
The Role of Pathogens and Anti-Infective Agents in Parkinson’s Disease, from Etiology to Therapeutic Implications
Abstract
Parkinson’s disease is a debilitating neurodegenerative disorder whose etiology is still unclear, hampering the development of effective treatments. There is an urgent need to identify the etiology and provide further effective treatments. Recently, accumulating evidence has indicated that infection may play a role in the etiology of Parkinson’s disease. The infective pathogens may act as a trigger for Parkinson’s disease, the most common of which are hepatitis C virus, influenza virus, and Helicobacter pylori. In addition, gut microbiota is increasingly recognized to influence brain function through the gut-brain axis, showing an important role in the pathogenesis of Parkinson’s disease. Furthermore, a series of anti-infective agents exhibit surprising neuroprotective effects via various mechanisms, such as interfering with α-synuclein aggregation, inhibiting neuroinflammation, attenuating oxidative stress, and preventing from cell death, independent of their antimicrobial effects. The pleiotropic agents affect important events in the pathogenesis of Parkinson’s disease. Moreover, most of them are less toxic, clinically safe and have good blood-brain penetrability, making them hopeful candidates for the treatment of Parkinson’s disease. However, the use of antibiotics and subsequent gut dysbiosis may also play a role in Parkinson’s disease, making the long-term effects of anti-infective drugs worthy of further consideration and exploration. This review summarizes the current evidence for the association between infective pathogens and Parkinson’s disease and subsequently explores the application prospects of anti-infective drugs in Parkinson’s disease treatment, providing novel insights into the pathogenesis and treatment of Parkinson’s disease.
INTRODUCTION
Parkinson’s disease (PD) is the second most common neurodegenerative disease worldwide, ranking only after Alzheimer’s disease [1]. The incidence of PD in people aged 65 years and older reaches 2% and increases rapidly with age [2]. Pathologically, PD is characterized by the loss of nigrostriatal dopamine neurons and the formation of Lewy bodies [3]. However, the etiology and underlying pathogenesis remain unknown. A range of genetic and environmental factors appear to play a role in the pathogenesis of PD [4]. It is generally believed that oxidative stress, α-synuclein (α-syn) aggregation, neuroinflammation, and mitochondrial dysfunction are implicated in the pathogenesis [5–7]. However, to date, there is no cure or disease-modifying agent available for PD. The current drugs for the treatment of PD mainly alleviate motor symptoms, but they cannot slow the progression of the disease [8]. Therefore, it is imperative to determine the actual etiology and develop drugs that can cure the disease at its source.
Accumulating evidence has revealed that infection may play a critical role in neurological diseases, such as herpes simplex virus in Alzheimer’s disease [9], Enterovirus [10] and human herpesvirus [11] in amyotrophic lateral sclerosis, and Zika virus in microcephaly [12]. Since the outbreak of encephalitic lethargica (EL) and post-encephalitic Parkinsonism (PEP) after the 1918 H1N1 influenza pandemic [13, 14], the possibility of an infective etiology for PD has been intensely discussed [15] (Fig. 1). Apart from this, many recent studies have reported that traditional anti-infective drugs exhibit novel neuroprotective effects, which are separate from their antimicrobial effects, showing a promising potential in treating PD [16, 17].
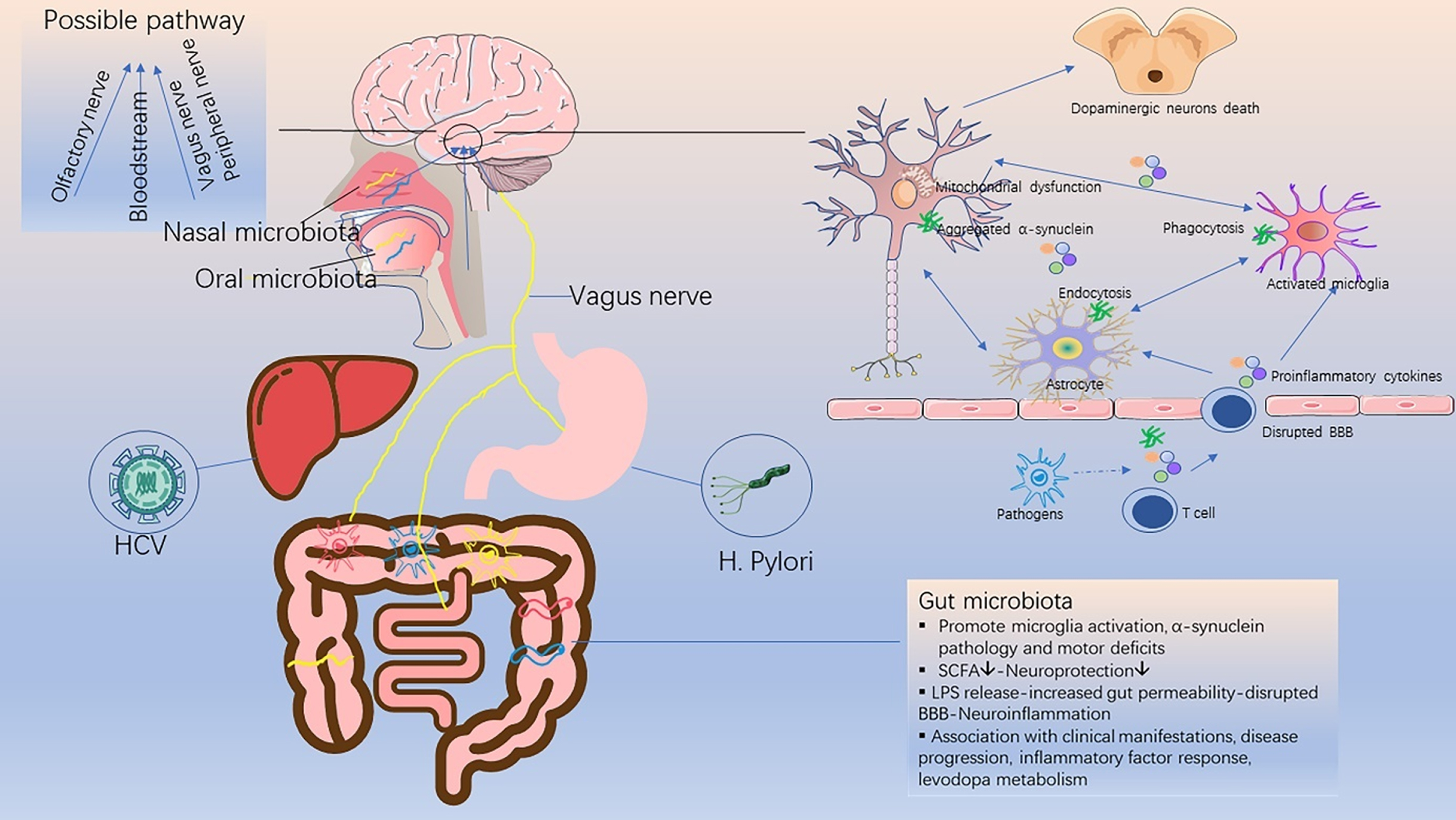
Fig. 1
Schematic diagram of the relationship between infective pathogens and Parkinson’s disease. Infective pathogens enter the central nervous system through the bloodstream or nerves, causing inflammatory response and blood-brain barrier disruption through the release of pro-inflammatory cytokines. This subsequently leads to a series of glial activation, neuroinflammation, α-synuclein accumulation and neuronal death, which trigger or accelerate the onset of PD. SCFA, short chain fatty acids; LPS, lipopolysaccharide; BBB, blood-brain barrier.
This review aims to summarize the current evidence for the association between infective pathogens and PD (Table 1), highlight recent studies with anti-infective agents in PD (Table 2), and provide new insights into the pathogenesis and treatment of PD.
Table 1
Association of infective pathogens in the development of Parkinson’s disease and possible pathogenesis
| Infective pathogens | The association between infective pathogens and PD | Indicated role in PD pathogenesis |
| HCV | •Patients with HCV infection have a significantly increased risk of developing PD [20–25] | •Essential HCV receptors were expressed on the brain microvascular endothelial cells, resulting in the viral entry and disruption in CNS [30] |
| •Interferon-based antiviral treatment against HCV is associated with a reduced risk of PD [28, 29] | •The neurotoxic effects of HCV on dopaminergic neurons [20] | |
| •The detection of HCV RNA sequence in postmortem brain tissue samples [31] | •The release of substantial inflammatory cytokines caused by HCV infection [20] | |
| •The alteration of dopaminergic neurotransmission in HCV-infected patients [32] | ||
| Influenza virus | •The outbreak of EL and PEP following the 1918 H1N1 influenza pandemic [13] | •Activating the innate immune response [39] |
| •The risk of developing parkinsonism is associated with recent influenza, influenza episodes number, and preceding influenza infections severity [34] | •Permanent glial activation [40] | |
| •The synergistic effect of influenza and MPTP in the dopaminergic neuron loss could be eliminated by influenza vaccination or treatment with oseltamivir carboxylate [35] | •The release of elevated inflammatory cytokines [38] | |
| •Degeneration of dopaminergic neurons [39] | ||
| •Promoting phosphorylation and aggregation of α-synuclein [41] | ||
| VZV | •A higher incidence of PD in VZV-infected patients by 2 recent epidemiological studies [42, 43] | •Neuroinflammation and immunological changes |
| •Chickenpox infection in childhood was found to be inversely correlated with PD [33] | ||
| JEV | •JEV infection could lead to a transient form of parkinsonism [45] | •Neuronal loss with gliosis [46] |
| •Noticeable bradykinesia, decreased dopamine levels and neuropathologic features were observed in JEV-induced rat model [47] | •Severe structural damage to the thalamus, basal ganglia, and brainstem [48] | |
| •Catecholamine levels alteration [47, 48] | ||
| WNV | •WNV could cause transient Parkinsonian manifestations [49] | •Stimulating α-synuclein production and inducing dopaminergic neuronal death [52] |
| •Increased α-syn expression in WNV-infected primary neurons [52] | ||
| WEEV | •Parkinsonism cases following encephalitis with WEEV infection [54] | •Persistent activation of microglia and astrocytes, selective dopaminergic neurons loss, and α-synuclein aggregation [55, 56] |
| •WEEV infection could induce persistent microgliosis and astrogliosis, selective dopaminergic neurons loss, and α-synuclein aggregation in vivo | ||
| HIV | •People infected with HIV often exhibit motor disorders such as bradykinesia, postural instability, gait abnormalities [57] | •Chronic inflammatory infiltrates, glial activation in basal ganglia [58, 60, 61] |
| •Decreased levels of dopamine and its metabolites in AIDS group [59] | •An effect of HIV on PD-related proteins, like DJ-1 and LRRK2 [63, 64] | |
| •A higher frequency of α-synuclein in the brain of HIV-infected patients [62] | ||
| SARS-CoV-2 | •Conjecture, need further evidence | •Uncertain |
| •SARS-CoV-2 could cause neurological symptoms and neuropathological damage [67, 68] | •Maybe invading CNS through nasal cavity and causing neuroinflammation, triggering or accelerating the early pathogenesis of PD [70] | |
| •Anosmia, an early sign of PD, is a common early symptom of COVID-19 [69] | ||
| •Motor and nonmotor symptoms significantly worsened in the COVID-19 group [74] | ||
| •Parkinsonism cases after SARS-CoV2 infection [71-73] | ||
| H. pylori | •PD patients have a higher prevalence of H. pylori infection [79, 80] | •Toxins produced by H. pylori |
| •H. pylori-infected PD patients display worse motor functions [81] | •Disruption of gut microbiota [85] | |
| •Eradication of H. pylori ameliorates motor symptoms in PD patients [82, 84] | •Substantial proinflammatory cytokines release, microglial activation, BBB dysfunction, resulting in neuroinflammation [86, 87] | |
| •Eradication of H. pylori improves levodopa absorption in PD patients [82, 83] | •The pharmacokinetic effects of H. pylori on levodopa [90–92] | |
| •Molecular mimicry between H. pylori and proteins essential for normal neurological functions [88, 89] |
VZV, varicella zoster virus; JEV, Japanese encephalitis virus; WNV, West Nile virus; BBB, blood-brain barrier; CNS, central nervous system; EL, encephalitic lethargica; PEP, post encephalitic Parkinsonism.
Table 2
Neuroprotective effects and possible mechanisms of anti-infective agents in Parkinson’s disease
| Anti -infective agents | Neuroprotective effects | The possible mechanisms/pathways | Derivatives |
| Anti-microbial agents | |||
| Ceftriaxone | Attenuating oxidative stress and neuroinflammation [123, 124] | Suppressing NF-κB/JNK/c-Jun signaling [126] | – |
| upregulating GLT-1 expression and glutamate uptake, reducing striatal tyrosine hydroxylase loss [125] | |||
| restoring BDNF levels [124] | |||
| inhibiting dopaminergic degeneration [128] | |||
| improving motor and memory deficits [127] | |||
| binding to α-synuclein with good affinity and blocking its polymerization [129] | |||
| Doxycycline | Protection against nigral dopaminergic degeneration [132] | Downregulation of MMP-3; | – |
| inhibiting microglial and astrocyte expression [130, 131] | inhibiting p38 MAPK and NF-κB signaling pathways [131] | ||
| anti-apoptotic and anti-inflammatory effects [132] | |||
| inhibiting α-synuclein aggregation and seeding of new oligomers [133] | |||
| Minocycline | Preventing nigrostriatal dopaminergic neurodegeneration [135] | Inhibiting the activation of p38 MAPK [138] | – |
| inhibiting glial activation [136, 138] | |||
| inhibiting IL-1β, NADPH-oxidase and iNOS [136] | |||
| reducing tyrosine hydroxylase-positive cell loss, increasing nigral cell size and fiber density [137] | |||
| reducing NMDA toxicity [138] | |||
| Rifampicin | Inhibiting α-synuclein fibrillation and disaggregating existing fibrils [144, 145] | Suppressing NF-κB pathway; decreasing the phosphorylation of MAPKs; | Rifampicin quinone (RifQ) [151] |
| scavenging free radical [146] | improving autophagy flux and lysosomal function [149, 150] | ||
| inhibiting glial activation and the pro-inflammatory mediator production [148] | inhibition of both PI3K- and non-PI3K-dependent signaling events [151] | ||
| increasing the surviving dopaminergic neuron numbers [142] | |||
| Geldanamycin | Reducing α-synuclein induced neurotoxicity [152, 155] | Inhibiting Hsp90, inducing Hsp70 [155] | 17-AAG, 17-DMAG; SNX-9114 [156] |
| Anti-fungal agents | |||
| Rapamycin | Decreasing dopaminergic neurons loss [159] | Blocking translation of RTP801 [157]; | – |
| reducing α-synuclein accumulation [158] | Via the mTOR-Akt-NF-κB cascade, partially the JAK2/STAT3 pathway [160] | ||
| protecting mitochondria against oxidative stress and apoptosis [159] | |||
| Glial and anti-inflammatory effects: upregulating glutamate transporter and IL-6 expression [160] | |||
| Anti-viral agents | |||
| Interferon | Interferon treatment can reduce the risk of PD in patients with HCV infection [28, 29] | Unknown | – |
| Amantadine | Clinical discovery: a female patient with PD improved motor symptoms after taking amantadine for flu [112, 113] | Enhancing dopamine release from presynaptic terminals [116], blocking NMDA receptors [117] and the mild anticholinergic effect [118] | ADS-5102 [119, 120] |
| improving dyskinesia effectively, especially in the early stage [115] | |||
| Anti-parasitic agents | |||
| Niclosamide | Activating PINK1 in cells through the reversible impairment of mitochondrial membrane potential [163] | Activating PINK1 [163] | AM85 [163] |
INFECTIVE PATHOGENS ASSOCIATED WITH PARKINSON’S DISEASE
Viral infection and Parkinson’s disease
Hepatitis C virus (HCV)
HCV is an enveloped, single-stranded RNA virus that causes a major public health problem worldwide. In addition to liver injury, chronic HCV infection also causes a series of extrahepatic manifestations, including fatigue, depression, cognitive dysfunction, diabetes, atherosclerosis, and stroke [18, 19].
Recently, emerging data from epidemiological studies has indicated that HCV infection is associated with PD as a risk factor. The HCV-infected patients in most of these studies usually had a significantly increased risk of developing PD [20–25], except a study using the U.S. Medicare database [26]. Furthermore, an extensive meta-analysis involving five studies with 323, 974 participants found a higher risk of PD among HCV-infected patients [27]. Remarkably, interferon-based antiviral therapy has recently been shown to reduce the risk of developing PD in patients with HCV infection [28, 29].
It was demonstrated that essential HCV receptors were expressed on brain microvascular endothelial cells, a major component of the blood-brain barrier (BBB), which could provide a gateway for HCV to invade the central nervous system (CNS) and cause neuroinflammation [30]. This notion was supported by the detection of HCV RNA sequences in postmortem brain tissue from infected patients [31]. In a previous study, it was found that both serotonergic and dopaminergic neurotransmissions were altered in HCV-infected patients with chronic fatigue and cognitive impairment [32]. In addition, it was HCV, not HBV, that was found to induce 60% dopaminergic neuron death in the midbrain neuron-glia coculture system in rats, suggesting that HCV had neurotoxic effects on dopaminergic neurons [20]. Furthermore, HCV infection could induce the release of substantial inflammatory cytokines, indicating that neuroinflammation may play a role in PD pathogenesis [20].
Influenza virus
According to clinical discovery, influenza virus may be related to PD, but this relationship is still controversial. The causal link between influenza virus and PD stems from the outbreak of EL and PEP following the 1918 H1N1 influenza pandemic [13]. It was also reported that there was a positive relationship between severe influenza and PD, as well as an inverse relationship between influenza vaccination and PD (although the latter was not statistically significant) [33]. In a large observational study, influenza infection was shown to have a link with PD-like symptoms, such as tremor or gait disturbances, especially in the first few weeks after the diagnosis or documentation of influenza infection [34]. In addition, the risk of developing parkinsonism symptoms was associated with recent influenza, number of influenza episodes, and severity of preceding influenza infections, which indicated that influenza-related neuronal damage may be a cumulative process [34]. What’s more, a synergistic effect of influenza and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) induced the loss of dopaminergic neurons in mice, which could be eliminated by influenza vaccination or treatment with oseltamivir carboxylate [35]. This further supports the link between influenza virus and PD.
However, the pathology and manifestations of PEP differ from those of idiopathic PD [36], and influenza genes were not detected in the brains of EL patients from 1916 to 1920 [37], which did not support the role of influenza virus as a causative agent of PD.
On the other hand, an array of proinflammatory mediators were elevated in patients with influenza virus-associated encephalopathy, indicating the role of inflammation in the pathogenesis of PEP [38]. An animal study found that the highly pathogenic H5N1 influenza virus could enter the CNS from the peripheral nervous system and activate the innate immune response in the brain, leading to a significant increase in phosphorylation and aggregation of α-synuclein and dopaminergic neuron degeneration [39]. Although the transient loss of tyrosine hydroxylase-positive (TH+) dopaminergic neurons was found to have largely recovered by 90 days post-infection, a long-lasting inflammatory response, especially permanent microglia activation, remained in H5N1-infected mouse models [40]. Moreover, a recent study demonstrated that H1N1 influenza A viral infection and replication could lead to the formation of α-synuclein aggregates [41].
Varicella zoster virus (VZV)
The initial infection of VZV causes varicella, which then lies dormant in the body and causes herpes zoster when reactivated. Early studies have suggested that there was no link between herpes zoster and PD. However, small sample sizes and bias due to self-reported infection history may limit the results. Recently, two cohort studies found a positive correlation between herpes zoster and the risk of PD, whether in elderly or midlife patients [42, 43]. Neuroinflammation and immunological changes involved in both conditions may explain the association. On the other hand, chickenpox infection was reported to have no association with PD [44]. However, in one study, chickenpox infection in childhood was found to be negatively correlated with PD, showing a possible protective effect [33]. The reverse relationship between childhood chickenpox infection and PD is difficult to explain, likely because the immune response activated at a younger age is important in reducing the possibility of viral infection that may damage dopaminergic neurons.
Japanese encephalitis virus (JEV)
Encephalitis caused by JEV is endemic throughout Asia each year, which could cause a high frequency of movement disorders. Japanese encephalitis (JE) can lead to a transient form of parkinsonism, which is characterized by varying severity of rigidity, hypokinesia, masking face, prominent hypophonia and a lower frequency of tremor [45]. Neuronal loss with gliosis in the substantia nigra pars compacta was observed in Fischer rats infected with JEV, which was similar to the pathological changes in PD [46]. Additionally, these rats showed noticeable bradykinesia and improved behavior after L-dopa administration. In addition, decreased dopamine levels and neuropathological characteristics with aging were also observed in JEV-induced PD rat models [47]. A significant decrease in the concentrations of norepinephrine, dopamine, and homovanillic acid in the cerebrospinal fluid was also noticed in JE patients [48], which may be caused by severe structural damage to the thalamus, basal ganglia, and brainstem, as detected on magnetic resonance imaging (MRI).
West Nile virus (WNV)
Movement disorders, especially tremor, myoclonus, and parkinsonism, may be unrecognized manifestations of acute WNV disease [49]. Parkinsonian features associated with acute WNV disease were transient in most cases and diminished over time, with a generally good prognosis [50]. On rare occasions, WNV infection can spread to the CNS in humans, which mechanisms are not yet completely understood. WNV encephalitis could result in immune responses in the brain and neuronal apoptosis. MRI showed abnormalities in the bilateral basal ganglia, thalamus, and pons in patients with severe persistent parkinsonism symptoms [51]. In addition, an increased expression of α-syn was observed in WNV-infected primary neurons. α-syn knockout mice also showed increased viral titers, increased neuronal damage, and accelerated mortality following the introduction of WNV [52]. Moreover, it was demonstrated that α-synuclein expression in intestinal neurons increased with gastrointestinal infection and induced leukocyte migration and dendritic cell maturation, suggesting the role of α-synuclein in gastrointestinal immunity [53]. The aforementioned aspects unveil the novel and significant role of α-synuclein in suppressing viral infection. It also further indicates that the acute onset of parkinsonism during WNV encephalitis may be due to WNV-induced dopaminergic neuronal death.
Western equine encephalitis virus (WEEV)
WEEV is a mosquito-borne arbovirus that can cause fatal encephalitis in humans and horses. Early studies have reported that WEEV infection could cause parkinsonian sequelae following encephalitis in humans [54]. The intranasal inoculation of recombinant WEEV in outbred CD-1 mice could cause significant neural invasion, resulting in the dissemination of the virus from the olfactory bulb to the basal midbrain [55]. The non-lethal encephalitic infection with WEEV led to sustained loss of dopaminergic neurons, persistent glial cell activation, formation of proteinase k-resistant α-synuclein aggregates, and gene expression profiles consistent with a neurodegenerative phenotype in mice. A subsequent study built on this result found that astrocytes played a key role in the initiation of PD-like pathology via the innate immune inflammatory pathway following WEEV infection [56].
Human immunodeficiency virus (HIV)
HIV, also known as acquired immunodeficiency syndrome (AIDS) virus, mainly attacks CD4 + T lymphocytes and macrophages in the human body. People infected with HIV often develop CNS disorders, commonly referred to as AIDS dementia complex (ADC). Psychomotor slowing, apathy, and motor disorders, such as bradykinesia, postural instability, gait abnormalities, and hypomimetic facies, were common manifestations of ADC [57]. It was found that HIV was preferentially present in inflammatory infiltrates and glial cells of the basal ganglia, including the substantia nigra [58]. Additionally, both dopamine and its major metabolite homovanillic acid were significantly reduced in the AIDS group [59]. This finding was consistent with the loss of dopaminergic neurons in the substantia nigra, which may be the basis of motor dysfunction in AIDS patients [60, 61]. Besides, an autopsy study found that the frequency of α-synuclein in the brains of HIV-infected patients was significantly higher than that of the healthy group, indicating that HIV patients may be more prone to develop PD [62]. In addition, HIV had an effect on PD-related proteins, such as DJ1 [63] and LRRK2 [64]. Moreover, a decrease in AIDS-related parkinsonism was observed after highly active antiretroviral therapy [65].
SARS-CoV-2
A serious threat to global health has been posed with the emergence of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection, coronavirus disease (COVID-19). Although respiratory symptoms are a prominent feature of COVID-19, SARS-CoV-2 can also invade other organs and affect multiple systems in the body. According to recent studies, SARS-CoV-2 could invade and attack the nervous system [66], causing neurological symptoms [67] and neuropathological damage [68]. Anosmia is a common early symptom of COVID-19 [69], and is also considered an early sign of PD. It is still unknown if this reflects an effect of SARS-CoV-2 on the peripheral nervous system, such as the olfactory nerve, or SARS-CoV-2 could further infect the CNS through the nasal cavity, a potential pathway into the CNS [70].
Compared with the 1918 influenza pandemic and avian influenza, reports of SARS-CoV-2-related encephalopathy have been slow to emerge. Until now, there have been relatively few reports of parkinsonism following SARS-CoV-2 infection. A 45-year-old man [71] and a 35-year-old woman [72] developed new asymmetric parkinsonism symptoms after SARS-CoV-2 infection. Both patients also showed radiographic abnormalities of dopamine dysfunction in the presynaptic striatum and responded well to levodopa. Interestingly, a 58-year-old man developed acute hypokinetic-rigid syndrome after SARS-CoV-2 infection and showed spontaneous improvement after 14 days [73]. According to an Italian community-based PD cohort study, motor and non-motor symptoms of the COVID-19 group significantly worsened [74]. However, unlike WNV, SARS-CoV-2 infection did not cause upregulation of serum and cerebrospinal fluid α-synuclein levels, regardless of the presence or absence of neurological symptoms in these COVID-19 patients [75].
Considering the far-reaching impact of SARS-CoV-2, further longitudinal studies are urgently needed.
Bacterial infection and Parkinson’s disease
Helicobacter pylori (H. pylori)
H. pylori is a helically-shaped, microaerophilic, gram-negative bacterium affecting approximately half of the world’s population. It is well-known that gastrointestinal symptoms precede motor symptoms in PD patients [76]. Besides, a follow-up study demonstrated that PD patients had a higher incidence of ulcers, which preceded parkinsonian symptoms by approximately 10 years [77]. Therefore, the relationship between H. pylori infection and PD has recently been extensively explored [78].
Increasing evidence demonstrated that PD patients had a higher prevalence of H. pylori infection. In one study, PD patients had a three-fold elevated risk of H. pylori seropositivity, compared with the control group [79]. Interestingly, their siblings were also more likely to suffer from parkinsonism symptoms and H. pylori positivity. An extensive meta-analysis involving 33, 125 participants revealed that a 1.5–2 fold increased risk of developing PD in H. pylori-infected persons [80]. Besides, it has been repeatedly reported that there existed a positive correlation between H. pylori infection and worse motor function in PD patients [81]. What’s more, eradication of H. pylori could ameliorate motor symptoms in PD patients, which was recommended in PD patients treated with levodopa because it may improve drug bioavailability and reduce motor fluctuations [82]. With the clearance of H. pylori, the levodopa onset time, ON duration, motor severity, and life quality all got improved [83]. A subsequent comprehensive meta-analysis found significant associations between H. pylori infection and higher Unified Parkinson’s Disease Rating Scale (UPDRS) scores, as well as a significantly lower UPDRS score in H. pylori-eradicated PD patients [84].
Overall, these results further confirm the view that H. pylori infection may play a key role in the course or even in the pathogenesis of PD [78]. However, the exact mechanism remains unclear. It is generally believed that possible pathogenic mechanisms include toxins produced by H. pylori, the disrupted gut microbiota [85], substantial proinflammatory cytokines [86, 87], the molecular mimicry between H. pylori and proteins essential for normal neurological functions [88, 89], in addition to the pharmacokinetic effects of H. pylori on levodopa [90–92].
Microbiota and Parkinson’s disease
Gut microbiota
Gut microbiota is composed of complex microbial communities. Recently, it has been increasingly recognized that gut microbiota affects brain function through the gut-brain axis. Emerging evidence suggests that there is a link between gut microbiota and PD. Gut microbiota was necessary for motor deficits, microglial activation, and α-synuclein pathology using α-synuclein overexpression mouse model [93]. Orally taken specific microbial metabolites promoted neuroinflammation and motor dysfunction in germ-free mice. The transplantation of gut microbiota from PD patients into germ-free α-syn overexpression mouse models exacerbated the motor deficits in mice, while antibiotic treatment reversed these effects, suggesting the critical role of gut microbiota in the pathogenesis of PD.
A number of studies have reported differences in the composition of the gut microbiota between PD patients and healthy controls [94]. It has also been shown that not only the concentration of beneficial fecal short chain fatty acids (SCFA), but also the abundance of SCFA-producing bacteria were lower in the PD group than in the control group [95, 96]. Bacteria that produce SCFA, particularly butyrate, may be an important factor in the pathogenesis and progression of PD. A link was found between PD clinical characteristics and the gut microbiota [97]. In this study, the genera Escherichia/Shigella were negatively associated with disease duration. The genera Butyricicoccus and Clostridium XlVb were associated with cognitive impairment. The genera Dorea and Phascolarcto bacterium were negatively associated with levodopa equivalent doses. In a two-year follow-up study, a more common Firmicutes-dominant enterotype and a lower abundance of Prevotella genus were observed in faster-progressing PD patients, compared with slower-progressing patients and controls, indicating a connection between gut microbiota and disease progression [98]. Altered gut microbiota was also found to be associated with the plasma cytokine response, in addition to clinical phenotype and severity [99]. Intriguingly, gut microbiota not only affects the health status of the host, but also the effectiveness of drug treatment [100, 101].
Oral microbiota
Comparing the oral microbiota of 72 PD patients and 76 healthy controls using 16S rRNA gene amplicon sequencing, significant differences were observed in the beta diversity and abundance of individual bacterial taxa [102]. Additionally, it was found that patients with periodontal inflammatory disease (PID) had a significantly higher risk of developing PD [103], while dental scaling over five consecutive years could reduce the risk of developing PD in individuals without PID, which emphasized the value of early and consecutive dental scaling in preventing the development of PD [104]. Moreover, PD patients tended to have poorer oral health, lower frequency of daily tooth-brushing, longer gaps between dental visits, and reduced salivary flow [105]. Even if confounding factors were controlled, PD patients still had more missing teeth, caries, dental plaque, cariogenic bacteria in saliva, and poorer periodontal health [106]. Individuals with poor oral health also tended to have higher Hoehn and Yahr scores and lower MMSE scores [107].
Nasal microbiota
The olfactory bulb was affected by the pathology of α-synuclein in the early stage, presenting as hyposmia. As assumed in the “dual-hit” hypothesis, the nasal cavity acts as an important portal for pathogens to spread to the CNS, which may be involved in the pathogenesis of PD [108].
After comparing the nasal microbiota using the 16S rRNA gene amplicon sequencing approach, no significant difference in alpha or beta diversity was observed between 69 PD patients and 67 healthy controls in one study [102]. Similar results were found in another study [109]. However, owing to the limited number of studies and patients, we cannot provide a definitive answer on whether the nasal microbiota is related to PD.
ANTI-INFECTIVE AGENTS AND PARKINSON’S DISEASE
Antiviral agents and Parkinson’s disease
Interferon
Wangensteen et al. identified that two patients developed PD in the short term during treatment of hepatitis C with interferon alpha [110]. Intriguingly, two recent epidemiological studies have shown that interferon treatment could reduce the risk of PD in patients with HCV infection [28, 29]. One study demonstrated that the reduced PD risk appeared to be obvious at five years after antiviral therapy and became more statistically significant at the end of follow-up [28]. Another study found that after treatment with PegIFN/RBV antiviral therapy, the risk of developing parkinsonism decreased by 38% [29]. One possible explanation for this discrepancy is that the two patients with hepatitis C in the case reports were already in the premotor stage, and antiviral treatment could not prevent their eventual progression to PD.
Amantadine
From 1966 to the present, amantadine experienced a transition from an anti-flu drug to an anti-parkinsonian agent [111]. In 1969, it was accidentally discovered that a female patient with PD improved motor symptoms after taking amantadine for flu, while her symptoms worsened after stopping the medication [112, 113]. Amantadine seemed to be most effective in the early stages of PD and had the greatest impact on tremors [114]. The effect of amantadine on dyskinesia has also been confirmed in multiple studies [115]. The mechanism of amantadine in treating PD may involve enhancing dopamine release from presynaptic terminals [116] and blocking N-methyl-D-aspartate (NMDA) receptors [117]. In addition, its mild anticholinergic effect has been reported [118].
ADS-5102, the amantadine extended-release capsule, has been demonstrated in several pivotal trials as an effective treatment for reducing OFF-time and dyskinesia [119]. Thus, it has recently been approved for the treatment of dyskinesia in PD patients by FDA [120].
Antimicrobial agents and Parkinson’s disease
Ceftriaxone
Ceftriaxone, a third-generation cephalosporin in the β-lactam antibiotic group, is a well-tested and safe drug that has been used for the treatment of a number of bacterial infections for several decades. Recently, the neuroprotective effects of ceftriaxone were observed in a wide range of neurological disorders, including Alzheimer’s disease, PD, amyotrophic lateral sclerosis, Huntington’s disease, cerebral ischemia, seizure, pain, and spinal cord injury [121].
It was proved that ceftriaxone could reduce L-dopa-induced dyskinesia severity in a 6-hydroxydopamine (6-OHDA) PD model [122]. In MPTP rat models, post-treatment with ceftriaxone significantly improved motor deficits and attenuated oxidative damage, as well as pro-inflammatory cytokines [123]. Additionally, ceftriaxone was shown to significantly restore the decreased activity of brain-derived neurotrophic factor (BDNF) in the striatum of MPTP-treated rats [124]. In a 6-OHDA PD rat model, ceftriaxone was reported to increase glutamate uptake, upregulate GLT-1 expression and reduce striatal tyrosine hydroxylase loss [125]. And it has been shown that ceftriaxone protected astrocytes from 1-methyl-4-phenylpyridinium (MPP+)-induced neurotoxicity by suppressing the NF-κB/JNK/c-Jun pathway [126]. In an MPTP-induced rat model of PD dementia, ceftriaxone not only reversed behavioral deficits but also prevented the loss of neurogenesis [127]. Another study concluded that ceftriaxone had an effect on behavioral and neuronal changes, which could prevent hippocampal cell loss and improve cognitive function in a PD rat model [128]. Furthermore, ceftriaxone could bind to α-synuclein with good affinity and block its polymerization in vitro.
Doxycycline
As a member of the tetracycline antibiotic family, doxycycline is used to treat a variety of bacterial infections, such as acne and intestinal infections. In clinical practice, it is widely used for its protective property with clinical safety records, increased lipid solubility, and excellent penetration of the BBB.
It has been demonstrated that doxycycline exhibits neuroprotective effects on PD both in vivo and in vitro, with MPTP and 6-OHDA animal models. In a 6-OHDA PD model, doxycycline blocked 6-OHDA neurotoxicity and conferred neuroprotection by inhibiting glial activation [130], the mechanism of which may be mediated by suppressing the p38 MAPK and NF-κB signaling pathways [131]. Doxycycline was neuroprotective against nigral dopaminergic degeneration by a dual mechanism, anti-apoptotic and anti-inflammatory mechanisms involving the downregulation of matrix metalloproteinase-3 (MMP-3) [132]. In addition, doxycycline not only inhibited the fibril formation of amyloidogenic proteins such as Aβ peptide, PrP peptide, and β-microglobulin, but also affected the aggregation of α-synuclein and the seeding of new oligomers. It was determined that doxycycline reshaped α-synuclein oligomers into off-pathway species, with parallel β-sheet content and a less hydrophobic surface, which did not evolve into fibril formation [133]. Furthermore, it is suggested that doxycycline at sub-antibiotic doses would be sufficient to exert anti-inflammatory activity and neuroprotective effects without changing bacterial susceptibility to antibiotics [134].
Minocycline
Minocycline, a semisynthetic second-generation tetracycline, exerts anti-inflammatory properties that are independent of its antimicrobial effects. Minocycline was shown to prevent nigrostriatal dopaminergic neurodegeneration. In the MPTP mouse model of PD, minocycline treatment blocked the MPTP-induced loss of striatal dopamine and its metabolites by inhibiting MPTP-induced glial iNOS, caspase 1 expression in vivo and NO-induced neurotoxicity in vitro [135]. In another study, minocycline inhibited MPTP-induced microglia activation and prevented the production of microglial-derived deleterious mediators, thereby reducing the loss of nigrostriatal dopaminergic neurons and the formation of nitrotyrosine produced by MPTP [136]. The behavioral and morphological effects of minocycline on PD have also been reported in the 6-OHDA rat model. Rats receiving minocycline displayed lower rotations, reduced tyrosine hydroxylase-positive cell loss, and increased nigral cell size and fiber density [137]. Minocycline conferred neuroprotection against NMDA neurotoxicity by inhibiting microglia. And the neuroprotective effect was associated with the inhibition of p38 MAPK in microglial cells [138].
However, beyond these promising results, the deleterious effects of minocycline on PD have also been reported [139–141]. Minocycline significantly exacerbated MPTP-induced damage to dopaminergic neurons, although it inhibited microglial activation [139]. This differs from previous studies in which minocycline inhibited microglial activation to exert neuroprotective effects [136]. The discrepancy may be attributed to the fact that minocycline may have variable or even deleterious effects depending on the route of administration, drug concentration or dosing interval. In addition, inhibiting microglia is not always beneficial since this may compromise its protective and reparative functions at the same time.
Rifampicin
Rifampicin, a semi-synthetic derivative of rifamycins, is widely used for the treatment of tuberculosis and leprosy. Rifampicin was able to prevent neuronal degeneration and significantly increase the surviving dopaminergic neuron numbers after MPP+intoxication in vitro [142]. Moreover, it was proved to attenuate the MPTP-induced neurotoxicity in the mouse brain [143]. Rifampicin has also been shown to inhibit α-synuclein fibrillation and disaggregate existing fibrils in a concentration-dependent manner [144]. This is consistent with a subsequent study in which rifampicin pretreatment caused a concentration-dependent increase in cell viability and a reduction in α-synuclein expression [145]. In addition, rifampicin-induced neuroprotection was suggested to be associated with its free radical scavenging activity [146]. It was found that rifampicin pretreatment could protect PC12 cells against cell death by suppressing rotenone-induced apoptosis and ameliorating mitochondrial oxidative stress [147]. It was also found that rifampicin could significantly inhibit the production of pro-inflammatory mediators in lipopolysaccharide-stimulated BV2 cells [148]. Moreover, the anti-inflammatory effects of rifampicin were mediated by the inhibition of NF-κB via the regulation of the IκB pathways as well as the suppression of MAPKs phosphorylation. Additionally, a recent study using human microglia cells demonstrated that rifampicin attenuated rotenone-induced microglial inflammation by improving autophagy flux and lysosomal function [149, 150].
Furthermore, rifampicin quinone, the oxidized form of rifampicin, is proved more potent than the parent compound itself, especially in reducing microglial inflammatory responses [151]. And the anti-inflammatory effects of these two drugs were related to the inhibition of PI3K-dependent and independent signaling events.
Geldanamycin (GA)
GA, a benzoquinone antibiotic isolated from the bacterium Streptomyces hygroscopicus, specifically binds to and inhibits the function of the molecular chaperone heat shock protein 90 (Hsp90), which promotes proteasomal degradation. GA has been shown to protect against α-synuclein-induced dopaminergic neuronal loss in the Drosophila model of PD [152]. Interestingly, previous studies have also shown that the molecular chaperones could suppress protein misfolding and aggregation, thus exerting protective effects against neurodegenerative diseases, including Huntington’s disease and spinal-cerebellar ataxia [153, 154]. It has been demonstrated that GA could induce Hsp70, prevent α-synuclein aggregation, and protect against α-synuclein-induced toxicity in vitro [155]. Although GA could selectively inhibit Hsp90, upregulate Hsp70, and reduce α-synuclein-induced neurotoxicity, its application is limited due to its hepatotoxicity and poor brain permeability. However, its structural analogs, such as 17-(allylamino)-17-demethoxygeldanamycin (17-AAG) and 17-dimethylaminoethylamino-17-demethoxy-geldanamycin (17-DMAG) exhibited greater potency, reduced toxicity, and improved brain permeability. Additionally, SNX-9114, a novel Hsp90 inhibitor, was reported to significantly rescue dopamine levels in the striatum after chronic treatment in a rat model of parkinsonism, although it failed to protect against α-synuclein-induced nigrostriatal toxicity [156].
Anti-fungal agents and Parkinson’s disease
Rapamycin
As an inhibitor of mTOR, rapamycin can effectively initiate the autophagy process and remove the accumulated denaturing proteins and damaged organelles of cells, which play a crucial role in the pathogenesis of neurodegenerative diseases.
Rapamycin has been verified to confer neuroprotection against PD toxins both in vitro and in vivo [157]. It could protect against neuronal death by blocking mTORC1–dependent translation of the pro-cell death protein RTP801. Rapamycin was reported to reduce α-synuclein accumulation and promote oligomer clearance via enhanced autophagy [158]. In addition, pretreatment with rapamycin could improve animal behaviors and decrease dopaminergic neuron loss in the 6-OHDA-induced rat model, which might be due to the property of rapamycin on protecting mitochondria from oxidative stress and apoptosis [159]. The effects of rapamycin on glia and anti-inflammation have been further verified in a study [160], wherein the astrocytic upregulation of glutamate transporters and improvement in IL-6 expression were observed in the MPTP models. Furthermore, the activation of the mTOR-Akt-NF-κB cascade, in part the JAK2/STAT3 pathway, appeared to be crucial in mediating the biological effects of rapamycin.
These findings suggest that rapamycin could be an effective and promising neuroprotective candidate for the treatment of PD (Fig. 2). However, the adverse effects of rapamycin as an immunosuppressive agent for long-term treatment should not be ignored. Long-term use of rapamycin could lead to lung toxicity [161] and increase the risk of type 2 diabetes [162].
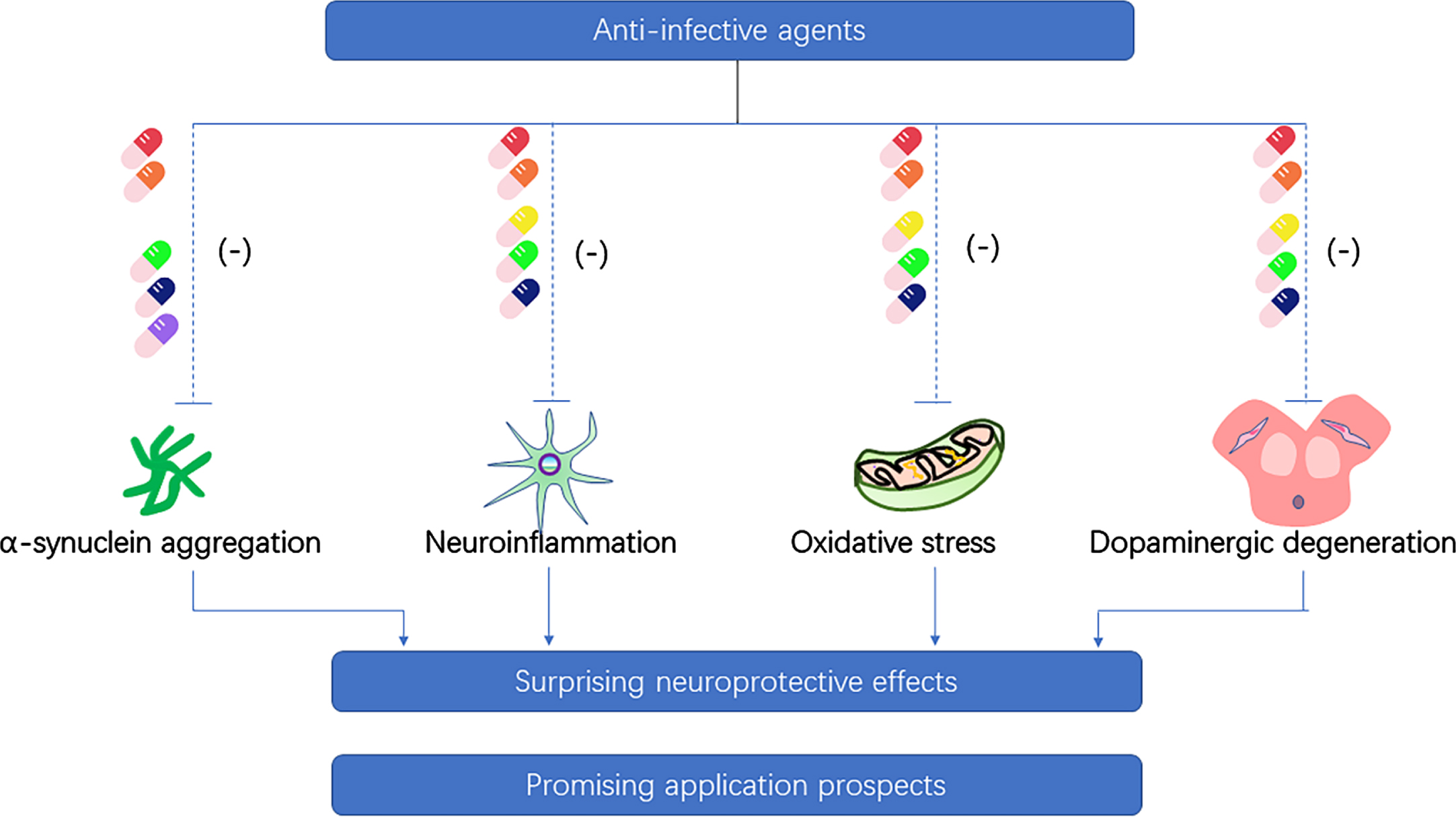
Fig. 2
Neuroprotective property of anti-infective agents on the pathogenesis of Parkinson’s disease. (Ceftriaxone-red pill, doxycycline-orange pill, minocycline-yellow pill, rifampicin-green pill, rapamycin-dark blue pill, geldanamycin-purple pill).
Anti-parasitic agents and Parkinson’s disease
Niclosamide
A recent study has shown that the anthelminthic drug, niclosamide, and its analog, AM85 can activate PINK1 in cells through the reversible impairment of mitochondrial membrane potential, which are expected to be a strategy for the treatment of PD [163]. In addition, niclosamide has been used safely to treat parasitic infections for a long time with no obvious serious adverse reactions.
CONCLUSIONS AND FUTURE PERSPECTIVES
In this review, we present the current evidence for the possibility of infective pathogens in the etiology of PD and the potential use of antibiotics as neuroprotective agents in PD. In the past few years, people’s attention on PD has mainly focused on α-synuclein. However, the etiology of PD remains elusive, hampering the development of effective treatments. Obviously, it is of great importance to identify the etiology and provide further corresponding treatments. Infection provides a fascinating hypothesis for the etiology of PD. As early as the 1920s, PD was suspected to be an infectious disease because it often appeared after EL [13]. In a recent study, it was discovered that the bacterial endotoxin lipopolysaccharide could transform asymptomatic Pink1–/–mice into a fully penetrant PD model, suggesting that intestinal infection may act as a triggering event in PD [164]. These studies highlight that chronic infection with bacterial and viral pathogens may be a trigger for PD. As an initiating factor, infection may cause a range of gut microbiota dysbiosis, glial activation, neuroinflammation, and α-synuclein accumulation, which may elicit or accelerate the pathogenesis of PD and lead to its occurrence. The underlying cause for PD with infection is unknown. Controversy still exists as to the potential pathogenesis of infection with PD. In addition, whether PD patients become ill as a result of infecting pathogens or simply because they are more susceptible to infective factors requires further research.
Currently, there are no cure or disease-modifying drugs that can stop or slow the progression of PD. However, a growing body of research suggests that anti-infective agents appear to be promising candidates for the treatment of PD. These agents affect crucial events in the pathogenesis of PD, such as interfering with α-synuclein aggregation, inhibiting neuroinflammation, attenuating oxidative stress, and preventing from cell death, thereby effectively hindering disease progression at multiple levels and showing promising application prospects. Although the potential for antibiotics in the treatment of PD is encouraging, few clinical trials have been conducted so far. As is known to all, several important idiopathic diseases have been found to be associated with previously unsuspected infections, such as H. pylori in gastric ulcer and gastric cancer [165], human papillomavirus in cervical cancer [166], Epstein-Barr virus in nasopharyngeal cancer [167], and hepatitis B virus in liver cancer [168]. These discoveries have led to new and effective treatments for the disease. If the role of infection in PD can be confirmed, it will provide a new direction and target for the treatment of PD. Besides, drug repurposing can quickly introduce drugs with well-documented safety to new patient populations, significantly speeding up the drug development process [169]. Recently, a nationwide case-control study was carried out, suggesting that early overexposure to antibiotics, particularly antianaerobics and broad-spectrum antibiotics, is associated with subsequent PD, with a delay of 10 to 15 years, which is consistent with the proposed duration of a prodromal period [170]. And this correlation is largely due to alteration in gut microbiota [171, 172]. Further research is needed to clarify the relationship between anti-infective agents and PD. In addition, if further research supports the infective etiology hypothesis for PD, a balance between efficacy and adverse effects should be taken into account. The choice of antibiotics, when and how to use them, the possible antibiotic resistance problem, and the potential impact of antimicrobial prescription on gut microbiota also need further consideration.
Taken together, results from recent studies suggest that infection may play a novel role in the pathogenesis of PD and highlight anti-infective agents as potential therapeutic targets for the treatment of PD. Further epidemiological and experimental studies are needed to confirm this hypothesis.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China to Dr. Chang-he Shi [grant number 81974211, 81771290, 82171247]; the National Key R&D Program of China to Dr. Yu-ming Xu [2017YFA0105000]; the National Natural Science Foundation of China to Dr. Yu-ming Xu [grant numbers U1904207, 91849115] and the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences [2020-PT310-01].
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Kalia LV , Lang AE ((2015) ) Parkinson’s disease. Lancet 386: , 896–912. |
[2] | Ascherio A , Schwarzschild MA ((2016) ) The epidemiology of Parkinson’s disease: risk factors and prevention. Lancet Neurol 15: , 1257–1272. |
[3] | Dauer W , Przedborski S ((2003) ) Parkinson’s disease: mechanisms and models. Neuron 39: , 889–909. |
[4] | Jankovic J , Tan EK ((2020) ) Parkinson’s disease: etiopathogenesis and treatment. J Neurol Neurosurg Psychiatry 91: , 795–808. |
[5] | Marino BLB , de Souza LR , Sousa KPA , Ferreira JV , Padilha EC , da Silva C , Taft CA , Hage-Melim LIS ((2020) ) Parkinson’s disease: a review from pathophysiology to treatment. Mini Rev Med Chem 20: , 754–767. |
[6] | Eriksen JL , Wszolek Z , Petrucelli L ((2005) ) Molecular pathogenesis of Parkinson disease. Arch Neurol 62: , 353–357. |
[7] | Lotharius J , Brundin P ((2002) ) Pathogenesis of Parkinson’s disease: dopamine, vesicles and alpha-synuclein. Nat Rev Neurosci 3: , 932–942. |
[8] | Fox SH , Katzenschlager R , Lim SY , Barton B , de Bie RMA , Seppi K , Coelho M , Sampaio C ((2018) ) International Parkinson and movement disorder society evidence-based medicine review: Update on treatments for the motor symptoms of Parkinson’s disease. Mov Disord 33: , 1248–1266. |
[9] | Panza F , Lozupone M , Solfrizzi V , Watling M , Imbimbo BP ((2019) ) Time to test antibacterial therapy in Alzheimer’s disease. Brain 142: , 2905–2929. |
[10] | Xue YC , Feuer R , Cashman N , Luo H ((2018) ) Enteroviral infection: the forgotten link to amyotrophic lateral sclerosis? Front Mol Neurosci 11: , 63. |
[11] | Cermelli C , Vinceti M , Beretti F , Pietrini V , Nacci G , Pietrosemoli P , Bartoletti A , Guidetti D , Sola P , Bergomi M , Vivoli G , Portolani M ((2003) ) Risk of sporadic amyotrophic lateral sclerosis associated with seropositivity for herpesviruses and echovirus-7. Eur J Epidemiol 18: , 123–127. |
[12] | Mlakar J , Korva M , Tul N , Popović M , Poljšak-Prijatelj M , Mraz J , Kolenc M , Resman Rus K , Vesnaver Vipotnik T , Fabjan Vodušek V , Vizjak A , Pižem J , Petrovec M , Avšič Županc T ((2016) ) Zika virus associated with microcephaly. N Engl J Med 374: , 951–958. |
[13] | Ravenholt RT , Foege WH ((1982) ) 1918 influenza, encephalitis lethargica, parkinsonism. Lancet 2: , 860–864. |
[14] | Taubenberger JK ((2006) ) The origin and virulence of the 1918 “Spanish” influenza virus. Proc Am Philos Soc 150: , 86–112. |
[15] | Limphaibool N , Iwanowski P , Holstad MJV , Kobylarek D , Kozubski W ((2019) ) Infectious etiologies of parkinsonism: pathomechanisms and clinical implications. Front Neurol 10: , 652. |
[16] | Reglodi D , Renaud J , Tamas A , Tizabi Y , Socías SB , Del-Bel E , Raisman-Vozari R ((2017) ) Novel tactics for neuroprotection in Parkinson’s disease: Role of antibiotics, polyphenols and neuropeptides. Prog Neurobiol 155: , 120–148. |
[17] | Socias SB , González-Liz F , Avila CL , Vera C , Acuña L , Sepulveda-Diaz JE , Del-Bel E , Raisman-Vozari R , Chehin RN ((2018) ) Exploiting the therapeutic potential of ready-to-use drugs: Repurposing antibiotics against amyloid aggregation in neurodegenerative diseases. Prog Neurobiol 162: , 17–36. |
[18] | Gill K , Ghazinian H , Manch R , Gish R ((2016) ) Hepatitis C virus as a systemic disease: reaching beyond the liver. Hepatol Int 10: , 415–423. |
[19] | Negro F , Forton D , Craxì A , Sulkowski MS , Feld JJ , Manns MP ((2015) ) Extrahepatic morbidity and mortality of chronic hepatitis C. Gastroenterology 149: , 1345–1360. |
[20] | Wu WY , Kang KH , Chen SL , Chiu SY , Yen AM , Fann JC , Su CW , Liu HC , Lee CZ , Fu WM , Chen HH , Liou HH ((2015) ) Hepatitis C virus infection: a risk factor for Parkinson’s disease. J Viral Hepat 22: , 784–791. |
[21] | Tsai HH , Liou HH , Muo CH , Lee CZ , Yen RF , Kao CH ((2016) ) Hepatitis C virus infection as a risk factor for Parkinson disease: A nationwide cohort study. Neurology 86: , 840–846. |
[22] | Kim JM , Jang ES , Ok K , Oh ES , Kim KJ , Jeon B , Jo HJ , Ki M , Jeong SH ((2016) ) Association between hepatitis C virus infection and Parkinson’s disease. Mov Disord 31: , 1584–1585. |
[23] | Pakpoor J , Noyce A , Goldacre R , Selkihova M , Mullin S , Schrag A , Lees A , Goldacre M ((2017) ) Viral hepatitis and Parkinson disease: A national record-linkage study. Neurology 88: , 1630–1633. |
[24] | Choi HY , Mai TH , Kim KA , Cho H , Ki M ((2020) ) Association between viral hepatitis infection and Parkinson’s disease: A population-based prospective study. J Viral Hepat 27: , 1171–1178. |
[25] | Goldstein L , Fogel-Grinvald H , Steiner I ((2019) ) Hepatitis B and C virus infection as a risk factor for Parkinson’s disease in Israel-A nationwide cohort study. J Neurol Sci 398: , 138–141. |
[26] | Golabi P , Otgonsuren M , Sayiner M , Arsalla A , Gogoll T , Younossi ZM ((2017) ) The Ppevalence of Parkinson disease among patients with hepatitis C infection. Ann Hepatol 16: , 342–348. |
[27] | Wijarnpreecha K , Chesdachai S , Jaruvongvanich V , Ungprasert P ((2018) ) Hepatitis C virus infection and risk of Parkinson’s disease: a systematic review and meta-analysis. Eur J Gastroenterol Hepatol 30: , 9–13. |
[28] | Lin WY , Lin MS , Weng YH , Yeh TH , Lin YS , Fong PY , Wu YR , Lu CS , Chen RS , Huang YZ ((2019) ) Association of antiviral therapy with risk of Parkinson disease in patients with chronic hepatitis C virus infection. JAMA Neurol 76: , 1019–1027. |
[29] | Su TH , Yang HC , Tseng TC , Chou SW , Lin CH , Liu CH , Liu CJ , Chen CL , Kao JH ((2019) ) Antiviral therapy in patients with chronic hepatitis C is associated with a reduced risk of parkinsonism. Mov Disord 34: , 1882–1890. |
[30] | Fletcher NF , Wilson GK , Murray J , Hu K , Lewis A , Reynolds GM , Stamataki Z , Meredith LW , Rowe IA , Luo G , Lopez-Ramirez MA , Baumert TF , Weksler B , Couraud PO , Kim KS , Romero IA , Jopling C , Morgello S , Balfe P , McKeating JA ((2012) ) Hepatitis C virus infects the endothelial cells of the blood-brain barrier. Gastroenterology 142: , 634–643.e636. |
[31] | Radkowski M , Wilkinson J , Nowicki M , Adair D , Vargas H , Ingui C , Rakela J , Laskus T ((2002) ) Search for hepatitis C virus negative-strand RNA sequences and analysis of viral sequences in the central nervous system: evidence of replication. J Virol 76: , 600–608. |
[32] | Weissenborn K , Ennen JC , Bokemeyer M , Ahl B , Wurster U , Tillmann H , Trebst C , Hecker H , Berding G ((2006) ) Monoaminergic neurotransmission is altered in hepatitis C virus infected patients with chronic fatigue and cognitive impairment. Gut 55: , 1624–1630. |
[33] | Harris MA , Tsui JK , Marion SA , Shen H , Teschke K ((2012) ) Association of Parkinson’s disease with infections and occupational exposure to possible vectors. Mov Disord 27: , 1111–1117. |
[34] | Toovey S , Jick SS , Meier CR ((2011) ) Parkinson’s disease or Parkinson symptoms following seasonal influenza. Influenza Other Respir Viruses 5: , 328–333. |
[35] | Sadasivan S , Sharp B , Schultz-Cherry S , Smeyne RJ ((2017) ) Synergistic effects of influenza and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) can be eliminated by the use of influenza therapeutics: experimental evidence for the multi-hit hypothesis. NPJ Parkinsons Dis 3: , 18. |
[36] | Rail D , Scholtz C , Swash M ((1981) ) Post-encephalitic Parkinsonism: current experience. J Neurol Neurosurg Psychiatry 44: , 670–676. |
[37] | Lo KC , Geddes JF , Daniels RS , Oxford JS ((2003) ) Lack of detection of influenza genes in archived formalin-fixed, paraffin wax-embedded brain samples of encephalitis lethargica patients from 1916 to 1920. Virchows Arch 442: , 591–596. |
[38] | Sun G , Ota C , Kitaoka S , Chiba Y , Takayanagi M , Kitamura T , Yamamoto K , Fujie H , Mikami H , Uematsu M , Hino-Fukuyo N , Munakata M , Kure S , Haginoya K ((2015) ) Elevated serum levels of neutrophil elastase in patients with influenza virus-associated encephalopathy. J Neurol Sci 349: , 190–195. |
[39] | Jang H , Boltz D , Sturm-Ramirez K , Shepherd KR , Jiao Y , Webster R , Smeyne RJ ((2009) ) Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc Natl Acad Sci U S A 106: , 14063–14068. |
[40] | Jang H , Boltz D , McClaren J , Pani AK , Smeyne M , Korff A , Webster R , Smeyne RJ ((2012) ) Inflammatory effects of highly pathogenic H5N1 influenza virus infection in the CNS of mice. J Neurosci 32: , 1545–1559. |
[41] | Marreiros R , Müller-Schiffmann A , Trossbach SV , Prikulis I , Hänsch S , Weidtkamp-Peters S , Moreira AR , Sahu S , Soloviev I , Selvarajah S , Lingappa VR , Korth C ((2020) ) Disruption of cellular proteostasis by H1N1 influenza A virus causes α-synuclein aggregation. Proc Natl Acad Sci U S A 117: , 6741–6751. |
[42] | Lai SW , Lin CH , Lin HF , Lin CL , Lin CC , Liao KF ((2017) ) Herpes zoster correlates with increased risk of Parkinson’s disease in older people: A population-based cohort study in Taiwan. Medicine (Baltimore) 96: , e6075. |
[43] | Cheng CM , Bai YM , Tsai CF , Tsai SJ , Wu YH , Pan TL , Chen TJ , Chen MH ((2020) ) Risk of Parkinson’s disease among patients with herpes zoster: a nationwide longitudinal study. CNS Spectr 25: , 797–802. |
[44] | Vlajinac H , Dzoljic E , Maksimovic J , Marinkovic J , Sipetic S , Kostic V ((2013) ) Infections as a risk factor for Parkinson’s disease: a case-control study. Int J Neurosci 123: , 329–332. |
[45] | Misra UK , Kalita J ((2002) ) Prognosis of Japanese encephalitis patients with dystonia compared to those with parkinsonian features only. Postgrad Med J 78: , 238–241. |
[46] | Ogata A , Tashiro K , Nukuzuma S , Nagashima K , Hall WW ((1997) ) A rat model of Parkinson’s disease induced by Japanese encephalitis virus. J Neurovirol 3: , 141–147. |
[47] | Hamaue N , Ogata A , Terado M , Ohno K , Kikuchi S , Sasaki H , Tashiro K , Hirafuji M , Minami M ((2006) ) Brain catecholamine alterations and pathological features with aging in Parkinson disease model rat induced by Japanese encephalitis virus. Neurochem Res 31: , 1451–1455. |
[48] | Misra UK , Kalita J , Pandey S , Khanna VK , Babu GN ((2005) ) Cerebrospinal fluid catecholamine levels in Japanese encephalitis patients with movement disorders. Neurochem Res 30: , 1075–1078. |
[49] | Sejvar JJ , Haddad MB , Tierney BC , Campbell GL , Marfin AA , Van Gerpen JA , Fleischauer A , Leis AA , Stokic DS , Petersen LR ((2003) ) Neurologic manifestations and outcome of West Nile virus infection. JAMA 290: , 511–515. |
[50] | Robinson RL , Shahida S , Madan N , Rao S , Khardori N ((2003) ) Transient parkinsonism in West Nile virus encephalitis. Am J Med 115: , 252–253. |
[51] | Sejvar JJ ((2014) ) Clinical manifestations and outcomes of West Nile virus infection. Viruses 6: , 606–623. |
[52] | Beatman EL , Massey A , Shives KD , Burrack KS , Chamanian M , Morrison TE , Beckham JD ((2015) ) Alpha-synuclein expression restricts RNA viral infections in the brain. J Virol 90: , 2767–2782. |
[53] | Stolzenberg E , Berry D , Yang D , Lee EY , Kroemer A , Kaufman S , Wong GCL , Oppenheim JJ , Sen S , Fishbein T , Bax A , Harris B , Barbut D , Zasloff MA ((2017) ) A role for neuronal alpha-synuclein in gastrointestinal immunity. J Innate Immun 9: , 456–463. |
[54] | Schultz DR , Barthal JS , Garrett G ((1977) ) Western equine encephalitis with rapid onset of parkinsonism. Neurology 27: , 1095–1096. |
[55] | Bantle CM , Phillips AT , Smeyne RJ , Rocha SM , Olson KE , Tjalkens RB ((2019) ) Infection with mosquito-borne alphavirus induces selective loss of dopaminergic neurons, neuroinflammation and widespread protein aggregation. NPJ Parkinsons Dis 5: , 20. |
[56] | Bantle CM , Rocha SM , French CT , Phillips AT , Tran K , Olson KE , Bass TA , Aboellail T , Smeyne RJ , Tjalkens RB ((2021) ) Astrocyte inflammatory signaling mediates α-synuclein aggregation and dopaminergic neuronal loss following viral encephalitis. Exp Neurol 346: , 113845. |
[57] | Koutsilieri E , Sopper S , Scheller C , ter Meulen V , Riederer P ((2002) ) Parkinsonism in HIV dementia. J Neural Transm (Vienna) 109: , 767–775. |
[58] | Brew BJ , Rosenblum M , Cronin K , Price RW ((1995) ) AIDS dementia complex and HIV-1 brain infection: clinical-virological correlations. Ann Neurol 38: , 563–570. |
[59] | Sardar AM , Czudek C , Reynolds GP ((1996) ) Dopamine deficits in the brain: the neurochemical basis of parkinsonian symptoms in AIDS. Neuroreport 7: , 910–912. |
[60] | Zauli G , Secchiero P , Rodella L , Gibellini D , Mirandola P , Mazzoni M , Milani D , Dowd DR , Capitani S , Vitale M ((2000) ) HIV-1 Tat-mediated inhibition of the tyrosine hydroxylase gene expression in dopaminergic neuronal cells. J Biol Chem 275: , 4159–4165. |
[61] | Bennett BA , Rusyniak DE , Hollingsworth CK ((1995) ) HIV-1 gp120-induced neurotoxicity to midbrain dopamine cultures. Brain Res 705: , 168–176. |
[62] | Khanlou N , Moore DJ , Chana G , Cherner M , Lazzaretto D , Dawes S , Grant I , Masliah E , Everall IP ((2009) ) Increased frequency of alpha-synuclein in the substantia nigra in human immunodeficiency virus infection. J Neurovirol 15: , 131–138. |
[63] | Roy U , Atluri VS , Agudelo M , Yndart A , Huang Z , Nair M ((2015) ) DJ1 expression downregulates in neuroblastoma cells (SK-N-MC) chronically exposed to HIV-1 and cocaine. Front Microbiol 6: , 749. |
[64] | Puccini JM , Marker DF , Fitzgerald T , Barbieri J , Kim CS , Miller-Rhodes P , Lu SM , Dewhurst S , Gelbard HA ((2015) ) Leucine-rich repeat kinase 2 modulates neuroinflammation and neurotoxicity in models of human immunodeficiency virus 1-associated neurocognitive disorders. J Neurosci 35: , 5271–5283. |
[65] | Rosso AL , Mattos JP , Correa RB , Nicaretta DH , Novis SA ((2009) ) Parkinsonism and AIDS: a clinical comparative study before and after HAART. Arq Neuropsiquiatr 67: , 827–830. |
[66] | Song E , Zhang C , Israelow B , Lu-Culligan A , Prado AV , Skriabine S , Lu P , Weizman OE , Liu F , Dai Y , Szigeti-Buck K , Yasumoto Y , Wang G , Castaldi C , Heltke J , Ng E , Wheeler J , Alfajaro MM , Levavasseur E , Fontes B , Ravindra NG , Van Dijk D , Mane S , Gunel M , Ring A , Kazmi SAJ , Zhang K , Wilen CB , Horvath TL , Plu I , Haik S , Thomas JL , Louvi A , Farhadian SF , Huttner A , Seilhean D , Renier N , Bilguvar K , Iwasaki A ((2021) ) Neuroinvasion of SARS-CoV-2 in human and mouse brain. J Exp Med 218: , e20202135. |
[67] | Mao L , Jin H , Wang M , Hu Y , Chen S , He Q , Chang J , Hong C , Zhou Y , Wang D , Miao X , Li Y , Hu B ((2020) ) Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol 77: , 1–9. |
[68] | Reichard RR , Kashani KB , Boire NA , Constantopoulos E , Guo Y , Lucchinetti CF ((2020) ) Neuropathology of COVID-19: a spectrum of vascular and acute disseminated encephalomyelitis (ADEM)-like pathology. Acta Neuropathol 140: , 1–6. |
[69] | Gane SB , Kelly C , Hopkins C ((2020) ) Isolated sudden onset anosmia in COVID-19 infection. A novel syndrome? Rhinology 58: , 299–301. |
[70] | Cain MD , Salimi H , Diamond MS , Klein RS ((2019) ) Mechanisms of pathogen invasion into the central nervous system. Neuron 103: , 771–783. |
[71] | Cohen ME , Eichel R , Steiner-Birmanns B , Janah A , Ioshpa M , Bar-Shalom R , Paul JJ , Gaber H , Skrahina V , Bornstein NM , Yahalom G ((2020) ) A case of probable Parkinson’s disease after SARS-CoV-2 infection. Lancet Neurol 19: , 804–805. |
[72] | Faber I , Brandão PRP , Menegatti F , de Carvalho Bispo DD , Maluf FB , Cardoso F ((2020) ) Coronavirus disease 2019 and parkinsonism: a non-post-encephalitic case. Mov Disord 35: , 1721–1722. |
[73] | Méndez-Guerrero A , Laespada-García MI , Gómez-Grande A , Ruiz-Ortiz M , Blanco-Palmero VA , Azcarate-Diaz FJ , Rábano-Suárez P , Álvarez-Torres E , de Fuenmayor-Fernández de la Hoz CP , Vega Pérez P , Rodríguez-Montalbán R , Pérez-Rivilla A , Sayas Catalán J , Ramos-González A , González de la Aleja J ((2020) ) Acute hypokinetic-rigid syndrome following SARS-CoV-2 infection. Neurology 95: , e2109–e2118. |
[74] | Cilia R , Bonvegna S , Straccia G , Andreasi NG , Elia AE , Romito LM , Devigili G , Cereda E , Eleopra R ((2020) ) Effects of COVID-19 on Parkinson’s disease clinical features: a community-based case-control study. Mov Disord 35: , 1287–1292. |
[75] | Blanco-Palmero VA , Azcárate-Díaz FJ , Ruiz-Ortiz M , Laespada-García MI , Rábano-Suárez P , Méndez-Guerrero A , Aramendi-Ramos M , Eguiburu JL , Pérez-Rivilla A , Marchán-López A , Rubio-Fernández M , Carro E , González de la Aleja J ((2021) ) Serum and CSF alpha-synuclein levels do not change in COVID-19 patients with neurological symptoms. J Neurol 268: , 3116–3124. |
[76] | Cersosimo MG , Raina GB , Pecci C , Pellene A , Calandra CR , Gutiérrez C , Micheli FE , Benarroch EE ((2013) ) Gastrointestinal manifestations in Parkinson’s disease: prevalence and occurrence before motor symptoms. J Neurol 260: , 1332–1338. |
[77] | Strang RR ((1965) ) The association of gastro-duodenal ulceration and Parkinson’s disease. Med J Aust 1: , 842–843. |
[78] | McGee DJ , Lu XH , Disbrow EA ((2018) ) Stomaching the possibility of a pathogenic role for Helicobacter pylori in Parkinson’s disease. J Parkinsons Dis 8: , 367–374. |
[79] | Charlett A , Dobbs RJ , Dobbs SM , Weller C , Brady P , Peterson DW ((1999) ) Parkinsonism: siblings share Helicobacter pylori seropositivity and facets of syndrome. Acta Neurol Scand 99: , 26–35. |
[80] | Shen X , Yang H , Wu Y , Zhang D , Jiang H ((2017) ) Meta-analysis: Association of Helicobacter pylori infection with Parkinson’s diseases. Helicobacter 22: , e12398. |
[81] | Tan AH , Mahadeva S , Marras C , Thalha AM , Kiew CK , Yeat CM , Ng SW , Ang SP , Chow SK , Loke MF , Vadivelu JS , Ibrahim N , Yong HS , Tan CT , Fox SH , Lang AE , Lim SY ((2015) ) Helicobacter pylori infection is associated with worse severity of Parkinson’s disease. Parkinsonism Relat Disord 21: , 221–225. |
[82] | Pierantozzi M , Pietroiusti A , Brusa L , Galati S , Stefani A , Lunardi G , Fedele E , Sancesario G , Bernardi G , Bergamaschi A , Magrini A , Stanzione P , Galante A ((2006) ) Helicobacter pylori eradication and l-dopa absorption in patients with PD and motor fluctuations. Neurology 66: , 1824–1829. |
[83] | Hashim H , Azmin S , Razlan H , Yahya NW , Tan HJ , Manaf MR , Ibrahim NM ((2014) ) Eradication of Helicobacter pylori infection improves levodopa action, clinical symptoms and quality of life in patients with Parkinson’s disease. PLoS One 9: , e112330. |
[84] | Dardiotis E , Tsouris Z , Mentis AA , Siokas V , Michalopoulou A , Sokratous M , Dastamani M , Bogdanos DP , Deretzi G , Kountouras J ((2018) ) H. pylori and Parkinson’s disease: Meta-analyses including clinical severity. Clin Neurol Neurosurg 175: , 16–24. |
[85] | Noto JM , Peek RM Jr. ((2017) ) The gastric microbiome, its interaction with Helicobacter pylori, and its potential role in the progression to stomach cancer. PLoS Pathog 13: , e1006573. |
[86] | Deretzi G , Kountouras J , Polyzos SA , Zavos C , Giartza-Taxidou E , Gavalas E , Tsiptsios I ((2011) ) Gastrointestinal immune system and brain dialogue implicated in neuroinflammatory and neurodegenerative diseases. Curr Mol Med 11: , 696–707. |
[87] | Kountouras J , Zavos C , Polyzos SA , Deretzi G , Vardaka E , Giartza-Taxidou E , Katsinelos P , Rapti E , Chatzopoulos D , Tzilves D , Stergiopoulos C , Christodoulou K ((2012) ) Helicobacter pylori infection and Parkinson’s disease: apoptosis as an underlying common contributor. Eur J Neurol 19: , e56. |
[88] | Suwarnalata G , Tan AH , Isa H , Gudimella R , Anwar A , Loke MF , Mahadeva S , Lim SY , Vadivelu J ((2016) ) Augmentation of autoantibodies by Helicobacter pylori in Parkinson’s disease patients may be linked to greater severity. PLoS One 11: , e0153725. |
[89] | Appelmelk BJ , Negrini R , Moran AP , Kuipers EJ ((1997) ) Molecular mimicry between Helicobacter pylori and the host. Trends Microbiol 5: , 70–73. |
[90] | Niehues M , Hensel A ((2009) ) In-vitro interaction of L-dopa with bacterial adhesins of Helicobacter pylori: an explanation for clinicial differences in bioavailability? J Pharm Pharmacol 61: , 1303–1307. |
[91] | Lahner E , Annibale B , Delle Fave G ((2009) ) Systematic review: Helicobacter pylori infection and impaired drug absorption. Aliment Pharmacol Ther 29: , 379–386. |
[92] | Pierantozzi M , Pietroiusti A , Galante A , Sancesario G , Lunardi G , Fedele E , Giacomini P , Stanzione P ((2001) ) Helicobacter pylori-induced reduction of acute levodopa absorption in Parkinson’s disease patients. Ann Neurol 50: , 686–687. |
[93] | Sampson TR , Debelius JW , Thron T , Janssen S , Shastri GG , Ilhan ZE , Challis C , Schretter CE , Rocha S , Gradinaru V , Chesselet MF , Keshavarzian A , Shannon KM , Krajmalnik-Brown R , Wittung-Stafshede P , Knight R , Mazmanian SK ((2016) ) Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 167: , 1469–1480.e1412. |
[94] | Elfil M , Kamel S , Kandil M , Koo BB , Schaefer SM ((2020) ) Implications of the gut microbiome in Parkinson’s disease. Mov Disord 35: , 921–933. |
[95] | Unger MM , Spiegel J , Dillmann KU , Grundmann D , Philippeit H , Bürmann J , Faßbender K , Schwiertz A , Schäfer KH ((2016) ) Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat Disord 32: , 66–72. |
[96] | Nuzum ND , Loughman A , Szymlek-Gay EA , Hendy A , Teo WP , Macpherson H ((2020) ) Gut microbiota differences between healthy older adults and individuals with Parkinson’s disease: A systematic review. Neurosci Biobehav Rev 112: , 227–241. |
[97] | Qian Y , Yang X , Xu S , Wu C , Song Y , Qin N , Chen SD , Xiao Q ((2018) ) Alteration of the fecal microbiota in Chinese patients with Parkinson’s disease. Brain Behav Immun 70: , 194–202. |
[98] | Aho VTE , Pereira PAB , Voutilainen S , Paulin L , Pekkonen E , Auvinen P , Scheperjans F ((2019) ) Gut microbiota in Parkinson’s disease: Temporal stability and relations to disease progression. EBioMedicine 44: , 691–707. |
[99] | Lin CH , Chen CC , Chiang HL , Liou JM , Chang CM , Lu TP , Chuang EY , Tai YC , Cheng C , Lin HY , Wu MS ((2019) ) Altered gut microbiota and inflammatory cytokine responses in patients with Parkinson’s disease. J Neuroinflammation 16: , 129. |
[100] | van Kessel SP , Frye AK , El-Gendy AO , Castejon M , Keshavarzian A , van Dijk G , El Aidy S ((2019) ) Gut bacterial tyrosine decarboxylases restrict levels of levodopa in the treatment of Parkinson’s disease. Nat Commun 10: , 310. |
[101] | Maini Rekdal V , Bess EN , Bisanz JE , Turnbaugh PJ , Balskus EP ((2019) ) Discovery and inhibition of an interspecies gut bacterial pathway for Levodopa metabolism. Science 364: , eaau6323. |
[102] | Pereira PAB , Aho VTE , Paulin L , Pekkonen E , Auvinen P , Scheperjans F ((2017) ) Oral and nasal microbiota in Parkinson’s disease. Parkinsonism Relat Disord 38: , 61–67. |
[103] | Chen CK , Wu YT , Chang YC ((2017) ) Periodontal inflammatory disease is associated with the risk of Parkinson’s disease: a population-based retrospective matched-cohort study. PeerJ 5: , e3647. |
[104] | Chen CK , Huang JY , Wu YT , Chang YC ((2018) ) Dental scaling decreases the risk of Parkinson’s disease: a nationwide population-based nested case-control study. Int J Environ Res Public Health 15: , 1587. |
[105] | Müller T , Palluch R , Jackowski J ((2011) ) Caries and periodontal disease in patients with Parkinson’s disease. Spec Care Dentist 31: , 178–181. |
[106] | Einarsdóttir ER , Gunnsteinsdóttir H , Hallsdóttir MH , Sveinsson S , Jónsdóttir SR , Olafsson VG , Bragason TH , Saemundsson SR , Holbrook WP ((2009) ) Dental health of patients with Parkinson’s disease in Iceland. Spec Care Dentist 29: , 123–127. |
[107] | Hanaoka A , Kashihara K ((2009) ) Increased frequencies of caries, periodontal disease and tooth loss in patients with Parkinson’s disease. J Clin Neurosci 16: , 1279–1282. |
[108] | Hawkes CH , Del Tredici K , Braak H ((2007) ) Parkinson’s disease: a dual-hit hypothesis. Neuropathol Appl Neurobiol 33: , 599–614. |
[109] | Heintz-Buschart A , Pandey U , Wicke T , Sixel-Döring F , Janzen A , Sittig-Wiegand E , Trenkwalder C , Oertel WH , Mollenhauer B , Wilmes P ((2018) ) The nasal and gut microbiome in Parkinson’s disease and idiopathic rapid eye movement sleep behavior disorder. Mov Disord 33: , 88–98. |
[110] | Wangensteen KJ , Krawitt EL , Hamill RW , Boyd JT ((2016) ) Parkinsonism in Patients with chronic hepatitis C treated with interferons: case reports and review of the literature. Clin Neuropharmacol 39: , 1–5. |
[111] | Hubsher G , Haider M , Okun MS ((2012) ) Amantadine: the journey from fighting flu to treating Parkinson disease. Neurology 78: , 1096–1099. |
[112] | Schwab RS , England AC Jr. , Poskanzer DC , Young RR ((1969) ) Amantadine in the treatment of Parkinson’s disease. JAMA 208: , 1168–1170. |
[113] | Schwab RS , England AC Jr. ((1969) ) Amantadine HCL (Symmetrel) and its relation to Levo-Dopa in the treatment of Parkinson’s disease. Trans Am Neurol Assoc 94: , 85–90. |
[114] | Rezak M ((2007) ) Current pharmacotherapeutic treatment options in Parkinson’s disease. Dis Mon 53: , 214–222. |
[115] | Sawada H , Oeda T , Kuno S , Nomoto M , Yamamoto K , Yamamoto M , Hisanaga K , Kawamura T ((2010) ) Amantadine for dyskinesias in Parkinson’s disease: a randomized controlled trial. PLoS One 5: , e15298. |
[116] | Bailey EV , Stone TW ((1975) ) The mechanism of action of amantadine in Parkinsonism: a review. Arch Int Pharmacodyn Ther 216: , 246–262. |
[117] | Chase TN , Bibbiani F , Oh JD ((2003) ) Striatal glutamatergic mechanisms and extrapyramidal movement disorders. Neurotox Res 5: , 139–146. |
[118] | Nastuk WL , Su P , Doubilet P ((1976) ) Anticholinergic and membrane activities of amantadine in neuromuscular transmission. Nature 264: , 76–79. |
[119] | Müller T , Kuhn W , Möhr JD ((2019) ) Evaluating ADS5102 (amantadine) for the treatment of Parkinson’s disease patients with dyskinesia. Expert Opin Pharmacother 20: , 1181–1187. |
[120] | Elmer LW , Juncos JL , Singer C , Truong DD , Criswell SR , Parashos S , Felt L , Johnson R , Patni R ((2018) ) Pooled analyses of Phase III studies of ADS-5102 (Amantadine) extended-release capsules for dyskinesia in Parkinson’s disease. CNS Drugs 32: , 387–398. |
[121] | Tai CH , Bellesi M , Chen AC , Lin CL , Li HH , Lin PJ , Liao WC , Hung CS , Schwarting RK , Ho YJ ((2019) ) A new avenue for treating neuronal diseases: Ceftriaxone, an old antibiotic demonstrating behavioral neuronal effects. Behav Brain Res 364: , 149–156. |
[122] | Chotibut T , Meadows S , Kasanga EA , McInnis T , Cantu MA , Bishop C , Salvatore MF ((2017) ) Ceftriaxone reduces L-dopa-induced dyskinesia severity in 6-hydroxydopamine parkinson’s disease model. Mov Disord 32: , 1547–1556. |
[123] | Bisht R , Kaur B , Gupta H , Prakash A ((2014) ) Ceftriaxone mediated rescue of nigral oxidative damage and motor deficits in MPTP model of Parkinson’s disease in rats. Neurotoxicology 44: , 71–79. |
[124] | Kaur B , Prakash A ((2017) ) Ceftriaxone attenuates glutamate-mediated neuro-inflammation and restores BDNF in MPTP model of Parkinson’s disease in rats. Pathophysiology 24: , 71–79. |
[125] | Chotibut T , Davis RW , Arnold JC , Frenchek Z , Gurwara S , Bondada V , Geddes JW , Salvatore MF ((2014) ) Ceftriaxone increases glutamate uptake and reduces striatal tyrosine hydroxylase loss in 6-OHDA Parkinson’s model. Mol Neurobiol 49: , 1282–1292. |
[126] | Zhang Y , Zhang X , Qu S ((2015) ) Ceftriaxone protects astrocytes from MPP(+) via suppression of NF-κB/JNK/c-Jun signaling. Mol Neurobiol 52: , 78–92. |
[127] | Hsieh MH , Meng WY , Liao WC , Weng JC , Li HH , Su HL , Lin CL , Hung CS , Ho YJ ((2017) ) Ceftriaxone reverses deficits of behavior and neurogenesis in an MPTP-induced rat model of Parkinson’s disease dementia. Brain Res Bull 132: , 129–138. |
[128] | Ho SC , Hsu CC , Pawlak CR , Tikhonova MA , Lai TJ , Amstislavskaya TG , Ho YJ ((2014) ) Effects of ceftriaxone on the behavioral and neuronal changes in an MPTP-induced Parkinson’s disease rat model. Behav Brain Res 268: , 177–184. |
[129] | Ruzza P , Siligardi G , Hussain R , Marchiani A , Islami M , Bubacco L , Delogu G , Fabbri D , Dettori MA , Sechi M , Pala N , Spissu Y , Migheli R , Serra PA , Sechi G ((2014) ) ceftriaxone blocks the polymerization of α-synuclein and exerts neuroprotective effects. ACS Chem Neurosci 5: , 30–38. |
[130] | Lazzarini M , Martin S , Mitkovski M , Vozari RR , Stühmer W , Bel ED ((2013) ) Doxycycline restrains glia and confers neuroprotection in a 6-OHDA Parkinson model. Glia 61: , 1084–1100. |
[131] | Santa-Cecília FV , Socias B , Ouidja MO , Sepulveda-Diaz JE , Acuña L , Silva RL , Michel PP , Del-Bel E , Cunha TM , Raisman-Vozari R ((2016) ) Doxycycline suppresses microglial activation by inhibiting the p38 MAPK and NF-kB signaling pathways. Neurotox Res 29: , 447–459. |
[132] | Cho Y , Son HJ , Kim EM , Choi JH , Kim ST , Ji IJ , Choi DH , Joh TH , Kim YS , Hwang O ((2009) ) Doxycycline is neuroprotective against nigral dopaminergic degeneration by a dual mechanism involving MMP-3. Neurotox Res 16: , 361–371. |
[133] | González-Lizárraga F , Socías SB , Ávila CL , TorresBugeau CM , Barbosa LR , Binolfi A , Sepúlveda-Díaz JE , Del-Bel E , Fernandez CO , Papy-Garcia D , Itri R , Raisman Vozari R , Chehín RN ((2017) ) Repurposing doxycycline for synucleinopathies: remodelling of α-synuclein oligomers towards non-toxic parallel beta-sheet structured species. Sci Rep 7: , 41755. |
[134] | Di Caprio R , Lembo S , Di Costanzo L , Balato A , Monfrecola G ((2015) ) Anti-inflammatory properties of low and high doxycycline doses: an study. Mediators Inflamm 2015: , 329418. |
[135] | Du Y , Ma Z , Lin S , Dodel RC , Gao F , Bales KR , Triarhou LC , Chernet E , Perry KW , Nelson DL , Luecke S , Phebus LA , Bymaster FP , Paul SM ((2001) ) Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. Proc Natl Acad Sci U S A 98: , 14669–14674. |
[136] | Wu DC , Jackson-Lewis V , Vila M , Tieu K , Teismann P , Vadseth C , Choi DK , Ischiropoulos H , Przedborski S ((2002) ) Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci 22: , 1763–1771. |
[137] | Quintero EM , Willis L , Singleton R , Harris N , Huang P , Bhat N , Granholm AC ((2006) ) Behavioral and morphological effects of minocycline in the 6-hydroxydopamine rat model of Parkinson’s disease. Brain Res 1093: , 198–207. |
[138] | Tikka TM , Koistinaho JE ((2001) ) Minocycline provides neuroprotection against N-methyl-D-aspartate neurotoxicity by inhibiting microglia. J Immunol 166: , 7527–7533. |
[139] | Yang L , Sugama S , Chirichigno JW , Gregorio J , Lorenzl S , Shin DH , Browne SE , Shimizu Y , Joh TH , Beal MF , Albers DS ((2003) ) Minocycline enhances MPTP toxicity to dopaminergic neurons. J Neurosci Res 74: , 278–285. |
[140] | Sriram K , Miller DB , O’Callaghan JP ((2006) ) Minocycline attenuates microglial activation but fails to mitigate striatal dopaminergic neurotoxicity: role of tumor necrosis factor-alpha. J Neurochem 96: , 706–718. |
[141] | Diguet E , Fernagut PO , Wei X , Du Y , Rouland R , Gross C , Bezard E , Tison F ((2004) ) Deleterious effects of minocycline in animal models of Parkinson’s disease and Huntington’s disease. Eur J Neurosci 19: , 3266–3276. |
[142] | Kilic U , Kilic E , Lingor P , Yulug B , Bähr M ((2004) ) Rifampicin inhibits neurodegeneration in the optic nerve transection model and after 1-methyl-4-phenylpyridinium intoxication. Acta Neuropathol 108: , 65–68. |
[143] | Oida Y , Kitaichi K , Nakayama H , Ito Y , Fujimoto Y , Shimazawa M , Nagai H , Hara H ((2006) ) Rifampicin attenuates the MPTP-induced neurotoxicity in mouse brain. Brain Res 1082: , 196–204. |
[144] | Li J , Zhu M , Rajamani S , Uversky VN , Fink AL ((2004) ) Rifampicin inhibits alpha-synuclein fibrillation and disaggregates fibrils. Chem Biol 11: , 1513–1521. |
[145] | Xu J , Wei C , Xu C , Bennett MC , Zhang G , Li F , Tao E ((2007) ) Rifampicin protects PC12 cells against MPP+-induced apoptosis and inhibits the expression of an alpha-Synuclein multimer. Brain Res 1139: , 220–225. |
[146] | Tomiyama T , Shoji A , Kataoka K , Suwa Y , Asano S , Kaneko H , Endo N ((1996) ) Inhibition of amyloid beta protein aggregation and neurotoxicity by rifampicin. Its possible function as a hydroxyl radical scavenger. J Biol Chem 271: , 6839–6844. |
[147] | Chen S , Sun Y , Zeng Z , Tao E ((2010) ) Rifampicin inhibits apoptosis in rotenone-induced differentiated PC12 cells by ameliorating mitochondrial oxidative stress. Neural Regen Res 5: , 251–256. |
[148] | Bi W , Zhu L , Wang C , Liang Y , Liu J , Shi Q , Tao E ((2011) ) Rifampicin inhibits microglial inflammation and improves neuron survival against inflammation. Brain Res 1395: , 12–20. |
[149] | Liang Y , Zheng D , Peng S , Lin D , Jing X , Zeng Z , Chen Y , Huang K , Xie Y , Zhou T , Tao E ((2020) ) Rifampicin attenuates rotenone-treated microglia inflammation via improving lysosomal function. Toxicol In Vitro 63: , 104690. |
[150] | Liang Y , Zhou T , Chen Y , Lin D , Jing X , Peng S , Zheng D , Zeng Z , Lei M , Wu X , Huang K , Yang L , Xiao S , Liu J , Tao E ((2017) ) Rifampicin inhibits rotenone-induced microglial inflammation via enhancement of autophagy. Neurotoxicology 63: , 137–145. |
[151] | Acuña Hamadat L , Corbalán NS , González-Lizárraga F , Dos-Santos-Pereira M , Rocca J , Díaz JS , Del-Bel E , Papy-García D , Chehín RN , Michel PP , Raisman Vozari R ((2019) ) Rifampicin and its derivative rifampicin quinone reduce microglial inflammatory responses and neurodegeneration induced by α-synuclein fibrillary aggregates. Cells 8: , 776. |
[152] | Auluck PK , Bonini NM ((2002) ) Pharmacological prevention of Parkinson disease in Drosophila. Nat Med 8: , 1185–1186. |
[153] | Chan HY , Warrick JM , Gray-Board GL , Paulson HL , Bonini NM ((2000) ) Mechanisms of chaperone suppression of polyglutamine disease: selectivity, synergy and modulation of protein solubility in Drosophila. Hum Mol Genet 9: , 2811–2820. |
[154] | Cummings CJ , Mancini MA , Antalffy B , DeFranco DB , Orr HT , Zoghbi HY ((1998) ) Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat Genet 19: , 148–154. |
[155] | McLean PJ , Klucken J , Shin Y , Hyman BT ((2004) ) Geldanamycin induces Hsp70 and prevents alpha-synuclein aggregation and toxicity. Biochem Biophys Res Commun 321: , 665–669. |
[156] | McFarland NR , Dimant H , Kibuuka L , Ebrahimi-Fakhari D , Desjardins CA , Danzer KM , Danzer M , Fan Z , Schwarzschild MA , Hirst W , McLean PJ ((2014) ) Chronic treatment with novel small molecule Hsp90 inhibitors rescues striatal dopamine levels but not α-synuclein-induced neuronal cell loss. PLoS One 9: , e86048. |
[157] | Malagelada C , Jin ZH , Jackson-Lewis V , Przedborski S , Greene LA ((2010) ) Rapamycin protects against neuron death in and models of Parkinson’s disease. J Neurosci 30: , 1166–1175. |
[158] | Liu K , Shi N , Sun Y , Zhang T , Sun X ((2013) ) Therapeutic effects of rapamycin on MPTP-induced Parkinsonism in mice. Neurochem Res 38: , 201–207. |
[159] | Jiang J , Jiang J , Zuo Y , Gu Z ((2013) ) Rapamycin protects the mitochondria against oxidative stress and apoptosis in a rat model of Parkinson’s disease. Int J Mol Med 31: , 825–832. |
[160] | Zhang Y , He X , Wu X , Lei M , Wei Z , Zhang X , Wen L , Xu P , Li S , Qu S ((2017) ) Rapamycin upregulates glutamate transporter and IL-6 expression in astrocytes in a mouse model of Parkinson’s disease. Cell Death Dis 8: , e2611. |
[161] | Chhajed PN , Dickenmann M , Bubendorf L , Mayr M , Steiger J , Tamm M ((2006) ) Patterns of pulmonary complications associated with sirolimus. Respiration 73: , 367–374. |
[162] | Johnston O , Rose CL , Webster AC , Gill JS ((2008) ) Sirolimus is associated with new-onset diabetes in kidney transplant recipients. J Am Soc Nephrol 19: , 1411–1418. |
[163] | Barini E , Miccoli A , Tinarelli F , Mulholland K , Kadri H , Khanim F , Stojanovski L , Read KD , Burness K , Blow JJ , Mehellou Y , Muqit MMK ((2018) ) The anthelmintic drug niclosamide and its analogues activate the Parkinson’s disease associated protein kinase PINK1. Chembiochem 19: , 425–429. |
[164] | Matheoud D , Cannon T , Voisin A , Penttinen AM , Ramet L , Fahmy AM , Ducrot C , Laplante A , Bourque MJ , Zhu L , Cayrol R , Le Campion A , McBride HM , Gruenheid S , Trudeau LE , Desjardins M ((2019) ) Intestinal infection triggers Parkinson’s disease-like symptoms in Pink1(-/-) mice. Nature 571: , 565–569. |
[165] | Wroblewski LE , Peek RM Jr. , Wilson KT ((2010) ) Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin Microbiol Rev 23: , 713–739. |
[166] | Crosbie EJ , Einstein MH , Franceschi S , Kitchener HC ((2013) ) Human papillomavirus and cervical cancer. Lancet 382: , 889–899. |
[167] | Tsao SW , Tsang CM , Lo KW ((2017) ) Epstein-Barr virus infection and nasopharyngeal carcinoma. Philos Trans R Soc Lond B Biol Sci 372: . |
[168] | Tu T , Bühler S , Bartenschlager R ((2017) ) Chronic viral hepatitis and its association with liver cancer. Biol Chem 398: , 817–837. |
[169] | Stott SRW , Wyse RK , Brundin P ((2021) ) Drug repurposing for Parkinson’s disease: The International Linked Clinical Trials experience. Front Neurosci 15: , 653377. |
[170] | Mertsalmi TH , Pekkonen E , Scheperjans F ((2020) ) Antibiotic exposure and risk of Parkinson’s disease in Finland: A nationwide case-control study. Mov Disord 35: , 431–442. |
[171] | Feng Y , Huang Y , Wang Y , Wang P , Song H , Wang F ((2019) ) Antibiotics induced intestinal tight junction barrier dysfunction is associated with microbiota dysbiosis, activated NLRP3 inflammasome and autophagy. PLoS One 14: , e0218384. |
[172] | Kelly LP , Carvey PM , Keshavarzian A , Shannon KM , Shaikh M , Bakay RA , Kordower JH ((2014) ) Progression of intestinal permeability changes and alpha-synuclein expression in a mouse model of Parkinson’s disease. Mov Disord 29: , 999–1009. |


