
Genetics of Progressive Supranuclear Palsy: A Review
Abstract
Progressive supranuclear palsy (PSP) is an atypical parkinsonism with prominent 4R-tau neuropathology, and the classical clinical phenotype is characterized by vertical supranuclear gaze palsy, unprovoked falls, akinetic-rigid syndrome and cognitive decline. Though PSP is generally regarded as sporadic, there is increasing evidence suggesting that a series of common and rare genetic variants impact on sporadic and familial forms of PSP. To date, more than 10 genes have been reported to show a potential association with PSP. Among these genes, the microtubule-associated protein tau (MAPT) is the risk locus with the strongest effect size on sporadic PSP in the case-control genome-wide association studies (GWAS). Additionally, MAPT mutations are the most common cause of familial PSP while the leucine-rich repeat kinase 2 (LRRK2) is a rare monogenic cause of PSP, and several other gene mutations may mimic the PSP phenotype, like the dynactin subunit 1 (DCTN1). In total, 15 MAPT mutations have been identified in cases with PSP, and the mean age at onset is much earlier than in cases carrying LRRK2 or DCTN1 mutations. GWAS have further identified several risk loci of PSP, proposing molecular pathways related to PSP. The present review focused on genetic studies on PSP and summarized genetic factors of PSP, which may help to elucidate the underlying pathogenesis and provide new perspectives for therapeutic strategies.
INTRODUCTION
Progressive supranuclear palsy (PSP), a rare neurodegenerative disorder with a prevalence of approximately 5–7 per 100000 [1], is traditionally considered to be one of the most common atypical parkinsonian syndromes and increasingly recognized to involve a range of motor, behaviour and language abnormalities [2]. PSP is clinically heterogeneous and presents as different phenotypes [3, 4], among which the most classic phenotype is Richardson’s syndrome (PSP-RS, also known as Steele–Richardson–Olszewski syndrome), which was first described as a clinicopathological entity in 1964 [5, 6]. New clinical diagnostic criteria of PSP (MDS-PSP) were published in 2017 [7], improving the sensitivity of PSP, in particular the variant PSP clinical presentations [8], and proposing four degrees of diagnostic certainty, namely, definite PSP, probable PSP, possible PSP and suggestive of PSP [7].
The pathological features of PSP are a predominance of 4-repeat (4R) tau inclusions in the form of neurofibrillary tangles, neuropil threads, tufted astrocytes and oligodendroglial coiled bodies in basal ganglia, diencephalon and brainstem, mainly affecting the globus pallidus, subthalamic nucleus and substantia nigra, in addition to neuronal loss and gliosis [9, 10]. PSP is the most common primary 4R-tauopathy, and the neuropathological 4R-tau begins to abnormally accumulate during the presymptomatic phase [2]. The localization of tau pathology is a major drive of clinical heterogeneity [4], and the distribution and severity of tau pathology vary in different clinical phenotypes, suggesting the significance of further studies into the pathological processes related to PSP [11, 12].
Although PSP is generally recognized as a sporadic syndrome, there are familial forms of PSP [13–15] and few pedigrees with PSP-like phenotypes [16, 17], and a pattern of autosomal dominant inheritance has been proposed [14, 15]. A case-control study has observed more first-degree relatives with parkinsonism or dementia in patients with PSP than in controls, demonstrating familial aggregation in PSP [18]. In contrast, genome-wide association studies (GWAS) in large cohorts over the past few years have identified several loci significantly associated with PSP [19–22], prompting studies on PSP genetics. Though a series of questions remain to be solved, genetics play an important role in PSP. This review focused on genetic studies and summarized genetic factors associated with PSP, especially recent additions, aiming to better understand PSP at the genetic level.
SINGLE-GENE MUTATIONS ASSOCIATED WITH PSP
MAPT in PSP
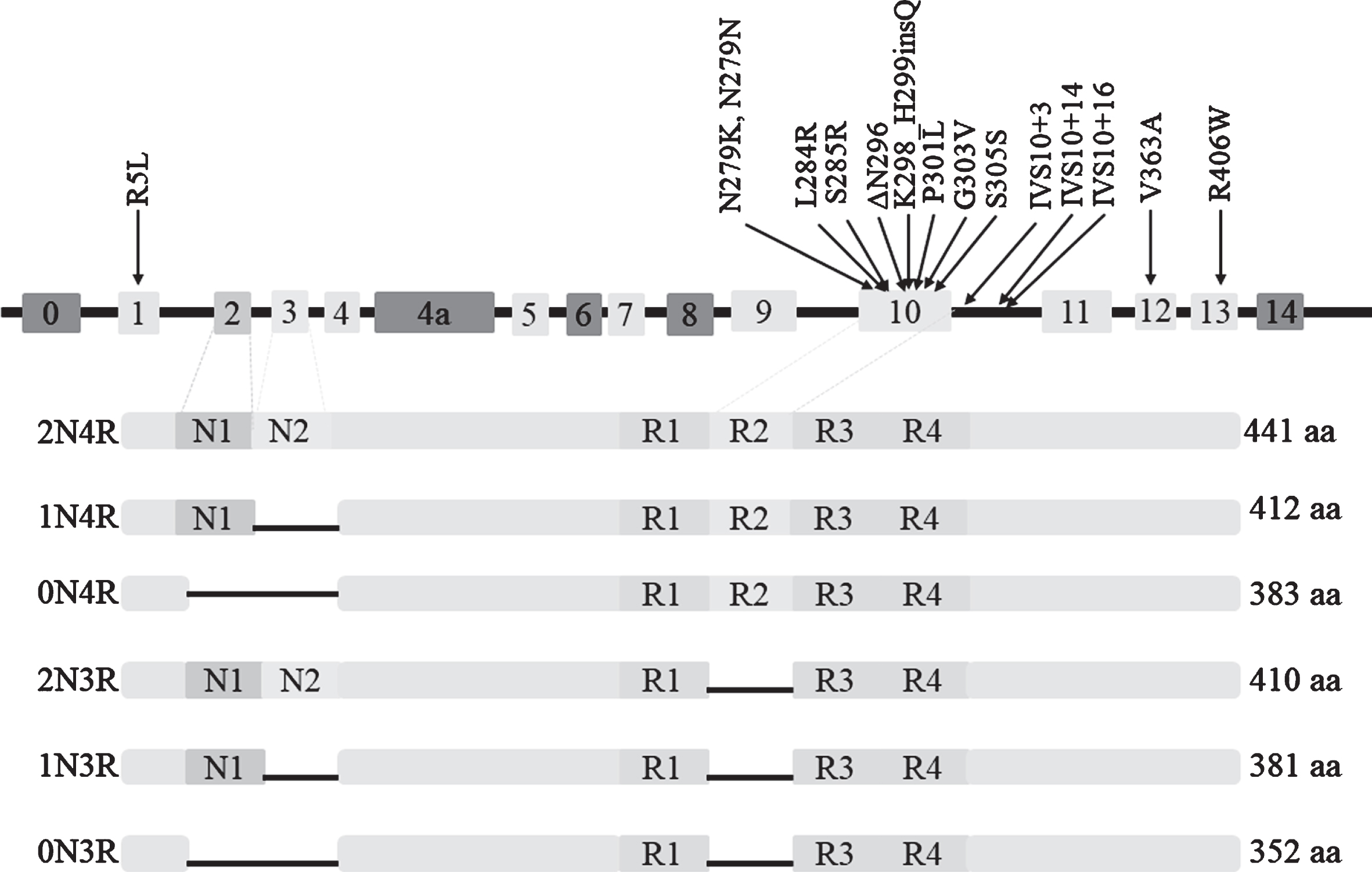
The microtubule-associated protein tau (MAPT) gene, encoding the tau protein, is located on chromosome 17q21.31 and consists of 16 exons [23]. Among the exons, exons E0 and 14 are non-coding, and exons 4a, 6 and 8 are not transcribed or expressed in human brain [24]. Alternative splicing of exon 10 produces two major isoforms of tau protein, namely 4R-tau and 3R-tau, with four and three microtubule binding repeats, respectively. In adult human brain, the ratio of 4R to 3R is roughly equal to 1 [25]. Zero, one or two N-terminal inserts result from alternative splicing of exon 2 or exon 2 and 3 together, and each N-terminal insert contains 29 amino acids. Therefore, there are six major tau isoforms with different lengths in the human brain (Fig. 1) [24]. MAPT mutations were first reported in families of frontotemporal dementia (FTD) and parkinsonism linked to chromosome 17 (FTDP-17) in 1998 [26]. Since then, more than 60 mutations in MAPT have been identified, mostly characterized by behavioural changes and/or clinical parkinsonism [27]. MAPT is involved in a series of neurodegenerative disorders [28] and the case-control GWAS of PSP has identified that MAPT is the risk locus with the strongest effect size [19].
Fig. 1
Six isoforms of protein tau and 15 mutations of MAPT associated with PSP.
MAPT haplotypes and PSP
A case-control study first observed that the homozygous A0 alleles containing 11 TG repeats in intron 9 of MAPT are overrepresented in PSP, identifying a dinucleotide repeat polymorphism linked with PSP and providing direct genetic evidence for the association between PSP and MAPT [29], which has been validated by several groups [30–32]. Baker et al. [33] subsequently found that the polymorphic dinucleotide marker is inherited in complete disequilibrium linkage with other eight common single nucleotide polymorphisms (SNPs). These researchers initially described two extended haplotypes (called H1 and H2) covering the entire MAPT and observed that haplotype H1 with 238 bp in intron 9 is overrepresented in PSP patients compared to controls. Various studies on Caucasian populations have emphasized the significant association between MAPT haplotype H1 and PSP [34, 35]. A GWAS directly compared rs8070723 to H1/H2 as a proxy and observed that the odds ratio (OR) for PSP in H1/H1 carriers in the GWAS is 5.46 (p = 1.5×10–116) [19], and the GWAS suggested that the H2 haplotype had a protective role [36].
The structure of the H1 haplotype has been explored, and over 20 common subhaplotypes have been identified. Among which, MAPT H1c is significantly overrepresented in patients with PSP and in patients with corticobasal degeneration (CBD) [36] and Alzheimer’s disease (AD) [37]. H1c increases MAPT expression, especially 4R-containing transcripts [38], but underlying mechanisms remain unclear. Using a large series of autopsy-confirmed PSP cases, Heckman et al. identified three other MAPT H1 subhaplotypes associated with PSP, namely H1g, H1d and H1o, and they also proposed that several subhaplotypes may play roles in the severity of tau pathology in PSP [39], further advancing the understanding of the H1 haplotype.
MAPT mutations and PSP
The frequency of PSP cases carrying MAPT mutations is various with a range from 0.6% to 14.3% [16, 18, 40, 41]. In total, 15 different mutations of MAPT have been described in cases presenting as PSP (clinical or neuropathological diagnosis), as shown in Table 1 and Fig. 1. Except for the R5L mutation in exon1, the V363A mutation in exon12, and the R406W mutation in exon13, other causative mutations exist in exon 10 and its splicing regions, resulting in an increased ratio of 4R/3R. The most common MAPT mutation causing PSP is located at codon 279, involving 11 cases. The p.K298_H299insQ in exon 10 has been identified in three patients with familial PSP through genetic sequencing of 165 cases with possible tauopathies, and this mutation is the first insertion mutation reported in MAPT. Yasuda et al. identified a N279K mutation in a Japanese patient with pallido-nigro-luysian degeneration (PNLD) [42], which is pathologically distinctive from typical PSP and is considered a variant of PSP [43].
Table 1
MAPT mutations causing PSP-like syndrome
| Mutation | Region | Reference | No. | Initial features | AAO years | Clinical diagnosis | Pathological diagnosis |
| R5L | Ex1 | Poorkaj et al. [46] | 1 | falls, dysarthria, and micrographia; | 62 | PSP | PSP |
| N279K | Ex10 | Yasuda et al. [42] | 1 | parkinsonism; | 41 | PNLDf | PSP-like |
| Delisle et al. [47] | 2 | apathy, memory disorder, parkinsonism; | 40 | FTDP-17f | PSP-like | ||
| indifference, attention disturbances; | 41 | FTDP-17f | NA | ||||
| Soliveri et al. [48] | 1 | personality and behaviour changes; | 47 | PSPf | NA | ||
| Ogaki et al. [49, 50] | 6 | parkinsonism and micrographia; | 42 | PSPf | PSP-like | ||
| parkinsonism and oscillopsia; | 44 | PSPf | NA | ||||
| parkinsonism; | 46 | PSPf | NA | ||||
| unstable gait and character changes; | 40 | PSP | NA | ||||
| shuffling gait and bradykinesia; | 41 | PSPf | NA | ||||
| walking difficulty; | 43 | PSPf | NA | ||||
| N279N | Ex10 | Ogaki et al. [49] | 1 | parkinsonism; | 44 | PSPf | NA |
| L284R | Ex10 | Rohrer et al. [51] | 1 | personality change and falls; | 43 | PSPf | NA |
| S285R | Ex10 | Ogaki et al. [49] | 1 | speaking and breathing difficulty; | 46 | PSP | NA |
| Fujioka et al. [14] | 2 | dystonia and supranuclear gaze palsy; | 40 | PSPf | PSP-AD | ||
| gait changes and bradykinesia; | 41 | PSPf | PSP | ||||
| ΔN296 | Ex10 | Pastor et al. [45] | 1 | speaking and memory problems; | 38 | Atypical PSPf | NA |
| Rossi et al. [44] | 1 | antecollis, dysarthria and fall; | 36 | PSP-likef | NA | ||
| K298_H299insQ | Ex10 | Nakayama et al. [41] | 3 | neck stiffness and postural instability; | 60 | PSPf | NA |
| gait disturbance; | NA | PSPf | NA | ||||
| gait disturbance and cognitive decline | NA | NA | NA | ||||
| P301L | Ex10 | Bird et al. [52] | 1 | tremor and articulation difficulty; | 41 | APDf | PSP-like |
| Kaat et al. [18] | 1 | NA | NA | PSPf | NA | ||
| G303V | Ex10 | Ros et al. [53] | 1 | parkinsonism, falls, micrographia, dysarthria, and ocular motor dysfunction; | 37 | PSPf | PSP |
| S305S | Ex10 | Stanford et al. [54] | 1 | dystonia, dysarthria, falls, bradykinesia; | 48 | PSPf | PSP |
| IVS10 + 3G>A | IVS10 | Spina et al. [55] | 2 | dizziness and neck stiffness; | 52 | Atypical PSPf | PSP-like |
| dizziness and neck stiffness; | 47 | Atypical PSPf | tauopathy | ||||
| IVS10 + 14C>T | IVS10 | Omoto et al. [56] | 1 | clumsiness, tremor and apathy; | 44 | Perry syndromef | PSP-like |
| IVS10 + 16C>T | IVS10 | Morris et al. [57] | 1 | fatigue, micrographia, and withdrawal; | 40 | PSPf | tauopathy |
| V363A | Ex12 | Rossi et al. [58] | 1 | diplopia, falls and bradykinesia; | 53 | PSPf | NA |
| R406W | Ex13 | Ygland et al. [17] | 2 | personality change; | 53 | ADf | PSP-like |
| dyscalculia, social withdrawal, apathy; | 50 | ADf | PSP-like |
No., number; AAO, age at onset; NA, not available; PSP, progressive supranuclear palsy; PNLD, pallido-nigro-luysian degeneration; FTDP-17, frontotemporal dementia and parkinsonism linked to chromosome 17; AD, Alzheimer’s disease; PSP-AD, PSP with concomitant AD. “PSP-like” pathology means that tau accumulations exist in brain but do not meet the pathological diagnostic criteria of PSP. The letter “f” in clinical diagnosis indicates the case with a family history of parkinsonism, dementia, or other neurodegenerative diseases.
The onset of PSP is insidious, presenting with different symptoms, which highlights the clinical heterogeneity and complexity of PSP as well as the necessity of follow-up. The mean age at onset (AAO) in PSP with MAPT mutations is approximately 44.8 years (range of 36–62 years) with a peak in the early 40s. Two families [44, 45] carrying the ΔN296 mutation had an earlier age (<40 years) when first PSP-related symptoms occurred compared to cases carrying other mutations. Moreover, Pastor et al. [45] identified a patient carrying the homozygous ΔN296 mutation, which is the first case carrying a pathogenic homozygous mutation in MAPT, resulting in a more severe phenotype compared to heterozygous mutations, the latter causing a milder condition with reduced penetrance.
Except for the R5L mutation described in a sporadic PSP case, Table 1 shows that the majority of cases with other MAPT mutations have a family history with parkinsonism, dementia, or other neurodegenerative disorders, further supporting the familial aggregation in PSP, which is consistent with the results reported by Kaat et al. [18]. This phenomenon indicates the importance to exclude MAPT mutations when such a family history exists.
Recently, Chen et al. observed 2 patients with autopsy-confirmed PSP harboring duplications spanning the entire MAPT locus (both copy number = 3) through genome-wide survey of copy number variants (CNVs) and proposed that MAPT duplications may be a genetic cause of PSP, which provides genetic evidence for the hypothesis that CNVs are associated with PSP [22] and indicates that there are more potential associations between MAPT and PSP.
LRRK2: a very rare monogenic cause of PSP pathology
Leucine-rich repeat kinase 2 (LRRK2) is considered as one of the most common genetic causes of Parkinson’s disease (PD), and genetic evidence has shown more than 5 LRRK2 mutations linked with both familial and sporadic PD [59–61]. Although several studies have detected negative results when screening LRRK2 in PSP cohorts [62–65], 5 mutations have been identified in patients presenting a PSP phenotype (Table 2). A recent study identified that common variation at the LRRK2 locus is a genetic determinant of PSP survival (Jabbari et al. unpublished data), adding further evidence for the association between LRRK2 and PSP.
Table 2
LRRK2 mutations in cases with PSP phenotype
| Mutation | R1441C | R1441H | G2019S | T2310M | A1413T | |||
| cDNA | c.4321C > T | c.4322G > A | c. 6055G > A | c.6928 C > T | c.4237 G > A | |||
| Region | Exon 31 | Exon 31 | Exon 41 | Exon 47 | Exon 30 | |||
| Year of publication | 2004 [66, 73] | 2006 [67] | 2006 [68] | 2011 [69] | 2017 [59] | 2019 [70] | 2015 [72] | 2017 [59] |
| Gender | Female | Male | Male | Male | Female | Female | NA | Male |
| Nationality | American | Greek | English | Italian | white Jewish | NA | Korean | Filipino |
| Familial history | + | + | + | – | – | + | NA | – |
| AAO, y | 78 | 61 | 78 | 72 | 73 | 80s | NA | 72 |
| Duration of disease, y | 11 | 11 | 7 | 12 | 7 | NA | NA | 7 |
| Clinical features | parkinsonism and supranuclear gaze palsy | parkinsonism, bulbar dysfunction and dementia | parkinsonism | tremor and micrographia | bulbar dysfunction, tremor, vertical gaze palsy and retrocollis | NA | NA | memory problems, falls and apraxia of eyelid opening |
| Response to L-dopa | Good | Good | Minimal | poor | NA | NA | NA | Poor |
| MAPT haplotype | H1/H1 | NA | H1/H1 | H1/H1 | H1/H1 | NA | NA | H1/H1 |
| Clinical diagnosis | PD | PSP | PD | PSP | PSP | PD | PSP | PSP |
| Pathology | Parkinsonism due to tauopathy | NA | PSP and early AD | PSP and early AD | PSP and early AD | AD and PSP | NA | PSP-like |
PSP, progressive supranuclear palsy; AD, Alzheimer’s disease; AAO, age at onset; NA, not available; “+”, present; “–”, absent.
The R1441C mutation has been reported in a large kindred named family D with pleomorphic pathology and within 10 affected members who are clinically characterized by parkinsonism, but only one member has pathological changes similar to PSP. Thus, the researchers speculated that LRRK2 might contribute to tauopathy in addition to synucleinopathy [66]. The R1441H mutation has been identified in a patient originally diagnosed as typical PD but transiting to PSP 8 years later. This unusual case indicates that the R1441H is involved in the PSP-Parkinsonism (PSP-P) [67], but the involvement of environmental factors in this process remains unknown. G2019S has been observed in 4 cases with PSP from different studies [59, 68–70], and functional in vitro studies have observed that the G2019S mutation increases kinase activity, potentially explaining how LRRK2 causes neurodegeneration [71]. Trinh et al. [72] observed the T2310M mutation in a patient with PSP when sequencing LRRK2, and they also identified 27 other rare nonsynonymous variants in the cohorts. A novel p.A1413T mutation has been identified in a case-control study containing 1039 PSP and 145 CBD patients, and this mutation has been predicted to be “disease damaging” by several in silico predictive algorithms [59].
Therefore, we conclude that the LRRK2 gene is a rare pathologic gene associated with PSP [59] despite inconsistent results in some studies. More association studies in larger cohorts and various populations are needed to further elucidate how these LRRK2 mutations lead to tauopathy and whether other mutations increase the risk for PSP.
Single-gene mutations in cases mimicking PSP
DCTN1 mutations in cases as PSP look-alike syndromes
Dynactin subunit 1 (DCTN1), which encodes p150glued, is the largest subunit of dynactin complex, and it is involved in microtubule binding and molecular transport [74]. DCTN1 mutations have been identified in families with motor neuron disease [75] and Perry syndrome [76], which is a rare autosomal dominant neurodegenerative disease. Unlike tauopathy in PSP, the underlying pathology in DCTN1 mutation carriers is transactive response DNA-binding protein of 43 kDa (TDP-43) proteinopathy [77]. To advance the understanding of clinical phenotype spectrum related to DCTN1 mutations, we summarized 3 DCTN1 mutations reported in patients with a PSP look-alike syndrome (Table 3). However, the p.R1261Q mutation in one PSP-PS case and the p.L896V mutation in one PSP-FTD case both detected by Yabe et al. [16] are not included in Table 3 due to lack of clinical data.
Table 3
DCTN1 mutations associated with PSP look-alike syndromes
| Mutation | G71E | K56R | F52L | |
| cDNA | c.212G > A | c.167A > G | c.156T > G | |
| Region | Exon 2 | Exon 2 | Exon 2 | |
| Reference | Caroppo et al. [78] | Gustavsson et al. [79] | Honda et al. [80] | |
| Number | 1 | 2 | 1 | |
| Gender | Male | Male | Male | Female |
| Nationality | French | Chinese-Canadian | Chinese | Japanese |
| Familial history | + | + | NA | NA |
| AAO, y | 59 | 61 | 57 | 48 |
| Duration of disease, y | 6 | NA | NA | 26 |
| Clinical features | parkinsonism, frontal signs and oculomotor disorders | parkinsonism and oculomotor disorders | parkinsonism and memory decline | parkinsonism, central hypoventilation |
| Imaging findings | mild mid brain and frontal atrophy | NA | diffuse cortical atrophy | frontal atrophy |
| Clinical diagnosis | PSP | PSP | PSP | Perry syndrome |
| Pathology | NA | NA | NA | PSP-like |
PSP, progressive supranuclear palsy; AAO, age at onset; NA, not available; “+”, present.
Table 3 shows that the mean AAO among these cases is 56.3 years with a large range of disease duration. All patients presented parkinsonism as a major clinical feature, and symmetrical frontal atrophy was obvious in more than half of the cases. DCTN1 mutations play a role in susceptibility to PSP. Due to the low frequency and limited knowledge, however, the underlying mechanism between DCTN1 and tauopathy remains unclear and needs further validation.
Other genes and PSP look-alike syndromes
As genetic studies on neurodegenerative disorders develop, more genes have shown potential links with PSP. A new series of gene mutations or polymorphisms have been identified in patients with a PSP look-alike phenotype, including but not limited to the NPC1 gene [81, 82], the C9orf72 gene [40, 83], the parkin gene (PARK2) [84–86], the transactivation response element DNA-binding protein gene (TARDBP) [87, 88], the progranulin gene (GRN) [16, 89], the TANK-binding kinase 1 gene (TBK1) [90], and the bassoon gene (BSN) [16] (Table 4). Some of these genes have been considered as causative genes in certain neurodegenerative diseases [91–94]. Considering the wide phenotype spectrum of monogenic mutations and the overlap among clinical features of neurodegenerative diseases [95], there may be common mechanisms and pathways in the pathogenesis of parkinsonian syndromes.
Table 4
Single-genes associated with PSP look-alike syndromes
| Genes | Mutations | Number | Authors and year |
| DCTN1 | G71E | 1 | Caroppo et al. 2014 [78] |
| K56R | 1 | Gustavsson et al. 2016 [79] | |
| F52L | 1 | Honda et al. 2018 [80] | |
| L896V | 1 | Yabe et al. 2018 [16] | |
| R1261Q | 1 | Yabe et al. 2018 [16] | |
| NPC1 | K576R | 1 | Godeiro-Junior et al. 2006 [81] |
| P1007A and F1221fsX20* | 1 | Godeiro-Junior et al. 2006 [81] | |
| F284LfsX26 | 1 | Cupidi et al. 2017 [82] | |
| C9orf72 | >60 repeat units | 1 | Lesage et al. 2013 [83] |
| 1 | Leber et al. 2013 [40] | ||
| >50 repeat units | 1 | Wilke et al. 2016 [96] | |
| PARK2 | C212Y | 1 | Morales et al. 2002 [84] |
| Sanchez et al. 2002 [85] | |||
| TARDBP | A382T | 2 | Cannas et al. 2014 [87] |
| I239V | 1 | Yabe et al. 2016 [88] | |
| GRN | Thr272fs | 2 | Tremolizzo et al. 2011 [89] |
| N118del | 1 | Yabe et al. 2018 [16] | |
| V500I | 1 | Yabe et al. 2018 [16] | |
| C221S | 1 | Yabe et al. 2018 [16] | |
| TBK1 | E643del | 1 | Wilke et al. 2018 [90] |
| BSN | P2855L | 1 | Yabe et al. 2018 [16] |
| R3146C | 2 | Yabe et al. 2018 [16] | |
| G3627V | 1 | Yabe et al. 2018 [16] | |
| P3866A | 3 | Yabe et al. 2018 [16] |
*The case carried a compound heterozygous mutation (P1007A and F1221fsX20).
In summary, cases carrying MAPT mutations have an earlier AAO (mean: 44.8 years), and the initial features are variable, mainly presenting parkinsonism, unstable walking and frontal cognitive/behavioural symptoms. In addition, a positive family history exists in most cases. The AAOs in patients with LRRK2 mutations are much older (mean: 72.3 years), and the main clinical presentation is parkinsonism with a baseline clinical diagnosis of PD in some cases. Similarly, the typical symptoms in cases carrying DCTN1 mutations are parkinsonism (mean AAO: 56.3 years) and brain atrophy. The relationships between genes and phenotypes may help clinicians to correctly diagnose. Although the associations between genes and PSP require more validations and investigations, further genetic screening is necessary when MAPT mutations are absent, especially in familial cases reminiscent of PSP.
COMMON VARIANTS IDENTIFIED THROUGH GENOME-WIDE APPROACHES
Melquist et al. carried out a pooled genome-wide scan of 500288 SNPs in 2007 and identified chromosome 11p12-p11 as another major risk locus for PSP following the MAPT haplotype H1. They further narrowed the locus to rs901746, the top ranked SNP, which mainly encompasses two genes (DNA damage-binding protein 2 (DDB2) and lysosomal acid phosphatase 2 (ACP2)) [97].
GWAS have identified thousands of genes and SNPs that contribute to complex diseases in humans [98] since the first GWAS was published in 2005 [99], introducing a new perspective on genetic studies and showing promising values in clinical applications [100]. the first large PSP GWAS revealed that MAPT rs8070723 and rs242557 are strongly associated with PSP (1.5×10–116 and 4.2×10–70, respectively), and it uncovered 3 novel risk loci of PSP (shown in Table 4) [19], highlighting new directions and strategies for PSP studies. Subsequent GWAS consisting of European cohorts identified additional variants (Table 5).
Table 5
Risk loci identified through GWAS
| Chr band | SNP | Nearest gene | OR (95% CI) | p | Cohorts | Method | Ref. |
| 17q21.31 | rs8070723 | MAPT | 5.46 | 1.5×10–116 | |||
| (4.72 –6.31) | |||||||
| 17q21.31 | rs242557 | MAPT | 0.51 | 4.2×10–70 | |||
| (0.47 –0.55) | |||||||
| 1q25.3 | rs1411478 | STX6 | 0.79 | 2.3×10–10 | PSP:2165 | Two-stage | Hoglinger |
| (0.74 –0.85) | NC:6807 | GWAS | et al. [19] | ||||
| 2p11.2 | rs7571971 | EIF2AK3 | 0.75 | 3.2×10–13 | |||
| (0.69 –0.81) | |||||||
| 3p22.1 | rs1768208 | MOBP | 0.72 | 1.0×10–16 | |||
| (0.67 –0.78) | |||||||
| 1q41 | rs6687758 | DUSP10 | 0.80 | 1.1×10–8 | PSP:2698 | Meta-analysis | Sanchez-Contreras |
| (0.74–0.86) | NC:8019 | of two GWAS | et al. [20] | ||||
| 12p12.1 | rs11568563 | SLCO1A2 | 0.67 | 5.3×10–10 | |||
| (0.59–0.76) | |||||||
| 6p21.1 | rs35740963 | RUNX2 | 0.77 | 1.8×10–8 | PSP:1646 | Joint | Chen |
| (NA) | NC:10662 | GWAS | et al. [21] |
Chr, chromosome; SNP, single nucleotide polymorphism; OR, odds ratio; CI, confidence interval; NA, not available; PSP, progressive supranuclear palsy; Ref., reference; NC, normal controls; GWAS, genome-wide association study; STX6, Syntaxin 6; EIF2AK3, Eukaryotic translation initiation factor 2 alpha kinase 3; MOBP, Myelin-associated oligodendrocyte basic protein; DUSP10, Dual specificity phosphatase 10; SLCO1A2, Solute carrier organic anion transporter family member 1A2; TRIM11, tripartite motif-containing protein 11; RUNX2, runt-related transcription factor 2.
GWAS have not only identified genetic factors likely increasing the risk for PSP but have also identified several molecular pathways contributing to PSP pathogenesis [19, 101]. Eukaryotic translation initiation factor 2 alpha kinase 3 (EIF2AK3) encodes the pancreatic endoplasmic reticulum kinase (PERK) protein, which is involved in the endoplasmic reticulum unfolded protein response (UPR) [102]. Activated UPR is present in regions affected in PSP, and UPR activation is related to tau [103]. Furthermore, loss of PERK function due to EIF2AK3 mutations causes neurodegeneration-like changes, including tau pathology [104]. Because tau normally does not traffic through the endoplasmic reticulum, the mechanisms involved in PERK, UPR and tauopathies are unknown [19]. Surprisingly, the increased representation of rs1768208 with the minor T-allele in PSP cases is more closely linked with the expression of the SLC25A38/Appoptosin gene, though its location is nearer to the myelin-associated oligodendrocyte basic protein (MOBP) gene [19, 105]. Zhao and his collaborators showed that appoptosin overexpression contributes to tau cleavage via caspase activation, which results in aggregation of insoluble tau, disruption of synaptic structures, and deficits of motor function in tau transgenic mice [105], confirming the associations among rs1768208, appoptosin and PSP as well as suggesting a potential diagnostic biomarker for tauopathies.
A GENETIC DETERMINANT OF PSP PHENOTYPE
Jabbari and colleagues carried out a clinical phenotype GWAS through comparing PSP-RS cases to non-RS cases in PSP cohorts, and suggested that rs564309, an intronic variant of the tripartite motif-containing protein 11 gene (TRIM11), may be a genetic modifier of clinical phenotype in PSP [106]. This is the first clinical phenotype GWAS in PSP and opens up new directions for the roles that genetics play in PSP phenotypes. On the other hand, as previous study has proposed that TRIM11 has a critical role in the clearance of misfolded proteins via ubiquitin proteasome system (UPS) [107], Jabbari’s study further proves that UPS is involved in tau pathology, which may provide a target for PSP therapy [106].
CONCLUSION
As a complex disorder involving multiple factors, PSP is challenging due to the inexplicit pathogenesis, lack of effective medications and poor prognosis. The genetic background of PSP has gained growing attention, and many data have suggested that genetics play a role in the susceptibility to PSP and that mutations of certain genes (such as MAPT) directly lead to PSP pathology. Faced with a series of risk factors and biological pathways associated with PSP, larger cohorts are required to validate these associations in addition to identify more novel loci. Functional studies are urgently needed to further elucidate underlying mechanisms, thus introducing new perspectives for diagnostic biomarkers and therapeutic targets for PSP and other tauopathies.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
This study was supported by the National Natural Science Foundation of China (No. 81671075, No. 81971029 and No. 81701134), the National Key R&D Program of China (No. 2017YFC0840100 and 2017YFC0840104), the Provincial Key Plan for Research and Development of Hunan (No. 2017SK2031).
REFERENCES
[1] | Kawashima M , Miyake M , Kusumi M , Adachi Y , Nakashima K ((2004) ) Prevalence of progressive supranuclear palsy in Yonago, Japan. Mov Disord 19: , 1239–1240. |
[2] | Boxer AL , Yu JT , Golbe LI , Litvan I , Lang AE , Höglinger GU ((2017) ) Advances in progressive supranuclear palsy: New diagnostic criteria, biomarkers, and therapeutic approaches. Lancet Neurol 16: , 552–563. |
[3] | Ling H ((2016) ) Clinical approach to progressive supranuclear palsy. J Mov Disord 9: , 3–13. |
[4] | Armstrong MJ ((2018) ) Progressive supranuclear palsy: An update. Curr Neurol Neurosci Rep 18: , 12. |
[5] | Steele JC , Richardson JC , Olszewski J ((1964) ) Progressive supranuclear palsy. A heterogeneous degeneration involving the brain stem, basal ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. Arch Neurol 10: , 333–359. |
[6] | Litvan I , Agid Y , Calne D , Campbell G , Dubois B , Duvoisin RC , Goetz CG , Golbe LI , Grafman J , Growdon JH , Hallett M , Jankovic J , Quinn NP , Tolosa E , Zee DS ((1996) ) Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): Report of the NINDS-SPSP international workshop. Neurology 47: , 1–9. |
[7] | Höglinger GU , Respondek G , Stamelou M , Kurz C , Josephs KA , Lang AE , Mollenhauer B , Müller U , Nilsson C , Whitwell JL , Arzberger T , Englund E , Gelpi E , Giese A , Irwin DJ , Meissner WG , Pantelyat A , Rajput A , van Swieten JC , Troakes C , Antonini A , Bhatia KP , Bordelon Y , Compta Y , Corvol JC , Colosimo C , Dickson DW , Dodel R , Ferguson L , Grossman M , Kassubek J , Krismer F , Levin J , Lorenzl S , Morris HR , Nestor P , Oertel WH , Poewe W , Rabinovici G , Rowe JB , Schellenberg GD , Seppi K , van Eimeren T , Wenning GK , Boxer AL , Golbe LI , Litvan I ((2017) ) Clinical diagnosis of progressive supranuclear palsy: The Movement Disorder Society criteria. Mov Disord 32: , 853–864. |
[8] | Ali F , Martin PR , Botha H , Ahlskog JE , Bower JH , Masumoto JY , Maraganore D , Hassan A , Eggers S , Boeve BF , Knopman DS , Drubach D , Petersen RC , Dunkley ED , van Gerpen J , Uitti R , Whitwell JL , Dickson DW , Josephs KA ((2019) ) Sensitivity and specificity of diagnostic criteria for progressive supranuclear palsy. Mov Disord 34: , 1144–1153. |
[9] | Hauw JJ , Daniel SE , Dickson D , Horoupian DS , Jellinger K , Lantos PL , mckee A , Tabaton M , Litvan I ((1994) ) Preliminary NINDS neuropathologic criteria for Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy). Neurology 44: , 2015–2019. |
[10] | Dickson DW , Rademakers R , Hutton ML ((2007) ) Progressive supranuclear palsy: Pathology and genetics. Brain Pathol 17: , 74–82. |
[11] | Williams DR , Holton JL , Strand C , Pittman A , de Silva R , Lees AJ , Revesz T ((2007) ) Pathological tau burden and distribution distinguishes progressive supranuclear palsy-parkinsonism from Richardson’s syndrome. Brain 130: , 1566–1576. |
[12] | Williams DR , Lees AJ ((2009) ) Progressive supranuclear palsy: Clinicopathological concepts and diagnostic challenges. Lancet Neurol 8: , 270–279. |
[13] | Rojo A , Pernaute RS , Fontán A , Ruíz PG , Honnorat J , Lynch T , Chin S , Gonzalo I , Rábano A , Martínez A , Daniel S , Pramstaller P , Morris H , Wood N , Lees A , Tabernero C , Nyggard T , Jackson AC , Hanson A , de Yébenes JG , Pramsteller P ((1999) ) Clinical genetics of familial progressive supranuclear palsy. Brain 122 (Pt 7): , 1233–1245. |
[14] | Fujioka S , Sanchez Contreras MY , Strongosky AJ , Ogaki K , Whaley NR , Tacik PM , van Gerpen JA , Uitti RJ , Ross OA , Wszolek ZK , Rademakers R , Dickson DW ((2015) ) Three sib-pairs of autopsy-confirmed progressive supranuclear palsy. Parkinsonism Relat Disord 21: , 101–105. |
[15] | Ros R , Gómez Garre P , Hirano M , Tai YF , Ampuero I , Vidal L , Rojo A , Fontan A , Vazquez A , Fanjul S , Hernandez J , Cantarero S , Hoenicka J , Jones A , Ahsan RL , Pavese N , Piccini P , Brooks DJ , Perez-Tur J , Nyggard T , de Yébenes JG ((2005) ) Genetic linkage of autosomal dominant progressive supranuclear palsy to 1q31.1. Ann Neurol 57: , 634–641. |
[16] | Yabe I , Yaguchi H , Kato Y , Miki Y , Takahashi H , Tanikawa S , Shirai S , Takahashi I , Kimura M , Hama Y , Matsushima M , Fujioka S , Kano T , Watanabe M , Nakagawa S , Kunieda Y , Ikeda Y , Hasegawa M , Nishihara H , Ohtsuka T , Tanaka S , Tsuboi Y , Hatakeyama S , Wakabayashi K , Sasaki H ((2018) ) Mutations in bassoon in individuals with familial and sporadic progressive supranuclear palsy-like syndrome. Sci Rep 8: , 819. |
[17] | Ygland E , van Westen D , Englund E , Rademakers R , Wszolek ZK , Nilsson K , Nilsson C , Landqvist Waldö M , Alafuzoff I , Hansson O , Gustafson L , Puschmann A ((2018) ) Slowly progressive dementia caused by MAPT R406W mutations: Longitudinal report on a new kindred and systematic review. Alzheimers Res Ther 10: , 2. |
[18] | Donker Kaat L , Boon AJW , Azmani A , Kamphorst W , Breteler MMB , Anar B , Heutink P , van Swieten JC ((2009) ) Familial aggregation of parkinsonism in progressive supranuclear palsy. Neurology 73: , 98–105. |
[19] | Hoglinger GU , Melhem NM , Dickson DW , Sleiman PM , Wang LS , Klei L , Rademakers R , de Silva R , Litvan I , Riley DE , van Swieten JC , Heutink P , Wszolek ZK , Uitti RJ , Vandrovcova J , Hurtig HI , Gross RG , Maetzler W , Goldwurm S , Tolosa E , Borroni B , Pastor P , Cantwell LB , Han MR , Dillman A , van der Brug MP , Gibbs JR , Cookson MR , Hernandez DG , Singleton AB , Farrer MJ , Yu CE , Golbe LI , Revesz T , Hardy J , Lees AJ , Devlin B , Hakonarson H , Muller U , Schellenberg GD ((2011) ) Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet 43: , 699–705. |
[20] | Sanchez-Contreras MY , Kouri N , Cook CN , Serie DJ , Heckman MG , Finch NA , Caselli RJ , Uitti RJ , Wszolek ZK , Graff-Radford N , Petrucelli L , Wang LS , Schellenberg GD , Dickson DW , Rademakers R , Ross OA ((2018) ) Replication of progressive supranuclear palsy genome-wide association study identifies SLCO1A2 and DUSP10 as new susceptibility loci. Mol Neurodegener 13: , 37. |
[21] | Chen JA , Chen Z , Won H , Huang AY , Lowe JK , Wojta K , Yokoyama JS , Bensimon G , Leigh PN , Payan C , Shatunov A , Jones AR , Lewis CM , Deloukas P , Amouyel P , Tzourio C , Dartigues JF , Ludolph A , Boxer AL , Bronstein JM , Al-Chalabi A , Geschwind DH , Coppola G ((2018) ) Joint genome-wide association study of progressive supranuclear palsy identifies novel susceptibility loci and genetic correlation to neurodegenerative diseases. Mol Neurodegener 13: , 41. |
[22] | Chen Z , Chen JA , Shatunov A , Jones AR , Kravitz SN , Huang AY , Lawrence L , Lowe JK , Lewis CM , Payan CAM , Lieb W , Franke A , Deloukas P , Amouyel P , Tzourio C , Dartigues JF , Ludolph A , Bensimon G , Leigh PN , Bronstein JM , Coppola G , Geschwind DH , Al-Chalabi A ((2019) ) Genome-wide survey of copy number variants finds MAPT duplications in progressive supranuclear palsy. Mov Disord 34: , 1049–1059. |
[23] | Spillantini MG , Goedert M ((2013) ) Tau pathology and neurodegeneration. Lancet Neurol 12: , 609–622. |
[24] | Rademakers R , Cruts M , van Broeckhoven C ((2004) ) The role of tau (MAPT) in frontotemporal dementia and related tauopathies. Hum Mutat 24: , 277–295. |
[25] | Wang Y , Mandelkow E ((2016) ) Tau in physiology and pathology. Nat Rev Neurosci 17: , 5–21. |
[26] | Hutton M , Lendon CL , Rizzu P , Baker M , Froelich S , Houlden H , Pickering-Brown S , Chakraverty S , Isaacs A , Grover A , Hackett J , Adamson J , Lincoln S , Dickson D , Davies P , Petersen RC , Stevens M , de Graaff E , Wauters E , van Baren J , Hillebrand M , Joosse M , Kwon JM , Nowotny P , Che LK , Norton J , Morris JC , Reed LA , Trojanowski J , Basun H , Lannfelt L , Neystat M , Fahn S , Dark F , Tannenberg T , Dodd PR , Hayward N , Kwok JB , Schofield PR , Andreadis A , Snowden J , Craufurd D , Neary D , Owen F , Oostra BA , Hardy J , Goate A , van Swieten J , Mann D , Lynch T , Heutink P ((1998) ) Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393: , 702–705. |
[27] | Shafei R , Woollacott IOC , Mummery CJ , Bocchetta M , Guerreiro R , Bras J , Warren JD , Lashley T , Jaunmuktane Z , Rohrer JD ((2020) ) Two pathologically confirmed cases of novel mutations in the MAPT gene causing frontotemporal dementia. Neurobiol Aging 87: , 141.e115–141.e120. |
[28] | Rossi G , Tagliavini F ((2015) ) Frontotemporal lobar degeneration: Old knowledge and new insight into the pathogenetic mechanisms of tau mutations. Front Aging Neurosci 7: , 192. |
[29] | Conrad C , Andreadis A , Trojanowski JQ , Dickson DW , Kang D , Chen X , Wiederholt W , Hansen L , Masliah E , Thal LJ , Katzman R , Xia Y , Saitoh T ((1997) ) Genetic evidence for the involvement of tau in progressive supranuclear palsy. Ann Neurol 41: , 277–281. |
[30] | Bennett P , Bonifati V , Bonuccelli U , Colosimo C , De Mari M , Fabbrini G , Marconi R , Meco G , Nicholl DJ , Stocchi F , Vanacore N , Vieregge P , Williams AC ((1998) ) Direct genetic evidence for involvement of tau in progressive supranuclear palsy. European Study Group on Atypical Parkinsonism Consortium. Neurology 51: , 982–985. |
[31] | Higgins JJ , Litvan I , Pho LT , Li W , Nee LE ((1998) ) Progressive supranuclear gaze palsy is in linkage disequilibrium with the tau and not the alpha-synuclein gene. Neurology 50: , 270–273. |
[32] | Morris HR , Janssen JC , Bandmann O , Daniel SE , Rossor MN , Lees AJ , Wood NW ((1999) ) The tau gene A0 polymorphism in progressive supranuclear palsy and related neurodegenerative diseases. J Neurol Neurosurg Psychiatry 66: , 665–667. |
[33] | Baker M , Litvan I , Houlden H , Adamson J , Dickson D , Perez-Tur J , Hardy J , Lynch T , Bigio E , Hutton M ((1999) ) Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum Mol Genet 8: , 711–715. |
[34] | Pastor P , Ezquerra M , Tolosa E , Muñoz E , Martí MJ , Valldeoriola F , Molinuevo JL , Calopa M , Oliva R ((2002) ) Further extension of the H1 haplotype associated with progressive supranuclear palsy. Mov Disord 17: , 550–556. |
[35] | Rademakers R , Melquist S , Cruts M , Theuns J , Del-Favero J , Poorkaj P , Baker M , Sleegers K , Crook R , De Pooter T , Bel Kacem S , Adamson J , Van den Bossche D , Van den Broeck M , Gass J , Corsmit E , De Rijk P , Thomas N , Engelborghs S , Heckman M , Litvan I , Crook J , De Deyn PP , Dickson D , Schellenberg GD , Van Broeckhoven C , Hutton ML ((2005) ) High-density SNP haplotyping suggests altered regulation of tau gene expression in progressive supranuclear palsy. Hum Mol Genet 14: , 3281–3292. |
[36] | Pittman AM , Myers AJ , Abou-Sleiman P , Fung HC , Kaleem M , Marlowe L , Duckworth J , Leung D , Williams D , Kilford L , Thomas N , Morris CM , Dickson D , Wood NW , Hardy J , Lees AJ , de Silva R ((2005) ) Linkage disequilibrium fine mapping and haplotype association analysis of the tau gene in progressive supranuclear palsy and corticobasal degeneration. J Med Genet 42: , 837–846. |
[37] | Myers AJ , Kaleem M , Marlowe L , Pittman AM , Lees AJ , Fung HC , Duckworth J , Leung D , Gibson A , Morris CM , de Silva R , Hardy J ((2005) ) The H1c haplotype at the MAPT locus is associated with Alzheimer’s disease. Hum Mol Genet 14: , 2399–2404. |
[38] | Myers AJ , Pittman AM , Zhao AS , Rohrer K , Kaleem M , Marlowe L , Lees A , Leung D , mckeith IG , Perry RH , Morris CM , Trojanowski JQ , Clark C , Karlawish J , Arnold S , Forman MS , Van Deerlin V , de Silva R , Hardy J ((2007) ) The MAPT H1c risk haplotype is associated with increased expression of tau and especially of 4 repeat containing transcripts. Neurobiol Dis 25: , 561–570. |
[39] | Heckman MG , Brennan RR , Labbé C , Soto AI , Koga S , deture MA , Murray ME , Petersen RC , Boeve BF , van Gerpen JA , Uitti RJ , Wszolek ZK , Rademakers R , Dickson DW , Ross OA ((2019) ) Association of MAPT subhaplotypes with risk of progressive supranuclear palsy and severity of tau pathology. JAMA Neurol 76: , 710–717. |
[40] | Le Ber I , Camuzat A , Guillot-Noel L , Hannequin D , Lacomblez L , Golfier V , Puel M , Martinaud O , Deramecourt V , Rivaud-Pechoux S , Millecamps S , Vercelletto M , Couratier P , Sellal F , Pasquier F , Salachas F , Thomas-Antérion C , Didic M , Pariente J , Seilhean D , Ruberg M , Wargon I , Blanc F , Camu W , Michel B-F , Berger E , Sauvée M , Thauvin-Robinet C , Mondon K , Tournier-Lasserve E , Goizet C , Fleury M , Viennet G , Verpillat P , Meininger V , Duyckaerts C , Dubois B , Brice A ((2013) ) C9ORF72 repeat expansions in the frontotemporal dementias spectrum of diseases: A flow-chart for genetic testing. J Alzheimers Dis 34: , 485–499. |
[41] | Nakayama S , Shimonaka S , Elahi M , Nishioka K , Oji Y , Matsumoto S-E , Li Y , Yoshino H , Mogushi K , Hatano T , Sato T , Ikura T , Ito N , Motoi Y , Hattori N ((2019) ) Tau aggregation and seeding analyses of two novel MAPT variants found in patients with motor neuron disease and progressive parkinsonism. Neurobiol Aging 84: , 240.e213–240.e222. |
[42] | Yasuda M , Kawamata T , Komure O , Kuno S , D’Souza I , Poorkaj P , Kawai J , Tanimukai S , Yamamoto Y , Hasegawa H , Sasahara M , Hazama F , Schellenberg GD , Tanaka C ((1999) ) A mutation in the microtubule-associated protein tau in pallido-nigro-luysian degeneration. Neurology 53: , 864–868. |
[43] | Ahmed Z , Josephs KA , Gonzalez J , delledonne A , Dickson DW ((2008) ) Clinical and neuropathologic features of progressive supranuclear palsy with severe pallido-nigro-luysial degeneration and axonal dystrophy. Brain 131: , 460–472. |
[44] | Rossi G , Gasparoli E , Pasquali C , Di Fede G , Testa D , Albanese A , Bracco F , Tagliavini F ((2004) ) Progressive supranuclear palsy and Parkinson’s disease in a family with a new mutation in the tau gene. Ann Neurol 55: , 448. |
[45] | Pastor P , Pastor E , Carnero C , Vela R , Garcia T , Amer G , Tolosa E , Oliva R ((2001) ) Familial atypical progressive supranuclear palsy associated with homozigosity for the deln296 mutation in the tau gene. Ann Neurol 49: , 263–267. |
[46] | Poorkaj P , Muma NA , Zhukareva V , Cochran EJ , Shannon KM , Hurtig H , Koller WC , Bird TD , Trojanowski JQ , Lee VM , Schellenberg GD ((2002) ) An R5L tau mutation in a subject with a progressive supranuclear palsy phenotype. Ann Neurol 52: , 511–516. |
[47] | Delisle MB , Murrell JR , Richardson R , Trofatter JA , Rascol O , Soulages X , Mohr M , Calvas P , Ghetti B ((1999) ) A mutation at codon 279 (N279K) in exon 10 of the Tau gene causes a tauopathy with dementia and supranuclear palsy. Acta Neuropathol 98: , 62–77. |
[48] | Soliveri P , Rossi G , Monza D , Tagliavini F , Piacentini S , Albanese A , Bugiani O , Girotti F ((2003) ) A case of dementia parkinsonism resembling progressive supranuclear palsy due to mutation in the tau protein gene. Arch Neurol 60: , 1454–1456. |
[49] | Ogaki K , Li Y , Takanashi M , Ishikawa K-I , Kobayashi T , Nonaka T , Hasegawa M , Kishi M , Yoshino H , Funayama M , Tsukamoto T , Shioya K , Yokochi M , Imai H , Sasaki R , Kokubo Y , Kuzuhara S , Motoi Y , Tomiyama H , Hattori N ((2013) ) Analyses of the MAPT, PGRN, and c9orf72 mutations in Japanese patients with FTLD, PSP, and CBS. Parkinsonism Relat Disord 19: , 15–20. |
[50] | Ogaki K , Motoi Y , Li Y , Tomiyama H , Shimizu N , Takanashi M , Nakanishi A , Yokoyama K , Hattori N ((2011) ) Visual grasping in frontotemporal dementia and parkinsonism linked to chromosome 17 (microtubule-associated with protein tau): A comparison of N-Isopropyl-p-[(123)I]-iodoamphetamine brain perfusion single photon emission computed tomography analysis with progressive supranuclear palsy. Mov Disord 26: , 561–563. |
[51] | Rohrer JD , Paviour D , Vandrovcova J , Hodges J , de Silva R , Rossor MN ((2011) ) Novel L284R MAPT mutation in a family with an autosomal dominant progressive supranuclear palsy syndrome. Neurodegener Dis 8: , 149–152. |
[52] | Bird TD , Nochlin D , Poorkaj P , Cherrier M , Kaye J , Payami H , Peskind E , Lampe TH , Nemens E , Boyer PJ , Schellenberg GD ((1999) ) A clinical pathological comparison of three families with frontotemporal dementia and identical mutations in the tau gene (P301L). Brain 122 (Pt 4): , 741–756. |
[53] | Ros R , Thobois S , Streichenberger N , Kopp N , Sanchez MP , Perez M , Hoenicka J , Avila J , Honnorat J , de Yebenes JG ((2005) ) A new mutation of the tau gene, G303V, in early-onset familial progressive supranuclear palsy. Arch Neurol 62: , 1444–1450. |
[54] | Stanford PM , Halliday GM , Brooks WS , Kwok JB , Storey CE , Creasey H , Morris JG , Fulham MJ , Schofield PR ((2000) ) Progressive supranuclear palsy pathology caused by a novel silent mutation in exon 10 of the tau gene: Expansion of the disease phenotype caused by tau gene mutations. Brain 123 (Pt 5): , 880–893. |
[55] | Spina S , Farlow MR , Unverzagt FW , Kareken DA , Murrell JR , Fraser G , Epperson F , Crowther RA , Spillantini MG , Goedert M , Ghetti B ((2008) ) The tauopathy associated with mutation+3 in intron 10 of Tau: Characterization of the MSTD family. Brain 131: , 72–89. |
[56] | Omoto M , Suzuki S , Ikeuchi T , Ishihara T , Kobayashi T , Tsuboi Y , Ogasawara J , Koga M , Kawai M , Iwaki T , Kanda T ((2012) ) Autosomal dominant tauopathy with parkinsonism and central hypoventilation. Neurology 78: , 762–764. |
[57] | Morris HR , Osaki Y , Holton J , Lees AJ , Wood NW , Revesz T , Quinn N ((2003) ) Tau exon 10+16 mutation FTDP-17 presenting clinically as sporadic young onset PSP. Neurology 61: , 102–104. |
[58] | Rossi G , Bastone A , Piccoli E , Morbin M , Mazzoleni G , Fugnanesi V , Beeg M , Del Favero E , Cantù L , Motta S , Salsano E , Pareyson D , Erbetta A , Elia AE , Del Sorbo F , Silani V , Morelli C , Salmona M , Tagliavini F ((2014) ) Different mutations at V363 MAPT codon are associated with atypical clinical phenotypes and show unusual structural and functional features. Neurobiol Aging 35: , 408–417. |
[59] | Sanchez-Contreras M , Heckman MG , Tacik P , Diehl N , Brown PH , Soto-Ortolaza AI , Christopher EA , Walton RL , Ross OA , Golbe LI , Graff-Radford N , Wszolek ZK , Dickson DW , Rademakers R ((2017) ) Study of LRRK2 variation in tauopathy: Progressive supranuclear palsy and corticobasal degeneration. Mov Disord 32: , 115–123. |
[60] | Deng H , Wang P , Jankovic J ((2018) ) The genetics of Parkinson disease. Ageing Res Rev 42: , 72–85. |
[61] | Zhang Y , Sun Q , Yi M , Zhou X , Guo J , Xu Q , Tang B , Yan X ((2017) ) Genetic analysis of R1628P in Parkinson’s disease in Asian populations. Parkinsons Dis 2017: , 8093124. |
[62] | Ross OA , Whittle AJ , Cobb SA , Hulihan MM , Lincoln SJ , Toft M , Farrer MJ , Dickson DW ((2006) ) Lrrk2 R1441 substitution and progressive supranuclear palsy. Neuropathol Appl Neurobiol 32: , 23–25. |
[63] | Tan EK , Skipper L , Chua E , Wong M-C , Pavanni R , Bonnard C , Kolatkar P , Liu J-J ((2006) ) Analysis of 14 LRRK2 mutations in Parkinson’s plus syndromes and late-onset Parkinson’s disease. Mov Disord 21: , 997–1001. |
[64] | Gaig C , Ezquerra M , Martí MJ , Valldeoriola F , Muñoz E , Lladó A , Rey MJ , Cardozo A , Molinuevo JL , Tolosa E ((2008) ) Screening for the LRRK2 G2019S and codon-1441 mutations in a pathological series of parkinsonian syndromes and frontotemporal lobar degeneration. J Neurol Sci 270: , 94–98. |
[65] | Madzar D , Schulte C , Gasser T ((2009) ) Screening for LRRK2 R1441 mutations in a cohort of PSP patients from Germany. Eur J Neurol 16: , 1230–1232. |
[66] | Zimprich A , Biskup S , Leitner P , Lichtner P , Farrer M , Lincoln S , Kachergus J , Hulihan M , Uitti RJ , Calne DB , Stoessl AJ , Pfeiffer RF , Patenge N , Carbajal IC , Vieregge P , Asmus F , Müller-Myhsok B , Dickson DW , Meitinger T , Strom TM , Wszolek ZK , Gasser T ((2004) ) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44: , 601–607. |
[67] | Spanaki C , Latsoudis H , Plaitakis A ((2006) ) LRRK2 mutations on Crete: R1441H associated with PD evolving to PSP. Neurology 67: , 1518–1519. |
[68] | Rajput A , Dickson DW , Robinson CA , Ross OA , Dächsel JC , Lincoln SJ , Cobb SA , Rajput ML , Farrer MJ ((2006) ) Parkinsonism, Lrrk2 G2019S, and tau neuropathology. Neurology 67: , 1506–1508. |
[69] | Ruffmann C , Giaccone G , Canesi M , Bramerio M , Goldwurm S , Gambacorta M , Rossi G , Tagliavini F , Pezzoli G ((2012) ) Atypical tauopathy in a patient with LRRK2-G2019S mutation and tremor-dominant Parkinsonism. Neuropathol Appl Neurobiol 38: , 382–386. |
[70] | Blauwendraat C , Pletnikova O , Geiger JT , Murphy NA , Abramzon Y , Rudow G , Mamais A , Sabir MS , Crain B , Ahmed S , Rosenthal LS , Bakker CC , Faghri F , Chia R , Ding J , Dawson TM , Pantelyat A , Albert MS , Nalls MA , Resnick SM , Ferrucci L , Cookson MR , Hillis AE , Troncoso JC , Scholz SW ((2019) ) Genetic analysis of neurodegenerative diseases in a pathology cohort. Neurobiol Aging 76: , 214.e211–214.e219. |
[71] | Greggio E , Cookson MR ((2009) ) Leucine-rich repeat kinase 2 mutations and Parkinson’s disease: Three questions. ASN Neuro 1: , e00002. |
[72] | Trinh J , Guella I , mckenzie M , Gustavsson EK , Szu-Tu C , Petersen MS , Rajput A , Rajput AH , mckeown M , Jeon BS , Aasly JO , Bardien S , Farrer MJ ((2015) ) Novel LRRK2 mutations in Parkinsonism. Parkinsonism Relat Disord 21: , 1119–1121. |
[73] | Wszolek ZK , Pfeiffer RF , Tsuboi Y , Uitti RJ , mccomb RD , Stoessl AJ , Strongosky AJ , Zimprich A , Müller-Myhsok B , Farrer MJ , Gasser T , Calne DB , Dickson DW ((2004) ) Autosomal dominant parkinsonism associated with variable synuclein and tau pathology. Neurology 62: , 1619–1622. |
[74] | Schroer TA ((2004) ) Dynactin. Annu Rev Cell Dev Biol 20: , 759–779. |
[75] | Puls I , Jonnakuty C , lamonte BH , Holzbaur ELF , Tokito M , Mann E , Floeter MK , Bidus K , Drayna D , Oh SJ , Brown RH , Ludlow CL , Fischbeck KH ((2003) ) Mutant dynactin in motor neuron disease. Nat Genet 33: , 455–456. |
[76] | Farrer MJ , Hulihan MM , Kachergus JM , Dächsel JC , Stoessl AJ , Grantier LL , Calne S , Calne DB , Lechevalier B , Chapon F , Tsuboi Y , Yamada T , Gutmann L , Elibol B , Bhatia KP , Wider C , Vilariño-Güell C , Ross OA , Brown LA , Castanedes-Casey M , Dickson DW , Wszolek ZK ((2009) ) DCTN1 mutations in Perry syndrome. Nat Genet 41: , 163–165. |
[77] | Konno T , Ross OA , Teive HAG , Sławek J , Dickson DW , Wszolek ZK ((2017) ) DCTN1-related neurodegeneration: Perry syndrome and beyond. Parkinsonism Relat Disord 41: , 14–24. |
[78] | Caroppo P , Le Ber I , Clot F , Rivaud-Péchoux S , Camuzat A , De Septenville A , Boutoleau-Bretonnière C , Mourlon V , Sauvée M , Lebouvier T , Bonnet A-M , Levy R , Vercelletto M , Brice A ((2014) ) DCTN1 mutation analysis in families with progressive supranuclear palsy-like phenotypes. JAMA Neurol 71: , 208–215. |
[79] | Gustavsson EK , Trinh J , Guella I , Szu-Tu C , Khinda J , Lin CH , Wu RM , Stoessl J , Appel-Cresswell S , mckeown M , Rajput A , Rajput AH , Petersen MS , Jeon BS , Aasly JO , Farrer MJ ((2016) ) DCTN1 p.K56R in progressive supranuclear palsy. Parkinsonism Relat Disord 28: , 56–61. |
[80] | Honda H , Sasagasako N , Shen C , Shijo M , Hamasaki H , Suzuki SO , Tsuboi Y , Fujii N , Iwaki T ((2018) ) DCTN1 F52L mutation case of Perry syndrome with progressive supranuclear palsy-like tauopathy. Parkinsonism Relat Disord 51: , 105–110. |
[81] | Godeiro-Junior C , Inaoka RJ , Barbosa MR , Silva MR , Aguiar Pde C , Barsottini O ((2006) ) Mutations in NPC1 in two Brazilian patients with Niemann-Pick disease type C and progressive supranuclear palsy-like presentation. Mov Disord 21: , 2270–2272. |
[82] | Cupidi C , Frangipane F , Gallo M , Clodomiro A , Colao R , Bernardi L , Anfossi M , Conidi ME , Vasso F , Curcio SA , Mirabelli M , Smirne N , Torchia G , Muraca MG , Puccio G , Di Lorenzo R , Zampieri S , Romanello M , Dardis A , Maletta RG , Bruni AC ((2017) ) Role of Niemann-Pick Type C disease mutations in dementia. J Alzheimers Dis 55: , 1249–1259. |
[83] | Lesage S , Le Ber I , Condroyer C , Broussolle E , Gabelle A , Thobois S , Pasquier F , Mondon K , Dion PA , Rochefort D , Rouleau GA , Dürr A , Brice A ((2013) ) c9orf72 repeat expansions are a rare genetic cause of parkinsonism. Brain 136: , 385–391. |
[84] | Morales B , Martínez A , Gonzalo I , Vidal L , Ros R , Gomez-Tortosa E , Rabano A , Ampuero I , Sánchez M , Hoenicka J , García De Yébenes J ((2002) ) Steele-Richardson-Olszewski syndrome in a patient with a single C212Y mutation in the parkin protein. Mov Disord 17: , 1374–1380. |
[85] | Sanchez MP , Gonzalo I , Avila J , De Yebenes JG ((2002) ) Progressive supranuclear palsy and tau hyperphosphorylation in a patient with a C212Y parkin mutation. J Alzheimers Dis 4: , 399–404. |
[86] | Ros R , Ampuero I , García de Yébenes J ((2008) ) Parkin polymorphisms in progressive supranuclear palsy. J Neurol Sci 268: , 176–178. |
[87] | Cannas A , Borghero G , Floris GL , Solla P , Chiò A , Traynor BJ , Calvo A , Restagno G , Majounie E , Costantino E , Piras V , Lavra L , Pani C , Orofino G , Di Stefano F , Tacconi P , Mascia MM , Muroni A , Murru MR , Tranquilli S , Corongiu D , Rolesu M , Cuccu S , Marrosu F , Marrosu MG ((2013) ) The p.A382T TARDBP gene mutation in Sardinian patients affected by Parkinson’s disease and other degenerative parkinsonisms. Neurogenetics 14: , 161–166. |
[88] | Yabe I , Nakano F , Shirai S , Matsushima M , Takahashi I , Sasaki H ((2016) ) Frontotemporal dementia and progressive supranuclear palsy-like syndrome with a novel TARDBP mutation. Neurol Clin Neurosc 4: , 76–77. |
[89] | Tremolizzo L , Bertola F , Casati G , Piperno A , Ferrarese C , Appollonio I ((2011) ) Progressive supranuclear palsy-like phenotype caused by progranulin p.Thr272fs mutation. Mov Disord 26: , 1964–1966. |
[90] | Wilke C , Baets J , De Bleecker JL , Deconinck T , Biskup S , Hayer SN , Zuchner S , Schule R , De Jonghe P , Synofzik M ((2018) ) Beyond ALS and FTD: The phenotypic spectrum of TBK1 mutations includes PSP-like and cerebellar phenotypes. Neurobiol Aging 62: , 244.e249–244.e213. |
[91] | Tang M , Gu X , Wei J , Jiao B , Zhou L , Zhou Y , Weng L , Yan X , Tang B , Xu J , Shen L ((2016) ) Analyses MAPT, GRN, and c9orf72 mutations in Chinese patients with frontotemporal dementia. Neurobiol Aging 46: , 235.e211–235.e215. |
[92] | Zhang Y , Shu L , Sun Q , Zhou X , Pan H , Guo J , Tang B ((2018) ) Integrated genetic analysis of racial differences of common variants in Parkinson’s disease: A meta-analysis. Front Mol Neurosci 11: , 43. |
[93] | Jiao B , Sun Q , Yuan Z , Wang J , Zhou L , Yan X , Tang B , Shen L ((2018) ) Rare variants in patients with frontotemporal dementia and amyotrophic lateral sclerosis in a Chinese cohort. Transl Neurodegener 7: , 31. |
[94] | Jiao B , Tang B , Liu X , Yan X , Zhou L , Yang Y , Wang J , Xia K , Shen L ((2014) ) Identification of c9orf72 repeat expansions in patients with amyotrophic lateral sclerosis and frontotemporal dementia in mainland China. Neurobiol Aging 35: , 936.e919–936.e922. |
[95] | Stamelou M , Quinn NP , Bhatia KP ((2013) ) “Atypical” atypical parkinsonism: New genetic conditions presenting with features of progressive supranuclear palsy, corticobasal degeneration, or multiple system atrophy-a diagnostic guide. Mov Disord 28: , 1184–1199. |
[96] | Wilke C , Pomper JK , Biskup S , Puskas C , Berg D , Synofzik M ((2016) ) Atypical parkinsonism in c9orf72 expansions: A case report and systematic review of 45 cases from the literature. J Neurol 263: , 558–574. |
[97] | Melquist S , Craig DW , Huentelman MJ , Crook R , Pearson JV , Baker M , Zismann VL , Gass J , Adamson J , Szelinger S , Corneveaux J , Cannon A , Coon KD , Lincoln S , Adler C , Tuite P , Calne DB , Bigio EH , Uitti RJ , Wszolek ZK , Golbe LI , Caselli RJ , Graff-Radford N , Litvan I , Farrer MJ , Dickson DW , Hutton M , Stephan DA ((2007) ) Identification of a novel risk locus for progressive supranuclear palsy by a pooled genomewide scan of 500,288 single-nucleotide polymorphisms. Am J Hum Genet 80: , 769–778. |
[98] | Huang Q ((2015) ) Genetic study of complex diseases in the post-GWAS era. J Genet Genomics 42: , 87–98. |
[99] | Klein RJ , Zeiss C , Chew EY , Tsai J-Y , Sackler RS , Haynes C , Henning AK , sangiovanni JP , Mane SM , Mayne ST , Bracken MB , Ferris FL , Ott J , Barnstable C , Hoh J ((2005) ) Complement factor H polymorphism in age-related macular degeneration. Science 308: , 385–389. |
[100] | Manolio TA ((2013) ) Bringing genome-wide association findings into clinical use. Nat Rev Genet 14: , 549–558. |
[101] | Ferrari R , Ryten M , Simone R , Trabzuni D , Nicolaou N , Hondhamuni G , Ramasamy A , Vandrovcova J , Weale ME , Lees AJ , Momeni P , Hardy J , de Silva R ((2014) ) Assessment of common variability and expression quantitative trait loci for genome-wide associations for progressive supranuclear palsy. Neurobiol Aging 35: , 1514.e1511–1512. |
[102] | Ron D , Walter P ((2007) ) Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8: , 519–529. |
[103] | Stutzbach LD , Xie SX , Naj AC , Albin R , Gilman S , Lee VMY , Trojanowski JQ , Devlin B , Schellenberg GD ((2013) ) The unfolded protein response is activated in disease-affected brain regions in progressive supranuclear palsy and Alzheimer’s disease. Acta Neuropathol Commun 1: , 31. |
[104] | Bruch J , Kurz C , Vasiljevic A , Nicolino M , Arzberger T , Höglinger GU ((2015) ) Early neurodegeneration in the brain of a child without functional PKR-like endoplasmic reticulum kinase. J Neuropathol Exp Neurol 74: , 850–857. |
[105] | Zhao Y , Tseng IC , Heyser CJ , Rockenstein E , Mante M , Adame A , Zheng Q , Huang T , Wang X , Arslan PE , Chakrabarty P , Wu C , Bu G , Mobley WC , Zhang Y-W , St George-Hyslop P , Masliah E , Fraser P , Xu H ((2015) ) Appoptosin-mediated caspase cleavage of tau contributes to progressive supranuclear palsy pathogenesis. Neuron 87: , 963–975. |
[106] | Jabbari E , Woodside J , Tan MMX , Shoai M , Pittman A , Ferrari R , Mok KY , Zhang D , Reynolds RH , de Silva R , Grimm MJ , Respondek G , Muller U , Al-Sarraj S , Gentleman SM , Lees AJ , Warner TT , Hardy J , Revesz T , Hoglinger GU , Holton JL , Ryten M , Morris HR ((2018) ) Variation at the TRIM11 locus modifies progressive supranuclear palsy phenotype. Ann Neurol 84: , 485–496. |
[107] | Chen L , Brewer MD , Guo L , Wang R , Jiang P , Yang X ((2017) ) Enhanced degradation of misfolded proteins promotes tumorigenesis. Cell Rep 18: , 3143–3154. |


