
Treating Parkinson’s Disease with Antibodies: Previous Studies and Future Directions
Abstract
Parkinson’s disease is a neurodegenerative disorder mainly characterized by the degeneration of dopaminergic neurons in the substantia nigra. Degenerating neurons contain abnormal aggregates called Lewy bodies, that are predominantly composed of the misfolded and/or mutated alpha-synuclein protein. Post-translational modifications, cellular stress, inflammation and gene mutations are thought to trigger its pathological misfolding and aggregation. With alpha-synuclein pathology being strongly associated with dopaminergic neuronal toxicity, strategies aimed to reduce its burden are expected to be beneficial in slowing disease progression. Moreover, multiple sources of evidence suggest a cell-to-cell transmission of pathological alpha-synuclein in a prion-like manner. Therefore, antibodies targeting extra- or intracellular alpha-synuclein could be efficient in limiting the aggregation and transmission. Several active and passive immunization strategies have been explored to target alpha-synuclein. Here, we summarize immunotherapeutic approaches that were tested in pre-clinical or clinical studies in the last two decades in an attempt to treat Parkinson’s disease.
INTRODUCTION
Parkinson’s disease (PD) is the second most common neurodegenerative disorder in the world after Alzheimer’s disease (AD). PD is characterized by the degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNpc), although other types of neurons are also affected [1]. Resulting symptoms are predominantly motor-related, including resting tremors, muscle rigidity, bradykinesia and postural instability [2, 3]. Several non-motor symptoms are also present and frequently appear several years before the onset of motor symptoms. They include sleep, olfactory and autonomic disorders, cognitive impairment, psychiatric symptoms, pain and fatigue (Fig. 1) [4, 5]. PD has a heterogenous clinical presentation and a complex pathogenesis. Around 15% of PD patients have family a history and 5–10% of cases are caused by monogenic mutations with Mendelian inheritance (SNCA, LRRK2, Parkin, VPS35, etc.) [6]. The remaining cases are sporadic with unknown etiology and are thought to occur following a combination of genetic and environmental risk factors [7, 8]. Despite that PD was first described more than 200 years ago [9], no disease modifying treatments are currently available. Existing therapies, solely focused on symptom management, are associated with incapacitating side effects [10].
Fig.1
Principal hallmarks of Parkinson’s disease. Parkinson’s disease is characterized by the loss of dopaminergic neurons in the substantia nigra. Affected neurons present insoluble aggregates in their cytoplasm and neurites called Lewy bodies and Lewy neurites, respectively. These inclusions are predominantly composed of the protein alpha-synuclein. Activated microglia releasing pro-inflammatory cytokines are also present and promote an inflammatory environment possibly contributing to neuronal loss. Dopaminergic neuron degeneration leads to the classical motor symptoms. It is hypothesized that the affection of non-dopaminergic neurons in other brain regions would be responsible for the various non-motor symptoms that also greatly affect quality of life. (Created with BioRender)
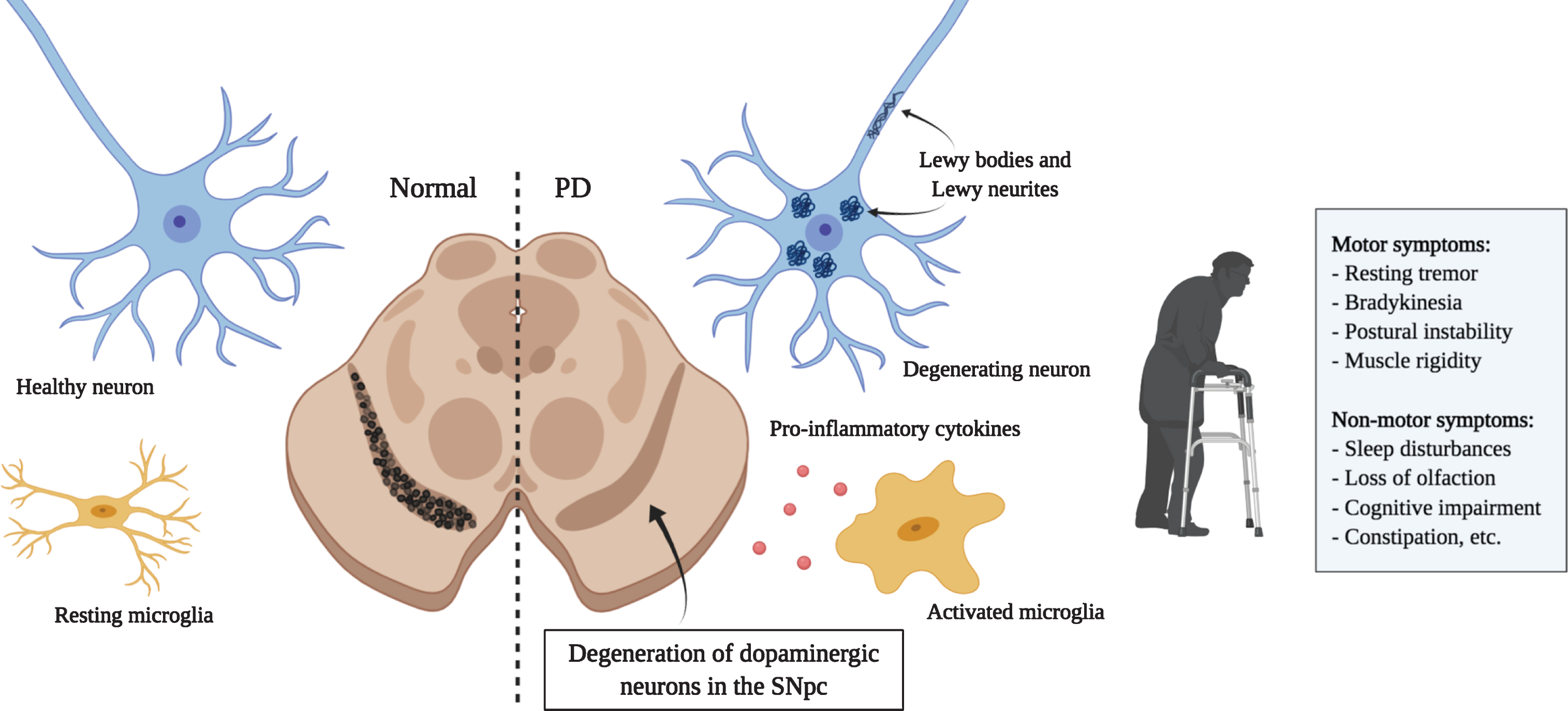
Parkinson’s disease, along with Huntington’s disease (HD) and amyotrophic lateral sclerosis (ALS), is a member of a class of proteinopathies characterized by the aberrant accumulation of disordered proteins. In PD, affected neurons present insoluble inclusions in the cytoplasm and neurites called Lewy bodies (LB) and Lewy neurites (LN) [11, 12]. These aggregates are composed of multiple misfolded proteins, membranous organelles, and lipids that interfere with several cellular processes and contribute to cell death [13]. Their main component is the protein alpha-synuclein (aSyn) [14, 15]. Post-translational modifications and cellular stress are thought to trigger the misfolding of this protein and its subsequent association in toxic oligomers and fibrils [16]. Other diseases including dementia with Lewy bodies (DLB) and multiple system atrophy (MSA), gathered under the name synucleinopathies, have been associated with the aberrant accumulation of aSyn [15]. Because aSyn aggregation strongly correlates with cellular toxicity [17–19], strategies aimed at reducing aSyn burden would likely be beneficial for the treatment of these diseases as well.
Aberrant proteins are an attractive substrate for antibody-based therapies because pathological strains or aberrantly folded proteins can be specifically bound and targeted for degradation. Moreover, aSyn was shown to be present in the blood and cerebrospinal fluid (CSF) in humans [20], indicating that it can be secreted by cells. This represents a pool of proteins that could be efficiently targeted extracellularly by antibodies. Numerous pre-clinical studies using immunotherapeutic strategies have been previously explored to treat various proteinopathies including: amyloid-β and Tau for AD, mutant huntingtin protein (mHtt) for HD, and TDP-43 for ALS [21–24]. Clinical trials involving antibodies have also been carried out, notably in AD and progressive supranuclear palsy (PSP) research, with variable performances. For AD, despite promising pre-clinical results and good target engagement, most immunotherapeutic agents tested in clinical trials have until now failed to generate significant clinical improvement [25, 26]. Several clinical trials are however still ongoing. In PSP, three monoclonal antibodies targeting extracellular Tau are currently under clinical investigation. Two failed to meet the primary endpoint in phase II (ABBV-8E12 [27, 28] and BIIB092 [29, 30]), while the third (UCB0107) showed favorable results in phase I [31] and will likely be evaluated in phase II [32]. Herein, we review the therapeutic relevance of targeting aSyn for PD treatment and the different active and passive immunization strategies that have been or are currently studied in various stages of pre-clinical and clinical studies. We further assess the challenges of these therapeutic approaches and the requirements for an effective transposition to clinic.
ALPHA-SYNUCLEIN
Alpha-synuclein is a 140 amino-acid protein encoded by the SNCA gene [33]. It is abundantly and ubiquitously expressed in the brain and localized mainly at the presynaptic compartment. The family of synucleins also includes β, and γ-synuclein, that are also located in synaptic terminals [34]. Several point mutations in the SNCA gene (for example A53T, A30P, E46K, etc.) or a multiplication of the whole locus (mostly triplication) are highly associated with PD [33, 35, 36]. Since aSyn is the main component of LB and LN, its involvement in the pathogenesis of the disease has been extensively studied. The aSyn protein can be divided in three main structural regions: a positively charged N-terminal region (residues 1–60); a central hydrophobic region (residues 61–95), called the non-amyloid-β component (NAC), which is responsible for β-sheet formation; and a negatively charged C-terminal region (residues 95–140) that is subject to numerous post-translational modifications [16].
aSyn exists in a dynamic equilibrium between a cytosolic and a membrane-bound form that influences its secondary structure. In its soluble state, aSyn is naturally unfolded and mostly monomeric. Studies have suggested that it also exists as a stable tetramer in the cytoplasm, and it is likely that both forms are present in physiological conditions [37, 38]. The N-terminal region adopts an alpha-helicoidal structure that allows it to associate to lipid membranes [39, 40]. This membrane association is necessary for the functions of the protein and prevents aggregation [41]. In a pathological context, aSyn tends to form β-sheets that aggregate, forms fibrils, and deposits in LBs [42, 43]. These toxic conformations most likely derive from the soluble and unfolded cytoplasmic protein [44]. In LBs and LNs, 90% of aSyn is phosphorylated at serine 129 (pS129) [45, 46], suggesting that this post-translational modification (PTM) plays a role in the pathological transformation of the protein. Indeed, only 4% of total aSyn is found phosphorylated in healthy individuals that lack LBs and LNs [46]. Several studies demonstrated that pS129-aSyn accelerates the formation of toxic inclusions and the loss of dopaminergic neurons in animal models [47–50]. Despite this, the role of pS129-aSyn in cell death is still debated [51]. Several other PTMs in aSyn are found in LBs, including phosphorylation at other residues, nitration, oxidation and truncation. Truncation mostly occurs in the C-terminal region of the protein and results in fragments that are more prone to fibrilization [52, 53].
The exact functions of aSyn are largely unknown. Its capacity to associate with highly curved membranes, interaction with other synaptic proteins, and localization to synaptic terminals suggest a role in neurotransmitter release [34]. Although it does not seem that aSyn is absolutely required for neurotransmission, studies showed that it regulates the clustering of synaptic vesicles at the membrane through its interaction with SNARE proteins [54]. aSyn was also found to modulate dopamine homeostasis through the inhibition of tyrosine hydroxylase (TH) expression, interaction with the dopamine transporter (DAT), and vesicular transporter VMAT2 which stores dopamine in synaptic vesicles [55–58].
PATHOLOGICAL SPREADING OF ALPHA-SYNUCLEIN
There are increasing numbers of evidences suggesting that pathologic forms of aSyn can propagate from cell to cell, and seed new LBs and LNs in a prion-like manner. According to this hypothesis, aSyn would spread following “permissive templating”— the process by which misfolded aSyn interacts with the endogenous protein, and induces its pathologic conformational change [59]. The first evidence of this process originates from the postmortem discovery of LBs in fetal tissue transplants more than 10 years after the graft in a PD patient [60, 61]. Since fetal tissue is composed of young and healthy cells, the presence of LBs suggested a transfer of toxic proteins from the recipient’s cells to the graft. As mentioned before, in humans, the presence of aSyn in plasma, brain interstitial fluid and CSF suggest that the protein can be released by neurons [20, 62, 63]. The observation that LB pathology spreads in humans from the brainstem to the neocortex in a succession of stages further supports the hypothesis of cell-to-cell transmission [64]. In addition, aSyn-positive inclusions have been observed outside the brain, notably in the gastrointestinal track early in the disease [65, 66]. This indicates that the pathology could also spread from the periphery to the CNS.
Cell-to-cell transmission of misfolded aSyn has now been extensively studied in vitro and in animal models. Internalization of aSyn monomers, oligomers and fibrils by neurons, astrocytes [67], microglia [68–70], and subsequent recruitment of intrinsic aSyn into LBs/LNs, all have been shown in cell cultures [71] and in the murine olfactory bulb [72, 73]. Similarly, the intracerebral injection of brain homogenates from mice with aSyn pathology triggers the development of LB/LN pathology in inoculated healthy animals [74–76]. Moreover, overexpression of aSyn in the vagus nerve and inoculation of pre-formed fibrils in the hind limb muscles have been shown to trigger the formation of pathological aggregates in the SNC, indicating that pathological aSyn can indeed propagate from peripheral sites to the brain [77, 78]. The most studied mechanism for aSyn uptake is dynamin-dependent endocytosis. Since the inhibition of this process substantially reduces fibril internalization, it is proposed to be the primary mechanism involved [67, 79]. The exact cell surface receptors implicated in aSyn binding are still being studied, but lymphocyte activation gene-3 (LAG-3), neurexin 1b, Aβ precursor-like protein 1 (APLP1), and connexin-21 (CX32) gap junction proteins have been proposed as possible interactors [80, 81]. The internalization of aSyn fibrils by macropinocytosis, a clathrin and actin-dependent process consisting in the engulfment of a large amount of extracellular fluid, has also been observed [82]. The binding of aSyn fibrils to heparan sulfate proteoglycans (HSPGs) was necessary to initiate the internalization process [82]. Following endocytosis, aSyn can be transported in the axons bidirectionally, although retrograde transport is predominant [83, 84]. It is then directed to late endosomal compartments and lysosomes [67, 85]. aSyn can be degraded by both chaperone-mediated autophagy (CMA) and macroautophagy. In pathological conditions, these processes are impaired, which contributes to aSyn accumulation within cells [85–90]. In the context of exposure of healthy tissue to pathology-inducing aSyn, it is not clear how internalized aSyn fibrils escape the endosomal pathway to interact with the cytoplasmic protein. It is possible that the seeding occurs partly inside the vesicles of the autophagic pathway and that abnormal aSyn can disrupt endosomal membranes afterwards [91, 92]. Despite this uncertainty, the colocalization of exogenous and intracellular aSyn have been reported [71, 93], indicating that misfolded aSyn can effectively act as a nucleating seed. Moreover, aSyn-KO neurons are resistant to this seeding process [94]. Finally, aSyn would be released in the extracellular space by unconventional exocytosis, although the exact process is yet to be determined [95, 96]. It is also possible that aSyn is released via exosomes. However, the presence of the fibrillar form of the protein in these vesicular packages is still debated [97, 98]. The secretion of aSyn occurs in physiological conditions but is significantly increased following cellular stress (mitochondrial, proteosomal and lysosomal dysfunction, as well as oxidative stress) [99, 100]. Lastly, it has been shown in different cell models that aSyn can also move from cell to cell via tunneling nanotubules (TNTs) [79, 101]. TNTs are long, thin and dynamic membranous tubes connecting the cytoplasm of neighboring cells. They are used for intercellular communication and exchange of cytosolic material [102]. The transfer of aSyn preformed fibrils (PFFs) enclosed in lysosomes through TNTs was observed in catecholaminergic (CAD) mouse neuron-like cells and primary cortical neurons [79]. aSyn was also shown to move via TNTs between SH-SY5H cells and primary brain pericytes from PD patients [101].
Multiple strategies could potentially be employed to reduce aSyn toxicity in vivo (Fig. 2). Antibody-based therapies typically target extracellular proteins. As mentioned before, several studies have shown evidence of a substantial amount of extracellular aSyn, particularly in a pathological environment [99, 100]. Because misfolded aSyn can propagate from one cell to another, targeting this extracellular pool could be beneficial to limit the spread. However, studies have not found an agreement on which species or assembly of aSyn protein is the most toxic for cells. Whether is it best to target monomeric, early oligomeric or late fibrillar forms of aSyn is still unknown [103–107]. Most studies have used antibodies against the oligomeric form in an attempt to act earlier in the disease progression [108–111]. Antibody binding have been shown to activate microglia-mediated degradation by internalization through the Fcγ receptor [112]. Another strategy to prevent the spread of the pathology is to target the receptors to which aSyn binds, therefore preventing by competition or obstruction its entry into cells [80]. Other teams have been using nanobodies that can be engineered to penetrate cells to target intracellular aSyn [113, 114]. Since the pathological process that leads to aSyn accumulation and toxicity happens intracellularly, the ability to reach this protein pool could show promise to slow disease progression. Intrabodies, that will be described further below, can stimulate degradation through intracellular degradation pathways and/or block oligomerization and fibrillization processes. Although not discussed in detail here, another possible immunotherapeutic strategy for PD treatment consists of targeting the immune system reactivity. Neuroinflammation has been strongly associated with PD pathogenesis and correlates with aSyn toxicity. Using antibodies targeting pro-inflammatory cytokines like IL-1β and TNF-α, or preventing microglia and monocyte activation could thus be beneficial in slowing the disease progression [115, 116].
Fig.2
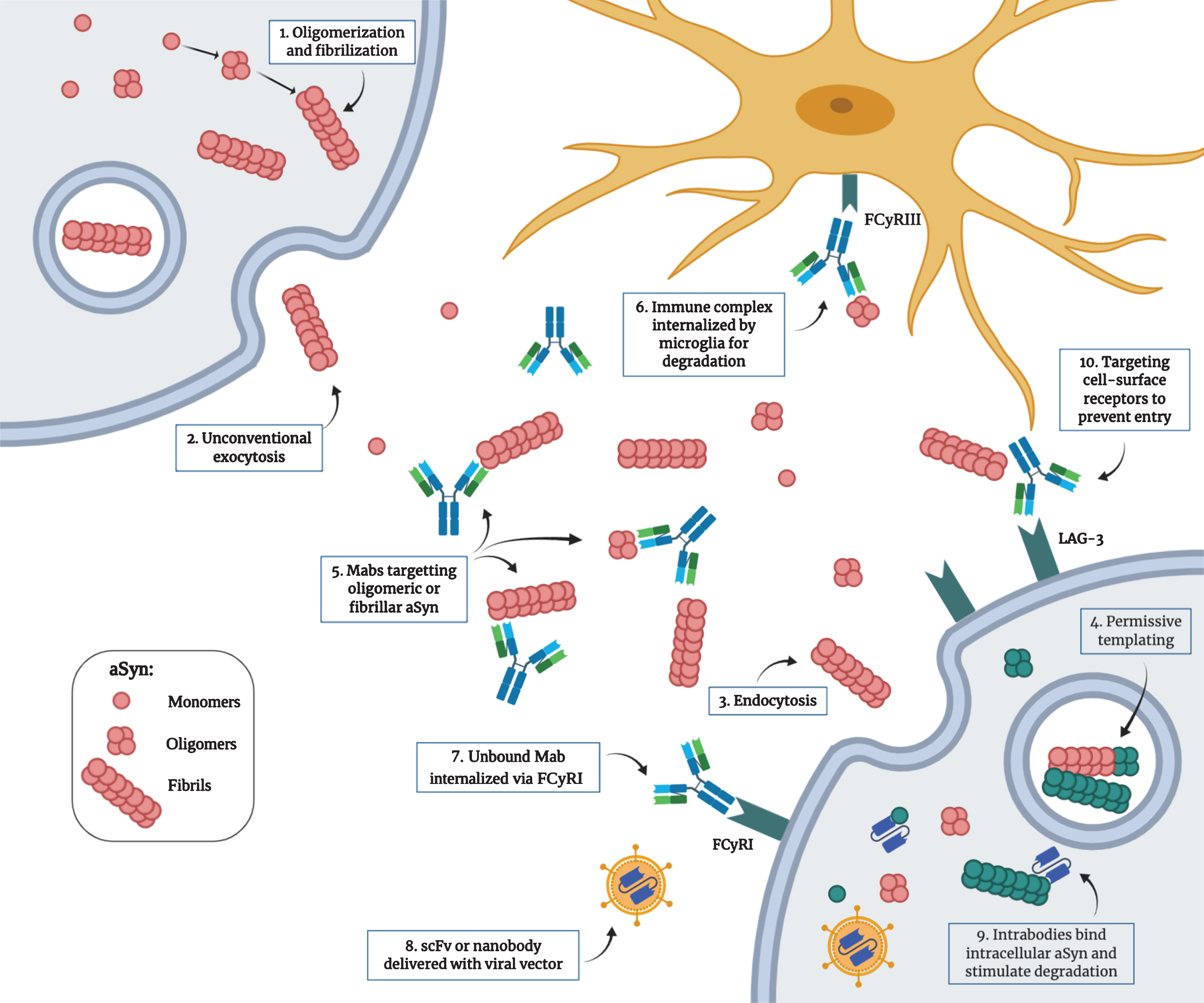
Proposed mechanism for the pathological transmission of aSyn and possible immunotherapeutic targets. 1) Post-translational modifications, cellular stress and mutations can trigger the misfolding and aggregation of aSyn into oligomers and fibrils. 2) Toxic forms of aSyn can be released in the extracellular space by unconventional exocytosis. 3) Once in the extracellular space, aggregated aSyn can be internalized by neighboring cells by endocytosis through binding of specific receptors. 4) Misfolded aSyn can act as a nucleating seed to trigger the misfolding of the endogenous protein. This process, called “permissive templating”, is thought to occur inside endosomes and lysosomes. 5) Monoclonal antibodies with different strain specificity can bind extracellular aSyn. 6) The immune complex can be internalized by microglia through the FCγRIII and is degraded intracellularly. 7) Unbound monoclonal antibodies can be internalized by neurons through the FCγRI and bind intracellular aSyn. 8) Single-chain variable fragment antibodies or single-domain antibodies (nanobodies) can be delivered as genes with viral vectors. 9) These intrabodies can target intracellular aSyn to prevent aggregation and can be engineered to be more efficient in stimulating degradation. 10) Antibodies can also target cell surface receptors used by aSyn to enter the cells (e.g., LAG3) to prevent entry. (Created with BioRender.com)
ACTIVE IMMUNIZATION
Active immunization refers to the classical vaccination strategy in which the administration of an antigen triggers the generation of specific antibodies by the immune system (humoral response). The advantage of this approach include a prolonged immune response that does not require repetitive drug administration, and is low-cost in a large-scale production [117]. However, PD is more prevalent in aged patients and immune responses are often diminished at a latter age [118]. Consequently, predicting the efficiency of an active immunization strategy in elderly patients is difficult, and variability in the response is also to be expected. Also, the active immunization against an endogenous protein has the risk of triggering an auto-immune response and neuroinflammation [119, 120]. Active immunization also implies a long-term or permanent response. Therefore, to ensure the safety of this approach, an extensive characterization of the immune response will be necessary. Studies regarding active immunization against aSyn that are discussed here are listed in Table 1. Agents that progressed to clinical trials are further described in Table 3.
Table 1
Summary of the active agents discussed in this review
| Name | Type | Target | Model (s) | Clinical trial | References |
| N/A | Purified recombinant haSyn | C-terminal | PDGFβ-aSyn mice | – | Masliah et al., 2005 [121] |
| N/A | Recombinant haSyn+Freund adjuvant | C-terminal | AAV-aSyn rats | – | Sanchez-Guajardo et al., 2013 [123] |
| AFFITOPE-PD01A | Peptide fragments | C-terminal | - mThy1-α-syn | Yes | Mandler et al., 2015 [125] |
| - PDGFβ-aSyn mice | (see Table 3) | ||||
| AFFITOPE-PD03A | Peptide fragments | C-terminal | - mThy1-α-syn | Yes | Mandler et al., 2015 [125] |
| -PDGFβ-aSyn mice | |||||
| α-Syn85-99-P30-MAP, αSyn109-126-P30-MAP, and α-Syn126-140-P30-MAP | Peptide fragments | C-terminal | Not tested in vivo | – | Ghochikyan et al., 2014 [127] |
| N/A | Peptide sensitized dendritic cells (PSDC) | C-terminal | Prnp-SNCA*A53T mice | – | Ugen et al., 2015 [192] |
| Qb-PD1, Qb-PD2, Qb-PD3 | Qbeta virus-like particles (VLP) peptide vaccine | Oligomers | SNCA-OVX mice | – | Doucet et al., 2017 [129] |
| GP+RAP/aSyn | Glucan particle vaccine+Rapamycin | N/A | PDGFβ-aSyn mice | – | Rockenstein et al., 2018 [130] |
In 2005, Masliah and colleagues were the first to provide evidence of the efficiency of an active immunization against extracellular aSyn in a model of synucleinopathy. In their study, administration of purified recombinant human aSyn (haSyn) to transgenic (tg) mice expressing haSyn in the brain under the control of the platelet-derived growth factor-β (PDGFβ) promoter, lead to the generation of antibodies against aSyn; this reduced the toxic accumulation in cell bodies and synapses, thus preserving synapses density [121]. Antibodies were mainly targeting C-terminal epitopes of the protein and were hypothesized to bind membranous oligomeric aSyn. Because the authors observed a colocalization of haSyn and lysosomal markers, they suggested that the antibody-aSyn complex was internalized and directed to lysosomal degradation [121]. In this study, the involvement of microglia in aSyn clearance was not assessed in detail, nor was the possible immune-mediated cellular response. The authors concluded that inflammation and a cellular immunity response were unlikely since they observed only a mild microglial activation and no lymphocyte infiltration [121]. In addition, motor symptoms and possible recovery was not evaluated. It is important to note that the mouse model used in this study displayed aSyn aggregates in the temporal cortex and hippocampus, accompanied by motor and cognitive deficits [122]. Thus, this model displayed more characteristics of a DLB model than a PD model.
In 2013, Sanchez-Guajardo et al. vaccinated rats with recombinant human aSyn combined with Freund adjuvant and then overexpressed haSyn in the substantia nigra by unilateral stereotaxic injection of an adeno-associated viral vector (AAV) encoding recombinant haSyn. This resulted in a high titer of antibodies against C-terminal aSyn, reducing the number of aSyn aggregates. The authors also observed an accumulation of MHC-II- and CD4-positive microglia and a modification of their morphology, a recruitment of CD4-positive T Cells and Foxp3-positive T cells (Tregs) in the nigrostriatal system. The conclusion drawn from these results was that a protective vaccination strategy should involve a long lasting Treg response and IgG production in order to prevent autoimmunity, and to delay dopaminergic degeneration [123]. However, a drawback from this study is that the vaccination was given 10 and 6 weeks before haSyn stereotaxic injection and before any cell loss, limiting the potential transfer of these results to humans. There is no biomarker currently available to detect PD prematurely; a diagnosis is only possible after the onset of symptoms, when more than 60% of dopaminergic neurons are already lost [124]. It remains to be proven if an anti-aSyn humoral response triggered after a moderate degeneration can still have a beneficial effect in delaying the pathology.
To prevent an auto-immune response against aSyn that would impair its physiological functions, immunization with peptide fragments in lieu of the whole protein has been favored. In clinical trials, the company Affiris is assessing the efficacy of two next generation AFFITOPE® vaccines (PD01A and PD03A) which are peptides that mimic the C-terminus of aSyn. Different versions were tested by Mandler and colleagues in pre-clinical studies [125]. The most promising peptide, AFF 1, was tested in two different transgenic models of synucleinopathy: PDGFβ-aSyn and Thy1-aSyn mice. A vaccination induced the production of antibodies specific for aSyn oligomers that were able to traffic into the CNS. In Thy1-aSyn mice, the authors observed a reduced accumulation of aSyn oligomers in the SN and striatum, and a diminution in motor deficits (body suspension test). In the PDGFβ-aSyn tg mice, aSyn levels were reduced in the neocortex and hippocampus. The immunized mice also performed better in the Morris water maze after vaccination. In the same model, AFF 1 administration reduced synaptic pathology and neuronal cell death, and promoted microglial aSyn clearance and anti-inflammatory cytokine production [125]. Following these results, phase I clinical trials of the two AFFITOPE® vaccines PD01A, and PD03A, which ended in February 2015, and August 2016, respectively, assessed the safety and tolerability in early PD patients with no serious adverse effects. The treatment resulted in a robust humoral response with target engagement [126]. This allowed the development of a phase II trial.
Several other studies that analyzed the effects of active immunization against aSyn were made. Ghochikyan et al. [127] evaluated the immunogenicity of three epitope vaccines against aSyn (α-Syn85-99-P30-MAP, αSyn109-126-P30-MAP, and α- Syn126-140-P30-MAP) in mice. All vaccines were composed of a B cell haSyn epitope associated with a “non-self” T helper (Th) epitope from tetanus toxin (P30). The Th epitope P30 was used to promote an efficient humoral response by B cells. All three peptides stimulated B-cells and generated antibodies specific for the three epitopes without activating autoreactive Th cells. The antibodies generated were also able to specifically detect LB and LN in human DLB samples. However, the efficiency of these vaccines in reducing aSyn pathology and motor deficits was not evaluated [127]. Ugen et al. evaluated the efficiency of a dendritic cell (DC) based vaccination in A53T-aSyn tg mice. This strategy uses antigen sensitized DCs as the vehicle for immunization. An advantage of this approach is that DCs can serve as self-adjuvant, thus does not require the addition of an adjuvant that can result in an over-activation of the immune system. In addition, DCs can be collected autologously, which eliminates the possibility of a rejection reaction. The immunization with a peptide or full-length aSyn sensitized DCs was able to generate specific antibodies against aSyn, that ameliorated motor symptoms in mice (rotarod), and reduced the level of the pro-inflammatory cytokine IL-1α [128]. Doucet and colleagues [129] tested a vaccine based on the RNA bacteriophage (Qbeta) virus-like particle associated with short peptides of haSyn in mice. Antibodies against the C-terminal region of aSyn were generated and could effectively recognize LBs in postmortem tissue from PD patients. However, in SNCA-OVX tg mice (a mouse model expressing high levels of haSyn in a mouse Snca–/– background), the vaccination was unable to lower oligomeric aSyn burden, nor attenuate the motor deficits [129]. Finally, Rockenstein et al. [130] evaluated a novel vaccination strategy that combined active humoral and cellular immunization. They used an antigen-presenting cell-targeting glucan particle (GP) vaccine associated with aSyn and administered rapamycin (RAP) concomitantly (GP+RAP/aSyn). The vaccination of PDGFβ-aSyn mice induced high titers of anti-aSyn antibodies and an increased number of CD25 (Treg), FoxP3 (Treg) and CD4 (T cells)- positive cells in the CNS, suggesting an immunomodulatory response. The RAP-containing vaccine increased the production of TGF-β1 by Tregs, which would mediate the conversion of microglia into a neuroprotective state. The authors noted a reduction in aSyn accumulation (total pS129 and proteinase K resistant aSyn), neuroinflammation, and neurodegeneration in this model. The combination of antibody production and an anti-inflammatory effect thus resulted in a more effective vaccine, compared to humoral or cellular immunization alone [130].
PASSIVE IMMUNIZATION
In order to eliminate any danger of an auto-immune response against physiologic aSyn or unspecific targets, many teams have explored passive immunization as a novel strategy. Passive immunization refers to the direct administration of laboratory generated antibodies. This approach does not involve a humoral response and the humanization of antibodies can prevent unwanted reactions against the non-self. The dose can be closely monitored, and cessation of the treatment is possible if adverse effects appear [128]. Furthermore, antibodies can be engineered to have specific characteristics that facilitate their functions. However, passive immunization requires repetitive administration of the antibodies for as long as the treatment is needed. Delivering them to the brain is another challenge due to the blood-brain barrier (BBB). Typically, 0.1% of monoclonal antibodies administered peripherally enters the brain [131]. Several monoclonal antibodies and intrabodies that target various species of aSyn are currently at different stages of development. Monoclonal antibodies and intrabodies discussed in this section are listed in Table 2. Agents that progressed to clinical trials are further described in Table 3.
Table 2
Summary of the passive agents discussed in this review
| Name | Type | Target | Model (s) | Clinical trial | References |
| 9E4/PRX002/Prasinezumab | Mab | C-terminal | PDGFβ-aSyn mice | Yes | Masliah et al., 2011 [132] |
| (see Table 3) | Schenk et al., 2017 [133] | ||||
| Jankovic et al., 2018 [134] | |||||
| 5C1, 1H7 | Mab | C-terminal | mThy1-aSyn mice | – | Games et al., 2014 [136] |
| 1H7 | Mab | C-terminal | LV-aSyn in mThy1-aSyn mice | – | Spencer et al., 2017 [138] |
| MEDI1341 | Mab | C-terminal (monomers and oligomers) | SH-SY5Y cells, LV-aSyn mice, rats, monkeys | Yes | Schofield et al., 2019 [139] |
| Lu AF82422 | Mab | N/A | N/A | Yes | Lundbeck |
| ABBV-0805 | Mab | N/A | N/A | Yes | BioArctic/Abbvie |
| BIIB054/Cinpanemab | Mab | N-terminal (oligomers and fibrils) | PFFs in M83 and WT mice | Yes | Weihofen et al., 2019 [140] |
| Brys et al., 2019 [141] | |||||
| Ab274 | Mab | C-terminal | BV-2 cells, PDGFβ-aSyn mice | – | Bae et al., 2012 [112] |
| Syn303 | Mab | N-terminal | Primary neurons, PFFs in WT mice | – | Tran et al., 2014 [142] |
| mAb47 | Mab | Oligomers/protofibrils | Thy1- hA30Pα-syn mice | – | Lindström et al., 2014 [108] |
| Syn-O1, -O4, -F1 | Mab | Full length fibrils | mThy1-aSyn | – | Vaikath et al., 2015 [144] |
| El-Agnaf et al., 2017 [143] | |||||
| AB1, AB2 | Mab | 1: N-terminal 2: C-terminal | AAV-aSyn in rats | – | Shahaduzzaman et al., 2015 [128] |
| C9B7W and 410C9 | Mab | LAG3 | PFFs in M83 mice | – | Mao et al., 2016 [80] |
| D10 | scFv | Monomers | HEK293 cells | – | Zhou et al., 2004 [155] |
| NAC32 | scFv | NAC region | ST14A cells | – | Lynch et al., 2008 [156] |
| Syn-10H | scFv | Oligomers | SH-SY5Y cells | – | Emadi et al., 2009 [110] |
| sD5-apoB | scFv | Oligomers | SH-SY5Y cells | – | Emadi et al., 2007 [109] |
| PDGFβ-aSyn mice | Spencer et al., 2014 [114] | ||||
| W20 | scFv | Amyloid assemblies | A53T-aSyn tg mice | – | Zha et al., 2016 [111] |
| NbSyn87 | Nanobody | C-terminal (oligomers and fibrils) | N/A | – | Iljina et al., 2017 [162] |
| NbSyn87*PEST VH14*EST | Nanobody | NbSyn87: C-terminal VH14: NAC region | AAV5-aSyn in rats | – | Chatterjee et al., 2018 [113] |
LV, lentivirus; PFF, preformed fibrils.
Table 3
Antibodies previously or currently involved in clinical trials for Parkinson’s disease treatment
| Type | Name | Sponsor | Phase | Clinical Trial Identifier | Start and End dates | Outcomes |
| Active | AFFITOPE-PD01A | Affiris | Ib | NCT02618941 | Feb 2016 | •Locally and systemically well tolerated |
| Feb 2017 | •No drug-related serious adverse events (SAE) detected or suspected unexpected serious adverse reactions (SUSAR) | |||||
| •Good immune response against the peptide | ||||||
| AFFITOPE-PD03A | Affiris | I | NCT02267434 | Dec 2014 | •Locally and systemically well tolerated | |
| Aug 2016 | •No drug-related SAE detected or SUSAR | |||||
| •Dose-dependent immune response | ||||||
| Passive | PRX002/Prasinezumab | Roche/Prothena | II | NCT03100149 | Jun 2017Apr 2026 | •No change from baseline in Movement Disorder Society-Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) total score after 52 weeks |
| •Improvement in other secondary endpoints | ||||||
| MEDI1341 | AstraZeneca | I | NCT03272165 | Oct 2017 | N/A | |
| Jan 2021 | ||||||
| Lu AF82422 | Lundbeck | I | NCT03611569 | Jul 2018 | N/A | |
| Dec 2020 | ||||||
| ABBV-0805 | AbbVie/ | I | NCT04127695 | Mar 2020 | N/A | |
| BioArctic | Jun 2020 | |||||
| BIIB054/Cinpanemab | Biogen | II | NCT03318523 | Jan 2018Jun 2021 | •Phase I showed no drug-related SAE and a good target engagement | |
| •No data available for phase II to this date |
Complete antibodies
As for active immunization, the exogenous administration of complete antibodies is thought to target mainly extracellular aSyn. These antibodies are almost all monoclonal IgGs selected from human or murine libraries. Masliah et al. [132] investigated the efficiency of the monoclonal antibody (Mab) 9E4 directed against the C-terminus of haSyn. PDGFβ-aSyn mice received weekly intraperitoneal (IP) injections of 9E4 for 6 months. 9E4 was able to reduce the accumulation of calpain-cleaved aSyn in axons and synapses, and consequently attenuate cognitive (water maze) and motor deficits (pole test and rotarod). The study showed that 9E4 travelled into the CNS and co-localized with aSyn in lysosomes, suggesting that aSyn clearance occurred via autophagy activation. The authors proposed that the antibody recognized aSyn aggregates lying on the cell surface; the complex was then internalized and directed to lysosomes [132]. A humanized version of 9E4, PRX002 (Prasinezumab), has been developed and tested in phase I and 1b clinical trials. In healthy and mild idiopathic PD patients, the antibody showed favorable safety, tolerability and pharmacokinetics, with no immunogenicity and serious adverse effects. A significant reduction in free serum aSyn was also observed [133, 134]. Following these favorable results, a phase II clinical trial led by Roche in collaboration with Prothena started in June 2017 (PASADENA study). The study is evaluating the effectiveness of high and low intravenous doses of PRX002 in early PD patients for 52 weeks. It is intended to conclude in February 2021. Prothena released an update on Part I of the study in April 2020. Although the study didn’t meet the primary endpoint (a significant improvement over one year in the Movement Disorder Society-Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) total score), several secondary clinical endpoints were improved [135]. Part II of the study is still ongoing and has a year extension in which the placebo group from Part I also receives Prasinezumab.
Because the antibody 9E4 was shown to decrease the amount of truncated aSyn and reduced behavioral deficits, other antibodies targeting the C-terminal region of aSyn were tested. Games et al. [136] immunized mThy1-aSyn tg mice with 1H7 and 5C1: two Mabs against the C-terminus of haSyn. The mice received weekly intraperitoneal injections for 6 months. The authors observed: a reduction in C-terminal truncated aSyn in axons and in the loss of tyrosine hydroxylase (TH) in the striatum; an improved motor performance (beam test); and a reduction in memory deficits (water maze). In vitro, they showed that the antibody could efficiently block the cleavage of aSyn and the propagation of full-length aSyn from cell to cell [136]. Because truncated aSyn is more prone to aggregate, blocking this event is thought to slow its accumulation and propagation [137]. The antibody 1H7 was further evaluated for its ability to block the propagation of aSyn in the axons and reduce neurodegeneration in a lentivirus (LV) mouse model [138]. Non-tg, aSyn-KO, and mThy1-aSyn tg mice were injected unilaterally in the hippocampus with a haSyn-lentivirus. The IP immunization was performed weekly for three months, starting the day after LV injection. 1H7 reduced aSyn axonal transport and accumulation contralaterally. Reduced axonal degeneration and attenuated behavioral deficits in the water maze were also observed. The antibody was thought to limit the spread by binding extracellular aSyn, reducing the internalization into neighboring neurons and its association in oligomers. The authors suggest that the complex would subsequently be degraded by microglia [138].
Another C-terminal-specific Mab (of human origin) that targets both monomeric and aggregated forms of haSyn (MEDI1341) is being evaluated in a phase I clinical trial, and is intended to terminate in November 2020. The pre-clinical development of the antibody was made by Schofield and colleagues [139] and the results were published in 2019. In neuroblastoma cells (SH-SY5Y), MEDI1341 was able to block cell-to-cell transmission of aSyn PFFs, presumably by sequestering them in the extracellular space. In rats and in monkeys, this antibody could significantly and, for a prolonged period of time, reduce the level of free aSyn in the CSF. In mice, a unilateral hippocampal injection with an aSyn-encoding lentivirus, a model of aSyn spreading [138], MEDI1341 reduced accumulation of aSyn in the ipsi- and contralateral side, as well as in the trans-hippocampal axonal connexions; this suggests a blockade of cell-to-cell propagation. The IP immunization was given weekly for 13 weeks, starting seven days after LV injection. Interestingly, a version of the antibody with a mutated Fc domain that prevents its interaction with Fc receptors was as effective as the non-mutated form. This indicates that the beneficial effects of this antibody are not due to Fc-mediated mechanisms, thus do not involve microglial activation. The authors proposed that the aSyn-Mab complexes could have been internalized by neurons and degraded by the proteasome, but the exact modalities were not studied [139]. The phase I clinical trial sponsored by AstraZeneca is being conducted with the effector-null version of the antibody, and the safety evaluation of a single ascending antibody dose in healthy volunteers is ongoing.
In July 2018, the Danish company Lundbeck started assessing the safety, tolerability, and pharmacokinetics of a single dose of a human Mab against aSyn (Lu AF82422) in healthy and PD patients. The phase I clinical trial is in progress until August 2020. Another humanized Mab targeting aSyn, ABBV-0805, was developed by the company BioArctic. A phase I clinical trial that started in March 2020 aims to evaluate the safety and tolerability of increasing antibody doses in participants with PD; it is financed by the company AbbVie. The completion of this first phase is intended for November 2021. No pre-clinical data is presently available for these two candidate antibodies.
Recently, a human-derived Mab against the N-terminus of aSyn (BIIB054), selective for oligomeric and fibrillar forms of aSyn, was described by Weihofen et al. [140]. When administered IP weekly to heterozygous M83 mice (bearing a hA53T-aSyn transgene under the Prp promoter) that received an intrastriatal injection of pre-formed fibrils (PFFs), this antibody delayed the onset of paralysis associated with aSyn spreading. In PFFs-inoculated WT mice, BIIB054 reduced the level of truncated aSyn and improved the performance in the wire hang test. Finally, in A53T-aSyn tg mice, the antibody significantly reduced the loss of DAT in the striatum. In these three experiments, immunization was given two or three times before PFFs injection, and weekly following the procedure [140]. BIIB054 (Cinpanemab) was evaluated for safety and tolerability by Biogen in a phase I clinical trial. In healthy and PD patients, the antibody efficiently formed complexes with aSyn in the plasma. Thus, BIIB054 showed favorable characteristics and no serious adverse effects, allowing the development of a phase II trial [141]. This second phase is currently ongoing (SPARK study), and the completion is intended for June 2021. The study is evaluating the dose-related safety, the pharmacodynamic effects on the integrity of nigrostriatal dopaminergic nerve terminals, the pharmacokinetic profile, and the immunogenicity of BIIB054.
Numerous other teams are conducting pre-clinical studies on Mabs against aSyn. Bae and colleagues [112] tried to clarify by which mechanism(s) antibodies stimulate the degradation of aSyn in vivo. They showed that antibodies targeting extracellular aSyn mainly activate microglial degradation through the Fcγ receptor, preventing its entry in other cells, including neurons and astrocytes. Weekly intraperitoneal injections of an aSyn-targeted Mab (274) in PDGFβ-aSyn mice reduced the accumulation of aSyn in the neocortex and hippocampus, limited neurodegeneration, and reduced motor deficits in the pole test and open field [112]. Tran and colleagues [142] proved the efficiency of another Mab in a now well-established in vivo model of aSyn propagation. Antibody Syn303 was administered IP weekly to WT mice injected intrastriatally with aSyn PFFs. The immunization started immediately after PFFs injection. Immunized mice showed less LB/LN pathology in the SN and amygdala, a reduction in dopaminergic neurons loss, and less motor impairment on the wire hang test [142]. In vitro, Syn303 prevented the formation of insoluble aggregates and limited synaptic and neuronal loss. The Mab blocked PFFs entry into the neurons and consequently prevented the transmission of pathologic aSyn aggregates. In this study, the authors proposed that the pathology reduction relied mostly on the blockade of PFFs internalization since they did not observe the PFF-Mab complex inside the cells [142].
In another study by Lindström et al. [108], a protofibril-specific Mab (mAb47) was administered IP weekly to Thy1-hA30P-aSyn tg mice. Their objective was to spare physiological and functional aSyn monomers while acting on early forms of aggregates. A lower level of soluble and membrane-bound aSyn protofibrils was detected in the spinal cord, and motor symptoms were reduced. However, the mechanism by which the antibody acted (intra- or extracellularly) was not studied and no significant beneficial effect was found in the brain [108]. El-Agnaf and colleagues [143, 144] developed multiple antibodies targeting oligomeric or fibrillar forms of aSyn. They proposed that more efficient and specific effects could be achieved with antibodies that can recognize selective species of aSyn. Their Mabs Syn-O1, -O4 (oligomers-specific) and -F1 (fibrils-specific) were the most effective in the reduction of aSyn oligomer accumulation (resistant to proteinase K), and the neurodegenerational prevention in a mThy1-aSyn tg mouse model. Weekly antibody treatment also preserved the colocalization between synapsin I and synaptophysin in synapses, which is affected in aSyn tg mice and often reduced in PD and DLB patients. These improvements were found in multiple brain regions, but unfortunately not in the SN [143]. Finally, Shahaduzzaman et al. [128] generated two novel antibodies against the N-terminal (AB1) and central region (AB2) of aSyn. In rats injected unilaterally with AAV9-WT-aSyn, both antibodies significantly reduced the level of total aSyn when administered IP every two weeks for 3 months, thus protecting DA neurons against degeneration. AB1 also induced a moderate rescue in motor deficits in the cylinder test [128].
In light of the results presented so far, it appears there may be more than one mechanism by which antibodies promote the clearance of extracellular aSyn. It is possible that the neuronal uptake of unbound monomeric antibodies through FcγRI can stimulate intracellular aSyn degradation prior to cell exodus. On the other hand, antibodies bound to extracellular aSyn may participate in the clearance of the protein by increasing the internalization of immunoglobulin (Ig)-aSyn complexes into microglia through FcγRIII. Different Fc receptors have different affinities for Ig species, as well as for their monomeric vs complexed forms. FcγRI is expressed on certain neurons and is able to bind monomeric IgGs while FcγRIII is expressed solely on glial cells and can bind only complexed IgGs [112, 145, 146].
Finally, Mao et al. [80] showed that aSyn PFFs are endocytosed by neurons through the binding to lymphocyte-activation gene 3 (LAG3) on the membrane. Treating SH-SY5Y cells exposed to PFFs with two antibodies that target LAG3 (C9B7W [147] and 410C9 [148]) significantly reduced aSyn toxicity (pS129-aSyn) and cell-to-cell transmission. The same effect was found with 410C9 in primary cortical cultures from M83 tg mice (A53T-aSyn) exposed to human PFFs, or brain homogenates from 10 month old symptomatic M83 mice [80]. Thus, it appears likely that multiple receptors are involved in aSyn internalization, and that at least some are specific for particular forms (monomers, oligomers or fibrils) of aSyn. Therefore, the target of one entry mode by aSyn mono- or multimers might only partially mitigate the disease in the human pathological process. Moreover, cell surface receptors frequently have several ligands; the obstruction of these ligands could induce off-target effects by the cellular inhibition of other molecules. A thorough examination of the in vivo effects will be crucial to ensure the effectiveness and safety of antibodies that target receptors.
Nanobodies
In most species, complete antibodies are large molecular complexes made up of two different gene products in various proportions [149]. As therapeutic molecules, this large size can impair tissue penetration and offer a considerable target for the patient’s own immune system [150]. In addition to their binding domains, complete antibodies also harbor other effector domains that may prove detrimental in vivo; for instance, regions involved in complement-mediated toxicity [151]. For all these reasons, researchers try to derive smaller, safer proteins that mimic antibodies in their antigen binding. The progress in the molecular cloning and gene engineering that encode the binding domains of antibodies, coupled with the discoveries of certain high affinity animal antibody species made from a single gene product have made these goals a reality [150]
Nanobodies are of two main types: single-chain variable fragment (scFv), and single-domain nanobodies. Both have the advantage of being substantially smaller than Mabs, facilitating the crossing of the BBB and the infiltration into the extracellular space. Their small size also means the encoding recombinant DNA is small. Thus, nanobodies can fit into most viral vectors for cell transfer [152], be functionalized by addition of other function-encoding regions, or even be multiplexed (ibid). Because they lack the Fc region of traditional antibodies, nanobodies are less immunogenic and pose no risk of engaging an unwanted immune response. However, their half-life (especially for scFvs) can be considerably shorter than that of a complete antibody [152]. The scFv is composed of a polypeptide made of the antigen-binding regions of both the light, and heavy chains of an IgG antibody, attached by a flexible linker region composed of a small stretch of polar or non-polar amino acids. Their size is usually around 250 amino acids (AA). Single-domain nanobodies are even smaller (from 125 to 140 AA) [152], and are derived from a single immunoglobulin chain from a single gene product that encodes a functional immunoglobulin binding domain. This binding domain can be selected in vitro from a library made of only the heavy chains of regular IgG antibodies, or cloned from immunoglobulins of animals (e.g., Camelids) that have natural classes of antibodies composed of only heavy chains [150, 153, 154].
Another major advantage of nanobodies is that their encoding sequence can be modified so that the nanobody can be made to be secreted like a regular antibody, or retained within the producing cell (as an intracellular antibody or “intrabody”). Cell-to-cell transmission of aSyn has only been documented in artificial models often overexpressing the protein, and clear evidence of this process has not been observed in human patients. Therefore, targeting intracellular aSyn could be an effective approach to slow down the disease progression by directly blocking aggregation and/or excretion of abnormal proteins. Intrabodies must be carefully tested for their affinity, stability, and solubility in vivo; the cellular cytoplasm is an unpredictable environment, particularly in neurons already affected by intracellular stress. Indeed, low intracellular solubility and stability is their principal disadvantage [152].
Zhou and colleagues [155] were amongst the first to test a scFv against aSyn in vitro. Their scFv, D10, binds to monomeric WT-aSyn. When transfected as a gene in HEK293 cells overexpressing WT-aSyn, D10 was able to efficiently bind monomeric aSyn intracellularly and prevent the formation of detergent-insoluble aSyn species. The antibody fragment also protected against the decrease in cell adhesion that is normally observed following aSyn overexpression [155]. Later, a scFv against the NAC region of aSyn was developed by Lynch et al. using a yeast surface display library of human scFv antibodies [156]. The antibody that showed the best affinity and viability, NAC32, was stably expressed in the cell line ST14A. When transfected with an aggregation-prone aSyn construct, cells that expressed NAC32 showed less pathological aggregation [156]. Emadi et al. [109] identified another scFv (syn-10H) from a phage display library specific to large oligomers of aSyn. The incubation of aSyn with syn-10H inhibited the aggregation of the protein, measured with a Thioflavin T assay. Added to the culture medium, it also reduced the cytotoxicity caused by aSyn aggregates in SH-SY5Y cells. Moreover, syn-10H was able to specifically recognize aSyn aggregates in human PD tissue when used as an immunohistochemical reagent [110].
The same group had previously identified from the same library another scFv binding specifically to oligomeric forms of aSyn (D5) [109]. D5 was further modified with the addition of the LDL receptor-binding domain of apoB (D5-apoB) to increase brain penetrance, cellular endocytosis, and degradation [114]. D5-apoB kept its previously described specificity for oligomeric forms of aSyn and reduced aggregation in rat neuroblastoma cells that overexpress WT-haSyn. The nanobody was encoded in a lentiviral vector and injected intraperitoneally in PDGFβ-aSyn mice. D5-apoB was thus secreted by the expressing cells. The addition of the apoB transport peptide significantly increased brain penetrance and colocalization with neuronal markers. D5-apoB decreased the accumulation of total and pS129-aSyn in the neocortex and hippocampus and reduced neurodegeneration. Accordingly, behavioral deficits were attenuated in mice that express the scFv (Morris water maze). Finally, the LDL receptor binding domain increased cellular uptake and degradation of aSyn by autophagy. The scFv-aSyn complex was imported into neurons through the endosomal sorting complex required for transport (ESCRT) pathway. The scFv colocalized with aSyn, LC3 (autophagosome marker), Rab5 (endosomal marker) and CHMP2B (ESCRT marker) [114]. This study showed strong evidence that scFvs can be effectively modified to enhance brain and neuronal penetrance as well as intracellular degradation.
Zha et al. [111] reported a conformation-specific scFv (W20) that can recognize various oligomeric assemblies of amyloid-β, aSyn, and prion protein. Purified W20 binds to a common epitope of various oligomeric structures assembled from different amyloids. Injection of the purified pan-amyloid oligomer-specific scFv could reduce motor (open field, rotarod and pole test) and cognitive (Morris water maze, object recognition test) impairments in A53T-aSyn tg mice. W20 also decreased the amount of total aSyn and pS129-aSyn, and maintained normal TH and DAT levels in the striatum [111]. These results suggest that targeting multiple abnormal proteins could be an effective strategy for neurodegenerative disorder treatments. Amyloid-β and Tau pathologies have been associated with PD dementia and also correlate with motor symptom severity, suggesting that these proteins can likely interact synergistically to worsen neurotoxicity [157–160]. Similarly, LB pathology is frequently observed in AD patients and sometimes leads to a double diagnosis [157]. Moreover, Gerson et al. demonstrated that targeting Tau oligomers with a Mab (TOMA) is protective against cognitive and motor deficits in A53T-aSyn tg mice. The reduction in Tau oligomers also prevented synaptotoxicity, and dopamine depletion in the olfactory bulb [161]. In light of these results, it would be particularly interesting to test a double-targeted strategy of Tau and aSyn in clinical trial for the treatment of PD.
Finally, Iljina et al. [162] described a camelid intrabody, NbSyn87, that binds to the C-terminal region of aSyn. Because it can bind to monomers and fibrils through the exposed C-terminus, NbSyn87 could both inhibit toxic fibril formation, and destabilize those that are already formed. Those effects could significantly reduce cellular damage [162]. NbSyn87, along with a human-derived intrabody that targets the NAC region of aSyn (VH14), was then modified with the addition of a proteasome targeting signal (PEST) [163]. The PEST motif, due to its negative charge, was used to increase cytoplasmic solubility and to stimulate degradation. When tested on different nanobodies (D5, 10H, D10, and VH14) the PEST motif was proven to be highly effective in enhancing steady-state soluble intrabody protein cellular levels, and specifically increase proteosomal degradation of the intrabody-aSyn complex [164]. In ST14A cells, both NbSyn87 and VH14 decreased aSyn levels and its related toxicity, while VH14*PEST showed a better efficiency [163]. NbSyn87*PEST and VH14*PEST were then tested in vivo in rats with a unilateral SN injection with AAV5-aSyn. The intrabodies were delivered as genes using AAV5 viral vectors three weeks after aSyn injection. Both significantly reduced the accumulation of pS129-aSyn in the SN. VH14*PEST also helped to preserve TH- and DAT-positive terminals in the striatum and showed a modest protection of DA neurons in the SN. VH14*PEST treated animals also showed a significant improvement of motor behavior in the stepping and cylinder tests compared to non-treated animals [113]. In this study, the NAC-targeted intrabody was more efficient than the C-terminus-targeted intrabody; suggesting that this region is important for aSyn aggregation. However, the two intrabodies are derived from different species (VH14 from human and NbSyn87 from camelid), which could partly explain the different outcomes.
SUMMARY AND CHALLENGES
Antibody-based therapies have been gaining increasing interest in the last few decades for the treatment of various diseases, including cancer and multiple sclerosis [165, 166]. Because antibodies can be selected and further modified to target specific protein species or conformations, they represent a promising therapeutic tool for the treatment of proteinopathies [24, 167]. Alpha-synuclein aggregation and accumulation in neurites and cytoplasm has been strongly associated with cellular toxicity in Parkinson’s disease [15, 16]. Moreover, several evidence point to a cell-to-cell transmission of misfolded aSyn through a “permissive templating” mechanism [59], which would explain the progression of the disease across the brain in distinct stages [64]. The presence of free aSyn in the CSF and plasma [20] further supports this hypothesis and has encouraged antibody development that can block extracellular transmission. Extracellular aSyn targeting should not interfere with aSyn physiological role. Both active and passive immunizations that use monoclonal antibodies or secreted nanobodies, are based on this approach. These methods have shown promising results in pre-clinical studies and some are being evaluated in phase I and II clinical trials. Active immunization has the advantage to be a long-term therapeutic option. However, the inability to accurately predict the immune response generated, especially in elderly patients, limits its applicability. Another drawback of this approach is the possible autoimmune response and neuroinflammation, as was observed in clinical trials for Alzheimer’s disease [168]. For this main reason, current studies mostly focus on passive immunization. This strategy does not pose the same level of immunogenic risk with the use of human or humanized antibodies (or nanobodies), and is easily reversible. How exactly these reagents promote toxic aSyn clearance is unknown, but multiple mechanisms are likely involved. Microglial activation has been shown in some cases. Internalization of the immune complex by neurons with subsequent intracellular degradation was also shown. It is possible that the binding specificity of the immunotool for certain mono, oligo, or higher-level assemblies of aSyn influences the type of degradation induced. In any case, a closer evaluation of the immune (cellular and humoral) response will be important to ensure the safety of any antibody-based therapy.
Studies that evaluated the prion-like transmission of aSyn were made in artificial models that overexpressed the normal or mutant protein; it is still unclear if this mode of transmission actually occurs in the human brain. Therefore, it is possible that intracellular aSyn would be a better target to achieve neuroprotection. Given that misfolding and toxic accumulation of aggregated aSyn originates inside the cells, blocking the initial protein oligomerization and fibrilization could provide a new strategy. Furthermore, intracellularly active nanobodies have the potential to stimulate the degradation through autophagy or proteasomal pathways. Along with their low immunogenicity, intrabodies are particularly attractive candidates for antibody-based therapies against aSyn. Their main disadvantage is the poor stability in the reduced environment of the cytoplasm. However, they can be easily modified to overcome this flaw with the incorporation of specific sequences [164].
The precise mechanisms underlying cell death in PD are still unclear. Mitochondrial, proteasomal, and autophagy dysfunctions are likely involved, but the sequence of events and the role of aSyn aggregates in these processes are not well established. aSyn has been shown to exist as a wide range of oligomeric and fibrillar assemblies. Typically, the protein initially forms an aggregation nuclei that acts as a seed for the elongation into protofibrils by the addition of monomers. These protofibrils then associate into mature amyloid fibrils [169, 170]. The term “oligomers” encompasses species with a variety of molecular weights and secondary structures. This heterogenicity, as well as their instability, renders them difficult to study, particularly in vivo. The term “fibrils”, on the other hand, refers to long, ordered, insoluble and compact fibril assemblies enriched in beta-sheet structures. aSyn fibrils can have various shapes (cylindrical-fibrils or flat-ribbons) and lengths [172–174]. Several studies have suggested that oligomers, rather than fibrils, could represent the major toxic assembly of aSyn rather than a simple intermediate in fibril formation [104, 175–177]. Lewy pathology is present in a surprisingly high proportion of postmortem brains from people without any neurological disorder [178, 179]. Also, the extent of LB deposition does not correlate with the severity of symptoms in several PD patients [180, 181]. Some patients with a clinical PD diagnosis do not even present Lewy pathology [182]. These elements suggest that the fibrils contained in LBs may not represent the major pathological strain of aSyn. However, several studies showed that fibrils are toxic to neurons as well [107, 183, 184]. Moreover, their capacity to act as nucleating seeds for the pathological misfolding and aggregation of aSyn and their role in the cell-to-cell transmission of the disease still make them a very toxic species [93, 94]. Thus, there is no consensus regarding which strain of aSyn is the most toxic. It is likely that a variety of species exert different toxic effects in cells and their combined actions result in cellular malfunction and death. Due to the persistent uncertainty of which aSyn species is the most harmful, it is unclear at what stage to intervene in the toxic assembly process for optimal neuroprotection. Researchers should pay particular attention to which strains of aSyn are present in their models, and in immunotherapeutic studies, against which strain is their antibody directed. This would allow a better understanding of aSyn cellular toxicity. Proximity-ligation assay against aSyn can be used to detect oligomers with a high specificity and probably represent the best technique to distinguish between LBs/fibrils and early oligomers [185]. Multiple models should also be used to test target engagement and behavioral outcomes.
Several pre-clinical antibodies have been shown to reduce the level of total aSyn in vitro and/or in vivo. Since some studies have shown that the knockout or subthreshold knockdown of this protein alters DA synthesis and release, and can induce DA neuron degeneration in the SN [186, 187], it will be important to decipher in detail the important biochemical role that aSyn plays in healthy DA neuron survival [186]. aSyn is also implicated in regulating the synaptic vesicle pool during neurotransmission [188]. Thereby, it will be crucial to ensure that nanobodies against intracellular aSyn do not prevent this role and negatively affect DA neurotransmission in surviving DA neurons, which could worsen the patient’s motor symptoms. The ability to exclusively target the pathological species of aSyn (oligomers, fibrils, pS129, or truncated aSyn) would be a very desirable property to avoid this outcome.
Finally, another challenge in the clinical development of antibody-based therapies against aSyn is the absence of a reliable diagnostic tool to assess the disease progression. PD diagnosis relies solely on clinical signs and symptoms, medical history and neurological and physical examinations [10]. A good diagnostic marker would be important for two main reasons: to ensure that patients enrolled in clinical trials are at an expected stage of disease progression, and to evaluate if the treatment leads to a sufficient target engagement. First, since clinical signs and symptoms are highly variable between patients, the precise assessment of LB/aSyn pathology in the brain is challenging. Moreover, a small proportion of patients with clinical PD do not present Lewy pathology— a situation mainly seen with Parkin and LRRK2 mutations [10]. It is thus possible that patients that lack LBs could be included in clinical trials. Conversely, patients with severe aSyn burden and neuronal loss could be included as well. In both cases, antibody therapy may not prove any functional benefit, which would undesirably alter the clinical trial results. Also, the possibility to perform a genetic analysis could result in the exclusion of patients with specific mutations in the clinical trial recruitment. Additionally, a diagnostic tool that allows early diagnosis of PD, before extensive neurodegeneration, would permit a presumably more successful intervention in terms of disease progression, symptoms reduction, and quality of life improvement. Second, to properly evaluate if the antibody is to effectively engage its target and promote degradation, a reliable non-invasive method must exist to detect total aSyn levels and/or aggregates. Unlike amyloid-β and Tau, no validated PET-tracers currently exist to detect aSyn in the brain, despite various development efforts [189, 190]. Imaging modalities will demand careful evaluation for their resolution, i.e., their correlation with the extent of proteinopathy and strain specificity. Standard rating scales to assess PD symptoms, as the Unified Parkinson’s Disease Rating Scale (UPDRS), and striatal binding ratio with DATscan, are not adequately precise to determine the effectiveness of an antibody treatment. Finally, the detection of free aSyn in serum or CSF is commonly used in clinical trials but lacks a validated correlation with clinical outcomes. The ability to specifically detect aggregation-prone aSyn species in the CSF or brain biopsies would however be highly valuable. The Protein-Misfolding Cyclic Amplification (PMCA) and the Real-Time Quacking-Induced Conversion (RT-QuIC) are protein amplification assays that can detect misfolded protein aggregates. Both have shown a high sensitivity and specificity in detecting pre-clinical and clinical synucleinopathies in preliminary studies [191].
CONCLUSION
In conclusion, antibody-based therapies against aSyn promise a new therapeutic approach for Parkinson’s disease. Antibodies could be effective to reduce the aberrant accumulation of aSyn and limit the cell-to-cell transmission. Both passive and active immunization have shown enviable results in clinical trials so far, but clinical/functional recovery is yet to be proven. The development of a reliable imaging strategy or biochemical marker for the assessment of synucleinopathy will be important to monitor clinical trials effectiveness. Moreover, a better understanding of aSyn disorganization, aggregation and related toxicity, along with a clarification of its cell-to-cell transmission mechanism will greatly help developing efficient antibodies.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
A.-M.C. is supported by a scholarship from the Neuroscience Thematic Research Centre. M.L. receives salary support from Fonds de Recherche du Quebec-Sante, Chercheur-Boursier Juniors 2 in partnership with Parkinson Quebec (34974). We also thank Modesto R Peralta III for help in revising the manuscript.
REFERENCES
[1] | Dickson DW ((2012) ) Parkinson’s disease and parkinsonism: Neuropathology. Cold Spring Med Harb Perspect 2: , a009258. |
[2] | Gelb DJ , Oliver E , Gilman S ((1999) ) Diagnostic criteria for Parkinson disease. Arch Neurol 56: , 33–39. |
[3] | Gibb WRG , Lees AJ ((1988) ) The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J Neurol Neurosurg Psychiatry 51: , 745–752. |
[4] | Postuma RB , Aarsland D , Barone P , Burn DJ , Hawkes CH , Oertel W , Ziemssen T ((2012) ) Identifying prodromal Parkinson’s disease: Pre-Motor disorders in Parkinson’s disease. Mov Disord 27: , 617–626. |
[5] | Khoo TK , Yarnall AJ , Duncan GW , Coleman S , O’Brien JT , Brooks DJ , Barker RA , Burn DJ ((2013) ) The spectrum of nonmotor symptoms in early Parkinson disease. Neurology 80: , 276–281. |
[6] | Lesage S , Brice A ((2009) ) Parkinson’s disease: From monogenic forms to genetic susceptibility factors. Hum Mol Genet 18: , 48–59. |
[7] | Dauer W , Przedborski S ((2003) ) Parkinson’s disease: Mechanisms and models. Neuron 39: , 889–909. |
[8] | Deng H , Wang P , Jankovic J ((2018) ) The genetics of Parkinson disease. Ageing Res Rev 42: , 72–85. |
[9] | Parkinson J ((2002) ) An essay on the shaking palsy. 1817. J Neuropsychiatry Clin Neurosci 14: , 223–236; discussion 222. |
[10] | Kalia LV , Lang AE ((2015) ) Parkinson’s disease. Lancet 386: , 896–912. |
[11] | Forno LS ((1996) ) Neuropathology of Parkinson’s disease. J Neuropathol Exp Neurol 55: , 259–272. |
[12] | Lewy F ((1912) ) Paralysis agitans. I. Pathologische Anatomie. In Handbuch der Neurologie, Lewandowsky M, Abelsdorff G, eds. Springer-Verlag, Berlin, pp. 920-933. |
[13] | Mahul-Mellier AL , Burtscher J , Maharjan N , Weerens L , Croisier M , Kuttler F , Leleu M , Knott GW , Lashuel HA ((2020) ) The process of Lewy body formation, rather than simply α-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc Natl Acad Sci U S A 117: , 4971–4982. |
[14] | Spillantini MG , Schmidt ML , Lee VM , Trojanowski JQ , Jakes R , Goedert M ((1997) ) Alpha-synuclein in Lewy bodies. Nature 388: , 839–840. |
[15] | Goedert M , Spillantini MG , Del Tredici K , Braak H ((2013) ) 100 years of Lewy pathology.. Nat Rev Neurol 9: , 13–24. |
[16] | Stefanis L ((2012) ) α-Synuclein in Parkinson’s disease. Cold Spring Harb Perspect Med 2: , a009399. |
[17] | Lo Bianco C , Ridet J-L , Schneider BL , Dé N , Aebischer P ((2002) ) a-Synucleinopathy and selective dopaminergic neuron loss in a rat lentiviral-based model of Parkinson’s disease. Proc Natl Acad Sci U S A 99: , 10813–10818. |
[18] | Xu J , Kao SY , Lee FJS , Song W , Jin LW , Yankner BA ((2002) ) Dopamine-dependent neurotoxicity of α- synuclein: A mechanism for selective neurodegeneration in Parkinson disease. Nat Med 8: , 600–606 |
[19] | Zhou W , Schaack J , Zawada WM , Freed CR ((2002) ) Overexpression of human α-synuclein causes dopamine neuron death in primary human mesencephalic culture. Brain Res 926: , 42–50. |
[20] | El-Agnaf OM , Salem SA , Paleologou KE , Cooper LJ , Fullwood NJ , Gibson MJ , Curran MD , Court JA , Mann DM , Ikeda S , Cookson MR , Hardy J , Allsop D ((2003) ) α-Synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J 17: , 1945–1947. |
[21] | Pedersen JT , Sigurdsson EM ((2015) ) Tau immunotherapy for Alzheimer’s disease. Trends Mol Med 21: , 394–402. |
[22] | Panza F , Lozupone M , Seripa D , Imbimbo BP ((2019) ) Amyloid-β immunotherapy for alzheimer disease: Is it now a long shot?. Ann Neurol 85: , 303–315. |
[23] | Denis HL , David LS , Cicchetti F ((2019) ) Antibody-based therapies for Huntington’s disease: Current status and future directions. Neurobiol Dis 132: , 104569. |
[24] | Pozzi S , Thammisetty SS , Codron P , Rahimian R , Plourde KV , Soucy G , Bareil C , Phaneuf D , Kriz J , Gravel C , Julien JP ((2019) ) Virus-mediated delivery of antibody targeting TAR DNA-binding protein-43 mitigates associated neuropathology. J Clin Invest 129: , 1581–1595. |
[25] | Wang Y , Yan T , Lu H , Yin W , Lin B , Fan W , Zhang X , Fernandez-Funez P ((2017) ) Lessons from anti-amyloid-β immunotherapies in Alzheimer disease: Aiming at a moving target. Neurodegener Dis 17: , 242–250. |
[26] | Bittar A , Sengupta U , Kayed R ((2018) ) Prospects for strain-specific immunotherapy in Alzheimer’s disease and tauopathies. NPJ Vaccines 3: , 1–9. |
[27] | West T , Hu Y , Verghese PB , Bateman RJ , Braunstein JB , Fogelman I , Budur K , Florian H , Mendonca N , Holtzman DM ((2017) ) Preclinical and clinical development of ABBV-8E12, a humanized anti-tau antibody, for treatment of Alzheimer’s disease and other tauopathies. J Prev Alzheimers Dis 4: , 236–241. |
[28] | Alzforum.org (2019)AbbVie’sTau Antibody Flops in Progressive Supranuclear Palsy. https://www.alzforum.org/news/research-news/abbvies-tau-antibody-flops-progressive-supranuclear-palsy |
[29] | Boxer AL , Qureshi I , Ahlijanian M , Grundman M , Golbe LI , Litvan I , Honig LS , Tuite P , McFarland NR , O’Suilleabhain P , Xie T , Tirucherai GS , Bechtold C , Bordelon Y , Geldmacher DS , Grossman M , Isaacson S , Zesiewicz T , Olsson T , Muralidharan KK , Graham DL , O’Gorman J , Haeberlein SB , Dam T ((2019) ) Safety of the tau-directed monoclonal antibody BIIB092 in progressive supranuclear palsy: A randomised, placebo-controlled, multiple ascending dose phase 1b trial. Lancet Neurol 18: , 549–558. |
[30] | Biogen (2019) Biogen Reports Top-Line Results from Phase 2 Study in Progressive Supranuclear Palsy. https://investors.biogen.com/news-releases/news-release-details/biogen-reports-top-line-results-phase-2-study-progressive |
[31] | Press Releases, UCB presents UCB0107 anti-Tau immunotherapy Phase I study results atWorld Movement Disorders Conference®, Last updated 2019. https://www.ucb.com/stories-media/Press-Releases/article/UCB-presents-UCB0107-anti-Tau-immunotherapy-Phase-I-study-results-at-World-Movement-Disorders-Conference. |
[32] | Vaswani PA , Olsen AL ((2020) ) Immunotherapy in progressive supranuclear palsy. Curr Opin Neurol 33: , 527–533. |
[33] | Rocha EM , De Miranda B , Sanders LH ((2018) ) Alpha-synucle in: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol Dis 109: , 249–257. |
[34] | Burré J ((2015) ) The synaptic function of α-synuclein. J Parkinsons Dis 5: , 699–713. |
[35] | Singleton AB , Farrer M , Johnson J , Singleton A , Hague S , Kachergus J , Hulihan M , Peuralinna T ((2003) ) a-Synuclein locus triplication causes Parkinson’s disease. Science 302: , 841–850. |
[36] | Polymeropoulos MH , Lavedan C , Leroy E , Ide SE , Dehejia A , Dutra A , Pike B , Root H , Rubenstein J , Boyer R , Stenroos ES , Chandrasekharappa S , Athanassiadou A , Papapetropoulos T , Johnson WG , Lazzarini AM , Duvoisin RC , Di Iorio G , Golbe LI , Nussbaum RL ((1997) ) Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 276: , 2045–2047. |
[37] | Wang W , Perovic I , Chittuluru J , Kaganovich A , Nguyen LTT , Liao J , Auclair JR , Johnson D , Landeru A , Simorellis AK , Ju S , Cookson MR , Asturias FJ , Agar JN , Webb BN , Kang CH , Ringe D , Petsko GA , Pochapsky TC , Hoang QQ ((2011) ) A soluble α-synuclein construct forms a dynamic tetramer. Proc Natl Acad Sci U S A 108: , 17797–17802. |
[38] | Bartels T , Choi JG , Selkoe DJ ((2011) ) α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477: , 107–111. |
[39] | Bussell R , Eliezer D ((2003) ) A structural and functional role for 11-mer repeats in alpha-synuclein and other exchangeable lipid binding proteins.. J Mol Biol 329: , 763–78. |
[40] | Davidson WS , Jonas A , Clayton DF , George JM ((1998) ) Stabilization of α-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem 273: , 9443–9449. |
[41] | Zhu M , Fink AL ((2003) ) Lipid binding inhibits α-synuclein fibril formation. J Biol Chem 278: , 16873–16877. |
[42] | El-Agnaf OMA , Jakes R , Curran MD , Middleton D , Ingenito R , Bianchi E , Pessi A , Neill D , Wallace A ((1998) ) Aggregates from mutant and wild-type α-synuclein proteins and NAC peptide induce apoptotic cell death in human neuroblastoma cells by formation of β-sheet and amyloid-like filaments. FEBS Lett 440: , 71–75. |
[43] | Conway KA , Harper JD , Lansbury PT ((2000) ) Fibrils formed in vitro from α-synuclein and two mutant forms linked to Parkinson’s disease are typical amyloid. Biochemistry 39: , 2552–2563. |
[44] | Burré J , Sharma M , Südhof TC ((2015) ) Definition of a molecular pathway mediating α-synuclein neurotoxicity. J Neurosci 35: , 5221–5232. |
[45] | Okochi M , Walter J , Koyama A , Nakajo S , Baba M , Iwatsubo T , Meijer L , Kahle PJ , Haass C ((2000) ) Constitutive phosphorylation of the Parkinson’s disease associated α- synuclein. J Biol Chem 275: , 390–397. |
[46] | Fujiwara H , Hasegawa M , Dohmae N , Kawashima A , Masliah E , Goldberg MS , Shen J , Takio K , Iwatsubo T ((2002) ) α-synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol 4: , 160–164. |
[47] | Sugeno N , Takeda A , Hasegawa T , Kobayashi M , Kikuchi A , Mori F , Wakabayashi K , Itoyama Y ((2008) ) Serine 129 phosphorylation of α-synuclein induces unfolded protein response-mediated cell death. J Biol Chem 283: , 23179–23188. |
[48] | Wakamatsu M , Ishii A , Ukai Y , Sakagami J , Iwata S , Ono M , Matsumoto K , Nakamura A , Tada N , Kobayashi K , Iwatsubo T , Yoshimoto M ((2007) ) Accumulation of phosphorylated α-synuclein in dopaminergic neurons of transgenic mice that express human α-synuclein. J Neurosci Res 85: , 1819–1825. |
[49] | Chen L , Feany MB ((2005) ) α-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat Neurosci 8: , 657–663. |
[50] | Schell H , Hasegawa T , Neumann M , Kahle PJ ((2009) ) Nuclear and neuritic distribution of serine-129 phosphorylated α-synuclein in transgenic mice. Neuroscience 160: , 796–804. |
[51] | Oueslati A ((2016) ) Implication of alpha-synuclein phosphorylation at S129 in synucleinopathies: What have we learned in the last decade?. J Parkinsons Dis 6: , 39–51. |
[52] | Kim TD , Paik SR , Yang CH ((2002) ) Structural and functional implications of C-terminal regions of α-synuclein. Biochemistry 41: , 13782–13790. |
[53] | Li W , West N , Colla E , Pletnikova O , Troncoso JC , Marsh L , Dawson TM , Jäkälä P , Hartmann T , Price DL , Lee MK ((2005) ) Aggregation promoting C-terminal truncation of α-synuclein is a normal cellular process and is enhanced by the familial Parkinson’s disease-linked mutations. Proc Natl Acad Sci U S A 102: , 2162–2167. |
[54] | Burré J , Sharma M , Tsetsenis T , Buchman V , Etherton MR , Südhof TC ((2010) ) α-Synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329: , 1663–1667. |
[55] | Butler B , Saha K , Rana T , Becker JP , Sambo D , Davari P , Goodwin JS , Khoshbouei H ((2015) ) Dopamine transporter activity is modulated by α-synuclein. J Biol Chem 290: , 29542–29554. |
[56] | Baptista MJ , O’Farrell C , Daya S , Ahmad R , Miller DW , Hardy J , Farrer MJ , Cookson MR ((2003) ) Co-ordinate transcriptional regulation of dopamine synthesis genes by α-synuclein in human neuroblastoma cell lines. J Neurochem 85: , 957–968. |
[57] | Perez RG , Waymire JC , Lin E , Liu JJ , Guo F , Zigmond MJ ((2002) ) A role for α-synuclein in the regulation of dopamine biosynthesis. J Neurosci 22: , 3090–3099. |
[58] | Guo JT , Chen AQ , Kong Q , Zhu H , Ma CM , Qin C ((2008) ) Inhibition of vesicular monoamine transporter-2 activity in α-synuclein stably transfected SH-SY5Y cells. Cell Mol Neurobiol 28: , 35–47. |
[59] | Rodriguez L , Marano MM , Tandon A ((2018) ) Import and export of misfolded α-synuclein. Front Neurosci 12: , 1–9. |
[60] | Kordower JH , Chu Y , Hauser RA , Freeman TB , Olanow CW ((2008) ) Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med 14: , 504–506. |
[61] | Li JY , Englund E , Holton JL , Soulet D , Hagell P , Lees AJ , Lashley T , Quinn NP , Rehncrona S , Björklund A , Widner H , Revesz T , Lindvall O , Brundin P ((2008) ) Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med 14: , 501–503. |
[62] | Borghi R , Marchese R , Negro A , Marinelli L , Forloni G , Zaccheo D , Abbruzzese G , Tabaton M ((2000) ) Full length α-synuclein is present in cerebrospinal fluid from Parkinson’s disease and normal subjects. Neurosci Lett 287: , 65–67. |
[63] | Lee PH , Lee G , Park HJ , Bang OY , Joo IS , Huh K ((2006) ) The plasma alpha-synuclein levels in patients with Parkinson’s disease and multiple system atrophy. J Neural Transm 113: , 1435–1439. |
[64] | Braak H , Del Tredici K , Rüb U , De Vos RAI , Jansen Steur ENH , Braak E ((2003) ) Staging of brain pathology related to sporadic Parkinson’s disease.. Neurobiol Aging 24: , 197–211. |
[65] | Edwards LL , Quigley EMM , Pfeiffer RF ((1992) ) Gastrointestinal dysfunction in Parkinson’s disease: Frequency and pathophysiology. Neurology 42: , 726–732. |
[66] | Braak H , De Vos RAI , Bohl J , Del Tredici K ((2006) ) Gastric α-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci Lett 396: , 67–72. |
[67] | Lee HJ , Suk JE , Bae EJ , Lee JH , Paik SR , Lee SJ ((2008) ) Assembly-dependent endocytosis and clearance of extracellular α-synuclein. Int J Biochem Cell Biol 40: , 1835–1849. |
[68] | Choi I , Zhang Y , Seegobin SP , Pruvost M , Wang Q , Purtell K , Zhang B , Yue Z ((2020) ) Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nat Commun 11: , 1–14. |
[69] | Lee HJ , Suk JE , Bae EJ , Lee SJ ((2008) ) Clearance and deposition of extracellular α-synuclein aggregates in microglia. Biochem Biophys Res Commun 372: , 423–428. |
[70] | Kim C , Ho DH , Suk JE , You S , Michael S , Kang J , Lee SJ , Masliah E , Hwang D , Lee HJ , Lee SJ ((2013) ) Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun 4: , 1562. |
[71] | Volpicelli-Daley LA , Luk KC , Lee VMY ((2014) ) Addition of exogenous α-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous α-synuclein to Lewy body and Lewy neurite-like aggregates. Nat Protoc 9: , 2135–2146. |
[72] | Rey NL , Steiner JA , Maroof N , Luk KC , Madaj Z , Trojanowski JQ , Lee VMY , Brundin P ((2016) ) Widespread transneuronal propagation of α-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson’s disease. J Exp Med 213: , 1759–1778. |
[73] | Rey NL , Petit GH , Bousset L , Melki R , Brundin P ((2013) ) Transfer of human α-synuclein from the olfactory bulb to interconnected brain regions in mice. Acta Neuropathol 126: , 555–573. |
[74] | Luk KC , Kehm VM , Zhang B , O’Brien P , Trojanowski JQ , Lee VMY ((2012) ) Intracerebral inoculation of pathological a-synuclein initiates a rapidly progressive neurodegenerative a-synucleinopathy in mice.. J Exp Med 209: , 975–986. |
[75] | Masuda-Suzukake M , Nonaka T , Hosokawa M , Oikawa T , Arai T , Akiyama H , Mann DMA , Hasegawa M ((2013) ) Prion-like spreading of pathological α-synuclein in brain. Brain 136: , 1128–1138. |
[76] | Schweighauser M , Bacioglu M , Fritschi SK , Shimshek DR , Kahle PJ , Eisele YS , Jucker M ((2015) ) Formaldehyde-fixed brain tissue from spontaneously ill α-synuclein transgenic mice induces fatal α-synucleinopathy in transgenic hosts. Acta Neuropathol 129: , 157–159. |
[77] | Ulusoy A , Rusconi R , Pérez-Revuelta BI , Musgrove RE , Helwig M , Winzen-Reichert B , Di Monte DA ((2013) ) Caudo-rostral brain spreading of α-synuclein through vagal connections. EMBO Mol Med 5: , 1119–1127. |
[78] | Sacino AN , Brooks M , Thomas MA , McKinney AB , Lee S , Regenhardt RW , McGarvey NH , Ayers JI , Notterpek L , Borchelt DR , Golde TE , Giasson BI ((2014) ) Intramuscular injection of α-synuclein induces CNS α-synuclein pathology and a rapid-onset motor phenotype in transgenic mice. Proc Natl Acad Sci U S A 111: , 10732–10737. |
[79] | Abounit S , Bousset L , Loria F , Zhu S , Chaumont F , Pieri L , Olivo-Marin J , Melki R , Zurzolo C ((2016) ) Tunneling nanotubes spread fibrillar α-synuclein by intercellular trafficking of lysosomes. EMBO J 35: , 2120–2138. |
[80] | Mao X , Ou MT , Karuppagounder SS , Kam TI , Yin X , Xiong Y , Ge P , Umanah GE , Brahmachari S , Shin JH , Kang HC , Zhang J , Xu J , Chen R , Park H , Andrabi SA , Kang SU , Gonçalves RA , Liang Y , Zhang S , Qi C , Lam S , Keiler JA , Tyson J , Kim D , Panicker N , Yun SP , Workman CJ , Vignali DAA , Dawson VL , Ko HS , Dawson TM ((2016) ) Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 353: , aah3374. |
[81] | Reyes JF , Sackmann C , Hoffmann A , Svenningsson P , Winkler J , Ingelsson M , Hallbeck M ((2019) ) Binding of α-synuclein oligomers to Cx32 facilitates protein uptake and transfer in neurons and oligodendrocytes. Acta Neuropathol 138: , 23–47. |
[82] | Holmes BB , DeVos SL , Kfoury N , Li M , Jacks R , Yanamandra K , Ouidja MO , Brodsky FM , Marasa J , Bagchi DP , Kotzbauer PT , Miller TM , Papy-Garcia D , Diamond MI ((2013) ) Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci U S A 110: , E3138–3147. |
[83] | Brahic M , Bousset L , Bieri G , Melki R , Gitler AD ((2016) ) Axonal transport and secretion of fibrillar forms of α-synuclein, Aβ42 peptide and HTTExon 1. Acta Neuropathol 131: , 539–548. |
[84] | Bieri G , Gitler AD , Brahic M ((2018) ) Internalization, axonal transport and release of fibrillar forms of alpha-synuclein. Neurobiol Dis 109: , 219–225. |
[85] | Karpowicz RJ , Haney CM , Mihaila TS , Sandler RM , Petersson EJ , Lee VMY ((2017) ) Selective imaging of internalized proteopathic α-synuclein seeds in primary neurons reveals mechanistic insight into transmission of synucleinopathies. J Biol Chem 292: , 13482–13497. |
[86] | Vogiatzi T , Xilouri M , Vekrellis K , Stefanis L ((2008) ) Wild type α-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem 283: , 23542–23556. |
[87] | Mak SK , McCormack AL , Manning-Bog AB , Cuervo AM , Di Monte DA ((2010) ) Lysosomal degradation of α-synuclein in vivo. J Biol Chem 285: , 13621–13629. |
[88] | Cuervo AM , Stafanis L , Fredenburg R , Lansbury PT , Sulzer D ((2004) ) Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 305: , 1292–1295. |
[89] | Crews L , Spencer B , Desplats P , Patrick C , Paulino A , Rockenstein E , Hansen L , Adame A , Galasko D , Masliah E ((2010) ) Selective molecular alterations in the autophagy pathway in patients with lewy body disease and in models of α-synucleinopathy. PLoS One 5: , e9313. |
[90] | Tanik SA , Schultheiss CE , Volpicelli-Daley LA , Brunden KR , Lee VMY ((2013) ) Lewy body-like α-synuclein aggregates resist degradation and impair macroautophagy. J Biol Chem 288: , 15194–15210. |
[91] | Freeman D , Cedillos R , Choyke S , Lukic Z , McGuire K , Marvin S , Burrage AM , Sudholt S , Rana A , O’Connor C , Wiethoff CM , Campbell EM ((2013) ) Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS One 8: , e62143. |
[92] | Lee HJ , Bae EJ , Lee SJ ((2014) ) Extracellular α-synuclein-a novel and crucial factor in Lewy body diseases. Nat Rev Neurol 10: , 92–98. |
[93] | Luk KC , Song C , O’Brien P , Stieber A , Branch JR , Brunden KR , Trojanowski JQ , Lee VMY ((2009) ) Exogenous α-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A 106: , 20051–20056. |
[94] | Volpicelli-Daley LA , Luk KC , Patel TP , Tanik SA , Riddle DM , Stieber A , Meaney DF , Trojanowski JQ , Lee VMY ((2011) ) Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72: , 57–71. |
[95] | Lee HJ , Patel S , Lee SJ ((2005) ) Intravesicular localization and exocytosis of α-synuclein and its aggregates. J Neurosci 25: , 6016–6024. |
[96] | Lee JG , Takahama S , Zhang G , Tomarev SI , Ye Y ((2016) ) Unconventional secretion of misfolded proteins promotes adaptation to proteasome dysfunction in mammalian cells. Nat Cell Biol 18: , 765–776. |
[97] | Emmanouilidou E , Melachroinou K , Roumeliotis T , Garbis SD , Ntzouni M , Margaritis LH , Stefanis L , Vekrellis K ((2010) ) Cell-produced α-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci 30: , 6838–6851. |
[98] | Danzer KM , Kranich LR , Ruf WP , Cagsal-Getkin O , Winslow AR , Zhu L , Vanderburg CR , McLean PJ ((2012) ) Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol Neurodegener 7: , 1. |
[99] | Jang A , Lee HJ , Suk JE , Jung JW , Kim KP , Lee SJ ((2010) ) Non-classical exocytosis of α-synuclein is sensitive to folding states and promoted under stress conditions. J Neurochem 113: , 1263–1274. |
[100] | Lee HJ , Cho ED , Lee KW , Kim JH , Cho SG , Lee SJ ((2013) ) Autophagic failure promotes the exocytosis and intercellular transfer of α-synuclein. Exp Mol Med 45: , e22. |
[101] | Dieriks BV , Park TIH , Fourie C , Faull RLM , Dragunow M , Curtis MA ((2017) ) α-synuclein transfer through tunneling nanotubes occurs in SH-SY5Y cells and primary brain pericytes from Parkinson’s disease patients. Sci Rep 7: , 42984. |
[102] | Marzo L , Gousset K , Zurzolo C ((2012) ) Multifaceted roles of tunneling nanotubes in intercellular communication. Front Physiol 3: , 72. |
[103] | Fusco G , Chen SW , Williamson PTF , Cascella R , Perni M , Jarvis JA , Cecchi C , Vendruscolo M , Chiti F , Cremades N , Ying L , Dobson CM , De Simone A ((2017) ) Structural basis of membrane disruption and cellular toxicity by a-synuclein oligomers. Science 358: , 1440–1443. |
[104] | Winner B , Jappelli R , Maji SK , Desplats PA , Boyer L , Aigner S , Hetzer C , Loher T , Vilar M , Campioni S , Tzitzilonis C , Soragni A , Jessberger S , Mira H , Consiglio A , Pham E , Masliah E , Gage FH , Riek R ((2011) ) In vivo demonstration that α-synuclein oligomers are toxic. Proc Natl Acad Sci U S A 108: , 4194–4199. |
[105] | Lashuel HA , Overk CR , Oueslati A , Masliah E ((2013) ) The many faces of α-synuclein: From structure and toxicity to therapeutic target. Nat Rev Neurosci 14: , 38–48. |
[106] | Bousset L , Pieri L , Ruiz-Arlandis G , Gath J , Jensen PH , Habenstein B , Madiona K , Olieric V , Böckmann A , Meier BH , Melki R ((2013) ) Structural and functional characterization of two alpha-synuclein strains. Nat Commun 4: , 1–13. |
[107] | Pieri L , Madiona K , Bousset L , Melki R ((2012) ) Fibrillar α-synuclein and huntingtin exon 1 assemblies are toxic to the cells. Biophys J 102: , 2894–2905. |
[108] | Lindström V , Fagerqvist T , Nordström E , Eriksson F , Lord A , Tucker S , Andersson J , Johannesson M , Schell H , Kahle PJ , Möller C , Gellerfors P , Bergström J , Lannfelt L , Ingelsson M ((2014) ) Immunotherapy targeting α-synuclein protofibrils reduced pathology in (Thy-1)-h[A30P] α-synuclein mice. Neurobiol Dis 69: , 134–143. |
[109] | Emadi S , Barkhordarian H , Wang M. , Schulz P , Sierks M. ((2007) ) Isolation of a human single chain antibody fragment against oligomeric α-synuclein that inhibits aggregation and prevents α- synuclein induced toxicity. J Mol Biol 368: , 1132–1144. |
[110] | Emadi S , Kasturirangan S , Wang MS , Schulz P , Sierks MR ((2009) ) Detecting morphologically distinct oligomeric forms of α-synuclein. J Biol Chem 284: , 11048–11058. |
[111] | Zha J , Liu XM , Zhu J , Liu SY , Lu S , Xu PX , Yu XL , Liu RT ((2016) ) A scFv antibody targeting common oligomeric epitope has potential for treating several amyloidoses. Sci Rep 6: , 1–15. |
[112] | Bae EJ , Lee HJ , Rockenstein E , Ho DH , Park EB , Yang NY , Desplats P , Masliah E , Lee SJ ((2012) ) Antibody-aided clearance of extracellular α-synuclein prevents cell-to-cell aggregate transmission. J Neurosci 32: , 13454–13469. |
[113] | Chatterjee D , Bhatt M , Butler D , De Genst E , Dobson CM , Messer A , Kordower JH ((2018) ) Proteasome-targeted nanobodies alleviate pathology and functional decline in an α-synuclein-based Parkinson’s disease model. NPJ Parkinsons Dis 4: , 1–10. |
[114] | Spencer B , Emadi S , Desplats P , Eleuteri S , Michael S , Kosberg K , Shen J , Rockenstein E , Patrick C , Adame A , Gonzalez T , Sierks M , Masliah E ((2014) ) ESCRT-mediated uptake and degradation of brain-targeted α-synuclein single chain antibody attenuates neuronal degeneration in vivo. Mol Ther 22: , 1753–1767. |
[115] | Mosley RL , Hutter-Saunders JA , Stone DK , Gendelman HE ((2012) ) Inflammation and adaptive immunity in Parkinson’s disease. Cold Spring Harb Perspect Med 2: , a009381. |
[116] | Przedborski S ((2010) ) Inflammation and Parkinson’s disease pathogenesis. Mov Disord 25: , S55–S57. |
[117] | Schneeberger A , Tierney L , Mandler M ((2016) ) Active immunization therapies for Parkinson’s disease and multiple system atrophy. Mov Disord 31: , 214–224. |
[118] | Zimmermann P , Curtis N ((2019) ) Factors that influence the immune response to vaccination.e. Clin Microbiol Rev 32: , 00084–18. |
[119] | Valera E , Masliah E ((2013) ) Immunotherapy for neurodegenerative diseases: Focus on α- synucleinopathies. Pharmacol Ther 138: , 311–322. |
[120] | Lannfelt L , Relkin NR , Siemers ER ((2014) ) Amyloid-ß-directed immunotherapy for Alzheimer’s disease. J Intern Med 275: , 284–295. |
[121] | Masliah E , Rockenstein E , Adame A , Alford M , Crews L , Hashimoto M , Seubert P , Lee M , Goldstein J , Chilcote T , Games D , Schenk D ((2005) ) Effects of α-synuclein immunization in a mouse model of Parkinson’s disease. Neuron 46: , 857–868. |
[122] | Rockenstein E , Crews L , Masliah E ((2007) ) Transgenic animal models of neurodegenerative diseases and their application to treatment development. Adv Drug Deliv Rev 59: , 1093–1102. |
[123] | Sanchez-Guajardo V , Annibali A , Jensen PH , Romero-Ramos M ((2013) ) α-synuclein vaccination prevents the accumulation of Parkinson disease-like pathologic inclusions in striatum in association with regulatory T cell recruitment in a rat model. J Neuropathol Exp Neurol 72: , 624–645. |
[124] | Fearnley JM , Lees AJ ((1990) ) Striatonigral degeneration: A clinicopathological study. Brain 113: , 1823–1842. |
[125] | Mandler M , Valera E , Rockenstein E , Weninger H , Adame A , Santic R , Meindl S , Vigl B ((2015) ) Next-generation active immunization approach for synucleinopathies - implications for Parkinson’s disease clinical trials. Acta Neuropathol 127: , 861–879. |
[126] | Volc D , Poewe W , Kutzelnigg A , Lührs P , Thun-Hohenstein C , Schneeberger A , Galabova G , Majbour N , Vaikath N , El-Agnaf O , Winter D , Mihailovska E , Mairhofer A , Schwenke C , Staffler G , Medori R ((2020) ) Safety and immunogenicity of the α-synuclein active immunotherapeutic PD01A in patients with Parkinson’s disease: A randomised, single-blinded, phase 1 trial. Lancet Neurol 19: , 591–600. |
[127] | Ghochikyana Anahit , Petrushinab I , Davtyana H , Hovakimyan A , Saing T , Davtyana , Arpine , Cribbsb DH , Agadjanyan MG ((2014) ) Immunogenicity of epitope vaccines targeting different B cell antigenic determinants of human α-synuclein: Feasibility study. Neuroscience 560: , 86–91. |
[128] | Shahaduzzaman M , Nash K , Hudson C , Sharif M , Grimmig B , Lin X , Bai G , Liu H , Ugen KE , Cao C , Bickford PC ((2015) ) Anti-human α-synuclein N-terminal peptide antibody protects against dopaminergic cell death and ameliorates behavioral deficits in an AAV-α-synuclein rat model of Parkinson’s disease. PLoS One 10: , e0116841. |
[129] | Doucet M , El-Turabi A , Zabel F , Hunn BHM , Bengoa-Vergniory N , Cioroch M , Ramm M , Smith AM , Gomes AC , De Miranda GC , Wade-Martins R , Bachmann MF ((2017) ) Preclinical development of a vaccine against oligomeric alpha-synuclein based on virus-like particles. PLoS One 12: , 1–25. |
[130] | Rockenstein E , Ostroff G , Dikengil F , Rus F , Mante M , Florio J , Adame A , Trinh I , Kim C , Overk C , Masliah E , Rissman RA ((2018) ) Combined active humoral and cellular immunization approaches for the treatment of synucleinopathies. J Neurosci 38: , 1000–1014. |
[131] | George S , Brundin P ((2015) ) Immunotherapy in Parkinson’s disease: Micromanaging alpha-synuclein aggregation. J Parkinsons Dis 5: , 413–424. |
[132] | Masliah E , Rockenstein E , Mante M , Crews L , Spencer B , Adame A , Patrick C , Trejo M , Ubhi K , Rohn TT , Mueller-Steiner S , Seubert P , Barbour R , McConlogue L , Buttini M , Games D , Schenk D ((2011) ) Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS One 6: , e19338. |
[133] | Schenk DB , Koller M , Ness DK , Griffith SG , Grundman M , Zago W , Soto J , Atiee G , Ostrowitzki S , Kinney GG ((2017) ) First-in-human assessment of PRX002, an anti– α-synuclein monoclonal antibody, in healthy volunteers. Mov Disord 32: , 211–218. |
[134] | Jankovic J , Goodman I , Safirstein B , Marmon TK , Schenk DB , Koller M , Zago W , Ness DK , Griffith SG , Grundman M , Soto J , Ostrowitzki S , Boess FG , Martin-Facklam M , Quinn JF , Isaacson SH , Omidvar O , Ellenbogen A , Kinney GG ((2018) ) Safety and tolerability of multiple ascending doses of PRX002/RG7935, an anti-α-synuclein monoclonal antibody, in patients with Parkinson disease: A randomized clinical trial. JAMA Neurol 75: , 1206–1214. |
[135] | Nasdaq.com (2020) Update on Phase 2 PASADENA Study of Prasinezumab (PRX002/RG7935) in Parkinson’s Disease. https://www.nasdaq.com/press-release/update-on-phase-2-pasadena-study-of-prasinezumab-prx002-rg7935-in-parkinsons-disease |
[136] | Games D , Valera E , Spencer B , Rockenstein E , Mante M , Adame A , Patrick C , Ubhi K , Nuber S , Sacayon P , Zago W , Seubert P , Barbour R , Schenk D , Masliah E ((2014) ) Reducing C-terminal-truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson’s disease-like models. J Neurosci 34: , 9441–9454. |
[137] | Mishizen-Eberz AJ , Norris EH , Giasson BI , Hodara R , Ischiropoulos H , Lee VMY , Trojanowski JQ , Lynch DR ((2005) ) Cleavage of α-synuclein by calpain: Potential role in degradation of fibrillized and nitrated species of α- synuclein. Biochemistry 44: , 7818–7829. |
[138] | Spencer B , Valera E , Rockenstein E , Overk C , Mante M , Adame A , Zago W , Seubert P , Barbour R , Schenk D , Games D , Rissman RA , Masliah E ((2017) ) Anti-α-synuclein immunotherapy reduces α-synuclein propagation in the axon and degeneration in a combined viral vector and transgenic model of synucleinopathy. Acta Neuropathol Commun 5: , 7. |
[139] | Schofield DJ , Irving L , Calo L , Bogstedt A , Rees G , Nuccitelli A , Narwal R , Petrone M , Roberts J , Brown L , Cusdin F , Dosanjh B , Lloyd C , Dobson C , Gurrell I , Fraser G , McFarlane M , Rockenstein E , Spencer B , Masliah E , Spillantini MG , Tan K , Billinton A , Vaughan T , Chessell I , Perkinton MS ((2019) ) Preclinical development of a high affinity α-synuclein antibody, MEDI1341, that can enter the brain, sequester extracellular α-synuclein and attenuate α-synuclein spreading in vivo. Neurobiol Dis 132: , 104582. |
[140] | Weihofen A , Liu YT , Arndt JW , Huy C , Quan C , Smith BA , Baeriswyl JL , Cavegn N , Senn L , Su L , Marsh G , Auluck PK , Montrasio F , Nitsch RM , Hirst WD , Cedarbaum JM , Pepinsky RB , Grimm J , Weinreb PH ((2019) ) Development of an aggregate-selective, human-derived α-synuclein antibody BIIB054 that ameliorates disease phenotypes in Parkinson’s disease models. Neurobiol Dis 124: , 276–288. |
[141] | Brys M , Fanning L , Hung S , Ellenbogen A , Penner N , Yang M , Welch M , Koenig E , David E , Fox T , Makh S , Aldred J , Goodman I , Pepinsky B , Liu YT , Graham D , Weihofen A , Cedarbaum JM ((2019) ) Randomized phase I clinical trial of anti– α-synuclein antibody BIIB054. Mov Disord 34: , 1154–1163. |
[142] | Tran HT , Chung CHY , Iba M , Zhang B , Trojanowski JQ , Luk KC , Lee VMY ((2014) ) α-Synuclein immunotherapy blocks uptake and templated propagation of misfolded α-synuclein and neurodegeneration. Cell Rep 7: , 2054–2065. |
[143] | El-Agnaf O , Overk C , Rockenstein E , Mante M , Florio J , Adame A , Vaikath N , Majbour N , Lee S , Kim C , Masliah E , Rissman RA ((2017) ) Differential effects of immunotherapy with antibodies targeting α-synuclein oligomers and fibrils in a transgenic model of synucleinopathy. Neurobiol Dis 104: , 85–96. |
[144] | Vaikath NN , Majbour NK , Paleologou KE , Ardah MT , van Dam E , van de Berg WDJ , Forrest SL , Parkkinen L , Gai WP , Hattori N , Takanashi M , Lee SJ , Mann DMA , Imai Y , Halliday GM , Li JY , El-Agnaf OMA ((2015) ) Generation and characterization of novel conformation-specific monoclonal antibodies for α-synuclein pathology. Neurobiol Dis 79: , 81–99. |
[145] | Okun E , Mattson MP , Arumugam T V ((2010) ) Involvement of Fc receptors in disorders of the central nervous system. Neuromolecular Med 12: , 164–178. |
[146] | Fuller J , Stavenhagen J , Teeling JL ((2014) ) New roles for Fc receptors in neurodegeneration-the impact on immunotherapy for Alzheimer’s disease. Front Neurosci 8: , 235. |
[147] | Workman CJ , Rice DS , Dugger KJ , Kurschner C , Vignali DAA ((2002) ) Phenotypic analysis of the murine CD4-related glycoprotein, CD223 (LAG-3). Eur J Immunol 32: , 2255–2263. |
[148] | Woo SR , Li N , Bruno TC , Forbes K , Brown S , Workman C , Drake CG , Vignali DAA ((2010) ) Differential subcellular localization of the regulatory T-cell protein LAG-3 and the coreceptor CD4. Eur J Immunol 40: , 1768–1777. |
[149] | Pandey JP ((1988) ) Immunoglobulin genes. Clin Rheumatol 2: , 583. |
[150] | Khodabakhsh F , Behdani M , Rami A , Kazemi-Lomedasht F ((2018) ) Single-domain antibodies or nanobodies: A class of next-generation antibodies.. Int Rev Immunol 37: , 316–322. |
[151] | Sörman A , Zhang L , Ding Z , Heyman B ((2014) ) How antibodies use complement to regulate antibody responses. Mol Immunol 61: , 79–88. |
[152] | Messer A , Butler D ((2020) ) Optimizing intracellular antibodies (intrabodies/nanobodies) to treat neurodegenerative disorders. Neurobiol Dis 134: , 104619. |
[153] | Bannas P , Hambach J , Koch-Nolte F ((2017) ) Nanobodies and nanobody-based human heavy chain antibodies as antitumor therapeutics. Front Immunol 8: , 1603. |
[154] | Muyldermans S , Baral TN , Retamozzo VC , De Baetselier P , De Genst E , Kinne J , Leonhardt H , Magez S , Nguyen VK , Revets H , Rothbauer U , Stijlemans B , Tillib S , Wernery U , Wyns L , Hassanzadeh-Ghassabeh G , Saerens D ((2009) ) Camelid immunoglobulins and nanobody technology. Vet Immunol Immunopathol 128: , 178–183. |
[155] | Zhou C , Emadi S , Sierks MR , Messer A ((2004) ) A human single-chain Fv intrabody blocks aberrant cellular effects of overexpressed α-synuclein. Mol Ther 10: , 1023–1031. |
[156] | Lynch SM , Zhou C , Messer A ((2008) ) An scFv intrabody against the nonamyloid component of α-synuclein reduces intracellular aggregation and toxicity.. J Mol Biol 377: , 136–147. |
[157] | Irwin DJ , Lee VMY , Trojanowski JQ ((2013) ) Parkinson’s disease dementia: Convergence of α-synuclein, tau and amyloid-β pathologies. Nat Rev Neurosci 14: , 626–636. |
[158] | Masliah E , Rockenstein E , Veinbergs I , Sagara Y , Mallory M , Hashimoto M , Mucke L ((2001) ) β-Amyloid peptides enhance α-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer’s disease and Parkinson’s disease. Proc Natl Acad Sci U S A 98: , 12245–12250. |
[159] | Clinton LK , Blurton-Jones M , Myczek K , Trojanowski JQ , LaFerla FM ((2010) ) Synergistic interactions between Aβ, tau, and α-synuclein: Acceleration of neuropathology and cognitive decline. J Neurosci 30: , 7281–7289. |
[160] | Hall S , Surova Y , Öhrfelt A , Zetterberg H , Lindqvist D , Hansson O ((2015) ) CSF biomarkers and clinical progression of Parkinson disease. Neurology 84: , 57–63. |
[161] | Gerson JE , Farmer KM , Henson N , Castillo-Carranza DL , Carretero Murillo M , Sengupta U , Barrett A , Kayed R ((2018) ) Tau oligomers mediate α-synuclein toxicity and can be targeted by immunotherapy.. Mol Neurodegener 13: , 13. |
[162] | Iljina M , Hong L , Horrocks MH , Ludtmann MH , Choi ML , Hughes CD , Ruggeri FS , Guilliams T , Buell AK , Lee JE , Gandhi S , Lee SF , Bryant CE , Vendruscolo M , Knowles TPJ , Dobson CM , De Genst E , Klenerman D ((2017) ) Nanobodies raised against monomeric α-synuclein inhibit fibril formation and destabilize toxic oligomeric species. BMC Biol 15: , 1–14. |
[163] | Butler DC , Joshi SN , De Genst E , Baghel AS , Dobson CM , Messer A ((2016) ) Bifunctional anti-non-amyloid component α-Synuclein nanobodies are protective in situ. PLoS One 11: , e0165964. |
[164] | Joshi SN , Butler DC , Messer A ((2012) ) Fusion to a highly charged proteasomal retargeting sequence increases soluble cytoplasmic expression and efficacy of diverse anti-synuclein intrabodies. MAbs 4: , 686–693. |
[165] | Baecher-Allan C , Kaskow BJ , Weiner HL ((2018) ) Multiple sclerosis: Mechanisms and immunotherapy. Neuron 97: , 742–768. |
[166] | Weiner GJ ((2015) ) Building better monoclonal antibody-based therapeutics. Nat Rev Cancer 15: , 361–370. |
[167] | Patel P , Kriz J , Gravel M , Soucy G , Bareil C , Gravel C , Julien JP ((2014) ) Adeno-associated virus-mediated delivery of a recombinant single-chain antibody against misfolded superoxide dismutase for treatment of amyotrophic lateral sclerosis. Mol Ther 22: , 498–510. |
[168] | Menéndez-González M , Pérez-Piñera P , Martínez-Rivera M , Muñiz AL , Vega JA ((2011) ) Immunotherapy for Alzheimer’s disease: Rational basis in ongoing clinical trials. Curr Pharm Des 17: , 508–520. |
[169] | Buell AK , Galvagnion C , Gaspar R , Sparr E , Vendruscolo M , Knowles TPJ , Linse S , Dobson CM ((2014) ) Solution conditions determine the relative importance of nucleation and growth processes in α-synuclein aggregation. Proc Natl Acad Sci U S A 111: , 7671–7676. |
[170] | Wood SJ , Wypych J , Steavenson S , Louis JC , Citron M , Biere AL ((1999) ) α-Synuclein fibrillogenesis is nucleation-dependent: Implications for the pathogenesis of Parkinson’s disease. J Biol Chem 274: , 19509–19512. |
[171] | Villar-Piqué A , Lopes da Fonseca T , Outeiro TF ((2016) ) Structure, function and toxicity of alpha-synuclein: The Bermuda triangle in synucleinopathies. J Neurochem 139: , 240–255. |
[172] | Bousset L , Pieri L , Ruiz-Arlandis G , Gath J , Jensen PH , Habenstein B , Madiona K , Olieric V , Böckmann A , Meier BH , Melki R ((2013) ) Structural and functional characterization of two alpha-synuclein strains. Nat Commun 4: , 2575. |
[173] | Sweers KKM , Segers-Nolten IMJ , Bennink ML , Subramaniam V ((2012) ) Structural model for α-synuclein fibrils derived from high resolution imaging and nanomechanical studies using atomic force microscopy. Soft Matter 8: , 7215–7222. |
[174] | Serpell LC , Berriman J , Jakes R , Goedert M , Crowther RA ((2000) ) Fiber diffraction of synthetic α-synuclein filaments shows amyloid-like cross-β conformation. Proc Natl Acad Sci U S A 97: , 4897–4902. |
[175] | Karpinar DP , Balija MBG , Kügler S , Opazo F , Rezaei-Ghaleh N , Wender N , Kim HY , Taschenberger G , Falkenburger BH , Heise H , Kumar A , Riedel D , Fichtner L , Voigt A , Braus GH , Giller K , Becker S , Herzig A , Baldus M , Jäckle H , Eimer S , Schulz JB , Griesinger C , Zweckstetter M ((2009) ) Pre-fibrillar α-synuclein variants with impaired α-structure increase neurotoxicity in parkinson’s disease models. EMBO J 28: , 3256–3268. |
[176] | Luth ES , Stavrovskaya IG , Bartels T , Kristal BS , Selkoe DJ ((2014) ) Soluble, prefibrillar α-synuclein oligomers promote complex I-dependent, Ca2+-induced mitochondrial dysfunction. J Biol Chem 289: , 21490–21507. |
[177] | Pacheco CR , Morales CN , Ramírez AE , Muñoz FJ , Gallegos SS , Caviedes PA , Aguayo LG , Opazo CM ((2015) ) Extracellular α-synuclein alters synaptic transmission in brain neurons by perforating the neuronal plasma membrane. J Neurochem 132: , 731–741. |
[178] | Frigerio R , Fujishiro H , Ahn TB , Josephs KA , Maraganore DM , DelleDonne A , Parisi JE , Klos KJ , Boeve BF , Dickson DW , Ahlskog JE ((2011) ) Incidental Lewy body disease: Do some cases represent a preclinical stage of dementia with Lewy bodies?. Neurobiol Aging 32: , 857–863. |
[179] | Parkkinen L , Pirttilä T , Tervahauta M , Alafuzoff I ((2005) ) Widespread and abundant α-synuclein pathology in a neurologically unimpaired subject. Neuropathology 25: , 304–314. |
[180] | Parkkinen L , Pirttilä T , Alafuzoff I ((2008) ) Applicability of current staging/categorization of α-synuclein pathology and their clinical relevance.. Acta Neuropathol 115: , 399–407. |
[181] | Colosimo C , Hughes AJ , Kilford L , Lees AJ ((2003) ) Lewy body cortical involvement may not always predict dementia in Parkinson’s disease. J Neurol Neurosurg Psychiatry 74: , 852–856. |
[182] | Cookson MR , Hardy J , Lewis PA ((2008) ) Genetic neuropathology of Parkinson’s disease. Int J Clin Exp Pathol 1: , 217–31. |
[183] | Gustot A , Gallea JI , Sarroukh R , Celej MS , Ruysschaert JM , Raussens V ((2015) ) Amyloid fibrils are the molecular trigger of inflammation in Parkinson’s disease. Biochem J 471: , 323–333. |
[184] | Flavin WP , Bousset L , Green ZC , Chu Y , Skarpathiotis S , Chaney MJ , Kordower JH , Melki R , Campbell EM ((2017) ) Endocytic vesicle rupture is a conserved mechanism of cellular invasion by amyloid proteins. Acta Neuropathol 134: , 629–653. |
[185] | Roberts RF , Wade-Martins R , Alegre-Abarrategui J ((2015) ) Direct visualization of alpha-synuclein oligomers reveals previously undetected pathology in Parkinson’s disease brain. Brain 138: , 1642–1657. |
[186] | Gorbatyuk OS , Li S , Nash K , Gorbatyuk M , Lewin AS , Sullivan LF , Mandel RJ , Chen W , Meyers C , Manfredsson FP , Muzyczka N ((2010) ) In vivo RNAi-mediated α-synuclein silencing induces nigrostriatal degeneration. Mol Ther 18: , 1450–1457. |
[187] | Collier TJ , Eugene Redmond D , Steece-Collier K , Lipton JW , Manfredsson FP ((2016) ) Is alpha-synuclein loss-of-function a contributor to parkinsonian pathology? Evidence from non-human primates.. Front Neurosci 10: , 12. |
[188] | Fusco G , Pape T , Stephens AD , Mahou P , Costa AR , Kaminski CF , Kaminski Schierle GS , Vendruscolo M , Veglia G , Dobson CM , De Simone A ((2016) ) Structural basis of synaptic vesicle assembly promoted by α-synuclein. Nat Commun 7: , 12563. |
[189] | Catafau A , Bullich S ((2017) ) Non-amyloid PET imaging biomarkers for neurodegeneration: Focus on tau, alpha-synuclein and neuroinflammation. Curr Alzheimer Res 14: , 169–177. |
[190] | Verdurand M , Levigoureux E , Zeinyeh W , Berthier L , Mendjel-Herda M , Cadarossanesaib F , Bouillot C , Iecker T , Terreux R , Lancelot S , Chauveau F , Billard T , Zimmer L ((2018) ) In silico, in vitro, and in vivo evaluation of new candidates for α-synuclein PET imaging. Mol Pharm 15: , 3153–3166. |
[191] | Paciotti S , Bellomo G , Gatticchi L , Parnetti L ((2018) ) Are we ready for detecting α-synuclein prone to aggregation in patients? The case of “Protein-Misfolding Cyclic Amplification” and “Real-Time Quaking-Induced Conversion” as diagnostic tools. Front Neurol 9: , 415. |
[192] | Ugen KE , Lin X , Bai G , Liang Z , Cai J , Li K , Song S , Cao C , Sanchez-Ramos J ((2015) ) Evaluation of an α synuclein sensitized dendritic cell based vaccine in a transgenic mouse model of Parkinson disease. Hum Vaccines Immunother 11: , 922–930. |


