
Central and Peripheral Inflammation: Connecting the Immune Responses of Parkinson’s Disease
Abstract
Inflammation has increasingly become a focus of study in regards to Parkinson’s disease (PD). Moreover, both central and peripheral sources of inflammation have been implicated in the pathogenesis of PD. Central inflammation consisting of activated microglia, astroglia, and T cell responses within the PD central nervous system; and peripheral inflammation referring to activated innate cells and T cell signaling in the enteric nervous system, gastrointestinal tract, and blood. This review will highlight important work that further implicates central and peripheral inflammation in playing a role in PD. We also discuss how these two distant inflammations appear related and how that may be mediated by autoantigenic responses to α-syn.
INTRODUCTION
Historically, Parkinson’s disease (PD) has been studied as a disorder that primarily deteriorates the motor circuit of the individuals it affects. In the last 20 years however, several important studies have expanded the scope of PD dysfunction to other systems within the central nervous system (CNS) such as areas controlling cognition (reviewed in Aarsland et al. [1]) and neuropsychiatry (reviewed in Weintraub et al. [2]). Importantly, a growing body of evidence has shown that PD pathobiology clearly has an influence on regions outside of the central nervous system. These regions affected by PD include the peripheral enteric nervous system (ENS) [3], the gastrointestinal (GI) tract [4, 5], and peripheral blood [6]. One obvious link connecting the central and peripheral nervous system dysfunction observed in PD is the presence α-synuclein (α-syn) pathology in neurons of both locations. Another crucial connection is the growing evidence for chronic inflammation in central and peripheral nervous systems and in circulating blood of individuals with PD. Here we specifically aim to highlight the more recent works in the field of PD research detailing inflammation in the central nervous system (brain parenchyma, cerebrospinal fluid, and meninges) and inflammation in the peripheral body (ENS, GI tract, and blood)—as well as discussing how these two distant inflammations may relate to one another in the pathogenesis of PD.
INFLAMMATION IN THE CENTRAL NERVOUS SYSTEM ASSOCIATED WITH PD
CNS inflammation was first associated with PD through pathology studies detailing reactive microglia within the mesencephalon of postmortem brain samples from individuals with PD [7]. This initial observation of inflammation within the primary brain tissue afflicted in PD has only expanded and now includes several additional cell types within the parenchyma as well as the cerebrospinal fluid (CSF).
Microglial inflammation in PD
Microglia, a type of tissue resident macrophage, are the primary immune cell found throughout the CNS. In PD, microglia not only display morphologies indicative of activation, but also have been shown to express markers of inflammation such as HLA-DR [8], CD68 [9], TLR4 [10], and NLRP3 [11]. Further evidence that these molecules are involved in the inflammatory state has been established in animal models of PD, where their genetic knockout has been shown to reduce the neuroinflammation and neurodegeneration observed in those models [11–13]. More recently, triggering receptor expressed on myeloid cells 2 (TREM2), an immune receptor expressed on microglia and previously identified as harboring a genetic risk allele for PD [14], has been shown to be in disproportionate levels in the CSF of PD individuals [15]. Interestingly, overexpression of TREM2 in the CNS of mice undergoing 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) intoxication (a classical model of PD) was both anti-inflammatory and neuroprotective [16]. Generally, it appears that most of the immune molecules associated with microglia in PD are indicative of a cell type responding to environmental stress, cell death, and immune signaling—all things microglia are seemingly equipped to deal with [17]. With that said, it remains puzzling as to what factors initiate, sustain, and prevent the resolution of the pro-inflammatory microglial response in PD. One long-studied source contributing to this microglial dysfunction is the α-syn pathology harbored and propagated by neurons [18, 19]. Recently, a study by Scheiblich et al. [20] detailed an intricate system that microglia employ to cope with degrading excess amounts of α-syn pathology. One component being that “overloaded” microglia will directly transfer their excess α-syn to neighboring naïve microglia. Furthermore, they also showed that lower amounts of α-syn pathology was associated with lower inflammatory profiles within microglia. Taken together, these findings indicate that α-syn pathology is a driver of microglial activation and dysfunction in PD.
Astrocytic inflammation in PD
Although far less studied compared to microglia in the context of PD, astrocytes have increasingly been implicated in playing a role in the PD disease process. One major discovery being the characterization and identification of reactive, neurotoxic astrocytes in postmortem PD brain [21], as well as rodent models of the disease [22]. A key trigger responsible for this pro-inflammatory and neurotoxic [23] switch in astrocytes has already been identified: microglial derived pro-inflammatory factors (IL-1α, TNF, and complement) [21, 23]. This microglia-astrocytic activation axis has also been identified in multiple sclerosis [24], where it was shown that pro-inflammatory T cells interface with these innate cells as well. It seems a similar scenario is occurring in PD, with activated CNS innate cells (microglia/astrocytes) being in close proximity to responding T cells from the periphery.
CNS lymphatics and PD
The presence of T cells in the brain parenchyma of PD, while widely accepted now, was controversial at the time of its observation. Infiltration of CD4 and CD8 T cells in and around the substantia nigra [25] expanded the role of the immune system from the closed CNS and its cells to an interconnected immune response involving the periphery. This concept of neuroimmune interactions in PD has only been bolstered with the re-discovery of the CNS lymphatics [26]. Now CNS-peripheral immune interactions are better realized as the lymphatic trafficking system facilitating them is further characterized in both steady state and disease [27, 28]. In regards to the CNS lymphatics in PD, a recent publication showed that the meningeal lymphatic system itself appears impacted in individuals with PD, resulting in reduced lymphatic drainage/flow compared to age-matched healthy controls [29]. These observations lead to the hypothesis that the route in which T cells traffic through the CNS in PD is impaired, as well as the cells themselves.
CNS T cell inflammation in PD
A central remaining question surrounding the presence of T cells in the PD CNS is the nature of their inflammatory status. Are these cells providing anti-inflammatory responses to the neuronal damage or are they specifically responding with pro-inflammatory responses of their own? Evidence collected recently suggests the latter. In their latest study, Gate et al. [30] reaffirmed that CD3+ T cells can be found in close association with α-syn-laden nigral neurons of postmortem Parkinson’s disease dementia (PDD) and Lewy body dementia (LBD) brains. More importantly, they went on to detail how these parenchymal T cells had increased expression of the pro-inflammatory cytokine IL-17a. Complementary to these findings, the study also showed that T cells within the CSF of PDD/LBD patients had increased expression of a host of immune trafficking and activation markers including CXCR4 and CD69, respectively. The identification of pro-inflammatory T cells in the PD brain and CSF mirrors multiple works that have previously shown activated T cells and inflammation in PD peripheral blood [31–33] as well as in preclinical models of the disease [12, 25, 34]. Overall, this knowledge could further the potential for T cell targeting therapies in PD, with the idea being that there is a common link between central and peripheral responses and targeting one (peripheral) can benefit the other (central).
INFLAMMATION IN THE PD PERIPHERY
Though it took longer to appreciate, the field of PD research now recognizes that the inflammation associated with the disease is not just contained within the CNS. Rather, it appears that the inflammation also extends (or originates) to the ENS/GI tract as well as the blood.
ENS and GI tract inflammation in PD
Braak’s seminal work detailing the initial early staging of α-syn pathology within the olfactory nucleus and vagal nerve was a breakthrough in the study of PD-affected regions outside of the traditional nigrostriatal pathway of PD [35]. Braak then went on to expand on the vagal tract in PD by detailing α-syn inclusions with the myenteric and submucosal plexus of the ENS of postmortem PD patients [36]—providing substantial evidence that the origin of α-syn pathogenesis may occur in the gut, and then propagate into the CNS.
Alongside the α-syn pathology localized to the gut, evidence has begun to accrue which describes a dysfunctional [4] and inflamed [37] GI tract associated with PD. In addition to being impaired and pro-inflammatory, the PD GI tract is also associated with microbiome dysbiosis, with multiple studies detailing alterations in PD microbiota [38, 39] and one recent publication detailing an overabundance of opportunistic pathogens within the PD gut microbiome [40]. This evidence of a diseased GI/ENS in PD is also backed up by several findings in preclinical mouse models of the disease. For instance, several groups have shown that α-syn inoculation in the duodenal wall of mice is sufficient to seed and propagate α-syn pathology/dysfunction from the ENS into several relevant PD CNS sites [41, 42]. Remarkably, this gut-seeded α-syn also induced local GI inflammation (including heightened IL-6) [41] and that this prion propagation of α-syn dysfunction could be halted by vagotomy [42]. Focusing more on potential autoimmune-drivers, a preprint publication [43] reports gut inflammation and a loss of enteric neurons in mice after their immunization with an antigenic α-syn epitope (previously identified in human PD [32]). Notably, when the group depleted CD4 T cells during this α-syn immunization paradigm—enteric neurons were partially rescued. In terms of preclinical PD microbiota work, one study has shown that α-syn overexpressing mice reconstituted with PD microbiota had exacerbated α-syn pathology, microglial activation, and motor deficits compared to normal mouse flora [44]. Lastly, in regards to peripheral gut inflammation and PD, a potential link to inflammatory bowel disease (IBD) has been described. This association gained traction when LRRK2, a common genetic risk factor for PD, was also found to be associated with cases of ulcerative colitis [45]. Perhaps more compelling, it has since been observed that IBD patients are at a higher risk for developing PD [46], and among IBD patients, those taking immunomodulatory anti-TNF treatment had a reduced risk compared to those not on anti-TNF [47]. Taken together, it may be that α-syn begins its PD pathogenesis in ENS of the GI through the potentiation by environmental stress, pathogenic bacteria, and/or misguided immune responses. Now established in the gut, α-syn pathology propagates up the vagus nerve and into the CNS. One potential link between these two processes being the circulating blood immune responses that survey (and remember) both systems.
Blood inflammation in PD
Given the systemic inflammatory immune response observed in multiple PD tissues, it makes sense that this would also be captured in the major pathway for immune cell trafficking in the body, the blood. Indeed, several studies examining the blood of individuals with PD have noted increases in numerous pro-inflammatory cytokines including IL-6, TNF, and IL-2 (reviewed in [48]). In addition to this overabundance of pro-inflammatory cytokines, altered immune cell compositions have been observed in PD as well. For example, PD blood-monocyte populations have been shown to have more proliferative capacity compared to age-matched healthy controls [49]. This immune dysregulation in PD also includes the adaptive immune system. Not only has it been reported that there are increases in IL-17 [31, 33], IL-4 [32, 33], and IFNγ [32, 33] producing T cells, but the target of some of these T cell responses has been identified—α-syn. Indeed, it appears that the pathological hallmark protein of PD, is also the antigenic target of some pro-inflammatory T cells found in the blood [30, 32, 50, 51] and CSF [30, 52] of individuals with PD. Interestingly, the same study that showed increased IL-17a expression in blood T cells stimulated with α-syn peptide also reported increases of IL-17a expressing T cells in the substantia nigra (reviewed earlier) of PDD/LBD patients. While these experiments fall short of directly linking peripheral α-syn specific T cell responses with those T cell response occurring directly in the CNS, the implication is hard to ignore and should be the subject of future studies.
CONNECTING THE CENTRAL AND PERIPHERAL IMMUNE RESPONSES IN PD
The pathobiology of PD has expanded in scope several times since its initial description by James Parkinson in 1817. The disease process of PD is now recognized to affect non-motor regions of the brain as well as areas outside of the CNS—mainly the ENS and the GI tract it innervates. Two key characteristics that appear universally shared by these PD-affected regions are the pathological misfolding of α-syn and increased inflammation. And as we have reviewed here, α-syn can directly potentiate inflammatory responses from both the innate (macrophages/microglia/astrocytes) and adaptive (T cells) arms of the immune system. Another important connection between α-syn pathology and inflammatory immune responses is that both appear to be occurring in PD before the onset of overt motor symptoms. Longstanding work [35, 36] has suggested that α-syn pathology originates in the ENS early and spreads to the CNS later in disease. Newer work suggests that an aspect of the PD immune response may also be occurring long before the establishment of overt neurodegeneration. That is, α-syn specific T cell responses have been observed to be at their highest around the initial diagnosis of PD and wane with progression [50]. Perhaps more compelling, the same study also reported on a longitudinal case of PD and showed that α-syn specific T cell responses were increased and present many years before that individual displayed symptoms and was subsequently diagnosed.
One theory connecting these findings all together (and illustrated in Fig. 1) is that early pathological events taking place in the GI/ENS involve both α-syn dysfunction and inflammatory immune responses. More specifically, dopaminergic enteric neurons acquire initial α-syn pathology through a combination of environmental, genetic, microbiome, and immune factors. This misfolded α-syn species may now be the new target of immune responses from both surrounding macrophages as well as T cells—leading to excess inflammation and neuronal death. Important to note, these excessive PD immune responses also appear to have a genetic basis (similar to α-syn pathology), with several reports detailing PD related genes/mutations promoting inflammation [53]. For instance, the G2019S LRRK2 mutation [54] as well as L444P/N370S GBA mutations [55, 56] being associated with hyper-inflammatory states.
Fig. 1
Connecting central and peripheral immune responses in Parkinson’s disease. Inflammatory and damaging events in the gastrointestinal tract (duodenum) caused by a combination of environmental, genetic, microbial, and immune factors leads to α-synuclein (α-syn) pathology in the innervating enteric nervous system (ENS). Misfolded α-syn species within the ENS may become targets for resident macrophages (MØ) with subsequent antigen presentation to T cells that misrecognize self-peptide and possibly expand in the gut-draining mesenteric lymph nodes (MLN). This α-syn provoked inflammatory response promotes further α-syn pathology and eventual propagation to the central nervous system via the vagus nerve. Now α-syn pathology with neurons leads to similar inflammation as described in the gut. With microglia taking up pathogenic α-syn—leading to their activation (and subsequent activation of neurotoxic astrocytes) and presentation of α-syn antigen to cerebrospinal fluid (CSF) patrolling T cells. Another link between the two distant systems is also the bloodstream that connects them. T cells and other immune cells may be able to extravagate through the blood-brain barrier in response to heightened inflammation coming from the brain parenchyma/CSF. Lastly, similar to the MLN, T cells may be encountering and expanding to α-syn and other autoinflammatory neuroantigens within the central nervous system draining deep cervical lymph node (dCLN). DA, dopamine; cDC, classical dendritic cell.
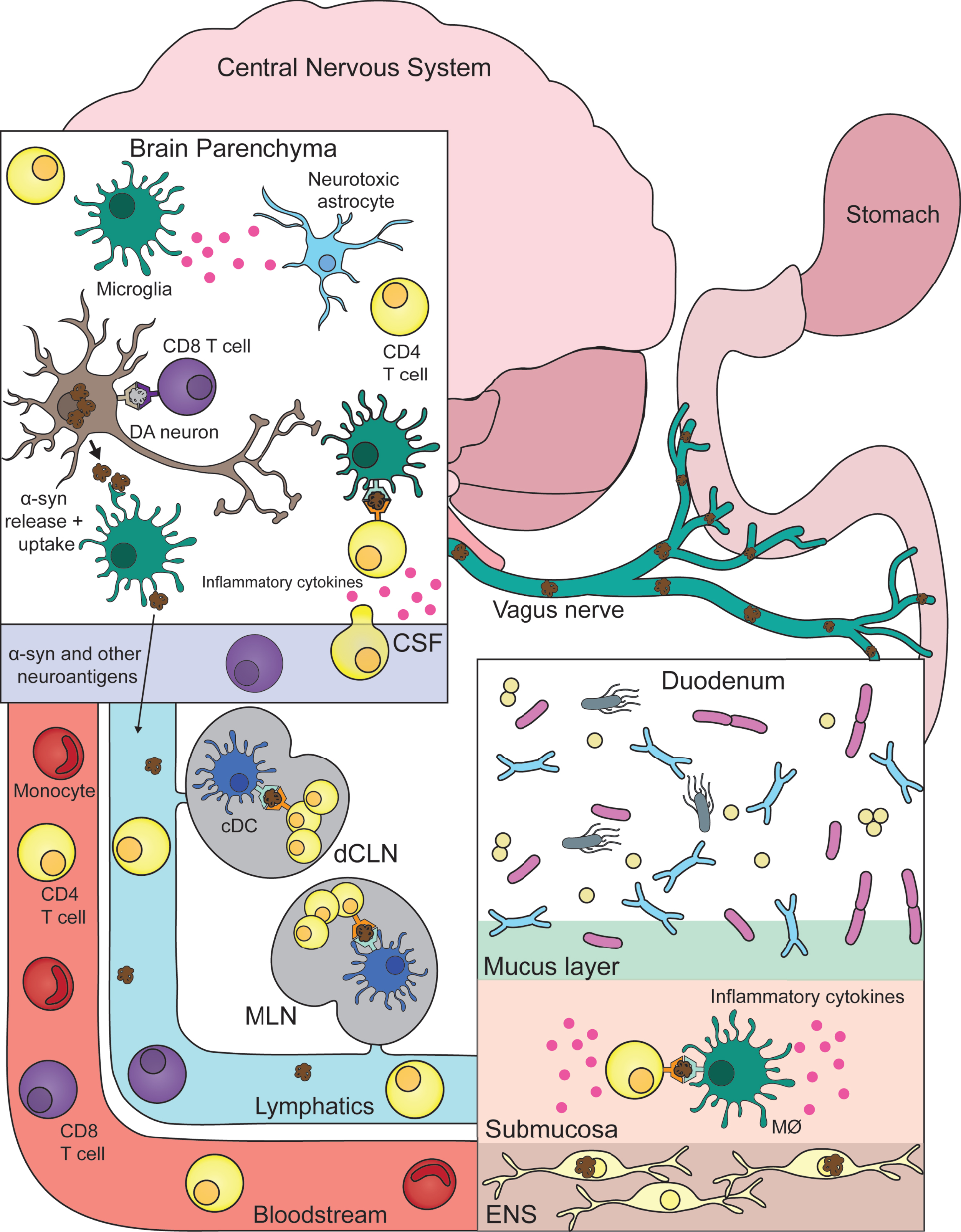
Over time, prion-like propagation of α-syn pathology ascends the vagus nerve into the CNS proper. Similar to ENS neurons, CNS dopaminergic neurons are now negatively affected by α-syn pathology and elicit heightened immune responses from microglia. Now, previously ENS primed circulating α-syn specific T cells produce their same pro-inflammatory response, but now leading to the destruction of CNS neurons. Obviously future work is needed to better realize and substantiate this theory but for now it serves as an aid in marrying peripheral and central inflammation to the pathogenesis of PD. One example of a pressing question that should be addressed in the aforementioned future work being: Are there additional neuroantigens being targeted by autoimmune T cells in PD? With multiple autoantigens indeed being the case for other autoimmune diseases such as multiple sclerosis [57] or lupus erythematosus [58].
In regards to viewing and studying PD as a potential inflammatory disorder, it is important to note and discuss the fact that PD clinical trials targeting immune features of PD have all virtually failed to achieve meaningful benefit for individuals with PD [53]. This could be in part due to their method and rationale for therapy, e.g., many trials are focused on immunization/antibody targeting of α-syn. In Alzheimer’s disease (AD), the targeting/clearance of amyloid-β (Aβ) has been a focus of an intense amount of research that has ultimately cast into doubt whether the reduction of amyloid load in AD patients is actually therapeutic [59]. The same may be true for α-syn in PD, or at the very least, this type of therapy as well as the other anti-inflammatory interventions require prodromal or preclinical PD patients to actually prevent or slow the disease. So even though previous immune-related PD clinical trials have been unsuccessful, it should be a priority in the field to learn and improve upon them to make way for novel immunotherapeutic approaches and patient populations.
CONCLUSION
In conclusion, there are distinct central and peripheral inflammatory responses observed in the brain, blood, and gut of individuals with PD. These inflammatory responses originate from several different cell types including microglia, astrocytes, and T cells. The target of many of these responses are associated with α-syn pathology and/or neurodegeneration. One potential link proposed here is that initial ENS α-syn pathology potentiates a destructive autoinflammatory immune response that then later affects the CNS. Future work is required to better understand these central/peripheral inflammations and how they might be leveraged in the development of immunotherapies for the treatment of PD.
ACKNOWLEDGMENTS
This study was supported by the joint efforts of The Michael J. Fox Foundation for Parkinson’s Research (MJFF) and the Aligning Science Across Parkinson’s (ASAP) initiative as well as the NIH T32AI125179 (GPW). MJFF administers the grant ASAP-000375 on behalf of the ASAP and itself.
CONFLICT OF INTEREST
The authors have no conflicts of interest to report.
REFERENCES
[1] | Aarsland D , Creese B , Politis M , Chaudhuri KR , Ffytche DH , Weintraub D , Ballard C ((2017) ) Cognitive decline in Parkinson disease. Nat Rev Neurol 13: , 217–231. |
[2] | Weintraub D , Aarsland D , Chaudhuri KR , Dobkin RD , Leentjens AF , Rodriguez-Violante M , Schrag A ((2022) ) The neuropsychiatry of Parkinson’s disease: Advances and challenges. Lancet Neurol 21: , 89–102. |
[3] | Chalazonitis A , Rao M ((2018) ) Enteric nervous system manifestations of neurodegenerative disease. Brain Res 1693: , 207–213. |
[4] | Fasano A , Visanji NP , Liu LW , Lang AE , Pfeiffer RF ((2015) ) Gastrointestinal dysfunction in Parkinson’s disease. Lancet Neurol 14: , 625–639. |
[5] | Romano S , Savva GM , Bedarf JR , Charles IG , Hildebrand F , Narbad A ((2021) ) Meta-analysis of the Parkinson’s disease gut microbiome suggests alterations linked to intestinal inflammation. NPJ Parkinsons Dis 7: , 27. |
[6] | Schonhoff AM , Williams GP , Wallen ZD , Standaert DG , Harms AS ((2020) ) Innate and adaptive immune responses in Parkinson’s disease. Prog Brain Res 252: , 169–216. |
[7] | Foix C , Nicolesco J ((1925) ) Anatomie cérébrale: Les noyaux gris centraux et la région mésencéphalo-sous-optique, suivie d’un appendice sur l’anatomie pathologique de la maladie de Parkinson, Masson et cie. |
[8] | McGeer PL , Itagaki S , Boyes BE , McGeer EG ((1988) ) Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38: , 1285–1291. |
[9] | Croisier E , Moran LB , Dexter DT , Pearce RK , Graeber MB ((2005) ) Microglial inflammation in the parkinsonian substantia nigra: Relationship to alpha-synuclein deposition. J Neuroinflammation 2: , 14. |
[10] | Drouin-Ouellet J , St-Amour I , Saint-Pierre M , Lamontagne-Proulx J , Kriz J , Barker RA , Cicchetti F ((2014) ) Toll-like receptor expression in the blood and brain of patients and a mouse model of Parkinson’s disease. Int J Neuropsychopharmacol 18: , pyu103. |
[11] | Gordon R , Albornoz EA , Christie DC , Langley MR , Kumar V , Mantovani S , Robertson AAB , Butler MS , Rowe DB , O’Neill LA , Kanthasamy AG , Schroder K , Cooper MA , Woodruff TM ((2018) ) Inflammasome inhibition prevents alpha-synuclein pathology and dopaminergic neurodegeneration in mice. Sci Transl Med 10: , eaah4066. |
[12] | Harms AS , Cao S , Rowse AL , Thome AD , Li X , Mangieri LR , Cron RQ , Shacka JJ , Raman C , Standaert DG ((2013) ) MHCII is required for alpha-synuclein-induced activation of microglia, CD4 T cell proliferation, and dopaminergic neurodegeneration. J Neurosci 33: , 9592–9600. |
[13] | Shao QH , Chen Y , Li FF , Wang S , Zhang XL , Yuan YH , Chen NH ((2019) ) TLR4 deficiency has a protective effect in the MPTP/probenecid mouse model of Parkinson’s disease. Acta Pharmacol Sin 40: , 1503–1512. |
[14] | Rayaprolu S , Mullen B , Baker M , Lynch T , Finger E , Seeley WW , Hatanpaa KJ , Lomen-Hoerth C , Kertesz A , Bigio EH , Lippa C , Josephs KA , Knopman DS , White CL , 3rd, Caselli R , Mackenzie IR , Miller BL , Boczarska-Jedynak M , Opala G , Krygowska-Wajs A , Barcikowska M , Younkin SG , Petersen RC , Ertekin-Taner N , Uitti RJ , Meschia JF , Boylan KB , Boeve BF , Graff-Radford NR , Wszolek ZK , Dickson DW , Rademakers R , Ross OA ((2013) ) TREM2 in neurodegeneration: Evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson’s disease. Mol Neurodegener 8: , 19. |
[15] | Wilson EN , Swarovski MS , Linortner P , Shahid M , Zuckerman AJ , Wang Q , Channappa D , Minhas PS , Mhatre SD , Plowey ED , Quinn JF , Zabetian CP , Tian L , Longo FM , Cholerton B , Montine TJ , Poston KL , Andreasson KI ((2020) ) Soluble TREM2 is elevated in Parkinson’s disease subgroups with increased CSF tau. Brain 143: , 932–943. |
[16] | Ren M , Guo Y , Wei X , Yan S , Qin Y , Zhang X , Jiang F , Lou H ((2018) ) TREM2 overexpression attenuates neuroinflammation and protects dopaminergic neurons in experimental models of Parkinson’s disease. Exp Neurol 302: , 205–213. |
[17] | Deczkowska A , Keren-Shaul H , Weiner A , Colonna M , Schwartz M , Amit I ((2018) ) Disease-associated microglia: A universal immune sensor of neurodegeneration. Cell 173: , 1073–1081. |
[18] | Burre J , Sharma M , Sudhof TC ((2018) ) Cell biology and pathophysiology of alpha-synuclein. Cold Spring Harb Perspect Med 8: , a024091. |
[19] | Ferreira SA , Romero-Ramos M ((2018) ) Microglia response during Parkinson’s disease: Alpha-synuclein intervention. Front Cell Neurosci 12: , 247. |
[20] | Scheiblich H , Dansokho C , Mercan D , Schmidt SV , Bousset L , Wischhof L , Eikens F , Odainic A , Spitzer J , Griep A , Schwartz S , Bano D , Latz E , Melki R , Heneka MT ((2021) ) Microglia jointly degrade fibrillar alpha-synuclein cargo by distribution through tunneling nanotubes. Cell 184: , 5089–5106 e5021. |
[21] | Liddelow SA , Guttenplan KA , Clarke LE , Bennett FC , Bohlen CJ , Schirmer L , Bennett ML , Munch AE , Chung WS , Peterson TC , Wilton DK , Frouin A , Napier BA , Panicker N , Kumar M , Buckwalter MS , Rowitch DH , Dawson VL , Dawson TM , Stevens B , Barres BA ((2017) ) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541: , 481–487. |
[22] | Yun SP , Kam TI , Panicker N , Kim S , Oh Y , Park JS , Kwon SH , Park YJ , Karuppagounder SS , Park H , Kim S , Oh N , Kim NA , Lee S , Brahmachari S , Mao X , Lee JH , Kumar M , An D , Kang SU , Lee Y , Lee KC , Na DH , Kim D , Lee SH , Roschke VV , Liddelow SA , Mari Z , Barres BA , Dawson VL , Lee S , Dawson TM , Ko HS ((2018) ) Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat Med 24: , 931–938. |
[23] | Guttenplan KA , Weigel MK , Prakash P , Wijewardhane PR , Hasel P , Rufen-Blanchette U , Munch AE , Blum JA , Fine J , Neal MC , Bruce KD , Gitler AD , Chopra G , Liddelow SA , Barres BA ((2021) ) Neurotoxic reactive astrocytes induce cell death via saturated lipids. Nature 599: , 102–107. |
[24] | Absinta M , Maric D , Gharagozloo M , Garton T , Smith MD , Jin J , Fitzgerald KC , Song A , Liu P , Lin JP , Wu T , Johnson KR , McGavern DB , Schafer DP , Calabresi PA , Reich DS ((2021) ) A lymphocyte-microglia-astrocyte axis in chronic active multiple sclerosis. Nature 597: , 709–714. |
[25] | Brochard V , Combadiere B , Prigent A , Laouar Y , Perrin A , Beray-Berthat V , Bonduelle O , Alvarez-Fischer D , Callebert J , Launay JM , Duyckaerts C , Flavell RA , Hirsch EC , Hunot S ((2009) ) Infiltration of CD4+lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest 119: , 182–192. |
[26] | Louveau A , Smirnov I , Keyes TJ , Eccles JD , Rouhani SJ , Peske JD , Derecki NC , Castle D , Mandell JW , Lee KS , Harris TH , Kipnis J ((2015) ) Structural and functional features of central nervous system lymphatic vessels. Nature 523: , 337–341. |
[27] | Louveau A , Herz J , Alme MN , Salvador AF , Dong MQ , Viar KE , Herod SG , Knopp J , Setliff JC , Lupi AL , Da Mesquita S , Frost EL , Gaultier A , Harris TH , Cao R , Hu S , Lukens JR , Smirnov I , Overall CC , Oliver G , Kipnis J ((2018) ) CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat Neurosci 21: , 1380–1391. |
[28] | Rustenhoven J , Drieu A , Mamuladze T , de Lima KA , Dykstra T , Wall M , Papadopoulos Z , Kanamori M , Salvador AF , Baker W , Lemieux M , Da Mesquita S , Cugurra A , Fitzpatrick J , Sviben S , Kossina R , Bayguinov P , Townsend RR , Zhang Q , Erdmann-Gilmore P , Smirnov I , Lopes MB , Herz J , Kipnis J ((2021) ) Functional characterization of the dural sinuses as a neuroimmune interface. Cell 184: , 1000–1016 e1027. |
[29] | Ding XB , Wang XX , Xia DH , Liu H , Tian HY , Fu Y , Chen YK , Qin C , Wang JQ , Xiang Z , Zhang ZX , Cao QC , Wang W , Li JY , Wu E , Tang BS , Ma MM , Teng JF , Wang XJ ((2021) ) Impaired meningeal lymphatic drainage in patients with idiopathic Parkinson’s disease. Nat Med 27: , 411–418. |
[30] | Gate D , Tapp E , Leventhal O , Shahid M , Nonninger TJ , Yang AC , Strempfl K , Unger MS , Fehlmann T , Oh H , Channappa D , Henderson VW , Keller A , Aigner L , Galasko DR , Davis MM , Poston KL , Wyss-Coray T ((2021) ) CD4(+) T cells contribute to neurodegeneration in Lewy body dementia. Science 374: , 868–874. |
[31] | Sommer A , Marxreiter F , Krach F , Fadler T , Grosch J , Maroni M , Graef D , Eberhardt E , Riemenschneider MJ , Yeo GW , Kohl Z , Xiang W , Gage FH , Winkler J , Prots I , Winner B ((2018) ) Th17 lymphocytes induce neuronal cell death in a human iPSC-based model of Parkinson’s disease. Cell Stem Cell 23: , 123–131 e126. |
[32] | Sulzer D , Alcalay RN , Garretti F , Cote L , Kanter E , Agin-Liebes J , Liong C , McMurtrey C , Hildebrand WH , Mao X , Dawson VL , Dawson TM , Oseroff C , Pham J , Sidney J , Dillon MB , Carpenter C , Weiskopf D , Phillips E , Mallal S , Peters B , Frazier A , Lindestam Arlehamn CS , Sette A ((2017) ) T cells from patients with Parkinson’s disease recognize alpha-synuclein peptides. Nature 546: , 656–661. |
[33] | Yan Z , Yang W , Wei H , Dean MN , Standaert DG , Cutter GR , Benveniste EN , Qin H ((2021) ) Dysregulation of the adaptive immune system in patients with early-stage Parkinson disease. Neurol Neuroimmunol Neuroinflamm 8: , e1036. |
[34] | Williams GP , Schonhoff AM , Jurkuvenaite A , Gallups NJ , Standaert DG , Harms AS ((2021) ) CD4 T cells mediate brain inflammation and neurodegeneration in a mouse model of Parkinson’s disease. Brain 144: , 2047–2059. |
[35] | Braak H , Del Tredici K , Rub U , de Vos RA , Jansen Steur EN , Braak E ((2003) ) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24: , 197–211. |
[36] | Braak H , de Vos RA , Bohl J , Del Tredici K ((2006) ) Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci Lett 396: , 67–72. |
[37] | Devos D , Lebouvier T , Lardeux B , Biraud M , Rouaud T , Pouclet H , Coron E , Bruley des Varannes S , Naveilhan P , Nguyen JM , Neunlist M , Derkinderen P ((2013) ) Colonic inflammation in Parkinson’s disease. Neurobiol Dis 50: , 42–48. |
[38] | Aho VTE , Pereira PAB , Voutilainen S , Paulin L , Pekkonen E , Auvinen P , Scheperjans F ((2019) ) Gut microbiota in Parkinson’s disease: Temporal stability and relations to disease progression. EBioMedicine 44: , 691–707. |
[39] | Barichella M , Severgnini M , Cilia R , Cassani E , Bolliri C , Caronni S , Ferri V , Cancello R , Ceccarani C , Faierman S , Pinelli G , De Bellis G , Zecca L , Cereda E , Consolandi C , Pezzoli G ((2019) ) Unraveling gut microbiota in Parkinson’s disease and atypical parkinsonism. Mov Disord 34: , 396–405. |
[40] | Wallen ZD , Appah M , Dean MN , Sesler CL , Factor SA , Molho E , Zabetian CP , Standaert DG , Payami H ((2020) ) Characterizing dysbiosis of gut microbiome in PD: Evidence for overabundance of opportunistic pathogens. NPJ Parkinsons Dis 6: , 11. |
[41] | Challis C , Hori A , Sampson TR , Yoo BB , Challis RC , Hamilton AM , Mazmanian SK , Volpicelli-Daley LA , Gradinaru V ((2020) ) Gut-seeded alpha-synuclein fibrils promote gut dysfunction and brain pathology specifically in aged mice. Nat Neurosci 23: , 327–336. |
[42] | Kim S , Kwon SH , Kam TI , Panicker N , Karuppagounder SS , Lee S , Lee JH , Kim WR , Kook M , Foss CA , Shen C , Lee H , Kulkarni S , Pasricha PJ , Lee G , Pomper MG , Dawson VL , Dawson TM , Ko HS ((2019) ) Transneuronal propagation of pathologic alpha-synuclein from the gut to the brain models Parkinson’s disease. Neuron 103: , 627–641 e627. |
[43] | Garretti F , Monahan C , Sloan N , Shariar S , Kim SW , Sette A , Cutforth T , Kanter E , Agalliu D , Sulzer D ((2022) ) Interaction of an α-synuclein epitope with HLA-DRB1*15:01 initiates early enteric features of Parkinson’s disease in humanized mice. bioRxiv, 2022.2002.2003.479014. |
[44] | Sampson TR , Debelius JW , Thron T , Janssen S , Shastri GG , Ilhan ZE , Challis C , Schretter CE , Rocha S , Gradinaru V , Chesselet MF , Keshavarzian A , Shannon KM , Krajmalnik-Brown R , Wittung-Stafshede P , Knight R , Mazmanian SK ((2016) ) Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 167: , 1469–1480 e1412. |
[45] | Franke A , McGovern DP , Barrett JC , Wang K , Radford-Smith GL , Ahmad T , Lees CW , Balschun T , Lee J , Roberts R , Anderson CA , Bis JC , Bumpstead S , Ellinghaus D , Festen EM , Georges M , Green T , Haritunians T , Jostins L , Latiano A , Mathew CG , Montgomery GW , Prescott NJ , Raychaudhuri S , Rotter JI , Schumm P , Sharma Y , Simms LA , Taylor KD , Whiteman D , Wijmenga C , Baldassano RN , Barclay M , Bayless TM , Brand S , Buning C , Cohen A , Colombel JF , Cottone M , Stronati L , Denson T , De Vos M , D’Inca R , Dubinsky M , Edwards C , Florin T , Franchimont D , Gearry R , Glas J , Van Gossum A , Guthery SL , Halfvarson J , Verspaget HW , Hugot JP , Karban A , Laukens D , Lawrance I , Lemann M , Levine A , Libioulle C , Louis E , Mowat C , Newman W , Panes J , Phillips A , Proctor DD , Regueiro M , Russell R , Rutgeerts P , Sanderson J , Sans M , Seibold F , Steinhart AH , Stokkers PC , Torkvist L , Kullak-Ublick G , Wilson D , Walters T , Targan SR , Brant SR , Rioux JD , D’Amato M , Weersma RK , Kugathasan S , Griffiths AM , Mansfield JC , Vermeire S , Duerr RH , Silverberg MS , Satsangi J , Schreiber S , Cho JH , Annese V , Hakonarson H , Daly MJ , Parkes M ((2010) ) Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet 42: , 1118–1125. |
[46] | Villumsen M , Aznar S , Pakkenberg B , Jess T , Brudek T ((2019) ) Inflammatory bowel disease increases the risk of Parkinson’s disease: A Danish nationwide cohort study 1977-2014. Gut 68: , 18–24. |
[47] | Peter I , Dubinsky M , Bressman S , Park A , Lu C , Chen N , Wang A ((2018) ) Anti-tumor necrosis factor therapy and incidence of Parkinson disease among patients with inflammatory bowel disease. JAMA Neurol 75: , 939–946. |
[48] | Qin XY , Zhang SP , Cao C , Loh YP , Cheng Y ((2016) ) Aberrations in peripheral inflammatory cytokine levels in Parkinson disease: A systematic review and meta-analysis. JAMA Neurol 73: , 1316–1324. |
[49] | Nissen SK , Shrivastava K , Schulte C , Otzen DE , Goldeck D , Berg D , Moller HJ , Maetzler W , Romero-Ramos M ((2019) ) Alterations in blood monocyte functions in Parkinson’s disease. Mov Disord 34: , 1711–1721. |
[50] | Lindestam Arlehamn CS , Dhanwani R , Pham J , Kuan R , Frazier A , Rezende Dutra J , Phillips E , Mallal S , Roederer M , Marder KS , Amara AW , Standaert DG , Goldman JG , Litvan I , Peters B , Sulzer D , Sette A ((2020) ) alpha-Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson’s disease. Nat Commun 11: , 1875. |
[51] | Lodygin D , Hermann M , Schweingruber N , Flugel-Koch C , Watanabe T , Schlosser C , Merlini A , Korner H , Chang HF , Fischer HJ , Reichardt HM , Zagrebelsky M , Mollenhauer B , Kugler S , Fitzner D , Frahm J , Stadelmann C , Haberl M , Odoardi F , Flugel A ((2019) ) beta-Synuclein-reactive T cells induce autoimmune CNS grey matter degeneration. Nature 566: , 503–508. |
[52] | Wang P , Yao L , Luo M , Zhou W , Jin X , Xu Z , Yan S , Li Y , Xu C , Cheng R , Huang Y , Lin X , Ma K , Cao H , Liu H , Xue G , Han F , Nie H , Jiang Q ((2021) ) Single-cell transcriptome and TCR profiling reveal activated and expanded T cell populations in Parkinson’s disease. Cell Discovery 7: , 52. |
[53] | Tansey MG , Wallings RL , Houser MC , Herrick MK , Keating CE , Joers V ((2022) ) Inflammation and immune dysfunction in Parkinson disease. Nat Rev Immunol, doi: 10.1038/s41577-022-00684-6 |
[54] | Dzamko N , Rowe DB , Halliday GM ((2016) ) Increased peripheral inflammation in asymptomatic leucine-rich repeat kinase 2 mutation carriers. Mov Disord 31: , 889–897. |
[55] | Chahine LM , Qiang J , Ashbridge E , Minger J , Yearout D , Horn S , Colcher A , Hurtig HI , Lee VM , Van Deerlin VM , Leverenz JB , Siderowf AD , Trojanowski JQ , Zabetian CP , Chen-Plotkin A ((2013) ) Clinical and biochemical differences in patients having Parkinson disease with vs without GBA mutations. JAMA Neurol 70: , 852–858. |
[56] | Panicker LM , Miller D , Park TS , Patel B , Azevedo JL , Awad O , Masood MA , Veenstra TD , Goldin E , Stubblefield BK , Tayebi N , Polumuri SK , Vogel SN , Sidransky E , Zambidis ET , Feldman RA ((2012) ) Induced pluripotent stem cell model recapitulates pathologic hallmarks of Gaucher disease. Proc Natl Acad Sci U S A 109: , 18054–18059. |
[57] | Riedhammer C , Weissert R ((2015) ) Antigen presentation, autoantigens, and immune regulation in multiple sclerosis and other autoimmune diseases. Front Immunol 6: , 322. |
[58] | Doyle HA , Yan J , Liang B , Mamula MJ ((2001) ) Lupus autoantigens: Their origins, forms, and presentation. Immunol Res 24: , 131–147. |
[59] | Panza F , Lozupone M , Logroscino G , Imbimbo BP ((2019) ) A critical appraisal of amyloid-beta-targeting therapies for Alzheimer disease. Nat Rev Neurol 15: , 73–88. |


