and in inflamed colonic tissue of Crohn’s disease patients [110]. Inflammatory triggers such as lipopolysaccharide or IFN-γ induce LRRK2 expression in several immune cells [111–113] implicating its role in inflammation. In the CNS, LRRK2 is highly expressed in microglia [111] and gain-of-function mutations found in PD such as G2019S may promote hyperactive or inappropriate microglia-mediated inflammation [114, 115]. When activated, microglia upregulate a variety of receptors and pro-inflammatory cytokines that stimulate the immune response and associated phagocytosis [116]. This is beneficial for the clearance of dead cells or debris, yet trouble arises when the microglia remain in an activated state for too long, as their activity also produces ROS that can damage neurons [5]. Evidence of activated microglia has been observed both in the aging brain [117, 118] and in the brain or cerebrospinal fluid of PD patients [119, 120] characterized by upregulation of pro-inflammatory cytokines such as IL-1β, IL-6, and TNFα, implying chronic innate immune activation. Additionally, microglia in both aging mammals and PD display an active (deramified) morphology [121, 122], increased phagocytic activity [121, 123] and inflammatory marker levels [124–127]. In PD, they are found in postmortem brain surrounding degenerating dopaminergic neurons [128] consistent with a potential role in neurodegeneration. While there is compelling evidence supporting microglial-induced inflammation in the brains of PD patients and aging mammals, it remains to be determined whether this is causal vs. consequence of age-related cognitive changes or PD onset and progression.
METABOLISM AT THE INTERSECTION OF AGING AND PD
The link between metabolism and aging has long been recognized. Over a century ago, the rate of living theory was postulated based on the observation that organismal lifespan shows a strong negative correlation with metabolic rate, indicating that metabolism influences longevity. Over the last few decades, a central role for metabolism as a driver of aging has emerged from invertebrate and rodent studies showing that reduced intake of calories, and in particular dietary protein is frequently associated with delayed mortality [129–132]. Further, numerous metabolic regulation genes have been identified whose modified expression in model organisms can extend lifespan [133]. The relationship between diet and longevity exists in primates and likely humans too. Calorie restriction can extend longevity in rhesus monkeys as well as delay the onset of multiple age-related pathologies [134] and human studies also support a relationship between protein intake and mortality [135].
A growing list of lifespan-regulating genes have been shown to influence PD-related neurodegeneration, thus strengthening the connection between aging and PD while uncovering potential molecular mediators. Informatively, many of these genes belong to major metabolic hubs such as insulin and mechanistic target of rapamycin (mTOR) signaling or Nicotinamide adenine dinucleotide (NAD+) metabolism, thus underscoring the impact of nutrient sensing and metabolic signaling to the neurodegeneration process. The role of these dietary and metabolic pathway factors in PD pathogenesis is discussed below.
Insulin/IGF-1 and mTOR signaling
A wealth of animal studies has demonstrated the conserved lifespan extending-effect of dietary restriction. At the cellular level, dietary restriction can promote adaptive cellular stress responses and reduce activity of conserved nutrient-sensing pathways such as insulin and IGF-1 signaling (IIS) and its targets in the forkhead box O (FOXO) transcription factor family and the connected mTOR pathway [136]. Roles for IIS and mTOR signaling pathways in the longevity effects of dietary restriction have been demonstrated through genetic epistasis studies and genetic perturbation of pathway constituents which can phenocopy the lifespan extending effects of dietary restriction [130, 137]. In agreement, genetic loss-of-function polymorphisms in the IGF-1 receptor (IGF-1 R), insulin receptor (INSR), Akt, and FOXO3A are also associated with longevity in humans and model organisms [138–141]. While activation of the mTOR signaling complex mTORC1 (mTOR complex 1) is promoted by insulin via PI3K/Akt mediators, mTORC1 also acts in response to nutrient amino acids and other growth factors to control a wide array of biosynthetic and metabolic functions including inhibition of autophagy. Thus, dietary restriction, and in particular amino acid restriction, can directly impact mTOR signaling which has been observed to extend lifespan similarly to mTOR pathway inhibitors such as rapamycin and other rapalogs.
Evidence collected over the last two decades suggests that diet can influence dopamine neuron health and viability, particularly when underlying neuronal insults related to toxin exposure or monogenic PD-causing mutations exist, and that impact on insulin and mTOR signaling as well as stress responses may play a role (Fig. 2). For example, reducing food consumption staves off motor deficits and nigral dopamine neuron loss in mouse and primate toxin models of PD [142, 143]. Subjecting rhesus monkeys to a low-calorie diet for 6 months bestowed protection from the dopaminergic neurotoxin MPTP [143]. The movement disorder and striatal dopamine deficits occurring upon MPTP exposure are substantially attenuated in monkeys fed low-calorie diets and this is associated with increased levels of striatal BDNF and GDNF levels, which at least for BDNF, have been shown to decline with age in several brain regions including striatum [144] and in serum [145]. Similarly, impairments in autonomic regulation of heart rate observed in mutant A53T α-synuclein transgenic mice can be reversed by a form of dietary restriction called alternate day fasting, and worsened by a high fat diet in young mice [146]. A high fat diet has also been shown to accelerate the onset of brainstem pathology and motor deficits in a separate line of α-synuclein mice [147]. Invertebrate and rodent genetic studies have been particularly useful in delineating the contribution of the insulin and TOR signaling pathways to aging and PD. For example, a loss-of-function mutation in daf-2 (the worm insulin-like growth factor 1 receptor) promotes organismal survival and partially rescues the loss of dopamine neuron viability in worms expressing human LRRK2 G2019S or α-synuclein A53T [148, 149]. FOXO transcription factors are major downstream mediators of PI3K/Akt that have been linked to lifespan regulation in humans (FOXO3) as well as model organisms (e.g., daf-16 in nematodes, dFOXO in Drosophila) [141, 150]. FOXO3A protein is reportedly found in lewy bodies in human PD brain [151] and fly dFOXO plays a role in protecting dopamine neurons against cellular stress and accordingly, a subset of these neurons are lost in dFOXO null mutants [152]. Silencing or overexpressing FOXO3 in rat substantia nigra dopamine neurons appears to protect them from cell death associated with human α-synuclein expression [153]. Together, these findings suggest that dietary restriction or genetic modulation of insulin signaling pathway components can have a prominent impact on dopamine neuron health and viability in the face of familial PD mutations or toxins. Whether these effects occur cell autonomously through altered insulin signaling in dopamine neurons, or whether glia are involved is currently unresolved. Additionally, while consumption of high calorie diets may promote aging and PD-related neurodegeneration in part through elevated insulin signaling, it should be noted that type 2 diabetes is associated with elevated PD risk [154–156] and that increased insulin resistance and decreased nigral insulin receptor mRNA are reported in PD patients compared to age-matched controls [157–159] raising the question of whether insulin resistance is a cause or consequence of PD-related neurodegeneration [160].
Fig. 2
Similar impact of insulin/IGF-1 and mTOR pathway manipulations on aging and PD-related neurodegeneration. Impact of dietary and genetic manipulations on several key nodes throughout the insulin/IGF-1 and connected mTOR signaling pathway on aging and PD-related neurodegeneration are shown. Green boxes denote beneficial effects (i.e., delayed/attenuated aging or PD neurodegeneration) and orange boxes denote deleterious effects (i.e., accelerated aging or PD neurodegeneration). Figure was created on BioRender.com.
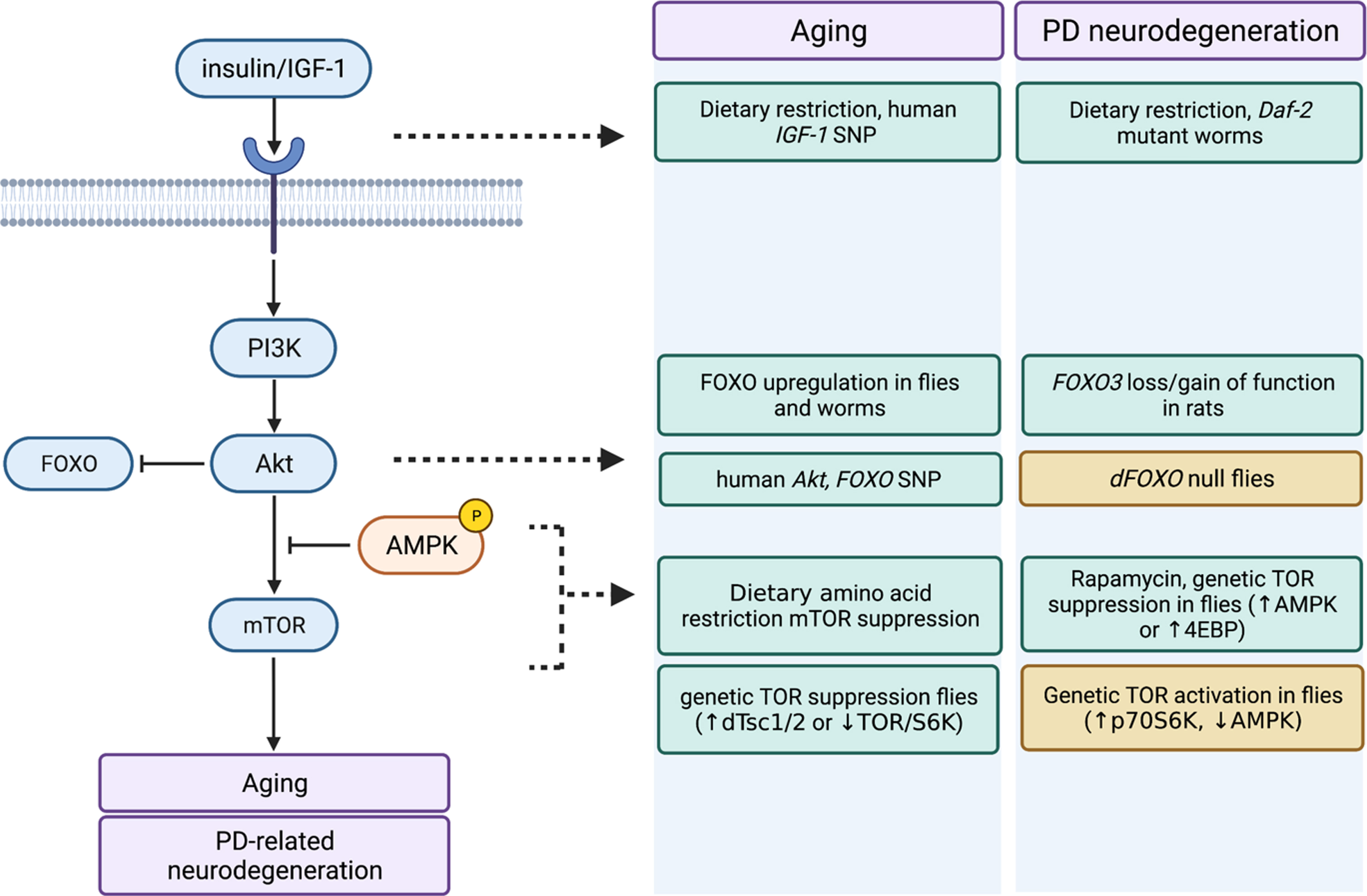
In Drosophila, rapamycin treatment or genetic suppression of TOR signaling through 4EBP overexpression can substantially extend longevity [130] and also appears to protect against dopamine neuron loss in Pink1 and parkin mutant flies [161], while overexpressing p70 S6 kinase (S6k) in a manner that putatively mimics TORC1 signaling exacerbates neuron loss in Pink1 mutant flies [162]. Activity of TOR complex 1 (TORC1) is negatively regulated by AMP-kinase (AMPK) signaling and there is evidence to suggest that AMPK phosphorylation, an index of its activity, is reduced in both aged mice and in Parkin-deficient mice [163]. Pharmacological or genetic activation of AMPK suppresses neurodegenerative phenotypes in LRRK2 G2019S-expressing and parkin mutant flies, while loss of AMPK exacerbates climbing defects in LRRK2 mutant flies [164]. It remains to be determined whether TOR inhibition plays a role in the protective impact of AMPK activation, since autophagy can be induced via AMPK signaling which is a feasible mechanism. Work from our own laboratory indicates that LRRK2 G2019S expressing flies chronically fed a moderately elevated amino acid diet across adulthood are protected from age-related loss of dopamine neurons and locomotor dysfunction observed in these flies on a standard diet in a mechanism that involves induction of AMPK activity by the fly Sestrin ortholog and consequent upregulation of autophagy [165]. TOR inhibition may be neuroprotective in the context of LRRK2 G2019S, since enhanced vulnerability to valinomycin-induced cytotoxicity seen in LRRK2 G2019S iPSC-derived neural cells from familial PD patients can be attenuated by treating them with rapamycin [166] consistent with TOR involvement.
Supporting the potential relevance of model organism findings to human PD, genetic variants in the mTOR pathway (specifically in the RPTOR and RPS6KA2 genes) have been shown to interact with the alpha-synuclein encoding gene, SNCA, to modify age at onset of PD [167]. It’s important to note, however, that whether and how these mTOR pathway variants affect signaling is not currently known. Another important caveat alongside the beneficial effects of dampened TOR signaling, is that strong TOR deficits such as those occurring in response to certain stress stimuli may promote cell death, e.g., via reduced Akt phosphorylation [168, 169]. RTP801 (a.k.a. REDD1) is a transcriptional target of HIF-1 and is rapidly upregulated under hypoxic and oxidative stress conditions [170–172]. RTP801 inactivates mTORC1 signaling through the tuberous sclerosis complex (TSC1/TSC2) and also appears to suppress Akt phosphorylation, possibly through dual mTORC2 inhibition [173, 174]. Supporting a role for RTP801 in PD, elevated RTP801 levels and reduced Akt Ser473/Thr308 phosphorylation are found in substantia nigra dopaminergic neurons from human idiopathic PD brain and RTP801 is induced in response to the PD-associated neurotoxin MPTP in mouse brain [173, 174]. Hence, while there is still much to be learned, preliminary evidence supports a model whereby an optimal window for dopamine neuron health exists where the activity of nutrient-sensing insulin and TOR pathways is sufficient to maintain robust Akt phosphorylation, while the anabolic and autophagy-suppressing effects of TOR signaling are not detrimentally high.
NAD+ and NAD+-dependent enzymes
NAD+ is consumed by many enzymes involved in metabolic function, redox homeostasis, DNA repair, and genomic stability that are implicated in cellular homeostasis and aging. It serves as a co-enzyme for redox reactions involving dehydrogenases important in glycolysis, citric acid cycle, fatty acid oxidation, and ATP generation. NAD+ is also an essential cofactor for NAD+-dependent enzymes including sirtuins and poly (ADP-ribose) polymerases (PARPs), involved in chromatin remodeling and DNA repair, respectively. Initial evidence from model organisms and humans suggests that aging is associated with a progressive decline in brain NAD+ levels [175–177], through unclear mechanisms. PARP is chronically activated upon aging in mice and nematodes [178], possibly as a result of age-related DNA damage, which could conceivably contribute to NAD+ depletion and impaired sirtuin activity in aged animals. Potentially tying the drop in NAD+ levels with lifespan, inducing levels of the NAD+ biosynthetic enzyme nicotinamidase are necessary and sufficient for the lifespan-extending effect of dietary restriction in yeast [179] and in flies, overexpression of orthologous D-NAAM also extends longevity [180]. The age-related decline in NAD+ and beneficial effects of enzymatically enhancing NAD+ production has generated interest in developing interventions to increase NAD+ levels. NAD+ precursors such as nicotinamide riboside (NR) show promise in restoring NAD+ levels as do PARP inhibitors that lower NAD+ consumption. In yeast and worms, exogenous administration of the NAD+ intermediate nicotinamide riboside to restore NAD+ levels enhances lifespan in a sirtuin-dependent manner [178, 181].
Altered NAD+ metabolism can be found in several PD genetic models where it appears to contribute to neurodegenerative phenotypes. Decreased NAD+ and its precursors NR and NMN are observed in fly parkin and Pink1 mutants, while dietary supplementation with the precursor NAM enhanced mitochondrial function and preserved viability of dopaminergic neurons in these flies [182, 183]. Additional support comes from recent work in PD patient tissues and fluids. Fibroblasts from PD patients with PRKN mutations display decreased NAD+/NADH ratio [184] as does blood from idiopathic PD patients compared to age-matched controls [185], while iPSC-derived neurons expressing mutant LRRK2 G2019S or mutant glucocerebrosidase (GBA) iPSC-derived neurons display evidence of reduced NAD+/NADH ratio or NAD+ levels as well as mitochondrial dysfunction [186, 187] which, in the case of GBA, could be attenuated by boosting NAD+ levels via nicotinamide riboside administration [186]. Lastly, NR which appears effective in extending lifespan may also protect against PD-related neurodegeneration. Mutant Transgenic flies expressing mutant GBA-N370S exhibit age-dependent dopaminergic neuron loss and climbing defects [188] which are prevented by administering NR via dietary supplementation during adulthood [186] suggesting that boosting NAD+ levels may preserve dopaminergic neurons in vivo.
As mentioned above, age-associated NAD+ depletion has been hypothesized to arise from age-related PARP1 activation [189]. There appears to be major competition between PARPs and sirtuins for NAD+ such that pharmacological blockade of PARP1 can increase NAD+ content and SIRT1 activity [190]. PARP1 activity may also influence NAD+ metabolism and neurodegeneration in genetic and toxin models of PD. Decreased NAD+ levels and increased protein PARylation (PARP-mediated post-translational modification that consumes NAD+) are observed in parkin null flies and consistent with lower NAD+ being PARP-dependent, flies expressing a PARP loss-of-function mutation exhibit increased NAD+ levels [182]. Further, loss of PARP1 expression or activity using PARP inhibitors is protective against dopamine neuron loss in rodents exposed to LPS [191] or MPTP [192] and decreased Parp expression ameliorates mitochondrial dysfunction, locomotor deficits and dopaminergic neuron loss in parkin or Pink1 mutant flies [182, 183]. More recently, pathologic alpha-synuclein has been shown to trigger PARP-1 activation, proposed to be a crucial driver of neurodegeneration [192]. PARP inhibitors or genetic deletion of PARP-1 were shown to block neurotoxicity associated with the presence of toxic α-synuclein pre-formed fibrils [192]. PARP1-dependent α-synuclein PARylation to form more neurotoxic strains is hypothesized to drive neurodegeneration [193], while a potential role for altered NAD+ bioavailability upon PARP1 inhibition in this model awaits further clarification. The activity of at least some sirtuins such as SIRT1 and SIRT6 are positively correlated with lifespan or healthy aging [194, 195] and are neuroprotective [196–199] with broad roles in DNA damage sensing [200], autophagy [201], and mitochondrial health [202]. Hence, modulation of NAD+-dependent sirtuin activity by rising NAD+ levels is one mechanism conjectured to account for the neuroprotective effect of NR, other NAD+ precursors or PARP silencing. Yet, whether this is the case in the context of PD is unclear and may depend on the specific sirtuin(s) activated. For example, sir2 activity has been shown to promote α-synuclein aggregation and pathology [203, 204] and accordingly, its genetic or pharmacological inhibition appears to be neuroprotective in preventing alpha-synuclein-mediated toxicity in models of PD [204].
CONCLUSIONS
Numerous hallmarks of aging occur in the brain [118, 136] and close inspection reveals that several of these have well established ties to PD, as described above. Since aging is the biggest risk factor for PD, it is reasonable to speculate that age-related declines in molecular/cellular function act as a major driver for PD neurodegeneration. Toward moving the aging-PD relationship beyond correlation, probably the best evidence to date supporting a causal role for aging in PD comes from studies in which manipulations that extend organismal lifespan or healthspan also impede the onset or progression of PD-related neurodegeneration. Here, a common influence of metabolic/nutrient sensing pathways on both aging and PD-related neurodegeneration in model organisms has begun to emerge. This evidence supports the causal hypothesis that aging is a pre-requisite for neurodegeneration to occur, as opposed to a distinct scenario in which PD-related phenotypes develop in a time-dependent manner and on a scale that simply coincides with the manifestation of aging. The robust relationship between PD and aging predicts a sharp rise in disease in the coming decades and generates compelling rationale for incorporating aging into studies into the pathogenesis and treatment of disease. When contemplating the inclusion of aging in PD disease modeling, important questions arise over how well the aging of commonly used model organisms recapitulates human aging. Despite the acknowledged caveats that differences in species lifespan and physiology exist comparing model organisms to humans, studies from worms to mice have revealed that lifespan regulating genes are often evolutionarily conserved which provides rationale for this approach. While the fact that aging is the biggest risk factor for PD makes disease modeling and therapeutic development challenging, future advances in delineating the biology of aging will offer a major opportunity for gaining novel insight into PD etiology and should be harnessed accordingly.
ACKNOWLEDGMENTS
The authors thank funding support for this work from The Parkinson’s Foundation, National Institutes of Health (R01NS119226 and R21AG075320 to I.M., T32AG055378 to C.C.) and The OHSU Foundation (I.M.).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Damier P , Hirsch E , Agid Y , Graybiel A ((1999) ) The substantia nigra of the human brain: , II.atterns of loss of dopamine-containing neurons in Parkinson’s disease, Brain 122: , 1437–1448 . |
[2] | Dexter D , Carter C , Wells F , Javoy-Agid F , Agid Y , Lees A , Jenner P , Marsden CD ((1989) ) Basal lipid peroxidation in substantia nigra is increased in Parkinson’s disease, J Neurochem 52: , 381–389. |
[3] | Schapira AH ((2008) ) Mitochondria in the aetiology and pathogenesis of Parkinson’s disease, Lancet Neurol 7: , 97–109. |
[4] | Jenner P ((2003) ) Oxidative stress in Parkinson’s disease, Ann Neurol 53: , S26–S38. |
[5] | Collins LM , Toulouse A , Connor TJ , Nolan YM ((2012) ) Contributions of central and systemic inflammation to the pathophysiology of Parkinson’s disease, Neuropharmacology 62: , 2154–2168. |
[6] | Gibb W , Lees A ((1988) ) The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease, J Neurol Neurosurg Psychiatry 51: , 745–752. |
[7] | Collier TJ , Kanaan NM , Kordower JH ((2011) ) Ageing as a primary risk factor for Parkinson’s disease: Evidence from studies of non-human primates, Nat Rev Neurosci 12: , 359–366. |
[8] | Hindle JV ((2010) ) Ageing, neurodegeneration and Parkinson’s disease, Age Ageing 39: , 156–161. |
[9] | Phillipson OT ((2014) ) Management of the aging risk factor for Parkinson’s disease, Neurobiol Aging 35: , 847–857. |
[10] | De Rijk M , Breteler M , Graveland G , Ott A , Grobbee D , Van der Meche F , Hofman A ((1995) ) Prevalence of Parkinson’s disease in the elderly: The Rotterdam Study, Neurology 45: , 2143–2146. |
[11] | Chiò A , Magnani C , Schiffer D ((1998) ) Prevalence of Parkinson’s disease in Northwestern Italy: Comparison of tracer methodology and clinical ascertainment of cases, Mov Disord 13: , 400–405. |
[12] | Kyrozis A , Ghika A , Stathopoulos P , Vassilopoulos D , Trichopoulos D , Trichopoulou A ((2013) ) Dietary and lifestyle variables in relation to incidence of Parkinson’s disease in Greece, Eur J Epidemiol 28: , 67–77. |
[13] | Benito-Leon J , Louis E , Bermejo-Pareja F ((2009) ) Risk of incident Parkinson’s disease and parkinsonism in essential tremor: A population based study, J Neurol Neurosurg Psychiatry 80: , 423–425. |
[14] | Perez F , Helmer C , Dartigues J-F , Auriacombe S , Tison F ((2010) ) A 15-year population-based cohort study of the incidence of Parkinson’s disease and dementia with Lewy bodies in an elderly French cohort, J Neurol Neurosurg Psychiatry 81: , 742–746. |
[15] | Ma C-l , Su L , Xie J-j , Long J-x , Wu P , Gu L ((2014) ) The prevalence and incidence of Parkinson’s disease in China: A systematic review and meta-analysis, J Neural Transm 121: , 123–134. |
[16] | Liou H , Tsai M , Chen C , Jeng J , Chang Y , Chen S , Chen R ((1997) ) Environmental risk factors and Parkinson’s disease: A case-control study in Taiwan, Neurology 48: , 1583–1588. |
[17] | Willis AW , Evanoff BA , Lian M , Criswell SR , Racette BA ((2010) ) Geographic and ethnic variation in Parkinson disease: A population-based study of US Medicare beneficiaries, Neuroepidemiology 34: , 143–151. |
[18] | Gorell JM , Peterson EL , Rybicki BA , Johnson CC ((2004) ) Multiple risk factors for Parkinson’s disease, J Neurol Sci 217: , 169–174. |
[19] | Tanner CM , Kamel F , Ross GW , Hoppin JA , Goldman SM , Korell M , Marras C , Bhudhikanok GS , Kasten M , Chade AR ((2011) ) Rotenone, paraquat, and Parkinson’s disease, Environ Health Perspect 119: , 866–872. |
[20] | Driver JA , Logroscino G , Gaziano JM , Kurth T ((2009) ) Incidence and remaining lifetime risk of Parkinson disease in advanced age, Neurology 72: , 432–438. |
[21] | World Health Organization (2015) World Report on Ageing and Health, World Health Organization. |
[22] | Zampese E , Surmeier DJ ((2020) ) Calcium, bioenergetics, and Parkinson’s disease, Cells 9: , 2045. |
[23] | Daiber A , Di Lisa F , Oelze M , Kröller-Schön S , Steven S , Schulz E , Münzel T ((2017) ) Crosstalk of mitochondria with NADPH oxidase via reactive oxygen and nitrogen species signalling and its role for vascular function, Br J Pharmacol 174: , 1670–1689. |
[24] | Redza-Dutor–doir M , Averill-Bates E , Schulz DA ,((2016) ) Activation of apoptosis signalling pathways by reactive oxygen species, Biochim Biophys Acta 1863: , 2977–2992. |
[25] | Sies H , Jones DP ((2020) ) Reactive oxygen species (ROS) as pleiotropic physiological signalling agents, Nat Rev Mol Cell Biol 21: , 363–383. |
[26] | Liguori I , Russo G , Curcio F , Bulli G , Aran L , Della-Morte D , Gargiulo G , Testa G , Cacciatore F , Bonaduce D ((2018) ) Oxidative stress, aging, and diseases, Clin Interv Aging 13: , 757. |
[27] | Golden TR , Hinerfeld DA , Melov S ((2002) ) Oxidative stress and aging: Beyond correlation, Aging Cell 1: , 117–123. |
[28] | Harman D ((1992) ) Free radical theory of aging, Mutat Res 275: , 257–266. |
[29] | Sohal RS , Orr WC ((2012) ) The redox stress hypothesis of aging, Free Radic Biol Med 52: , 539–555. |
[30] | Ramsay RR , Salach JI , Dadgar J , Singer TP ((1986) ) Inhibition of mitochondrial NADH dehydrogenase by pyridine derivatives and its possible relation to experimental and idiopathic parkinsonism, Biochem Biophys Res Commun 135: , 269–275. |
[31] | Ramsay RR , Salach JI , Singer TP ((1986) ) Uptake of the neurotoxin 1-methyl-4-phenylpyridine (MPP+) by mitochondria and its relation to the inhibition of the mitochondrial oxidation of NAD+-linked substrates by MPP+, Biochem Biophys Res Commun 134: , 743–748. |
[32] | Betarbet R , Sherer TB , MacKenzie G , Garcia-Osuna M , Panov AV , Greenamyre JT ((2000) ) Chronic systemic pesticide exposure reproduces features of Parkinson’s disease, Nat Neurosci 3: , 1301–1306. |
[33] | Horgan DJ , Singer TP , Casida J ((1968) ) Studies on the respiratory chain-linked reduced nicotinamide adenine dinucleotide dehydrogenase: XIII. Binding sites of rotenone, piericidin A, and amytal in the respiratory chain, J Biol Chem 243: , 834–843. |
[34] | Schapira A , Cooper J , Dexter D , Clark J , Jenner P , Marsden C ((1990) ) Mitochondrial complex I deficiency in Parkinson’s disease, J Neurochem 54: , 823–827. |
[35] | González-Rodríguez P , Zampese E , Stout KA , Guzman JN , Ilijic E , Yang B , Tkatch T , Stavarache MA , Wokosin DL , Gao L ((2021) ) Disruption of mitochondrial complex I induces progressive parkinsonism, Nature 599: , 650–656. |
[36] | Cornelissen T , Vilain S , Vints K , Gounko N , Verstreken P , Vandenberghe W ((2018) ) Deficiency of parkin and PINK1 impairs age-dependent mitophagy in Drosophila, Elife 7: , e35878. |
[37] | Dawson TM , Dawson VL ((2010) ) The role of parkin in familial and sporadic Parkinson’s disease, Mov Disord 25: , S32–S39. |
[38] | Tanaka K , Suzuki T , Hattori N , Mizuno Y ((2004) ) Ubiquitin, proteasome and parkin, Biochim Biophys Acta 1695: , 235–247. |
[39] | Bonello F , Hassoun S-M , Mouton-Liger F , Shin YS , Muscat A , Tesson C , Lesage S , Beart PM , Brice A , Krupp J ((2019) ) LRRK2 impairs PINK1/Parkin-dependent mitophagy via its kinase activity: Pathologic insights into Parkinson’s disease, Hum Mol Genet 28: , 1645–1660. |
[40] | Clark IE , Dodson MW , Jiang C , Cao JH , Huh JR , Seol JH , Yoo SJ , Hay BA , Guo M ((2006) ) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin, Nature 441: , 1162–1166. |
[41] | Greene JC , Whitworth AJ , Andrews LA , Parker TJ , Pallanck LJ ((2005) ) Genetic and genomic studies of Drosophila parkin mutants implicate oxidative stress and innate immune responses in pathogenesis, Hum Mol Genet 14: , 799–811. |
[42] | Hsieh C-H , Shaltouki A , Gonzalez AE , da Cruz AB , Burbulla LF , Lawrence ES , Schüle B , Krainc D , Palmer TD , Wang X ((2016) ) Functional impairment in miro degradation and mitophagy is a shared feature in familial and sporadic Parkinson’s disease, Cell Stem Cell 19: , 709–724. |
[43] | Korecka JA , Thomas R , Christensen DP , Hinrich AJ , Ferrari EJ , Levy SA , Hastings ML , Hallett PJ , Isacson O ((2019) ) Mitochondrial clearance and maturation of autophagosomes are compromised in LRRK2 G2019S familial Parkinson’s disease patient fibroblasts, Hum Mol Genet 28: , 3232–3243. |
[44] | Sanders LH , Laganière J , Cooper O , Mak SK , Vu BJ , Huang YA , Paschon DE , Vangipuram M , Sundararajan R , Urnov FD ((2014) ) LRRK2 mutations cause mitochondrial DNA damage in iPSC-derived neural cells from Parkinson’s disease patients: Reversal by gene correction, Neurobiol Dis 62: , 381–386. |
[45] | Angeles DC , Ho P , Chua LL , Wang C , Yap YW , Ng C , Zhou Zd , Lim K-L , Wszolek ZK , Wang HY ((2014) ) Thiol peroxidases ameliorate LRRK2 mutant-induced mitochondrial and dopaminergic neuronal degeneration in Drosophila, Hum Mol Genet 23: , 3157–3165. |
[46] | Niu J , Yu M , Wang C , Xu Z ((2012) ) Leucine-rich repeat kinase 2 disturbs mitochondrial dynamics via Dynamin-like protein, J Neurochem 122: , 650–658. |
[47] | Polymeropoulos MH , Lavedan C , Leroy E , Ide SE , Dehejia A , Dutra A , Pike B , Root H , Rubenstein J , Boyer R ((1997) ) Mutation in the α-synuclein gene identified in families with Parkinson’s disease, Science 276: , 2045–2047. |
[48] | Satake W , Nakabayashi Y , Mizuta I , Hirota Y , Ito C , Kubo M , Kawaguchi T , Tsunoda T , Watanabe M , Takeda A ((2009) ) Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease, Nat Genet 41: , 1303. |
[49] | Li W-W , Yang R , Guo J-C , Ren H-M , Zha X-L , Cheng J-S , Cai D-F ((2007) ) Localization of α-synuclein to mitochondria within midbrain of mice, Neuroreport 18: , 1543–1546. |
[50] | Nakamura K , Nemani VM , Azarbal F , Skibinski G , Levy JM , Egami K , Munishkina L , Zhang J , Gardner B , Wakabayashi J ((2011) ) Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein α-synuclein, J Biol Chem 286: , 20710–20726. |
[51] | Ordonez DG , Lee MK , Feany MB ((2018) ) α-synuclein induces mitochondrial dysfunction through spectrin and the actin cytoskeleton, Neuron 97: , 108–124. e106. |
[52] | Martin LJ , Pan Y , Price AC , Sterling W , Copeland NG , Jenkins NA , Price DL , Lee MK ((2006) ) Parkinson’s disease α-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death, J Neurosci 26: , 41–50. |
[53] | Canet-Avilés RM , Wilson MA , Miller DW , Ahmad R , McLendon C , Bandyopadhyay S , Baptista MJ , Ringe D , Petsko GA , Cookson MR ((2004) ) The Parkinson’s disease protein DJ-1 is neuroprotective due tocysteine-sulfinic acid-driven mitochondrial localization, ProcNatl Acad Sci U S A 101: , 9103–9108. |
[54] | Thomas KJ , McCoy MK , Blackinton J , Beilina A , van der Brug M , Sandebring A , Miller D , Maric D , Cedazo-Minguez A , Cookson MR ((2011) ) DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy, Hum Mol Genet 20: , 40–50. |
[55] | Bonifati V , Rizzu P , Van Baren MJ , Schaap O , Breedveld GJ , Krieger E , Dekker MC , Squitieri F , Ibanez P , Joosse M ((2003) ) Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism, Science 299: , 256–259. |
[56] | Cookson MR ((2010) ) DJ-1, PINK1, and their effects on mitochondrial pathways, Mov Disord 25: , S44–S48. |
[57] | Dolgacheva LP , Berezhnov AV , Fedotova EI , Zinchenko VP , Abramov AY ((2019) ) Role of DJ-1 in the mechanism of pathogenesis of Parkinson’s disease, J Bioenerg Biomembr 51: , 175–188. |
[58] | Paterna J-C , Leng A , Weber E , Feldon J , Büeler H ((2007) ) DJ-1 and Parkin modulate dopamine-dependent behavior and inhibit MPTP-induced nigral dopamine neuron loss in mice, Mol Ther 15: , 698–704. |
[59] | Goldberg MS , Pisani A , Haburcak M , Vortherms TA , Kitada T , Costa C , Tong Y , Martella G , Tscherter A , Martins A ((2005) ) Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1, Neuron 45: , 489–496. |
[60] | Kim RH , Smith PD , Aleyasin H , Hayley S , Mount MP , Pownall S , Wakeham A , You-Ten AJ , Kalia SK , Horne P ((2005) ) Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyrindine (MPTP) and oxidative stress, Proc Natl Acad Sci U S A 102: , 5215–5220. |
[61] | Mitsumoto A , Nakagawa Y , Takeuchi A , Okawa K , Iwamatsu A , Takanezawa Y ((2001) ) Oxidized forms of peroxiredoxins and DJ-1 on two-dimensional gels increased in response to sublethal levels of paraquat, Free Radic Res 35: , 301–310. |
[62] | Meulener MC , Xu K , Thomson L , Ischiropoulos H , Bonini NM ((2006) ) Mutational analysis of DJ-1 in Drosophila implicates functional inactivation by oxidative damage and aging, Proc Natl Acad Sci U S A 103: , 12517–12522. |
[63] | Khrapko K , Bodyak N , Thilly WG , Van Orsouw NJ , Zhang X , Coller HA , Perls TT , Upton M , Vijg J , Wei JY ((1999) ) Cell-by-cell scanning of whole mitochondrial genomes in aged human heart reveals a significant fraction of myocytes with clonally expanded deletions, Nucleic Acids Res 27: , 2434–2441. |
[64] | Ameur A , Stewart JB , Freyer C , Hagström E , Ingman M , Larsson N-G , Gyllensten U ((2011) ) Ultra-deep sequencing of mouse mitochondrial DNA: Mutational patterns and their origins, PLoS Genet 7: , e1002028. |
[65] | Trifunovic A , Wredenberg A , Falkenberg M , Spelbrink JN , Rovio AT , Bruder CE , Bohlooly-Y M , Gidlöf S , Oldfors A , Wibom R ((2004) ) Premature ageing in mice expressing defective mitochondrial DNA polymerase, Nature 429: , 417–423. |
[66] | Vermulst M , Wanagat J , Kujoth GC , Bielas JH , Rabinovitch PS , Prolla TA , Loeb LA ((2008) ) DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice, Nat Genet 40: , 392–394. |
[67] | Luoma P , Eerola J , Ahola S , Hakonen A , Hellström O , Kivistö KT , Tienari PJ , Suomalainen A ((2007) ) Mitochondrial DNA polymerase gamma variants in idiopathic sporadic Parkinson disease, Neurology 69: , 1152–1159. |
[68] | Ylönen S , Ylikotila P , Siitonen A , Finnilä S , Autere J , Majamaa K ((2013) ) Variations of mitochondrial DNA polymerase γ in patients with Parkinson’s disease, J Neurol 260: , 3144–3149. |
[69] | Bender A , Krishnan KJ , Morris CM , Taylor GA , Reeve AK , Perry RH , Jaros E , Hersheson JS , Betts J , Klopstock T ((2006) ) High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease, Nat Genet 38: , 515–517. |
[70] | Kraytsberg Y , Kudryavtseva E , McKee AC , Geula C , Kowall NW , Khrapko K ((2006) ) Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons, Nat Genet 38: , 518–520. |
[71] | Perier C , Bender A , García-Arumí E , Melia MJ , Bove J , Laub C , Klopstock T , Elstner M , Mounsey RB , Teismann P ((2013) ) Accumulation of mitochondrial DNA deletions within dopaminergic neurons triggers neuroprotective mechanisms, Brain 136: , 2369–2378. |
[72] | Clark-Matott J , Saleem A , Dai Y , Shurubor Y , Ma X , Safdar A , Beal MF , Tarnopolsky M , Simon DK ((2015) ) Metabolomic analysis of exercise effects in the POLG mitochondrial DNA mutator mouse brain, Neurobiol Aging 36: , 2972–2983. |
[73] | Hauser DN , Primiani CT , Langston RG , Kumaran R , Cookson MR ((2015) ) The Polg mutator phenotype does not cause dopaminergic neurodegeneration in DJ-1-deficient mice. Eneuro 2: , 2015ENEURO.0075-14. |
[74] | Dai Y , Clark J , Zheng K , Kujoth GC , Prolla TA , Simon DK ((2014) ) Somatic mitochondrial DNA mutations do not increase neuronal vulnerability to MPTP in young POLG mutator mice, Neurotoxicol Teratol 46: , 62–67. |
[75] | Samstag CL , Hoekstra JG , Huang C-H , Chaisson MJ , Youle RJ , KennedySR , Pallanck LJ ((2018) ) Deleterious mitochondrial DNA point mutationsare overrepresented in Drosophila expressing aproofreading-defective DNA polymerase γ, PLoS Genet 14: , e1007805. |
[76] | Shoghi-Jadid K , Small GW , Agdeppa ED , Kepe V , Ercoli LM , Siddarth P , Read S , Satyamurthy N , Petric A , Huang S-C ((2002) ) Localization of neurofibrillary tangles and beta-amyloid plaques in the brains of living patients with Alzheimer disease, Am J Geriatr Psychiatry 10: , 24–35. |
[77] | Calderwood SK , Murshid A , Prince T ((2009) ) The shock of aging: Molecular chaperones and the heat shock response in longevity and aging–a mini-review, Gerontology 55: , 550–558. |
[78] | Soti C , Csermely P ((2000) ) Molecular chaperones and the aging process, Biogerontology 1: , 225–233. |
[79] | Rizza S , Cardaci S , Montagna C , Di Giacomo G , De Zio D , Bordi M , Maiani E , Campello S , Borreca A , Puca AA ((2018) ) S-nitrosylation drives cell senescence and aging in mammals by controlling mitochondrial dynamics and mitophagy, Proc Natl Acad Sci U S A 115: , E3388–3397. |
[80] | Stadtman ER ((2006) ) Protein oxidation and aging, Free Radic Res 40: , 1250–1258. |
[81] | Chiti F , Dobson CM ((2006) ) Protein misfolding, functional amyloid, and human disease, Annu Rev Biochem 75: , 333–366. |
[82] | Ross CA , Poirier MA ((2004) ) Protein aggregation and neurodegenerative disease, Nat Med 10: , S10–S17. |
[83] | David DC , Ollikainen N , Trinidad JC , Cary MP , Burlingame AL , Kenyon C ((2010) ) Widespread protein aggregation as an inherent part of aging in C. elegans., PLoS Biol 8: , e1000450. |
[84] | Gidalevitz T , Ben-Zvi A , Ho KH , Brignull HR , Morimoto RI ((2006) ) Progressive disruption of cellular protein folding in models of polyglutamine diseases, Science 311: , 1471–1474. |
[85] | Mazzeo LEM , Dersh D , Boccitto M , Kalb RG , Lamitina T ((2012) ) Stress and aging induce distinct polyQ protein aggregation states, Proc Natl Acad Sci U S A 109: , 10587–10592. |
[86] | Brockmann K , Berg D ((2014) ) The significance of GBA for Parkinson’s disease, J Inherit Metab Dis 37: , 643–648. |
[87] | Tanase M , Urbanska AM , Zolla V , Clement CC , Huang L , Morozova K , Follo C , Goldberg M , Roda B , Reschiglian P ((2016) ) Role of carbonyl modifications on aging-associated protein aggregation, Sci Rep 6: , 19311. |
[88] | Conway KA , Harper JD , Lansbury PT ((1998) ) Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease, Nat Med 4: , 1318–1320. |
[89] | Miller D , Hague S , Clarimon J , Baptista M , Gwinn-Hardy K , Cookson M , Singleton A ((2004) ) α-synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication, Neurology 62: , 1835–1838. |
[90] | Ostrerova-Golts N , Petrucelli L , Hardy J , Lee JM , Farer M , Wolozin B ((2000) ) The A53T α-synuclein mutation increases iron-dependent aggregation and toxicity, J Neurosci 20: , 6048–6054. |
[91] | Mikolaenko I , Pletnikova O , Kawas CH , O’Brien R , Resnick SM , Crain B , Troncoso JC ((2005) ) Alpha-synuclein lesions in normal aging, Parkinson disease, and Alzheimer disease: Evidence from the Baltimore Longitudinal Study of Aging (BLSA), J Neuropathol Exp Neurol 64: , 156–162. |
[92] | Balestrino R , Tunesi S , Tesei S , Lopiano L , Zecchinelli AL , Goldwurm S ((2020) ) Penetrance of glucocerebrosidase (GBA) mutations in Parkinson’s disease: A Kin Cohort Study, Mov Disord 35: , 2111–2114. |
[93] | Li Y , Sekine T , Funayama M , Li L , Yoshino H , Nishioka K , Tomiyama H , Hattori N ((2014) ) Clinicogenetic study of GBA mutations in patients with familial Parkinson’s disease, Neurobiol Aging 35: , 935. e933–935. e938. |
[94] | Sidransky E , Nalls MA , Aasly JO , Aharon-Peretz J , Annesi G , Barbosa ER , Bar-Shira A , Berg D , Bras J , Brice A ((2009) ) Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease, N Engl J Med 361: , 1651–1661. |
[95] | Goker-Alpan O , Stubblefield BK , Giasson BI , Sidransky E ((2010) ) Glucocerebrosidase is present in α-synuclein inclusions in Lewy body disorders, Acta Neuropathol 120: , 641–649. |
[96] | Hallett PJ , Huebecker M , Brekk OR , Moloney EB , Rocha EM , Priestman DA , Platt FM , Isacson O ((2018) ) Glycosphingolipid levels and glucocerebrosidase activity are altered in normal aging of the mouse brain, Neurobiol Aging 67: , 189–200. |
[97] | Mazzulli JR , Xu Y-H , Sun Y , Knight AL , McLean PJ , Caldwell GA , Sidransky E , Grabowski GA , Krainc D ((2011) ) Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies, Cell 146: , 37–52. |
[98] | Cilia R , Tunesi S , Marotta G , Cereda E , Siri C , Tesei S , Zecchinelli AL , Canesi M , Mariani CB , Meucci N ((2016) ) Survival and dementia inGBA-associated Parkinson’s disease: The mutation matters, AnnNeurol 80: , 662–673. |
[99] | Winder-Rhodes SE , Evans JR , Ban M , Mason SL , Williams-Gray CH , Foltynie T , Duran R , Mencacci NE , Sawcer SJ , Barker RA ((2013) ) Glucocerebrosidase mutations influence the natural history of Parkinson’s disease in a community-based incident cohort, Brain 136: , 392–399. |
[100] | Dice JF ((2007) ) Chaperone-mediated autophagy, Autophagy 3: , 295–299. |
[101] | Cuervo AM , Wong E ((2014) ) Chaperone-mediated autophagy: Roles in disease and aging, Cell Res 24: , 92–104. |
[102] | Orenstein SJ , Kuo S-H , Tasset I , Arias E , Koga H , Fernandez-Carasa I , Cortes E , Honig LS , Dauer W , Consiglio A ((2013) ) Interplay of LRRK2 with chaperone-mediated autophagy, Nat Neurosci 16: , 394–406. |
[103] | Xilouri M , Brekk OR , Landeck N , Pitychoutis PM , Papasilekas T , Papadopoulou-Daifoti Z , Kirik D , Stefanis L ((2013) ) Boosting chaperone-mediated autophagy in vivo mitigates α-synuclein-induced neurodegeneration, Brain 136: , 2130–2146. |
[104] | Franceschi C , Garagnani P , Parini P , Giuliani C , Santoro A ((2018) ) Inflammaging: A new immune–metabolic viewpoint for age-related diseases, Nat Rev Endocrinol 14: , 576–590. |
[105] | Woods JA , Wilund KR , Martin SA , Kistler BM ((2012) ) Exercise, inflammation and aging, Aging Dis 3: , 130. |
[106] | Klingelhoefer L , Reichmann H ((2015) ) Pathogenesis of Parkinson disease— the gut–brain axis and environmental factors, Nat Rev Neurol 11: , 625–636. |
[107] | Hamza TH , Zabetian CP , Tenesa A , Laederach A , Montimurro J , Yearout D , Kay DM , Doheny KF , Paschall J , Pugh E , Kusel VI , Collura R , Roberts J , Griffith A , Samii A , Scott WK , Nutt J , Factor SA , Payami H ((2010) ) Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease, Nat Genet 42: , 781–785. |
[108] | Nalls MA , Pankratz N , Lill CM , Do CB , Hernandez DG , Saad M , DeStefano AL , Kara E , Bras J , Sharma M , Schulte C , Keller MF , Arepalli S , Letson C , Edsall C , Stefansson H , Liu X , Pliner H , Lee JH , Cheng R ; International Parkinson’s Disease Genomics Consortium (IPDGC); Parkinson’s Study Group (PSG) Parkinson’s Research: The Organized GENetics Initiative (PROGENI); 23andMe; GenePD; NeuroGenetics Research Consortium (NGRC); Hussman Institute of Human Genomics (HIHG); Ashkenazi Jewish Dataset Investigator; Cohorts for Health and Aging Research in Genetic Epidemiology (CHARGE); North American Brain Expression Consortium (NABEC); United Kingdom Brain Expression Consortium (UKBEC); Greek Parkinson’s Disease Consortium; Alzheimer Genetic Analysis Group Ikram MA , Ioannidis JP , Hadjigeorgiou GM , Bis JC , Martinez M , Perlmutter JS , Goate A , Marder K , Fiske B , Sutherland M , Xiromerisiou G , Myers RH , Clark LN , Stefansson K , Hardy JA , Heutink P , Chen H , Wood NW , Houlden H , Payami H , Brice A , Scott WK , Gasser T , Bertram L , Eriksson N , Foroud T , Singleton AB ((2014) ) Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease, Nat Genet 46: , 989–993. |
[109] | Dzamko N , Geczy CL , Halliday GM ((2015) ) Inflammation is genetically implicated in Parkinson’s disease, Neuroscience 302: , 89–102. |
[110] | Herrick MK , Tansey MG ((2021) ) Is LRRK2 the missing link between inflammatory bowel disease and Parkinson’s disease? NPJ Parkinsons Dis 7: , 26. |
[111] | Gardet A , Benita Y , Li C , Sands BE , Ballester I , Stevens C , Korzenik JR , Rioux JD , Daly MJ , Xavier RJ , Podolsky DK ((2010) ) LRRK2 is involved in the IFN-gamma response and host response to pathogens, J Immunol 185: , 5577–5585. |
[112] | Hakimi M , Selvanantham T , Swinton E , Padmore RF , Tong Y , Kabbach G , Venderova K , Girardin SE , Bulman DE , Scherzer CR , LaVoie MJ , Gris D , Park DS , Angel JB , Shen J , Philpott DJ , Schlossmacher MG ((2011) ) Parkinson’s disease-linked LRRK2 is expressed in circulating and tissue immune cells and upregulated following recognition of microbial structures, J Neural Transm (Vienna) 118: , 795–808. |
[113] | Kuss M , Adamopoulou E , Kahle PJ ((2014) ) Interferon-gamma induces leucine-rich repeat kinase LRRK2 via extracellular signal-regulated kinase ERK5 in macrophages, J Neurochem 129: , 980–987. |
[114] | Dwyer Z , Rudyk C , Thompson A , Farmer K , Fenner B , Fortin T , Derksen A , Sun H , Hayley S ((2020) ) Leucine-rich repeat kinase-2 (LRRK2) modulates microglial phenotype and dopaminergic neurodegeneration, Neurobiol Aging 91: , 45–55. |
[115] | Kim KS , Marcogliese PC , Yang J , Callaghan SM , Resende V , Abdel-Messih E , Marras C , Visanji NP , Huang J , Schlossmacher MG ((2018) ) Regulation of myeloid cell phagocytosis by LRRK2 via WAVE2 complex stabilization is altered in Parkinson’s disease, Proc Natl Acad Sci U S A 115: , E5164–E5173. |
[116] | McGeer PL , McGeer EG ((2004) ) Inflammation and neurodegeneration in Parkinson’s disease, Parkinsonism Relat Disord 10: , S3–S7. |
[117] | Cribbs DH , Berchtold NC , Perreau V , Coleman PD , Rogers J , Tenner AJ , Cotman CW ((2012) ) Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: A microarray study, J Neuroinflammation 9: , 179. |
[118] | Mattson MP , Arumugam TV ((2018) ) Hallmarks of brain aging: Adaptive and pathological modification by metabolic states, Cell Metab 27: , 1176–1199. |
[119] | Mogi M , Harada M , Narabayashi H , Inagaki H , Minami M , Nagatsu T ((1996) ) Interleukin (IL)-1 beta, IL-2, IL-4, IL-6 and transforming growth factor-alpha levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson’s disease, Neurosci Lett 211: , 13–16. |
[120] | Mogi M , Harada M , Riederer P , Narabayashi H , Fujita K , Nagatsu T ((1994) ) Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients, Neurosci Lett 165: , 208–210. |
[121] | Doorn KJ , Moors T , Drukarch B , van de Berg W , Lucassen PJ , van Dam AM ((2014) ) Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson’s disease patients, Acta Neuropathol Commun 2: , 90. |
[122] | Norden DM , Godbout JP ((2013) ) Review: Microglia of the aged brain: , rimed to be activated and resistant to regulation, Neuropathol Appl Neurobiol 39: , 19–34 . |
[123] | Croisier E , Moran LB , Dexter DT , Pearce RK , Graeber MB ((2005) ) Microglial inflammation in the parkinsonian substantia nigra: Relationship to alpha-synuclein deposition, J Neuroinflammation 2: , 14. |
[124] | Frank MG , Barrientos RM , Biedenkapp JC , Rudy JW , Watkins LR , Maier SF ((2006) ) mRNA up-regulation of MHC II and pivotal pro-inflammatory genes in normal brain aging, Neurobiol Aging 27: , 717–722. |
[125] | Loeffler DA , Camp DM , Conant SB ((2006) ) Complement activation in the Parkinson’s disease substantia nigra: An immunocytochemical study, J Neuroinflammation 3: , 29. |
[126] | Perry VH , Matyszak MK , Fearn S ((1993) ) Altered antigen expression of microglia in the aged rodent CNS, Glia 7: , 60–67. |
[127] | VanGuilder HD , Bixler GV , Brucklacher RM , Farley JA , Yan H , Warrington JP , Sonntag WE , Freeman WM ((2011) ) Concurrent hippocampal induction of MHC II pathway components and glial activation with advanced aging is not correlated with cognitive impairment, J Neuroinflammation 8: , 138. |
[128] | McGeer PL , Itagaki S , Akiyama H , McGeer EG ((1988) ) Rate of cell death in parkinsonism indicates active neuropathological process, Ann Neurol 24: , 574–576. |
[129] | Grandison RC , Piper MD , Partridge L ((2009) ) Amino-acid imbalance explains extension of lifespan by dietary restriction in Drosophila, Nature 462: , 1061–1064. |
[130] | Kapahi P , Zid BM , Harper T , Koslover D , Sapin V , Benzer S ((2004) ) Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway, Curr Biol 14: , 885–890. |
[131] | Orentreich N , Matias JR , DeFelice A , Zimmerman JA ((1993) ) Low methionine ingestion by rats extends life span, J Nutr 123: , 269–274. |
[132] | Solon-Biet SM , McMahon AC , Ballard JWO , Ruohonen K , Wu LE , Cogger VC , Warren A , Huang X , Pichaud N , Melvin RG ((2014) ) The ratio of macronutrients, not caloric intake, dictates cardiometabolic health, aging, and longevity in ad libitum-fed mice, Cell Metab 19: , 418–430. |
[133] | Gems D , Partridge L ((2013) ) Genetics of longevity in model organisms: Debates and paradigm shifts, Ann Rev Physiol 75: , 621–644. |
[134] | Colman RJ , Anderson RM , Johnson SC , Kastman EK , Kosmatka KJ , Beasley TM , Allison DB , Cruzen C , Simmons HA , Kemnitz JW ((2009) ) Caloric restriction delays disease onset and mortality in rhesus monkeys, Science 325: , 201–204. |
[135] | Levine ME , Suarez JA , Brandhorst S , Balasubramanian P , Cheng C-W , Madia F , Fontana L , Mirisola MG , Guevara-Aguirre J , Wan J ((2014) ) Low protein intake is associated with a major reduction in IGF-1, cancer, and overall mortality in the 65 and younger but not older population, Cell Metab 19: , 407–417. |
[136] | López-Otín C , Blasco MA , Partridge L , Serrano M , Kroemer G ((2013) ) The hallmarks of aging, Cell 153: , 1194–1217. |
[137] | Zid BM , Rogers AN , Katewa SD , Vargas MA , Kolipinski MC , Lu TA , Benzer S , Kapahi P ((2009) ) 4E-BP extends lifespan upon dietary restriction by enhancing mitochondrial activity in Drosophila, Cell 139: , 149–160. |
[138] | Kojima T , Kamei H , Aizu T , Arai Y , Takayama M , Nakazawa S , Ebihara Y , Inagaki H , Masui Y , Gondo Y , Sakaki Y , Hirose N ((2004) ) Association analysis between longevity in the Japanese population and polymorphic variants of genes involved in insulin and insulin-like growth factor 1 signaling pathways, Exp Gerontol 39: , 1595–1598. |
[139] | Pawlikowska L , Hu D , Huntsman S , Sung A , Chu C , Chen J , Joyner AH , Schork NJ , Hsueh WC , Reiner AP , Psaty BM , Atzmon G , Barzilai N , Cummings SR , Browner WS , Kwok PY , Ziv E , Study of Osteoporotic F ((2009) ) Association of common genetic variation in the insulin/IGF1 signaling pathway with human longevity, Aging Cell 8: , 460–472. |
[140] | Suh Y , Atzmon G , Cho MO , Hwang D , Liu B , Leahy DJ , Barzilai N , Cohen P ((2008) ) Functionally significant insulin-like growth factor I receptor mutations in centenarians, Proc Natl Acad Sci U S A 105: , 3438–3442. |
[141] | Willcox BJ , Donlon TA , He Q , Chen R , Grove JS , Yano K , Masaki KH , Willcox DC , Rodriguez B , Curb JD ((2008) ) FOXO3A genotype is strongly associated with human longevity, Proc Natl Acad Sci U S A 105: , 13987–13992. |
[142] | Bayliss JA , Lemus MB , Stark R , Santos VV , Thompson A , Rees DJ , Galic S , Elsworth JD , Kemp BE , Davies JS ((2016) ) Ghrelin-AMPK signaling mediates the neuroprotective effects of calorie restriction in Parkinson’s disease, J Neurosci 36: , 3049–3063. |
[143] | Maswood N , Young J , Tilmont E , Zhang Z , Gash DM , Gerhardt GA , Grondin R , Roth GS , Mattison J , Lane MA ((2004) ) Caloric restriction increases neurotrophic factor levels and attenuates neurochemical and behavioral deficits in a primate model of Parkinson’s disease, Proc Natl Acad Sci U S A 101: , 18171–18176. |
[144] | Katoh-Semba R , Semba R , Takeuchi IK , Kato K ((1998) ) Age-related changes in levels of brain-derived neurotrophic factor in selected brain regions of rats, normal mice and senescence-accelerated mice: A comparison to those of nerve growth factor and neurotrophin-3, Neurosci Res 31: , 227–234. |
[145] | Erickson KI , Prakash RS , Voss MW , Chaddock L , Heo S , McLaren M , Pence BD , Martin SA , Vieira VJ , Woods JA ((2010) ) Brain-derived neurotrophic factor is associated with age-related decline in hippocampal volume, J Neurosci 30: , 5368–5375. |
[146] | Griffioen KJ , Rothman SM , Ladenheim B , Wan R , Vranis N , Hutchison E , Okun E , Cadet JL , Mattson MP ((2013) ) Dietary energy intake modifies brainstem autonomic dysfunction caused by mutant α-synuclein, Neurobiol Aging 34: , 928–935. |
[147] | Rotermund C , Truckenmüller FM , Schell H , Kahle PJ ((2014) ) Diet-induced obesity accelerates the onset of terminal phenotypes in α-synuclein transgenic mice, J Neurochem 131: , 848–858. |
[148] | Cooper JF , Dues DJ , Spielbauer KK , Machiela E , Senchuk MM , Van Raamsdonk JM ((2015) ) Delaying aging is neuroprotective in Parkinson’s disease: A genetic analysis in C. elegans models, NPJ Parkinsons Dis 1: , 15022. |
[149] | Senchuk MM , Van Raamsdonk JM , Moore DJ ((2021) ) Multiple genetic pathways regulating lifespan extension are neuroprotective in a G2019S LRRK2 nematode model of Parkinson’s disease, Neurobiol Dis 151: , 105267. |
[150] | Timmers P , Wilson JF , Joshi PK , Deelen J ((2020) ) Multivariate genomic scan implicates novel loci and haem metabolism in human ageing, Nat Commun 11: , 3570. |
[151] | Su B , Liu H , Wang X , Chen SG , Siedlak SL , Kondo E , Choi R , Takeda A , Castellani RJ , Perry G , Smith MA , Zhu X , Lee HG ((2009) ) Ectopic localization of FOXO3a protein in Lewy bodies in Lewy body dementia and Parkinson’s disease, Mol Neurodegener 4: , 32. |
[152] | Tas D , Stickley L , Miozzo F , Koch R , Loncle N , Sabado V , Gnägi B , Nagoshi E ((2018) ) Parallel roles of transcription factors dFOXO and FER2 in the development and maintenance of dopaminergic neurons, PLoS Genet 14: , e1007271. |
[153] | Pino E , Amamoto R , Zheng L , Cacquevel M , Sarria JC , Knott GW , Schneider BL ((2014) ) FOXO3 determines the accumulation of α-synuclein and controls the fate of dopaminergic neurons in the substantia nigra, Hum Mol Genet 23: , 1435–1452. |
[154] | Biosa A , Outeiro TF , Bubacco L , Bisaglia M ((2018) ) Diabetes mellitus as a risk factor for Parkinson’s disease: A molecular point of view, Mol Neurobiol 55: , 8754–8763. |
[155] | Driver JA , Smith A , Buring JE , Gaziano JM , Kurth T , Logroscino G ((2008) ) Prospective cohort study of type 2 diabetes and the risk of Parkinson’s disease, Diabetes Care 31: , 2003–2005. |
[156] | Sun Y , Chang YH , Chen HF , Su YH , Su HF , Li CY ((2012) ) Risk of Parkinson disease onset in patients with diabetes: A 9-year population-based cohort study with age and sex stratifications, Diabetes Care 35: , 1047–1049. |
[157] | Duarte AI , Moreira PI , Oliveira CR ((2012) ) Insulin in central nervous system: More than just a peripheral hormone, J Aging Res 2012: , 384017. |
[158] | Morris JK , Vidoni ED , Perea RD , Rada R , Johnson DK , Lyons K , Pahwa R , Burns JM , Honea RA ((2014) ) Insulin resistance and gray matter volume in neurodegenerative disease, Neuroscience 270: , 139–147. |
[159] | Takahashi M , Yamada T , Tooyama I , Moroo I , Kimura H , Yamamoto T , Okada H ((1996) ) Insulin receptor mRNA in the substantia nigra in Parkinson’s disease, Neurosci Lett 204: , 201–204. |
[160] | Athauda D , Foltynie T ((2016) ) Insulin resistance and Parkinson’s disease: A new target for disease modification? Prog Neurobiol 145-146: , 98–120. |
[161] | Tain LS , Mortiboys H , Tao RN , Ziviani E , Bandmann O , Whitworth AJ ((2009) ) Rapamycin activation of 4E-BP prevents parkinsonian dopaminergic neuron loss, Nat Neurosci 12: , 1129–1135. |
[162] | Liu S , Lu B ((2010) ) Reduction of protein translation and activation of autophagy protect against PINK1 pathogenesis in Drosophila melanogaster, PLoS Genet 6: , e1001237. |
[163] | Hang L , Thundyil J , Goh GWY , Lim KL ((2019) ) AMP kinase activation is selectively disrupted in the ventral midbrain of mice deficient in parkin or PINK1 expression, Neuromolecular Med 21: , 25–32. |
[164] | Ng CH , Guan MS , Koh C , Ouyang X , Yu F , Tan EK , O’Neill SP , Zhang X , Chung J , Lim KL ((2012) ) AMP kinase activation mitigates dopaminergic dysfunction and mitochondrial abnormalities in Drosophila models of Parkinson’s disease, J Neurosci 32: , 14311–14317. |
[165] | Chittoor-Vinod VG , Villalobos-Cantor S , Roshak H , Shea K , Abalde-Atristain L , Martin I ((2020) ) Dietary amino acids impact LRRK2-induced neurodegeneration in Parkinson’s disease models, J Neurosci 40: , 6234–6249. |
[166] | Cooper O , Seo H , Andrabi S , Guardia-Laguarta C , Graziotto J , Sundberg M , McLean JR , Carrillo-Reid L , Xie Z , Osborn T , Hargus G , Deleidi M , Lawson T , Bogetofte H , Perez-Torres E , Clark L , Moskowitz C , Mazzulli J , Chen L , Volpicelli-Daley L , Romero N , Jiang H , Uitti RJ , Huang Z , Opala G , Scarffe LA , Dawson VL , Klein C , Feng J , Ross OA , Trojanowski JQ , Lee VM , Marder K , Surmeier DJ , Wszolek ZK , Przedborski S , Krainc D , Dawson TM , Isacson O ((2012) ) Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease., Sci Transl Med 4: , 141ra190. |
[167] | Fernández-Santiago R , Martín-Flores N , Antonelli F , Cerquera C , Moreno V , Bandres-Ciga S , Manduchi E , Tolosa E , Singleton AB , Moore JH , Martí MJ , Ezquerra M , Malagelada C ((2019) ) SNCA and mTOR pathway single nucleotide polymorphisms interact to modulate the age at onset of Parkinson’s disease, Mov Disord 34: , 1333–1344. |
[168] | Lamming DW , Mihaylova MM , Katajisto P , Baar EL , Yilmaz OH , Hutchins A , Gultekin Y , Gaither R , Sabatini DM ((2014) ) Depletion of Rictor, an essential protein component of mTORC2, decreases male lifespan, Aging Cell 13: , 911–917. |
[169] | Sarbassov DD , Ali SM , Sengupta S , Sheen JH , Hsu PP , Bagley AF , Markhard AL , Sabatini DM ((2006) ) Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB, Mol Cell 22: , 159–168. |
[170] | Ellisen LW , Ramsayer KD , Johannessen CM , Yang A , Beppu H , Minda K , Oliner JD , McKeon F , Haber DA ((2002) ) REDD1, a developmentally regulated transcriptional target of p63 and p53, links p63 to regulation of reactive oxygen species, Mol Cell 10: , 995–1005. |
[171] | Gupta I , Ganguly S , Rozanas CR , Stuehr DJ , Panda K ((2016) ) Ascorbate attenuates pulmonary emphysema by inhibiting tobacco smoke and Rtp801-triggered lung protein modification and proteolysis.E, Proc Natl Acad Sci U S A 113: , 4208–4217. |
[172] | Shoshani T , Faerman A , Mett I , Zelin E , Tenne T , Gorodin S , Moshel Y , Elbaz S , Budanov A , Chajut A , Kalinski H , Kamer I , Rozen A , Mor O , Keshet E , Leshkowitz D , Einat P , Skaliter R , Feinstein E ((2002) ) Identification of a novel hypoxia-inducible factor 1-responsive gene, RTP801, involved in apoptosis, Mol Cell Biol 22: , 2283–2293. |
[173] | Malagelada C , Jin ZH , Greene LA ((2008) ) RTP801 is induced in Parkinson’s disease and mediates neuron death by inhibiting Akt phosphorylation/activation, J Neurosci 28: , 14363–14371. |
[174] | Malagelada C , Ryu EJ , Biswas SC , Jackson-Lewis V , Greene LA ((2006) ) RTP801 is elevated in Parkinson brain substantia nigral neurons and mediates death in cellular models of Parkinson’s disease by a mechanism involving mammalian target of rapamycin inactivation, J Neurosci 26: , 9996–10005. |
[175] | Covarrubias AJ , Perrone R , Grozio A , Verdin E ((2021) ) NAD(+) metabolism and its roles in cellular processes during ageing, Nat Rev Mol Cell Biol 22: , 119–141. |
[176] | Stein LR , Imai S ((2014) ) Specific ablation of Nampt in adult neural stem cells recapitulates their functional defects during aging, EMBO J 33: , 1321–1340. |
[177] | Zhu XH , Lu M , Lee BY , Ugurbil K , Chen W ((2015) ) NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences, Proc Natl Acad Sci U S A 112: , 2876–2881. |
[178] | Mouchiroud L , Houtkooper RH , Moullan N , Katsyuba E , Ryu D , Cantó C , Mottis A , Jo YS , Viswanathan M , Schoonjans K , Guarente L , Auwerx J ((2013) ) The NAD(+)/Sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling, Cell 154: , 430–441. |
[179] | Anderson RM , Bitterman KJ , Wood JG , Medvedik O , Sinclair DA ((2003) ) Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae, Nature 423: , 181–185. |
[180] | Balan V , Miller GS , Kaplun L , Balan K , Chong ZZ , Li F , Kaplun A , VanBerkum MFA , Arking R , Freeman DC , Maiese K , Tzivion G ((2008) ) Life span extension and neuronal cell protection by Drosophila nicotinamidase, J Biol Chem 283: , 27810–27819. |
[181] | Belenky P , Racette FG , Bogan KL , McClure JM , Smith JS , Brenner C ((2007) ) Nicotinamide riboside promotes Sir2 silencing and extendslifespan via Nrk and Urh1/Pnp1/Meu1 pathways to NAD+, Cell 129: , 473–484. |
[182] | Lehmann S , Costa AC , Celardo I , Loh SH , Martins LM ((2016) ) Parp mutations protect against mitochondrial dysfunction and neurodegeneration in a PARKIN model of Parkinson’s disease, Cell Death Dis 7: , e2166. |
[183] | Lehmann S , Loh SH , Martins LM ((2017) ) Enhancing NAD(+) salvage metabolism is neuroprotective in a PINK1 model of Parkinson’s disease, Biol Open 6: , 141–147. |
[184] | Ferretta A , Gaballo A , Tanzarella P , Piccoli C , Capitanio N , Nico B , Annese T , Di Paola M , Dell’aquila C , De Mari M , Ferranini E , Bonifati V , Pacelli C , Cocco T ((2014) ) Effect of resveratrol on mitochondrial function: Implications in parkin-associated familiar Parkinson’s disease, Biochim Biophys Acta 1842: , 902–915. |
[185] | Wakade C , Chong R , Bradley E , Thomas B , Morgan J ((2014) ) Upregulation of GPR109A in Parkinson’s disease, PLoS One 9: , e109818. |
[186] | Schöndorf DC , Ivanyuk D , Baden P , Sanchez-Martinez A , De Cicco S , Yu C , Giunta I , Schwarz LK , Di Napoli G , Panagiotakopoulou V , Nestel S , Keatinge M , Pruszak J , Bandmann O , Heimrich B , Gasser T , Whitworth AJ , Deleidi M ((2018) ) The NAD+ precursor nicotinamide riboside rescues mitochondrial defects and neuronal loss in iPSC and fly models of Parkinson’s disease, Cell Rep 23: , 2976–2988. |
[187] | Schwab AJ , Sison SL , Meade MR , Broniowska KA , Corbett JA , Ebert AD ((2017) ) Decreased sirtuin deacetylase activity in LRRK2 G2019S iPSC-derived dopaminergic neurons, Stem Cell Rep 9: , 1839–1852. |
[188] | Sanchez-Martinez A , Beavan M , Gegg ME , Chau KY , Whitworth AJ , Schapira AH ((2016) ) Parkinson disease-linked GBA mutation effects reversed by molecular chaperones in human cell and fly models, Sci Rep 6: , 31380. |
[189] | Imai SI , Guarente L ((2016) ) It takes two to tango: NAD(+) and sirtuins in aging/longevity control, NPJ Aging Mech Dis 2: , 16017. |
[190] | Bai P , Cantó C , Oudart H , Brunyánszki A , Cen Y , Thomas C , Yamamoto H , Huber A , Kiss B , Houtkooper RH , Schoonjans K , Schreiber V , Sauve AA , Menissier-de Murcia J , Auwerx J ((2011) ) PARP-1inhibition increases mitochondrial metabolism through SIRT1activation, Cell Metab 13: , 461–468. |
[191] | Wu XL , Wang P , Liu YH , Xue YX ((2014) ) Effects of poly (ADP-ribose) polymerase inhibitor 3-aminobenzamide on blood-brain barrier and dopaminergic neurons of rats with lipopolysaccharide-induced Parkinson’s disease, J Mol Neurosci 53: , 1–9. |
[192] | Mandir AS , Przedborski S , Jackson-Lewis V , Wang ZQ , Simbulan-Rosenthal CM , Smulson ME , Hoffman BE , Guastella DB , Dawson VL , Dawson TM ((1999) ) Poly(ADP-ribose) polymerase activation mediates 1-methyl-4-phenyl-1, 2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism, Proc Natl Acad Sci U S A 96: , 5774–5779. |
[193] | Kam TI , Mao X , Park H , Chou SC , Karuppagounder SS , Umanah GE , Yun SP , Brahmachari S , Panicker N , Chen R , Andrabi SA , Qi C , Poirier GG , Pletnikova O , Troncoso JC , Bekris LM , Leverenz JB , Pantelyat A , Ko HS , Rosenthal LS , Dawson TM , Dawson VL ((2018) ) Poly(ADP-ribose) drives pathologic α-synuclein neurodegeneration in Parkinson’s disease, Science 362: , eaat8407. |
[194] | Herranz D , Muñoz-Martin M , Cañamero M , Mulero F , Martinez-Pastor B , Fernandez-Capetillo O , Serrano M ((2010) ) Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer, Nat Commun 1: , 3. |
[195] | Kanfi Y , Naiman S , Amir G , Peshti V , Zinman G , Nahum L , Bar-Joseph Z , Cohen HY ((2012) ) The sirtuin SIRT6 regulates lifespan in male mice, Nature 483: , 218–221. |
[196] | Cao K , Dong YT , Xiang J , Xu Y , Li Y , Song H , Yu WF , Qi XL , Guan ZZ ((2020) ) The neuroprotective effects of SIRT1 in mice carrying the APP/PS1 double-transgenic mutation and in SH-SY5Y cells over-expressing human APP670/671 may involve elevated levels of α7 nicotinic acetylcholine receptors, Aging (Albany NY) 12: , 1792–1807. |
[197] | Jeong H , Cohen DE , Cui L , Supinski A , Savas JN , Mazzulli JR , Yates JR , 3rd Bordone L , Guarente L , Krainc D ((2011) ) Sirt1 mediates neuroprotection from mutant huntingtin by activation of the TORC1 and CREB transcriptional pathway, Nat Med 18: , 159–165. |
[198] | Jiang M , Wang J , Fu J , Du L , Jeong H , West T , Xiang L , Peng Q , Hou Z , Cai H , Seredenina T , Arbez N , Zhu S , Sommers K , Qian J , Zhang J , Mori S , Yang XW , Tamashiro KL , Aja S , Moran TH , Luthi-Carter R , Martin B , Maudsley S , Mattson MP , Cichewicz RH , Ross CA , Holtzman DM , Krainc D , Duan W ((2011) ) Neuroprotective role of Sirt1 in mammalian models of Huntington’s disease through activation of multiple Sirt1 targets, Nat Med 18: , 153–158. |
[199] | Kaluski S , Portillo M , Besnard A , Stein D , Einav M , Zhong L , Ueberham U , Arendt T , Mostoslavsky R , Sahay A , Toiber D ((2017) ) Neuroprotective functions for the histone deacetylase SIRT6, Cell Rep 18: , 3052–3062. |
[200] | Onn L , Portillo M , Ilic S , Cleitman G , Stein D , Kaluski S , Shirat I , Slobodnik Z , Einav M , Erdel F , Akabayov B , Toiber D ((2020) ) SIRT6 is a DNA double-strand break sensor, Elife 9: , e51636. |
[201] | Lee IH , Cao L , Mostoslavsky R , Lombard DB , Liu J , Bruns NE , Tsokos M , Alt FW , Finkel T ((2008) ) A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy, Proc Natl Acad Sci U S A 105: , 3374–3379. |
[202] | Hafner AV , Dai J , Gomes AP , Xiao CY , Palmeira CM , Rosenzweig A , Sinclair DA ((2010) ) Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy, Aging (Albany NY) 2: , 914–923. |
[203] | de Oliveira RM , Vicente Miranda H , Francelle L , Pinho R , SzegöÉM , Martinho R , Munari F , Lázaro DF , Moniot S , Guerreiro P , Fonseca-Ornelas L , Marijanovic Z , Antas P , Gerhardt E , Enguita FJ , Fauvet B , Penque D , Pais TF , Tong Q , Becker S , Kügler S , Lashuel HA , Steegborn C , Zweckstetter M , Outeiro TF ((2017) ) The mechanism ofsirtuin 2-mediated exacerbation of alpha-synuclein toxicity inmodels of Parkinson disease, PLoS Biol 15: , e2000374. |
[204] | Outeiro TF , Kontopoulos E , Altmann SM , Kufareva I , Strathearn KE , Amore AM , Volk CB , Maxwell MM , Rochet JC , McLean PJ , Young AB , Abagyan R , Feany MB , Hyman BT , Kazantsev AG ((2007) ) Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease, Science 317: , 516–519. |


